
Abstract
Green analytical chemistry is one of the newest trends in analytical chemistry nowadays targeting the concept of green laboratory practices on chemists and environment. In this text, green practices are proposed in this work for the determination of sofosbuvir (SF) and velpatasvir (VP) in their pharmaceutical formulation. The analysis of SF in a binary mixture with VP represents an analytical challenge due to the complete overlapping of the UV spectrum of SF by that of VP. Therefore, the direct absorbance and derivative measurements cannot resolve such interference and failed to determine SF. In this paper, three direct and simple methods were developed for the analysis of SF without any interference from VP without sample pre-treatment. The proposed methods include measuring the second derivative amplitude of the ratio spectrum of the mixture using VP as a divisor, measuring the absorbance difference of the mixture in NaOH solution against its HCl solution, and using the derivative compensation technique. On the other hand, VP was determined specifically in presence of SF by two methods. Firstly, by its reaction with 4-chloro-7-nitrobenzofurazan (NBD-Cl) where the reaction product was measured spectrophotometrically and spectrofluorometrically and secondly through the reaction of VP with 3-methyl-2-benzothiazolinone hydrazone hydrochloride (MBTH). The calibration curves showed good correlation coefficient (r2 > 0.999). The developed methods were highly precise with RSD% values less than 2%. The method greenness profile was compared with other published methods by applying the eco-scale protocol. Assessment results proved that our analytical procedure is greener than other reported methods. Moreover, upon comparison with other methods, the proposed methods showed better or comparable sensitivity in addition to being inexpensive and ecofriendly. Accordingly, these methods could be readily applied for quality control purposes as an eco-friendly, simple and efficient analytical tool.
Similar content being viewed by others


Introduction
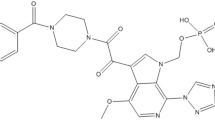
Hepatitis C is considered as one of the biggest problems in public health that leads to chronic liver disease (Williams, 2006). Liver cirrhosis, liver failure, and hepatocellular carcinoma occur as a consequence of hepatitis C, which is deemed in Europe and the USA as the main reason of liver transplantation (Nahon et al., 2017). The prevalence of HCV infection is estimated by WHO as more than 170 million people worldwide (Poynard et al., 2003). The effective treatment of HCV is required to achieve sustained viral response with 24 weeks of aviremia after completion of the treatment course (Poynard et al., 2003). Previously, ribavirin, a purine nucleoside analog, combined with pegylated-interferon alpha showed enhancement of the sustained viral response rates (Poynard et al., 2003). However, such combination had some limitations, as the lengthy treatment period, several contraindications, with dangerous side effects (Poynard et al., 2003). Direct-acting antiviral agents (DAAs) have been recently used either alone or in combination with each other for the treatment of HCV (Cheung et al., 2016). They offer simpler, shorter, and more efficient interferon-limiting therapies for different HCV genotypes (Cheung et al., 2016; Jakobsen et al., 2017). Moreover, most DAAs are considered as efficient and safe HCV treatment (Cheung et al., 2016). Preliminary results have shown that DAAs seem to eradicate HVC from the blood much more frequently, in addition to, the early improvements in liver function (Cheung et al., 2016; Jakobsen et al., 2017). Examples of the DAAs that are commonly administered alone or in combination are sofosbuvir, ledipasvir, daclatasvir, and velpatasvir (Cheung et al., 2016; Jakobsen et al., 2017). Combined sofosbuvir (SF) and velpatasvir (VP) therapy resulted in high rate of sustained virologic response in HCV patients with genotype 1, 2, 3, 4, 5, or 6 treated for 12 weeks with this regimen (Feld et al., 2015; Foster et al., 2015). The mixture of 400 mg SF and 100 mg VP, newly approved by the FDA, proved to be effective in a wide range of HCV patients and with compensated cirrhosis patients (Feld et al., 2015; Foster et al., 2015). SF (Fig. 1a) is a prodrug, acts as a nucleotide inhibitor of NS5B polymerase, and administered alone or in a mixture with other drugs (McQuaid et al., 2015), VP (Fig. 1b) is a second-generation inhibitor of NS5A protein which is fundamental for HCV replication (Greig, 2016).

(a) SF and (b) VP structure (c) absorption spectra of 40 μg/mL SF overlapped with 10 μg/mL VP
Literature revealed few articles on the simultaneous analysis of SF and VP in this novel mixture. These include RP-HPLC (Vanaja et al., 2018; Rani & Devanna, 2017; Tahir et al., 2018), LC-MS/MS (Elkady & Aboelwafa, 2018), HPTLC (Rezk et al., 2019a; Saraya et al., 2019), UPLC (Rezk et al., 2018; Moustapha et al., 2019; Kamal et al., 2019), spectrophotometry (Kamal et al., 2019; Rezk et al., 2019b; Attia et al., 2018), and spectrofluorimetry (El-Gamal et al., 2018; Omar et al., 2018).
Spectrophotometric and spectrofluorimetric methods are two of the greenest analytical methods available because of the minimum solvent consumption and instrumental hazards (Van Aken et al., 2006; Gałuszka et al., 2012). Also, they are widely available techniques in most of the analytical laboratories with minimum expenses. Therefore, they are commonly used for the determination of many analytical mixtures (Kamal et al., 2019; Rezk et al., 2019b; Attia et al., 2018; El-Gamal et al., 2018; Omar et al., 2018). However, the great overlap in the absorption spectra of the selected mixture of SF and VP (Fig. 1c) presents a challenge for their simultaneous spectrophotometric analysis. Thus, the goal of this study is to develop methods for the determination of each drug without interference of the other drug in the same mixture without any previous sample separation.
