
Abstract
Background
Despite the understanding of sepsis-induced extracellular vesicles (EVs), such as exosomes, and their role in intercellular communication during sepsis, little is known about EV contents such as microRNA (miRNA), which modulate important cellular processes contributing to sepsis in body fluids. This study aimed to analyze the differential expression of exosomal miRNAs in plasma samples collected from sepsis patients and healthy controls, and to identify potential miRNA regulatory pathways contributing to sepsis pathogenesis.
Methods
Quantitative real-time PCR-based microarrays were used to profile plasma exosomal miRNA expression levels in 135 patients with sepsis and 11 healthy controls from an ongoing prospective registry of critically ill adult patients admitted to the intensive care unit. The identified exosomal miRNAs were tested in an external validation cohort (35 sepsis patients and 10 healthy controls). And then, functional enrichment analyses of gene ontology, KEGG pathway analysis, and protein–protein interaction network and cluster analyses were performed based on the potential target genes of the grouped miRNAs. Finally, to evaluate the performance of the identified exosomal miRNAs in predicting in-hospital and 90-day mortalities of sepsis patients, receiver operating characteristic curve (ROC) and Kaplan–Meier analyses were performed.
Results
Compared with healthy controls, plasma exosomes from sepsis patients showed significant changes in 25 miRNAs; eight miRNAs were upregulated and 17 downregulated. Additionally, the levels of hsa-let-7f-5p, miR-331-3p miR-301a-3p, and miR-335-5p were significantly lower in sepsis patients than in healthy controls (p < 0.0001). These four miRNAs were confirmed in an external validation cohort. In addition, the most common pathway for these four miRNAs were PI3K-Akt and mitogen-activated protein kinase (MAPK) signaling pathways based on the KEGG analysis. The area under the ROC of hsa-let-7f-5p, miR-331-3p, miR-301a-3p, and miR-335-5p level for in-hospital mortality was 0.913, 0.931, 0.929, and 0.957, respectively (p < 0.001), as confirmed in an external validation cohort. Also, the Kaplan–Meier analysis showed a significant difference in 90-day mortality between sepsis patients with high and low miR-335-5p, miR-301a-3p, hsa-let-7f-5p, and miR-331-3p levels (p < 0.001, log-rank test).
Conclusion
Among the differentially-expressed miRNAs detected in microarrays, the top four downregulated exosomal miRNAs (hsa-let-7f-5p, miR-331-3p miR-301a-3p, and miR-335-5p) were identified as independent prognostic factors for in-hospital and 90-day mortalities among sepsis patients. Bioinformatics analysis demonstrated that these four microRNAs might provide a significant contribution to sepsis pathogenesis through PI3K-Akt and MAPK signaling pathway.
Similar content being viewed by others


Background
Sepsis is a life-threatening multisystemic dysfunction caused by a dysregulated host immune response to infection [1]. Given its significant global health burden, a tremendous effort to develop specific diagnoses of sepsis, but as yet these have not been successful [2]. Hence, there is a need to recognize sepsis early and more precisely categorize patients. In this regard, research into biological markers has not been so far successful [3]. Current sepsis biomarkers, including lactate, C-reactive protein (CRP), and procalcitonin (PCT) can be used to determine organ failure and evaluate the patient’s clinical course. However, these biomarkers have limited specificity and sensitivity in elucidating the complex regulatory mechanism of sepsis [4].
Exosomes are membrane-bound extracellular vesicles released from almost all cell types and are used as diagnostic markers in several diseases, including malignancy, cardiovascular disease, and chronic lung disease [5,6,7]. In addition, previous studies have shown the clinical importance of exosomes in sepsis especially based on their use as biomarkers [8, 9]. Recently, we demonstrated that plasma exosomes are highly predictive of cross-sectional and longitudinal sepsis outcomes [10, 11].
Exosomes contain cargo molecules including RNA, lipids, and proteins, which they transfer to recipient cells [12]. Among them, microRNAs (miRNAs) are a class of endogenous non-coding RNAs that mediate biological processes such as cell-to-cell communication via gene regulation by binding to their target messenger RNAs [13], cell proliferation, apoptosis, and differentiation [14]. Furthermore, miRNAs are extraordinarily stable in blood and reliably detected at low concentrations with currently available analytical methods in clinical laboratories [15]. Therefore, plasma miRNAs have been proposed as sepsis biomarkers for reflecting the pathophysiological changes at early stages [16]. However, the exosomal miRNAs related to sepsis are not fully understood [17]. In the present study, we compared the expression of sepsis-associated exosomal miRNAs in plasma collected from sepsis patients and healthy controls using miRNA PCR array profiling and validated the identified exosomal miRNAs in an independent samples from sepsis patients.
Methods
We analyzed clinical data and prospectively collected samples from the Samsung Medical Center Registry of Critical Illness (SMC RoCI), an ongoing prospective registry of critically ill adult patients from a tertiary referral center in Seoul, South Korea (Samsung Medical Center, 1989 beds, university affiliated). This cohort was initially recruited in April 2014 for the establishment of a human sample repository and discovery of new biological markers for critical illness [10]. Informed consent prior to enrollment including the research purpose, extraction of clinical data and blood specimens, and future reporting of collected data was obtained from all study participants or their legal representatives. This study was conducted according to the Declaration of Helsinki and approved by the Ethics Committee of Samsung Medical Center (IRB no. 2013-12-033).