Three spectrophotometric methods (Kamal et al., 2019; Rezk et al., 2019b; Attia et al., 2018) were published for the determination of SF and VP. Kamal et. al (Kamal et al., 2019) and Rezk et. al. (Rezk et al., 2019b) were able to determine SF in the mixture by applying the ratio difference (Kamal et al., 2019; Rezk et al., 2019b), first derivative of ratio spectra method (Kamal et al., 2019; Rezk et al., 2019b), dual-wavelength (Rezk et al., 2019b) and ratio subtraction (Rezk et al., 2019b). While Attia et. al (Attia et al., 2018) applied Savitsky–Golay filters signal processing to enable determination of SF in its binary mixture with VP. On the other hand, VP was determined in presence of SF by the first derivative of ratio spectra and ratio difference methods (Kamal et al., 2019) and by direct zero-order spectrophotometric method at 302.5 nm (Rezk et al., 2019b) and 339 nm (Attia et al., 2018). While the reported spectrofluorimetric methods, El-Gamal et. al. (El-Gamal et al., 2018) and Omar et. al. (Omar et al., 2018), have employed native fluorescence intensity measurements of VP in methanol at 385 nm and 400 nm after excitation at 295 nm (El-Gamal et al., 2018) and in methanol and various surface-active agents at 383 nm after excitation at 339 nm (Omar et al., 2018).
The objective of this study is to develop green sensitive and simple spectrophotometric methods that can displace the hazardous conventional methods for the determination of SF and VP. In this work, three spectrophotometric methods were suggested for the detection of SF in presence of VP. These methods include derivative ratio (2DD), delta absorbance (ΔA, ΔD1), and derivative compensation. Moreover, colorimetric methods (ElKimary & El-Yazbi, 2016; El-Yazbi, 2017; Ibrahim et al., 2017; El-Yazbi et al., 2019) can be used for the specific analysis of VP. In this study, two specific methods were proposed for the detection of VP by its specific reaction with NBD-Cl and MBTH reagents without interference from SF. The developed work offers simple, rapid, inexpensive methods for the analysis of SF and VP in their bulk and combined dosage form without any interference from each other. Analytical eco-scale protocol was also applied to evaluate the greenness of our developed method compared to some of the chromatographic reported methods where they did not consider any green analytical practices in terms of hazardous chemicals, energy consumption, and waste production (Van Aken et al., 2006; Gałuszka et al., 2012). Our results prove that although the spectrophotometric methods face the challenge of determining each drug in the presence of the other and not their simultaneous determination at the same time, such methods have the advantage of being greener than various chromatographic methods. Also, the ICH guidelines (International Conference on Harmonization; Validation of aAnalytical pProcedures, 2005) were applied on all the proposed methods to assure validation of the methods for routine work and quality assurance in pharmaceutical industries.
Experimental
Materials and reagents
SF and VP were kindly supplied as a gift sample by Amriya Pharmaceutical Industries, Alexandria, Egypt. Analytical grade chemicals were used (Methanol, Acetone, HCl), purchased from El-Nasr Pharmaceutical Co., Cairo, Egypt. A total of 0.2% methanolic solution of NBD-Cl (4-chloro-7-nitrobenzo-2-oxo-1,3-diazole) brought from Sigma Chemical Co., St. Louis, USA. Borate buffer pH 8 prepared from (boric acid and potassium chloride). A total of 0.01 M MBTH (3-methyl-2-benzothiazolinone hydrazone hydrochloride) (Fluka, Switzerland) prepared in 0.1 M HCl, 1% w/v Fe(III) chloride (Nice Chemical CO., Kerala, India) dissolved in 0.1 N HCl. Epclusa® tablets containing 400 mg SF and 100 mg VP were obtained from the local market.
Instrumentation
Perkin Elmer LS 45 Luminescence Spectrometer used for measuring the fluorescence spectra and intensity values, connected with a lamp 150 W Xenon, and data are recorded on a personal computer supplied with FL Win Lab Software.
All the spectrophotometric measurements were performed on Shimadzu UV Model 1800 spectrophotometer with a pair of 1 cm matched quartz cells. The pH of all the buffer solutions was measured using a pH-Meter Model pH 211 (Hanna instruments, USA), and an MLW type thermostatically controlled water bath was used.
Standard solutions
SF and VP standard stock solutions of 1 mg/ml (5 mg/ml for NBD-Cl method) were prepared in methanol and stored in the refrigerator.
Construction of the calibration graphs
Determination of SF in presence of VP
Derivative ratio method (2DD)
Different volumes of stock solution of SF were transferred into 10-mL volumetric flasks to prepare solutions in the range 5-50 μg/ml using methanol. Then the absorption spectrum of each solution together with that of a solution containing 10 μg/ml of VP was scanned within the range 200-400 nm. DD method is carried out by dividing, wavelength by wavelength, each SF absorption spectrum by that of 10 μg/mL VP as a divisor to obtain the ratio spectrum (Fig. 2a). Thereafter, the second derivative of the obtained ratio spectrum is calculated using Δλ = 6 (Fig. 2b). The second derivative amplitude at 254 nm is proportional to the concentration of SF; therefore, the calibration graph is used to determine the concentration of SF in the mixture.