Study population
The protocols of patient enrollment and data collection for the sample repository have been previously described [10]. Briefly, critically ill adult patients (≥ 19 years old) admitted to the medical intensive care unit (ICU) were prospectively enrolled, and baseline demographics and clinical details were collected. As this is an ongoing cohort, we included a total of 135 patients with sepsis between October 2015 and January 2020 for the discovery cohort. The diagnosis of sepsis was based on the guidelines of the third International Consensus Definitions for Sepsis and Septic Shock (Sepsis-3) [1]. Since enrollment for this study began in October 2015, patients enrolled before the new definition were reclassified. In addition, 11 healthy controls (≥ 19 years) donated blood specimens (5 mL each) for research purposes. Written informed consent was obtained from all healthy volunteers. Independent data of additional critically ill patients with sepsis (n = 35) between February 2020 and April 2021 and healthy controls (n = 10) were used as external validation cohort (Fig. 1).

Patient flow chart
Data collection
A trained study coordinator used patient hospital records to prepare a standardized case report form. Clinical data consisting of patient demographics, reasons for ICU admission, severity of illness, and laboratory data (PaO2/FiO2, lactic acid, CRP, PCT, interleukin-6 (IL-6), platelet count, albumin, total bilirubin, and creatinine levels) were obtained at enrollment. Severity of illness was assessed using the Acute Physiology and Chronic Health Evaluation II (APACHE II), Simplified Acute Physiology Score 3 (SAPS 3), and Sequential Organ Failure Assessment (SOFA) scores [18,19,20]. In addition to clinical data, 19 mL of whole blood were drawn from each patient within 48 h of enrollment.
Exosomal miRNAs analysis
Plasma preparation
Blood samples (≥ 15 mL) were collected for plasma preparation. Plasma was prepared by drawing peripheral blood into ethylenediaminetetraacetic acid tubes, followed by centrifugation at 480 g (Eppendorf Centrifuge 5810 No. 0012529-rotor A-4-81) for 10 min at 4 °C. Several plasma aliquots from each study participant were isolated and stored at − 80 °C until further analysis. Prior to exosome isolation and RNA purification, plasma samples were pre-filtered using 0.8 µm syringe filters, followed by additional centrifugation to eliminate residual cellular debris.
Exosome isolation
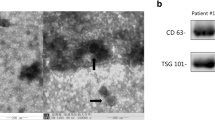
Exosomes were isolated from plasma samples using ExoQuick Plasma prep and Exosome isolation kit (System Biosciences, CA, USA). We modified the manufacturer’s protocol for sample-optimized exosome isolation. The modified protocol was intended to avoid unnecessary sample waste and remove residual impurities. An ExoQuick reagent was used to precipitate plasma exosomes, while the ExoRNeasy mini kit (Qiagen) was used to directly purify RNA from exosomes. To isolate exosomes, 250 µL of the filtered plasma was added to 63 µL of ExoQuick (System Biosciences, CA, USA) and incubated for 30 min at 4 °C. The ExoQuick/plasma mixture was centrifuged at 1500 g for 30 min, followed by supernatant removal and centrifugation of the EV pellet at 1500 g for 5 min to remove the residual ExoQuick solution. The EV pellet was resuspended in 200 µL of phosphate-buffered saline, and the precipitated exosomes were used immediately.
Characterization of exosomes
To analyze cell-specific plasma exosomes, the isolated exosomes were suspended in Phosphate-Buffered Saline containing 1% heat-inactivated fetal bovine serum (FACS buffer) and rested at 37 °C for 15 min. The exosomes were then incubated with 10 µg/ml of the appropriate primary antibody [CD235a (granulocyte), CD3 (lymphocytes), CD11b (monocytes), CD4L (platelet)] plus the exosomal marker CD63. After incubation, the exosomes were washed twice with ice-cold FACS buffer and then stained with 0.25 µg of a FITC-linked secondary antibody for 30 min at 4 °C. After washing with FACS buffer three times, the exosome counts were analyzed by gating the live cell population based on forward and side scatter properties. Flow cytometry data were obtained by a FACS Canto flow cytometer using FACSDiva v8.0 software (BD Biosciences) and analysed using FlowJo v10.0.5 (Tree Star, Ashland, OR). The exosome counts were proportional to the number of fluorescently labelled exosomes, CD63 (total exosomes), [CD235a, CD63]-granulocyte-specific exosomes, [CD3, CD63]-lymphocyte-specific exosomes, [CD11b, CD63]-monocyte-specific exosomes, and [CD4L, CD63]-platelet-specific exosomes.
The concentration and size distribution of the isolated exosomes were assessed by nanoparticle tracking analysis (NTA) using a Nanosight NS300 (NanoSight Ltd., Amesbury, UK). Protein quantification of plasma-derived exosome preparations were performed using Pierce BCA Protein Assay kit (Thermo Fisher Scientific, MA, USA).
Total RNA isolation and quality analysis
Total RNA was extracted from plasma-isolated exosomes using the exoRNeasy Midi Kit following manufacturer’s instructions (Qiagen, Valencia, CA, USA). Briefly, the filtered samples were washed and centrifuged using Buffer XBP and XWP. The isolated exosomes were eluted in 700 µL of QIAzol reagent (QIAGEN Cat. 79,306) and spiked with RNA Spike-In Control (QIAGEN Cat. 339,390) before proceeding with the extraction. Then, 90 µL of chloroform was added to the lysate for subsequent phase separation. The upper aqueous phase containing RNA was transferred to a new collection tube, and 400 µL of the aqueous phase mixed with 800 µL of 100% ethanol; the mixture was transferred into an RNeasy MinElute spin column in a 2 mL collection tube. After centrifugation, the RNA was adhered to the column membrane, and washed with Buffer RWT and RPE; then, DNase/RNase-Free water was added and centrifuged 1 ~ 2 min at 10,000 × g, the collected flow-through corresponded to the exosome total RNA. RNA quantity was measured by NanoDrop 2000 (Thermo Fisher Scientific, MA, USA). RNA purity was assayed by the assessing the 260/280 nm and 260/230 nm absorbance ratios. RNA integrity was assessed using an Agilent 2100 Bioanalyzer with an RNA 6000 Pico kit (Agilent Technologies, CA, USA) (see Additional file 1: Fig. S1). RNA libraries were constructed from 3 ng of total RNA using the SMARTer smRNA-Seq Kit for Illumina (Takara Bio, Shiga, Japan).