a Ratio spectrum of 40 μg/mL SF using a devisor of 10 μg/mL VP, b second derivative ratio spectrum of 40 μg/mL SF using 10 μg/mL VP divisor at Δλ = 6 nm, c second derivative spectrum of ratio spectra of 40 μg/mL SF using 10 μg/mL VP divisor superimposed with the synthetic mixture of 40 μg/mL SF and 10 μg/mL VP using 10 μg/mL VP divisor, d second derivative spectrum of ratio spectrum of 40 μg/mL SF using 10 μg/mL VP divisor at Δλ = 2 nm, e second derivative spectrum of ratio spectrum of 40 μg/mL SF using 10 μg/mL VP divisor at Δλ = 4 nm, f second derivative spectrum of ratio spectrum of 40 μg/mL SF using 10 μg/mL VP divisor at Δλ = 8 nm
Derivative compensation
The undesirable absorption during spectrophotometric analysis can be determined and removed by the non-mathematical method called the compensation method. Different volumes from the VP stock solution within the range 4-20 μg/mL were prepared in methanol and their absorption spectra were recorded and then the corresponding first derivative spectra were calculated. The D1 values of each solution were recorded at the specified λ and the corresponding D1 ratios for standard VP were calculated (Table 1). Fixed concentration of the mixture solution containing 40 μg/mL SF: 10 μg/mL VP was placed in the sample compartment while solutions of different concentrations of SF below or above the concentration present in the mixture were placed in the blank compartment. The first derivative spectrum (D1) of each solution was recorded and at each time the corresponding D1 ratios were calculated at the specified λ (Table 1). To detect the exact balance point, the D1 ratio of the mixture must equal that of pure VP (El-Yazbi et al., 2007) where the concentration of SF in the mixture compartment is the same as that in the blank compartment.
Graphical method
A graphical method is suggested to detect the exact balance point in order to avoid the several preparations of different concentrations of SF standard solutions. The compensation method steps were followed and a line was obtained (Fig. 3) by plotting the mixture derivative ratio calculated in each instance against the reference pure SF concentration. By replacing the ratio of pure VP from the graph the concentration of SF could be interpolated easily.

Graphical plot of the (a) (D1 273/D1 299) and (b) D1 273/D1 355 ratio of VP-SF mixture in the sample cell versus SF in the reference cell for the determination of SF
Delta absorbance method (Δ A)
Equal volumes of SF standard solution within the concentration range 5-50 μg/mL were transferred into two sets of 10-ml volumetric flasks. The first set was diluted to mark with 0.1 N NaOH and the second set was diluted with 0.1 N HCl. The absorption of the drug solutions in 0.1 N NaOH was measured against the corresponding drug solutions in 0.1 N HCl to obtain the Δ A spectra (Fig. 4c) and then its first derivative (Δ D1) spectra (Fig. 4d). The values of ΔA and Δ D1 amplitudes were measured at 232and 245 nm, respectively.

a Δ A absorption spectrum of 40 μg/mL SF and the synthetic mixture 0f 40 μg/mL SF and 10 μg/mL VP, b first derivative of Δ A overlapped absorption spectra of 40 μg/mL SF and synthetic mix of 40 μg/mL SF and 10 μg/mL VP, c Δ A absorbance spectra of SF in 0.1 N NaOH against SF of the same concentration in 0.1 N HCl, d first derivative of Δ A absorbance spectra of SF in 0.1 N NaOH against SF of the same concentration in 0.1 N HCl
Determination of VP in presence of SF
Spectrophotometric and spectroflurimetric determination of VP using 4-chloro-7-nitrobenzofurazan (NBD-Cl)
Spectrophotometric method
Into a set of 10-ml volumetric flasks, different volumes of stock VP solution in the concentration range 5-500 μg/ml were transferred. One milliliter of 0.2 M borate buffer pH 8 was added and then 3 ml of 0.2% methanolic solution of NBD-Cl. The solutions were heated in a thermostatically controlled water bath at 80 °C for 30 min and then cooled in an ice bath; thereafter, 1 ml of 0.2 M HCl was added. Finally, the solutions were completed to mark with acetone.
Each solution absorbance was measured at 436 nm. A calibration curve was obtained by plotting the absorbance values against the concentrations and the data of the regression equation recorded in Table 2.
Spectrofluorimetric method
Different volumes from a stock solution of VP within the range of 0.5-20 μg/mL were transferred into a set of 10 ml volumetric flasks, 1 ml of 0.2 M borate buffer pH 8 was added followed by 1 ml of 0.2% methanolic solution of NBD-Cl. The solutions were heated at 80 °C for 20 min in a thermostatically controlled water bath and then cooled in an ice bath; thereafter, 0.5 ml of 0.2 M HCl was added. Finally, the solutions were completed to volume with acetone. Then, the fluorescence intensity at λem/ex 550/480 nm was measured and recorded. The fluorescence intensity values were plotted against the concentrations and a calibration curve was obtained with the data of regression equation recorded in Table 2.
Spectrophotometric determination of VP using MBTH
Dilutions from the standard solution of VP were transferred to a set of 10 ml volumetric flasks within the range 5-150 μg/mL. To each flask, 2 mL of freshly prepared 0.01 M MBTH solution was added, followed by 3 ml of 1% w/v ferric (III) chloride solution. The contents were mixed very well and left to stand for 15 min where a deep blue color is produced, then completed to volume with 0.1 N HCl. Finally, the absorbance is recorded at 709 nm against the blank solution. A linear calibration curve is obtained by plotting the absorbance values against the corresponding concentrations. Data of the regression equation were mentioned in Table 2.
Analysis of pharmaceutical dosage form
Ten tablets of Epclusa®, tablets reported to contain 400 mg SF and 100 mg VP, were weighed and crushed into a fine powder, and an accurately weighed amount of powder equivalent to 40 mg of SF and 10 mg VP, was transferred to a 25-mL volumetric flask and dissolved in a small amount of methanol followed by sonication for 20 min. The flask was completed to volume with methanol, mixed, and filtered. The filtrate of the tablets was analyzed spectrophotometrically and spectrofluorimetrically under the conditions mentioned above in the construction of the calibration curve section.
Results and discussion
Direct spectrophotometric determination of both SF and VP simultaneously in their mixture is not feasible because of the high spectral overlap (Fig. 1c) between SF and VP which represents a real challenge in the analysis of such formulation. Therefore, the proposed study describes three methods for the specific determination of SF in presence of VP, and two specific colorimetric methods for the determination of VP in presence of SF in their bulk and pharmaceutical form.
Determination of SF in presence of VP
Derivative ratio method
Figure 1c shows that the UV absorption spectrum of SF is completely overlapped by that of VP.
This interference was resolved using the suggested derivative ratio spectrum method. SF was determined by dividing the absorption spectra of SF and the mixture solutions, wavelength by wavelength, by VP standard solution (10 μg/mL) in order to obtain the corresponding ratio spectra (Fig. 2c), the second derivative of the ratio spectrum was preferred over the first derivative as it produced a better resolution. Best results for the determination of SF concentration without any contribution of VP were obtained by measuring the 2DD amplitude at 254 nm. The derivative ratio method was optimized by testing the effect of variables as the Δλ and the divisor concentration. In order to obtain the optimum wavelength interval for the second derivative of the ratio spectra, the effect of different Δλ intervals (2, 4, 6, and 8 nm) were studied (Fig. 2b, d, e, f). The obtained results showed that Δλ = 6 obtained the best reproducibility for the determination of SF using this method. The influence of the divisor concentration on the calibration graph was also tested. The obtained results showed that the divisor concentration is proportional to the second derivative values, so increasing or decreasing the divisor concentration results in a decrease or increase in the second derivative values, in spite that, the maxima and minima remain at the same wavelengths. The optimum divisor concentration for VP is 10 μg/mL. The calibration graph gave a linear straight line by plotting the second derivative values at 254 nm against the corresponding concentrations. The regression equation data are found in Table 2.
Derivative compensation
The spectrophotometric derivative compensation method was established for the determination of SF in presence of VP. The D1 maximum ratios for VP were calculated and the mean of the ratio values for six determinations of VP were recorded in Table 1 and used for the determination of the exact balance point. The suggested method proved to have good reproducibility indicated by RSD% < 2.
SF concentration was detected by placing the mixture in the sample compartment and different concentrations of SF solutions in the reference compartment. In order to determine the exact balance point, the mixture D1 ratios were compared with the VP ratios. At that point where the mixture ratio is equal to that of the VP, the SF concentration in the mixture corresponded to that in the reference compartment (Wahbi et al., 1992).
Detailed steps in the compensation method can be eliminated by the graphical method. A straight line was achieved from the graphical method by plotting the mixture ratio calculated against the reference SF concentration (Fig. 3).
Delta absorbance method (Δ A)
The application of ΔA method is very beneficial as it removes the error due to any interfering absorbing substance. The aim of this method is to measure the absorption spectrum of SF in different solvents. The choice of the solvents is based on affecting SF structure in order to obtain two different absorption spectra in the two solvents (Ragab et al., 2018). Subtracting these two spectra would allow the selective determination of only SF and would overcome the interference of VP.
In this work, SF obtained different absorption spectra in 0.1 N NaOH and 0.1 N HCl. SF delta absorbance (Δ A) and its first derivative (Δ D1) spectra measured in 0.1 N NaOH against 0.1 N HCl are shown in Fig. 4. As shown in the figure, at 232 nm, the SF ΔA spectrum is superimposed with the mixture ΔA spectrum, indicating the ΔA method can successfully determine SF in its mixture with VP without any interference from VP. The ΔD1 (Fig. 4b) was obtained by examining different Δλ intervals and showed that optimum results were achieved with Δλ = 2. Determination of SF in presence of VP is carried out by measuring the Δ A value at 232 nm and Δ D1 with Δλ = 2 at 245 nm.
Determination of VP in presence of SF
NBD-Cl and MBTH are selected in order to selectively react with VP in presence of SF, without any interference from SF present in the mixture (Fig. 5a, c).

a Absorption spectra of the colored reaction product obtained from VP, SF, and SF-VP mixture reaction with 0.2% w/v NBD-Cl. b Fluorescence excitation (1) and emission (2) spectra of the reaction product of VP (5 μg/mL) with 0.2% w/v NBD-Cl. c Absorption spectra of the colored reaction product obtained from VP, SF, and SF-VP mixture reaction with MBTH in presence of the oxidant Fe(III) chloride
Determination of VP in presence of SF using NBD-Cl
In the NBD-Cl method, VP was found to react with NBD-Cl in borate buffer pH 8 yielding a highly fluorescent yellow product with maximum absorption at 436 nm and exhibiting strong fluorescence at 550 nm, after excitation at 480 nm (Fig. 5b) (Miyano et al., 1985; Walash et al., 2011; El-Yazbi et al., 2016; Darwish et al., 2009; Attia et al., 2017; Annenkov et al., 2015; Rageh et al., 2010; Omar et al., 2017; Mohamed et al., 2019). This reaction occurs as NBD-Cl reacts with the amine group in the VP under mild alkaline conditions yielding a highly fluorescent yellow colored derivative as shown in Scheme 1 (Miyano et al., 1985; Walash et al., 2011; El-Yazbi et al., 2016; Darwish et al., 2009; Attia et al., 2017; Annenkov et al., 2015; Rageh et al., 2010; Omar et al., 2017; Mohamed et al., 2019).