Reverse transcription and microRNA expression profiling
The miRCURY LNA™ miRNA Focus PCR panel was used for miRNA profiling of exosomal miRNAs from patients with sepsis and healthy controls (QIAGEN, Cat no. 339325, YAHS-106Y, Qiagen, Hilgen, Germany) (see Additional file 1: Fig. S2). The total extracted RNA including miRNAs (10 ng/µL concentration) was first reverse transcribed into first-strand cDNA using a single miRCURY LNA miRNA PCR assay following manufacturer’s instructions (QIAGEN, Cat no. 339306). Then, 1 µL cDNA per well was mixed with SYBR Green qPCR Master Mix and placed into two 96-well PCR-array plates containing a panel of 179 miRNAs sequences. One µL cDNA was brought to a 10 µL final volume reaction for real-time PCR analysis using an Applied Biosystems Step-One Plus Real-Time PCR system. Relative amounts were calculated using the ΔΔCT method. Samples without good RNA quality were excluded from statistical analysis.
Sequencing data analysis was performed using the GeneGlobe Data Analysis Center (Qiagen). GeneGlobe provides the fold-change (FC = miRNA expression of the nephrotoxicity group / miRNA expression of the non-nephrotoxicity group), FR (FR = FC, if FC ≥ 1 or FR = FC ≥ 1, if FC < 1) and p-value based on the Wald test for each miRNA. miRNA analysis was performed using the geNorm method to identify the best reference controls. The median miR-486-5p, miR-151a-5p, and hsa-miR-532-3p expression was used for data normalization [21]. The relative gene expression was calculated using the comparative cycle threshold (2−ΔΔCT) method [22].
Validation of differentially-expressed miRNAs by quantitative RT-PCR
Custom 96-wells Pick-&-Mix microRNA PCR plates (Qiagen) were used to validate teach miRNA candidate. qRT-PCR was performed to quantify the expression levels of candidate exosomal miRNAs using an miRCURY LNA miRNA PCR Starter Kit (Qiagen, No. 339320) and a miRCURY LNA SYBR Green PCR Kit (Qiagen, No. 339347). In addition, we used the Applied Biosystems QuantStudio 7 Flex Real-Time PCR System in a total volume of 10 μL. qRT-PCR conditions were 95 °C for 2 min, followed by 40 cycles of 95 °C for 10 s and 56 °C for 1 min, followed by melting curve analysis. Synthetic UniSp3 was analyzed as interplate calibrator and qRT-PCR control. Amplification curves were evaluated using QuantStudio Software v1.3 (Thermo Fisher Scientific, Massachusetts, USA). Quantification cycles (Cq) > 35 cycles were censored at the minimum level observed for each miRNA. cel-miR-39-3p levels were stable across samples. Relative quantification was performed using the 2−ΔCq method (ΔCq = CqmiRNA-Cqcel-miR-39-3p). Expression levels were log-transformed for statistical purposes [23].
Bioinformatics analysis
Differentially-expressed miRNAs with FR > 2.5 or FR < − 2.5 were selected for analysis. All selected miRNAs presented no comments or comment “A” and a p-value < 0.05. The miRWalk platform, which provides a list of predicted miRNA target genes according to 12 different algorithms, including TargetScan, were assessed to predict the target genes of differently expressed miRNAs (see Additional file 1: Fig. S3). Then, a protein–protein interaction (PPI) network of target genes was constructed using the STRING database (http://string-db.org). The PPI network was further visualized by Cytoscape v3.9.1. In addition, we used the Gene Ontology (GO) database (http://www.geneontology.org/) for function enrichment. The most statistically significant term within a cluster was chosen to represent the cluster. Only terms with p-value < 0.05 and gene numbers ≥ 1.5 were considered meaningful (≥ 1 gene) [24]. The Database for Annotation, Visualization and Integrated Discovery (DAVID) Bioinformatics resource was used to conduct functional enrichment analysis and delineate the underlying biological processes and pathways of aberrantly expressed intersecting genes based on the Kyoto Encyclopedia of Genes and Genomes (KEGG) database [25].
Based on the enriched pathways of target genes, the biological functions of the grouped miRNAs were predicted by miRsystem database, which interprets seven algorithms predicting miRNA targets (DIANA, miRanda, miRBridge, PicTar, PITA, rna22, and TargetScan) and two experimentally validated databases (TarBase and miRecords). The PPI network was established using the STRING database to annotate the functional interaction between target genes and other genes. Active interaction sources, including text mining, experiments, databases, and co-expression as well as species limited to Homo sapiens and an interaction score > 0.7 were applied to construct the PPI network, which showed the physical and functional interactions between genes [26]. We selected gene pairs with a total score > 0.9 for PPI network construction [27]. Then, the Cytoscape software was applied to merge the indirect PPI with driver genes’ PPI to discover interconnected and intersected functional modules and target the core genes. Eventually, we analyzed the interactions between differential expression of exosomal miRNAs and well-known sepsis mechanisms.