Proposed reaction pathway of VP and NBD-Cl
2.1.1.Optimization of reaction conditions
Different conditions were studied for the spectrophotometric and spectrofluorimetric development and stability of the colored product. The effects of variation in the reagent volume, temperature, pH, reaction time, etc., have been examined to optimize such conditions.
2.1.1.1.Effect of NBD-Cl concentration
The impact of the volume of NBD-Cl was studied using different volumes of 0.2% of the reagent in borate buffer, pH 8. The UV absorbance and fluorescence intensity increases proportionally as the volume of the reagent increases until reaching their maxima at 1 mL and 3 ml for the spectrofluorimetric and spectrophotometric methods, respectively, after that gradual decrease in the absorbance and the fluorescence intensity values occur. Therefore, 1 mL of 0.2% NBD-Cl solution was chosen as the optimal volume of the reagent for spectrofluorimetric method, while 3 mL was the optimal volume for spectrophotometric method (Fig. 6a, b).

Effect of NBD-Cl volume on absorbance a and on fluorescence intensity b of the reaction product. Effect of buffer pH on absorbance c and on fluorescence intensity d of the reaction product. Effect of buffer volume on absorbance e and fluorescence intensity f of the reaction product. Effect of heating temperature on absorbance g and fluorescence intensity h of the reaction product. Effect of heating time on absorbance i and fluorescence intensity j of the reaction product. Effect of HCl volume on absorbance k and fluorescence intensity l of the reaction product. For absorbance measurements, 50 μg/mL of VP was used with 0.2% w/v NBD-Cl and for fluorescence intensity measurements, 10 μg/mL of VP was used with 0.2% w/v NBD-Cl
2.1.1.2.Effect of buffer pH and volume
The influence of pH on the absorbance and the fluorescence intensity of the resulting products was studied. Maximum values were reached at pH 8 for both methods, after which both the absorbance and fluorescence intensity progressively decreased (Fig. 6c, d). This decrease in fluorescence was previously reported (El-Yazbi et al., 2016; Darwish et al., 2009; Attia et al., 2017) and was attributed to the formation of NBD-OH in the excess quantity of hydroxide ion that prohibits the reaction of NBD-Cl with the drug.
Phosphate buffer with the same pH was also tested and compared with 0.2 M borate buffer. It was found that the absorbance and fluorescence intensity were higher in borate buffer. Therefore, borate buffer is superior to phosphate buffer with the same pH and this is possibly due to the rate of hydrolysis of NBD-Cl to NBD-OH was much slower which agrees with Miyano et al. (Miyano et al., 1985).
The buffer volume was also examined where the absorbance and fluorescence intensity increased as the volume increased until reaching their maxima at 1 mL after which they decreased gradually. So, the optimum buffer volume in this study was 1 mL for both the spectrofluorimetric and spectrophotometric methods (Fig. 6e, f).
2.1.1.3.Effect of Heating temperature and time
Heating temperature and time are two main experimental conditions that must be optimized as the reaction is kinetically stimulated. Different temperature settings and time intervals were studied in a thermostatically controlled water bath (Fig. 6 g, h, i, j). The optimum conditions were found to be heating at 80 °C for 20 min for the spectrofluorimetric method, while for the spectrophotometric method the highest absorbance was obtained upon heating at 80 °C for 30 min.
2.1.1.4.Effect of HCl volume
The fluorescence intensity or the absorbance value of the obtained product of NBD-Cl, namely, 4-hydroxy7-nitrobenzo-2-oxa-1,3-diazole (NBD-OH) is produced by lowering the pH of the reaction medium to 1. Thus, before the measurement of the fluorescence intensity or absorbance value, the reaction mixture is acidified producing a noticeable decrease in the background fluorescence or absorbance due to the formation of NBD-OH without affecting the drug reagent adduct, consequently, the sensitivity was increased (Walash et al., 2011).
Therefore, different HCl volumes were investigated for the acidification of the reaction mixture. It was found that optimum acidified conditions were reached using 0.5 mL of 0.2 M HCl for the spectrofluorimetric method and using 1 mL of 0.2 M HCl for the spectrophotometric method (Fig. 6 k, l).
2.1.1.5.Effect of diluting solvent
Different diluting solvents were tested as methanol, borate buffer pH 8, acetonitrile, diethyl ether, water, ethanol, and acetone. The best fluorescence intensity and absorbance value was reached using acetone as a diluting solvent.
All the abovementioned optimum conditions were used for validating this method for the determination of VP in the presence of SF.