Statistical analysis
Clinical data is presented as numbers (percentages) for categorical variables, and as the median and interquartile range (IQR, 25th–75th percentiles) for continuous variables. Relative miRNA levels were obtained using 2DCT (2CT of reference miRNAs–CT of miRNA of interest) for each miRNA. The p-value of differential miRNA expression was calculated based on Poisson’s distribution [28] and the p-value threshold was determined by the false discovery rate [29]. Receiver operating characteristic (ROC) analysis was performed, and the area under the curve (AUC) reported to evaluate the performance of miRNAs in predicting in-hospital mortality of sepsis patients. In addition, patients were reclassified into two groups; high and low miRNA levels according to each optimal cutoff level calculated by Youden’s index [30]. Kaplan–Meier equation was used to determine the 90-day mortality curves according to each miRNA levels, which were then compared by the log-rank test. All tests were two-sided, and a p-value < 0.05 was considered statistically significant.
Results
Baseline characteristics of the study population
The baseline characteristics of study subjects on ICU admission are presented in Table 1. The median age was 66 years (58–74 years) and 67% were male. A total of 59 (44%) patients required mechanical ventilation and 70 (52%) patients needed vasopressor support. The median SAPS3 score on ICU admission was 55 (47–63); the median APACHE II and SOFA scores were 24 (20–30) and 9 (7–11), respectively. The interval between ICU admission to blood sampling was 30 (21–37) hours. The median serum CRP, PCT, and IL-6 were 12.5 (5.9–24.8) mg/dL, 6.06 (0.85–25.18) ng/mL, and 228 (65–807) pg/mL, respectively.
Characterization of exosomes
The analysis of flow cytometry experiments demonstrated that a significant portion of vesicles was positive for CD63 and CD11b in sepsis patients compared to healthy volunteers, suggesting an exosomal and monocyte origin for these vesicles. Taken together, our results strongly suggest that the major component of our vesicle pool was composed of monocyte-derived exosomes (Fig. S4, see Additional file 1).
The levels of exosomal protein isolates from 1 mL, 2.5 mL, and 5 mL of plasma were 663 (± 500) µg, 1440 (± 204) µg, and 2270 (± 182) µg, respectively. According to the amount of plasma, the levels of exosomal RNA were 2446 (± 770) ng in 1 mL, 3010 (± 780) ng in 2.5 mL, and 5779 (± 1020) ng in 5 mL. The RNA purity was determined spectrophotometrically by means of the 260/280 nm and 260/230 nm absorbance ratios. The mean of OD260/280 was 2.04 (± 0.07) (see Additional file 2). As shown in the additional file 2, the ratios were greater than or equal to 2.0 in most samples, which indicate a high-quality level of RNA. Bioanalyzer data revealed that plasma-derived exosomes contain a broad range of RNA sizes including a high amount of small RNAs (25–200 nucleotides). Among the small RNAs, transfer RNA (18.7%) and miRNAs (14.7%) were the most abundant. Other small exosomal RNAs included ling noncoding RNA (10.5%), Y RNA (4.8%), and ribosomal RNAs (3.9%).
Exosomal miRNA profiling
To analyze the miRNA content of plasma-derived exosomes, we performed miRNA profiling analysis in patients with sepsis and compared the results with those of healthy individuals; 179 miRNAs were differentially expressed compared with the controls. We found eight upregulated miRNAs (miR-223-3p, miR-122-5p, miR-30e-3p, miR-99a-5p, miR-155-5p, miR-125b-5p, miR-378a-3p, and miR-150-5p) and 17 downregulated miRNAs (miR-335-5p, hsa-let-7f-5p, miR-301a-3p, miR-331-3p, miR-495-3p, miR-374b-5p, miR-409-3p, miR-18a-5p, miR-133b, miR-28-5p, miR-148b-3p, miR-376c-3p, miR-133a-3p, miR-584-5p, miR-766-3p, miR-22-5p, and miR-376c-3p; Fig. 2).

miRNA expression patterns (A) and fold-change in plasma exosomal miRNAs (B) in patients with sepsis. Compared to healthy controls, eight miRNAs were significantly elevated and 17 miRNAs significantly decreased in patients with sepsis
Validation of differentially-expressed miRNAs
We identified 25 differentially-expressed miRNAs and selected the top four downregulated exosomal miRNAs for qRT-PCR validation: hsa-let-7f-5p, miR-335-5p, miR-331-3p, and miR-301a-3p, in accordance with the microarray screening results. As shown in Fig. 3, miR-335-5p, miR-301a-3p, hsa-let-7f-5p, and miR-331-3p levels were significantly decreased in sepsis patients compared with healthy controls (p < 0.0001). In addition, levels of the top four exosomal miRNAs were significantly decreased in septic shock compared to sepsis (Fig. 3).

Quantitative RT-PCR validation for four differentially-expressed miRNAs in patients with sepsis (n = 36) and septic shock (n = 99), and healthy controls (n = 11)
In the external validation cohort, we tested another 35 patients with sepsis and five healthy controls (see Additional file 1: Table S1) for analysis of differences in the top four downregulated exosomal miRNAs. Compared with healthy controls, the selected miRNAs were downregulated in sepsis (p < 0.0001; see Additional file 1: Fig. S5).
Bioinformatics analysis
KEGG pathways for grouped miRNAs
KEGG pathway analysis revealed that the target genes of these four miRNAs mainly belonged to 10 signaling pathways: mitogen-activated protein kinase (MAPK), phosphatidylinositol 3-kinase/protein kinase B (PI3K-Akt), mammalian target of rapamycin (mTOR), Ras, Wnt, forkhead box protein O (FoxO), insulin, transforming growth factor-β (TGF-β), longevity regulating, and advanced glycation end products/receptor for advanced glycation end products (AGE-RAGE) (Fig. 4). Additionally, the most common pathway for hsa-let-7f-5p, miR-331, miR-301a, and miR-335 was PI3K-Akt and MAPK signaling pathway (Table 2).

KEGG pathway analysis of four downregulated exosomal miRNAs in sepsis
GO enrichment for identifying potential target genes of the grouped miRNAs
The potential target genes of these four miRNAs were predicted by miRsystem database. A total of 1817 target genes for these four differentially-expressed miRNAs were predicted and used for GO function enrichment. The analysis of biological processes indicated that the selected miRNAs were mainly involved in cellular processes, biological regulation, and metabolic processes. With respect to molecular functions, it included binding, protein binding, and ion binding. Finally, at the cellular level, the selected miRNAs were related to cellular anatomical entity, intracellular, and organelles (see Additional file 1: Fig. S6).
PPI networks
To identify the core genes and interaction between genes from common KEGG pathways, we mapped 405 target genes of the grouped miRNAs to the STRING database. The PPI network consisted of 1224 nodes and 2429 edges. Enrichment pathway analysis showed that the genes were related to cancer pathways, PI3K-Akt signaling, and MAPK signaling (see Additional file 1: Fig. S7).
Cluster analysis of predicted genes
The cluster analysis of the predicted 405 genes was performed to discover groups of correlated genes potentially associated to disease or conditions. Finally, the results of the top 2 KEGG pathways and PPI networks were integrated as shown in Fig. 5.

Merged protein–protein interaction networks. Dots represent genes and lines represent interactions
Predictive value for in-hospital and 90-day mortalities
The results of ROC analysis and AUCs of the four selected miRNAs are shown in Fig. 6. The AUC of hsa-let-7f-5p, miR-335-5p, miR-331-3p, and miR-301a-3p level for in-hospital mortality was 0.913, 0.957, 0.931, and 0.929, respectively (p < 0.001). In the external validation cohort, their AUCs for in-hospital mortality were 0.920, 0.902, 0.907, and 0.914, respectively (p < 0.001) (see Additional file 1: Fig. S8). Furthermore, Kaplan–Meier survival estimation demonstrated a significant difference in 90-day survival between patients with high and low miR-335-5p, miR-301a-3p, hsa-let-7f-5p, and miR-331-3p levels, respectively (all p < 0.001, log-rank test) (Fig. 7).

Receiver operating characteristic curves of the predictive value of four miRNAs for in-hospital mortality in patients with sepsis

Kaplan–Meier survival estimation of sepsis patients with high and low miRNAs (A, miR-335-5p; B, miR-301a-3p; C, has-lef-7f-5p; D, miR-331-3p) for 90-day mortality
Discussion
We evaluated differentially-expressed miRNAs between sepsis patients and healthy controls based on the miRNA profile of plasma exosomes from patients with sepsis. A total of eight upregulated and 17 downregulated miRNAs were identified in sepsis patients. Among the differentially-expressed miRNAs detected in microarrays, the top four downregulated exosomal miRNAs were validated using qRT-PCR (miR-335-5p, miR-301a-3p, hsa-let-7f-5p, and miR-331-3p), and identified as prognostic factors for in-hospital and 90-day mortalities among sepsis patients. Based on the KEGG pathway analysis, these four miRNAs might provide a significant contribution to sepsis pathogenesis through MAPK and PI3K-Akt signaling pathway.
Sepsis is a complex syndrome that involves multiple organs and tissues; current biomarkers cannot detect its early phase [31]. To overcome these limitations, several studies investigated the pro-inflammatory role of miRNAs in sepsis [32]. Moreover, miRNA profiling has shown their potential as biomarker candidates in sepsis [33]. The miRNAs identified in the present study were evaluated in previous sepsis-related studies (Table 3). Furthermore, functional studies investigating their role revealed the effect of miR-223-3p on immune cell differentiation. miR-223 is significantly elevated during tissue inflammation, and regulates immune cell proliferation, differentiation, and polarization [34]. Moreover, Zhu et al. reported that miR-99a-5p mediates γδ T cell activation and apoptosis, serving as antigen-presenting cells in the adaptive immune response [35]. Chaudhuri et al. reported that miR-125b-5p regulates macrophage activation via IFN regulatory factor 4 levels and serves as an inflammatory response inhibitor [36]. Finally, Nie et al. reported that miR-331-3p regulates the inflammatory response via a nucleotide-binding oligomerization domain, leucine rich repeat, and pyrin domain-containing protein (NLRP) 6, which plays an important role in apoptosis, inflammation, and immune response [37]. The present results confirm that the identified miRNAs are related to sepsis pathophysiology.
Herein, we obtained actively released miRNAs of plasma exosome from 135 patients with sepsis and performed qRT-PCR-based microarray analysis of 179 different miRNAs to identify additional differentially-expressed miRNAs to those in previous studies. Among them, we established down-regulation of four miRNAs (miR-335-5p, miR-301a-3p, hsa-let-7f-5p, and miR-331-3p) in patients with sepsis. To better understand the interactions of target genes in the four miRNAs, KEGG analysis was performed. Our data showed that exosomal hsa-let-7f-5p, miR-335-5p, miR-331-3p, and miR-301a-3p were concentrated on 60 signaling pathways, the top two being MAPK and PI3K-Akt signaling. The signaling pathways related to these miRNAs and thus considered related to sepsis were identified. In past studies, the MAPK and PI3K-Akt pathways were associated with cell apoptosis [38]. MAPK and p38 are crucial intracellular signal transduction pathways that mediate endothelial inflammatory injury during sepsis [39]. These pro-survival pathways control the transcription of genes related to cell apoptosis, and can be activated by various inflammatory mediators and environmental stress, inducing the excessive release of pro-inflammatory cytokines, including TNF-a, IL-6 and other inducible enzymes [40]. Further, the PI3K-Akt signaling pathway was recently associated with negative signals in the hyper-inflammatory state of sepsis [41]. The FoxO and mTOR signaling pathways downstream of PI3K-Akt signaling are also related to the cell cycle [42]. Functional studies focusing on these miRNAs are required to provide additional information with potential therapeutic value.