Determination of VP in presence of SF using MBTH
For the MBTH method, VP reacts with MBTH in presence of Fe(III) as an oxidizing agent through oxidative coupling (Scheme 2) yielding a colored product with an absorption maximum at 709 nm (Fig. 5c) (Ribeiro et al., 2009; El-Yazbi et al., 1993a; El-Kommos, 1987; El-Yazbi et al., 1984; El-Yazbi et al., 1993b; El-Yazbi et al., 1999; El-Ragehy et al., 2001; Sowjanya et al., 2011; Anthon & Barrett, 2004; Granja et al., 2018). On oxidation of MBTH with the oxidant, Fe(III) chloride, MBTH loses two electrons and one proton resulting in the formation of the active coupling species called electrophilic intermediate (Ribeiro et al., 2009; El-Yazbi et al., 1993a; El-Kommos, 1987; El-Yazbi et al., 1984; El-Yazbi et al., 1993b; El-Yazbi et al., 1999; El-Ragehy et al., 2001; Sowjanya et al., 2011; Anthon & Barrett, 2004; Granja et al., 2018). The carbon atom with maximum electron density in VP is attacked by the electrophile (Scheme 2) yielding the deep blue product with a specific absorption maximum at 709 nm specific to VP (Ribeiro et al., 2009; El-Yazbi et al., 1993a; El-Kommos, 1987; El-Yazbi et al., 1984; El-Yazbi et al., 1993b; El-Yazbi et al., 1999; El-Ragehy et al., 2001; Sowjanya et al., 2011; Anthon & Barrett, 2004; Granja et al., 2018). Figure 5c demonstrates the absorption spectra for the deep blue product obtained from VP solution alone and in presence of SF, showing no interference from SF with the absorption maximum of VP-colored product.

Proposed reaction pathway of VP and MBTH
Optimization of reaction conditions
Different conditions were studied for the spectrophotometric development and stability of the colored product. The effects of different oxidizing agents, and their concentration, MBTH concentration temperature, reaction time, etc., have been examined to optimize such conditions.
Influence of different oxidizing agents
The procedure was applied using two oxidizing agents: Ce (IV) sulfate and Fe(III) chloride. Fe(III) offered the highest absorbance and the maximum color intensity with the reaction product as shown in Fig. 7a.

a Absorption spectra of the reaction product of VP with 0.01 M MBTH using 1% w/v Fe(III) chloride and Ce (IV) sulfate, b the effect of 1% w/v Fe(III) volume on the absorbance of the reaction product using VP (50 μg/mL), c effect of 0.01 M MBTH volume on the absorbance of the reaction product using VP (50 μg/mL), d effect of reaction time on the absorbance of the reaction product using VP (50 μg/mL) in presence of 0.01 M MBTH and 1% w/v Fe(III), e effect of diluting solvent on the absorbance of the reaction product using VP (50 μg/mL) in presence of 0.01 M MBTH and 1% w/v Fe(III)
Influence of ferric chloride volume
Maximum color intensity and stability was achieved using 3 mL of 1% w/v Fe(III) chloride in the above procedure (Fig. 7b). Chromogen oxidation or several oxidation products are expected to occur in higher concentrations of the oxidant leading to color fainting with time (El-Kommos, 1987).
Influence of MBTH volume
Different volumes (0.5-3 mL) of freshly prepared 0.01 M MBTH were tested, 2 mL of 0.01 M MBTH was found to give the optimum maximum color intensity and absorbance (Fig. 7c).
Influence of heating
The effect of heating was examined; however, the maximum absorbance was achieved at room temperature.
Influence of time
After optimization of MBTH and Fe(III) chloride conditions, the time for color development was studied at different time intervals (5-35 min). As shown in (Fig. 7d), maximum absorbance was achieved after 15 min.
Influence of the diluent
Different diluting solvents as methanol, acetonitrile, acetone, water, and HCl, were studied to decide the solvent used in this reaction. The deep blue colored product was obtained by water and HCl in contrast to the other solvents that gave deep green color. As shown in Fig. 7e, HCl is the diluting solvent chosen as it gave maximum absorbance.
Influence of the sequence of addition
The sequence of addition is very critical for the color development, maximum color intensity was obtained by the addition of VP, MBTH, and Fe(III) then the abovementioned procedure was followed.
All the abovementioned optimum conditions were used for validating this method for the determination of VP in the presence of SF.
Method validation
The method is validated according to the ICH guidelines (International Conference on Harmonization; Validation of aAnalytical pProcedures, 2005) regarding linearity, accuracy, precision, limit of detection (LOD), and limit of quantitation (LOQ).
Linearity
Six concentrations of SF and VP were analyzed using the suggested methods in order to evaluate linearity. SF solutions of 5-50 μg/mL concentrations were used for all the proposed spectrophotometric methods. While for VP, solutions of 5-500 and 0.5-20 μg/mL concentrations were used for the NBD-Cl spectrophotometric and spectrofluorimetric methods, respectively. Also, for the MBTH method, 5-150 μg/mL VP solutions were used. Results are demonstrated in Table 2 with R2 greater than 0.999 and RSD% less than 2, showing good linearity for all proposed methods.
Accuracy and precision
Three different concentrations of SF and VP mixture within the linearity range were determined in triplicates by the developed methods. Results show accepted mean% recoveries with low Er (%) indicating good accuracy (Table 3).
Intra-day precision was evaluated by measuring three different concentrations of SF and VP where each concentration is measured three times within the day using the developed methods. The inter-day precision was assessed in the same way but on various 3 days. The results of RSD% were less than 2 indicating the precision of the developed methods (Table 3).
LOD and LOQ
LOD and LOQ were calculated and evaluated (Table 2) by the use of the developed methods LOQ = 10σ/b and LOD = 3.3σ/b, where σ is the standard deviation of a blank replicate and b is the slope of the calibration graph. Results show that all the suggested methods have low LOD and LOQ indicating the sensitivity of the proposed methods.
Specificity
Determination of the drugs in the different ratios of the synthetic mixtures without any interference from any substance present in the mixture proved the selectivity of the suggested methods for the detection of SF and VP in presence of each other.
Standard solutions stability
Stock standard solutions under test were found to be stable for 2 weeks when stored at 4 °C in the refrigerator and through the day at room temperature.