Additionally, the Cq values in this study were significantly greater in the sepsis group than in healthy controls, indicating reduced expression, and supporting the potential of these miRNAs as sepsis biomarkers. Furthermore, we ascertained that the low expression of miR-335-5p, miR-301a-3p, hsa-let-7f-5p, and miR-331-3p in subjects with sepsis significantly correlates with in-hospital and 90-day mortalities. These miRNAs could improve risk prediction of hospital mortality beyond APACHE II (AUC of 0.721; 95% CI, 0.624–0.817) or SAPS 3 (AUC of 0.720; 95% CI, 0.617–0.823), commonly used to predict hospital mortality in clinical practice [18, 19].
Although this study provides additional information on the differentially-expressed miRNAs as biomarkers of the diagnosis and morality prediction of sepsis via molecular experiments and clinical information from a prospective cohort, our study has certain limitations that should be acknowledged. First, the study population was relatively small; thus, future bioinformatics studies are necessary to confirm our findings. In addition, we could not evaluate the dynamic profile of exosomal miRNAs during the progression of sepsis, since subsequent samples from the enrollment were not available in many patients with sepsis. Second, this study was conducted at a single referral center in Korea, which might limit the generalization of our results, especially to other institutions or ethnic groups. Third, multiple preexisting diseases, such as diabetes and malignancies, may confound the total RNA and miRNA in exosomes with pleiotropic effects. In this study, however, sepsis patients with diabetes or malignancy were also included for unbiased comparison with the general sepsis patients, and the top four downregulated exosomal miRNAs were not associated with these underlying diseases. In addition, additional analysis with sepsis patients who have no comorbidities revealed that the selected miRNAs were downregulated in sepsis compared with healthy controls (p < 0.0001; see Additional file 1: Fig. S9).
Conclusions
The present study identified plasma exosomal miRNAs with dysregulated expression in patients with sepsis compared with healthy controls; the differentially-expressed miRNAs have potential as biomarkers for sepsis. Moreover, we demonstrated their potential as early prognostic tools in sepsis. Further studies are required to elucidate the mechanisms by which these miRNAs affect intercellular communication during sepsis.
Availability of data and materials
The data that support the findings of this study are available on request from the corresponding author. The data are not publicly available due to privacy or ethical restrictions.
Abbreviations
- APACHE II:
-
Acute Physiology and Chronic Health Evaluation II
- AUC:
-
Area under the curve
- CRP:
-
C-reactive protein
- DAVID:
-
Database for Annotation, Visualization and Integrated Discovery
- EV:
-
Extracellular vesicle
- FoxO:
-
Forkhead box protein O
- GO:
-
Gene Ontology
- ICU:
-
Intensive care unit
- IQR:
-
Interquartile range
- KEGG:
-
Kyoto Encyclopedia of Genes and Genomes
- MAPK:
-
Mitogen-activated protein kinase
- miRNA:
-
MicroRNA
- mTOR:
-
Mammalian target of rapamycin
- PI3K-Akt:
-
Phosphatidylinositol 3-kinase/protein kinase B
- PCT:
-
Procalcitonin
- PPI:
-
Protein–protein interaction
- ROC:
-
Receiver operating characteristic
- SAPS 3:
-
Simplified Acute Physiology Score 3
- SOFA:
-
Sequential Organ Failure Assessment
References
Singer M, Deutschman CS, Seymour CW, Shankar-Hari M, Annane D, Bauer M, et al. The third international consensus definitions for sepsis and septic shock (Sepsis-3). JAMA. 2016;315(8):801–10.
Pierrakos C, Velissaris D, Bisdorff M, Marshall JC, Vincent JL. Biomarkers of sepsis: time for a reappraisal. Crit Care. 2020;24(1):287.
Vincent JL, Teixeira L. Sepsis biomarkers. Value and limitations. Am J Respir Crit Care Med. 2014;190(10):1081–2.
Tan M, Lu Y, Jiang H, Zhang L. The diagnostic accuracy of procalcitonin and C-reactive protein for sepsis: a systematic review and meta-analysis. J Cell Biochem. 2019;120(4):5852–9.
Cortés-Sempere M, Ibáñez de Cáceres I. microRNAs as novel epigenetic biomarkers for human cancer. Clin Transl Oncol. 2011;13(6):357–62.
Kim GH. MicroRNA regulation of cardiac conduction and arrhythmias. Transl Res. 2013;161(5):381–92.
Trappe A, Donnelly SC, McNally P, Coppinger JA. Role of extracellular vesicles in chronic lung disease. Thorax. 2021;76(10):1047–56.
Raeven P, Zipperle J, Drechsler S. Extracellular vesicles as markers and mediators in sepsis. Theranostics. 2018;8(12):3348–65.
Terrasini N, Lionetti V. Exosomes in critical illness. Crit Care Med. 2017;45(6):1054–60.
Im Y, Yoo H, Lee JY, Park J, Suh GY, Jeon K. Association of plasma exosomes with severity of organ failure and mortality in patients with sepsis. J Cell Mol Med. 2020;24(16):9439–45.
Im Y, Yoo H, Ko RE, Lee JY, Park J, Jeon K. Exosomal CD63 in critically ill patients with sepsis. Sci Rep. 2021;11(1):20300.
Zhang Y, Liu Y, Liu H, Tang WH. Exosomes: biogenesis, biologic function and clinical potential. Cell Biosci. 2019;9:19.
Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116(2):281–97.
Ambros V. The functions of animal microRNAs. Nature. 2004;431(7006):350–5.
Chen X, Ba Y, Ma L, Cai X, Yin Y, Wang K, et al. Characterization of microRNAs in serum: a novel class of biomarkers for diagnosis of cancer and other diseases. Cell Res. 2008;18(10):997–1006.
Essandoh K, Fan GC. Role of extracellular and intracellular microRNAs in sepsis. Biochim Biophys Acta. 2014;1842(11):2155–62.
Real JM, Ferreira LRP, Esteves GH, Koyama FC, Dias MVS, Bezerra-Neto JE, et al. Exosomes from patients with septic shock convey miRNAs related to inflammation and cell cycle regulation: new signaling pathways in sepsis? Crit Care. 2018;22(1):68.
Knaus WA, Draper EA, Wagner DP, Zimmerman JE. APACHE II: a severity of disease classification system. Crit Care Med. 1985;13(10):818–29.
Moreno RP, Metnitz PG, Almeida E, Jordan B, Bauer P, Campos RA, et al. SAPS 3—from evaluation of the patient to evaluation of the intensive care unit. Part 2: development of a prognostic model for hospital mortality at ICU admission. Intensive Care Med. 2005;31(10):1345–55.
Vincent JL, Moreno R, Takala J, Willatts S, De Mendonça A, Bruining H, et al. The SOFA (Sepsis-related Organ Failure Assessment) score to describe organ dysfunction/failure. On behalf of the Working Group on Sepsis-Related Problems of the European Society of Intensive Care Medicine. Intensive Care Med. 1996;22(7):707–10.
Vandesompele J, De Preter K, Pattyn F, Poppe B, Van Roy N, De Paepe A, et al. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 2002;3(7):RESEARCH0034.
Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25(4):402–8.
Rao X, Huang X, Zhou Z, Lin X. An improvement of the 2^(-delta delta CT) method for quantitative real-time polymerase chain reaction data analysis. Biostat Bioinform Biomath. 2013;3(3):71–85.
Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinform. 2008;9:559.
da Huang W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4(1):44–57.
Szklarczyk D, Morris JH, Cook H, Kuhn M, Wyder S, Simonovic M, et al. The STRING database in 2017: quality-controlled protein-protein association networks, made broadly accessible. Nucleic Acids Res. 2017;45(D1):D362–8.
Pan Q, Zhou R, Su M, Li R. The effects of plumbagin on pancreatic cancer: a mechanistic network pharmacology approach. Med Sci Monit. 2019;25:4648–54.
Audic S, Claverie JM. The significance of digital gene expression profiles. Genome Res. 1997;7(10):986–95.
Benjamini Y, Drai D, Elmer G, Kafkafi N, Golani I. Controlling the false discovery rate in behavior genetics research. Behav Brain Res. 2001;125(1–2):279–84.
Bewick V, Cheek L, Ball J. Statistics review 13: receiver operating characteristic curves. Crit Care. 2004;8(6):508–12.
Evans L, Rhodes A, Alhazzani W, Antonelli M, Coopersmith CM, French C, et al. Surviving sepsis campaign: international guidelines for management of sepsis and septic shock 2021. Crit Care Med. 2021;49(11):e1063–143.
Hashemian SM, Pourhanifeh MH, Fadaei S, Velayati AA, Mirzaei H, Hamblin MR. Non-coding RNAs and exosomes: their role in the pathogenesis of sepsis. Mol Ther Nucleic Acids. 2020;21:51–74.
Shen X, Zhang J, Huang Y, Tong J, Zhang L, Zhang Z, et al. Accuracy of circulating microRNAs in diagnosis of sepsis: a systematic review and meta-analysis. J Intensive Care. 2020;8(1):84.
Jiao P, Wang XP, Luoreng ZM, Yang J, Jia L, Ma Y, et al. miR-223: an effective regulator of immune cell differentiation and inflammation. Int J Biol Sci. 2021;17(9):2308–22.
Zhu Y, Zhang S, Li Z, Wang H, Li Z, Hu Y, et al. miR-125b-5p and miR-99a-5p downregulate human γδ T-cell activation and cytotoxicity. Cell Mol Immunol. 2019;16(2):112–25.
Chaudhuri AA, So AY, Sinha N, Gibson WS, Taganov KD, O’Connell RM, et al. MicroRNA-125b potentiates macrophage activation. J Immunol. 2011;187(10):5062–8.
Nie H, Hu Y, Guo W, Wang W, Yang Q, Dong Q, et al. miR-331-3p inhibits inflammatory response after intracerebral hemorrhage by directly targeting NLRP6. Biomed Res Int. 2020;2020:6182464.
McCubrey JA, Steelman LS, Chappell WH, Abrams SL, Wong EW, Chang F, et al. Roles of the Raf/MEK/ERK pathway in cell growth, malignant transformation and drug resistance. Biochim Biophys Acta. 2007;1773(8):1263–84.
Dong R, Xue Z, Fan G, Zhang N, Wang C, Li G, et al. Pin1 promotes NLRP3 inflammasome activation by phosphorylation of p38 MAPK pathway in septic shock. Front Immunol. 2021;12: 620238.
Chen J, Xue X, Cai J, Jia L, Sun B, Zhao W. Protective effect of taurine on sepsis-induced lung injury via inhibiting the p38/MAPK signaling pathway. Mol Med Rep. 2021. https://doi.org/10.3892/mmr.2021.12292.
Vergadi E, Ieronymaki E, Lyroni K, Vaporidi K, Tsatsanis C. Akt signaling pathway in macrophage activation and M1/M2 polarization. J Immunol. 2017;198(3):1006–14.
Darici S, Alkhaldi H, Horne G, Jørgensen HG, Marmiroli S, Huang X. Targeting PI3K/Akt/mTOR in AML: rationale and clinical evidence. J Clin Med. 2020. https://doi.org/10.3390/jcm9092934.
Wang JF, Yu ML, Yu G, Bian JJ, Deng XM, Wan XJ, et al. Serum miR-146a and miR-223 as potential new biomarkers for sepsis. Biochem Biophys Res Commun. 2010;394(1):184–8.