Analysis of SF and VP in pharmaceutical dosage form
Determination of SF and VP in their pharmaceutical tablets (Epclusa® tablets) was carried out by applying the proposed methods. The obtained results were statistically compared with those of the reported methods. This comparison was carried out using variance ratio F test and student’s t test. Table 4 shows the obtained results demonstrating no considerable difference between the execution of the two methods concerning accuracy and precision.
Comparison to other reported methods
Literature reveals few methods for the estimation of SF and VP in pharmaceutical dosage forms. Three spectrophotometric methods (Kamal et al., 2019; Rezk et al., 2019b; Attia et al., 2018) have been reported where SF was determined in the mixture by applying the ratio difference (Kamal et al., 2019; Rezk et al., 2019b), first derivative of ratio spectra method (Kamal et al., 2019; Rezk et al., 2019b), dual wavelength (Rezk et al., 2019b), ratio subtraction (Rezk et al., 2019b), and Savitsky–Golay filters signal processing (Attia et al., 2018). On the other hand, VP has been determined in presence of SF by the first derivative of ratio spectra and ratio difference methods (Kamal et al., 2019) and by direct zero-order spectrophotometric method at 302.5 nm (Rezk et al., 2019b) and 339 nm (Attia et al., 2018). While the reported spectrofluorimetric methods have employed native fluorescence intensity measurements of VP in methanol at 385 nm and 400 nm after excitation at 295 nm (El-Gamal et al., 2018) and in methanol and various surface-active agents at 383 nm after excitation at 339 nm (Omar et al., 2018).
Tables 5 and 6 present the comparison of the proposed methods with other reported spectrophotometric methods for the determination of SF and VP in presence of each other.
Results show that the derivative ratio (2DD), delta absorbance (ΔA, ΔD1), and derivative compensation methods, proposed in this work for the determination of SF in presence of VP, are of better or comparable sensitivity to other reported methods (Kamal et al., 2019; Rezk et al., 2019b; Attia et al., 2018). This is demonstrated by the LOD and LOQ values in Table 5. While for the determination of VP in presence of SF, the proposed NBD-Cl spectrofluorimetric method proves to be the most sensitive method compared to all the reported spectrophotometric methods (Kamal et al.,2019; Rezk et al., 2019b; Attia et al., 2018; El-Gamal et al., 2018). Moreover, our proposed colorimetric methods have comparable sensitivity to other reported methods (Kamal et al., 2019; Rezk et al., 2019b; Attia et al., 2018) but have wider linearity range which increase the scope of applicability of our proposed methods for the determination of the selected mixture in different matrixes (Table 6). Also, the use of inexpensive reagents and simple instrumentations promotes our proposed methods to be used for routine analysis of VP and SF pharmaceutical formulations in quality control laboratories.
Assessment of method greenness
Ideal green analysis is achieved by removing or minimizing the usage of hazardous chemicals, reducing energy consumption and the least amount of waste production. The greenness profile of an analytical method is evaluated by the use of an analytical eco-scale tool which is considered a semi-quantitative tool for the assessment of the method greenness (Van Aken et al., 2006). Penalty points are used for calculating the eco-scale score, where the high score value indicates a green economic analytical technique regarding reagents, hazards, waste produced, and energy used (Gałuszka et al., 2012). Total penalty points of the entire procedure is subtracted from the ideal score value, 100, to calculate the analytical eco-scale score (Mohamed & Lamie, 2016). The proposed methods eco-scale score was compared with other reported method scores (Vanaja et al., 2018; Elkady & Aboelwafa, 2018) as shown in Table 7. The proposed methods high eco-scale value > 75 indicate that our technique is an excellent green analysis compared to other reported methods (Table 7). Therefore, the proposed methods proved to be eco-friendly, simple, and accurate technique for the analysis of SF and VP without any interference from each other in the pharmaceutical formulation to be used in routine analysis and quality control laboratories. Moreover, our results prove that although the spectrophotometric methods face the challenge of determining each drug in the presence of the other and not their simultaneous determination at the same time, such methods have the advantage of being greener than various chromatographic methods.
Conclusion
The novel mixture of SF and VP was analyzed using inexpensive simple, accurate, rapid, and environment-friendly methods for the detection and determination of SF and VP in presence of each other without prior separation. The developed methods were identified to be specific, feasible, and reliable. Due to the high spectral overlap between SF and VP, SF was determined in presence of VP spectrophotometrically by the derivative ratio, the delta absorbance, and the derivative compensation methods. On the other hand, VP was analyzed in presence of SF spectrophotometrically and spectrofluorimetrically by the use of two colorimetric methods using two reagents: NBD-Cl and MBTH.
The suggested methods were applied on the synthetic mixture and the pharmaceutical tablet. Results of the percentage recovery and the relative standard deviation indicated good accuracy and precision.
When comparing our developed methods with reported methods, results showed no considerable difference with the other reported HPLC, HPTLC, and LC-MS/MS but have an advantage of being an excellent green analytical technique according to the analytical eco-scale protocol. The proposed work offers simple sample preparation, non-hazardous reagents, and inexpensive methods with no need for complicated instruments that require time for development, these advantages allow their routine application in quality assurance units in pharmaceutical industries.
Availability of data and materials
Data and materials are presented in details in the manuscript. For further information, please contact the authors.
References
Annenkov VV, Verkhozina ON, Shishlyannikova TA, Danilovtseva EN. Application of 4-chloro-7-nitrobenzo-2-oxa-1,3-diazole in analysis: fluorescent dyes and unexpected reaction with tertiary amines. Analytical Biochemistry. 2015;486:5–13.