Wang L, Wang HC, Chen C, Zeng J, Wang Q, Zheng L, et al. Differential expression of plasma miR-146a in sepsis patients compared with non-sepsis-SIRS patients. Exp Ther Med. 2013;5(4):1101–4.
Liu J, Shi K, Chen M, Xu L, Hong J, Hu B, et al. Elevated miR-155 expression induces immunosuppression via CD39(+) regulatory T-cells in sepsis patient. Int J Infect Dis. 2015;40:135–41.
Lan C, Shi X, Guo N, Pei H, Zhang H. Value of serum miR-155-5p and miR-133a-3p expression for the diagnosis and prognosis evaluation of sepsis. Zhonghua Wei Zhong Bing Ji Jiu Yi Xue. 2016;28(8):694–8.
Wu X, Yang J, Yu L, Long D. Plasma miRNA-223 correlates with risk, inflammatory markers as well as prognosis in sepsis patients. Medicine (Baltimore). 2018;97(27): e11352.
Zhang W, Jia J, Liu Z, Si D, Ma L, Zhang G. Circulating microRNAs as biomarkers for Sepsis secondary to pneumonia diagnosed via Sepsis 3.0. BMC Pulm Med. 2019;19(1):93.
Zhu X. MiR-125b but not miR-125a is upregulated and exhibits a trend to correlate with enhanced disease severity, inflammation, and increased mortality in sepsis patients. J Clin Lab Anal. 2020;34(3): e23094.
Acknowledgements
None.
Funding
This work was supported by Future Medicine 20*30 Project of the Samsung Medical Center (SMX1210751) and National Research Foundation of Korea grant funded by the Korean Government (MSIT) (NRF-2020R1F1A1074038).
Author information
Authors and Affiliations
Contributions
KJ conceived and designed the study; BS, JYL, YI, HY, and JP collected the data; BS, JYL, YI, HY, JP, JSL, KYL, and KJ conducted experiments and, analyzed and interpreted the data; BS, JYL, and KJ drafted the manuscript for intellectual content; BS, JYL, YI, HY, JP, JSL, KYL, and KJ revised the manuscript. All authors read and approved the final version of the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The study was performed in accordance with the Declaration of Helsinki. The Ethics Committee of Samsung Medical Center (IRB no. 2013-12-033) reviewed and approved the study. Written informed consent was obtained from patients or their legally authorized representative prior to enrolment.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional
file 1: Table S1. Demographic and clinical characteristics of the validation cohort with sepsis (N = 35). Figure S1. Bioanalyzer exosomal RNA quality control data. An ANA Agilent 2100 Bioanalyzer was used to examine exosomal RNA quality (Agilent Technologies, Inc. Santa Clara, CA, USA). The RNA ladder standard (in the first lane) contains six RNA fragments ranging between 0.2–6 kb. Representative bands of our sample’s RNA (in the second lane) showing 5S (120 nt), 18S (1,900 nt), and 28S rRNA (4,700 nt). For exoRNA bands, all samples showed an obvious band in the small RNA area. Figure S2. Hierarchical clustering of exosomal miRNA expression in a selected group of 135 sepsis patients utilizing 179 differentially-expressed miRNAs. Exosomal miRNA levels are shown as a heat map. Hierarchical clustering of aberrantly expressed miRNAs with significantly different expression was performed using Sabiosciences’ online data analysis tool. Figure S3. Identification of potential target genes for exosomal miRNAs. Venn diagrams showing the intersection between the predicted target genes of plasma exosomal miRNAs from TargetScan and miRDB, which provide a list of predicted miRNA target genes according to different algorithms. Figure S4. Characterization of exosomes in plasma of sepsis patients and healthy controls by flow cytometry. The graph depicts the percentage of positive events of 50,000 vesicles. Exosomes from sepsis patients and healthy controls were incubated with CD63 (exosome marker) and CD 11b (monocyte marker). Figure S5. Quantitative RT-PCR validation for four differentially-expressed microRNAs in patients with sepsis (n= 35) and healthy controls (n = 10) from the validation cohort. Figure S6. Cluster analysis of 405 predicted genes using STRING. Using miRsystem database, a total of 1817 target genes were identified for the four-miRNAs. Tabulated results of enriched pathways of miRNA target genes based on the consistency across multiple algorithms and observed/expected ratios. The analysis parameters in STRING were as follows: hit frequency = 3, observed to expected ratio ≥1, and matched pathways from the Kyoto Encyclopedia of Genes and Genomes database. The 934 target genes were entered into the STRING database to construct a protein–protein interaction (PPI) network, which shows the physical and functional interactions among genes. PPI networks were constructed based on important susceptibility genes. The networks were evaluated based on two topological parameters, combined scores, and degree. Degree ≥7 and a combined score ≥0.9 were cut-off criteria. Dots represent genes and lines represent interactions. Figure S7. KEGG pathway ranking for predicted target genes of four grouped miRNAs using miRSystem (Rank score ≥ 1). Figure S8. Receiver operating characteristic curves of the predictive value of four miRNAs for in-hospital mortality in patients with sepsis from the validation cohort. Figure S9. Quantitative RT-PCR validation for four differentially-expressed microRNAs in sepsis patients who had no comorbidities (n = 24) and healthy controls (n = 11) from the discovery cohort.
Additional
file 2: Quantity and quality of the exosomal RNA. Details are described in the Excel file.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Shin, B., Lee, J.Y., Im, Y. et al. Prognostic implication of downregulated exosomal miRNAs in patients with sepsis: a cross-sectional study with bioinformatics analysis. j intensive care 11, 35 (2023). https://doi.org/10.1186/s40560-023-00683-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40560-023-00683-2