Anthon GE, Barrett DM. Comparison of three colorimetric reagents in the determination of methanol with alcohol oxidase. Application to the Assay of Pectin Methylesterase. J. Agric. Food Chem. 2004;52:3749–53.
Attia K, Magdy N, Mohamed G. Validated spectrofluorimetric method for the determination of cefoxitin sodium in its pure form and powder for injection via derivatization with 4-chloro-7-nitrobenzo-2-oxa-1, 3-diazole (NBD-CL). J Adv Pharmacy Res. 2017;1(4):209–2015.
Attia KA, El-Abasawi NM, El-Olemy A, Abdelazim AH. Application of different analytical techniques for determination of velpatsvir and sofosbuvir in the pharmaceutical preparation. J Liquid Chromatography & Related Technologies. 2018;41:467–73.
Cheung MC, Walker AJ, Hudson BE, Verma S, McLauchlan J, Mutimer DJ, Brown A, Gelson WT, MacDonald DC, Agarwal K, Foster GR. Outcomes after successful direct-acting antiviral therapy for patients with chronic hepatitis C and decompensated cirrhosis. Journal of hepatology. 2016a;65:741–7.
Darwish IA, Amer SM, Abdine HH, Al-Rayes LI. New spectrophotometric and fluorimetric methods for determination of fluoxetine in pharmaceutical formulations. Int J Analytical Chemistry. 2009. https://doi.org/10.1155/2009/257306.
El-Gamal RM, Abdel-Gawad SA, Belal FF, Moustapha ME. Selective and sensitive spectrofluorimetric quantification of velpatasvir in presence of sofosbuvir. Application to their co-formulated tablet. RSC Adv. 2018;8:32909–15.
Elkady EF, Aboelwafa AA (2018) Rapid bioanalytical LC-MS/MS method for the simultaneous determination of sofosbuvir and velpatasvir in human plasma-application to a pharmacokinetic study in Egyptian volunteers. Journal of Chromatography B, 1102:116-124.
ElKimary EI, El-Yazbi AF. An eco-friendly stability-indicating spectrofluorimetric method for the determination of two anticancer stereoisomer drugs in their pharmaceutical preparations following micellar enhancement: application to kinetic degradation studies. Spectrochimica Acta Part A: Molecular and Biomolecular Spectroscopy. 2016;163:145–53.
El-Kommos ME. Spectrophotometric determination of dobutamine hydrochloride using 3-methylbenzothiazolin-2-one hydrazone. Analyst. 1987;112:101–3.
El-Ragehy NA, Abbas SS, El-Khateeb SZ. Stability indicating method for determination of nortriptyline hydrochloride using 3-methyl-2-benzothiazolinone hydrazone (MBTH). J. Pharm. Biomed. Analysis. 2001;25(1):143–51.
El-Yazbi AF. Comparative validation of the determination of sofosbuvir in pharmaceuticals by several inexpensive ecofriendly chromatographic, electrophoretic, and spectrophotometric methods. J AOAC Int. 2017;100:1000–7.
El-Yazbi AF, Guirguis KM, Bedair MM, Belal TS. Simple mix-and-read assays for the determination of baclofen in pharmaceutical formulation. Heliyon. 2019;5:e01854.
El-Yazbi AF, Wagih MM, Ibrahim F, Barary MA. Spectrofluorimetric determination of topiramate and levetiracetam as single components in tablet formulations and in human plasma and simultaneous fourth derivative synchronous fluorescence determination of their co-administered mixture in human plasma. J Fluorescence. 2016;26(4):1225–38.
El-Yazbi FA, Abdine HH, Shaalan RA. Spectrophotometric method for the determination of benazepril hydrochloride in its single and multicomponent dosage forms. J. Pharm. Biomed. Anal. 1999;20:343–50.
El-Yazbi FA, Galal SM, Blaih SM. Determination of some 1, 4-benzodiazepines by oxidative coupling of their acid induced degradation products. Bulletin des Sociétés Chimiques Belges. 1993a;102:233–7.
El-Yazbi FA, Hammud HH, Sonji GM. Determination of tTelmisartan and Hydrochlorothiazide in binary mixture. ULTRA SCIENTIST OF PHYSICAL SCIENCES. 2007;19:93.
El-Yazbi FA, Korany MA, Abdel-Hay MA. Colorimetric determination of carazolol and pindolol using 3- methyl- 2 benzothiazolone hydrazone hydrochloride. Indian. J. Pharm. Sci. 1984;46:183–5.
El-Yazbi FA, Mahgoub H, Barary MH. Spectrophotometric method for the determination of metoclopramide. Bull. Fac. Pharm. Cairo Univ. 1993b;31(1):63–5.
Feld JJ, Jacobson IM, Hézode C, Asselah T, Ruane PJ, Gruener N, Abergel A, Mangia A, Lai CL, Chan HL, Mazzotta F. Sofosbuvir and velpatasvir for HCV genotype 1, 2, 4, 5, and 6 infection. New Engl J Med. 2015;373:2599–607.
Foster GR, Afdhal N, Roberts SK, Bräu N, Gane EJ, Pianko S, Lawitz E, Thompson A, Shiffman ML, Cooper C, Towner WJ. Sofosbuvir and velpatasvir for HCV genotype 2 and 3 infection. New Engl J Med. 2015;373:2608–17.
Gałuszka A, Migaszewski ZM, Konieczka P, Namieśnik J. Analytical Eco-Scale for assessing the greenness of analytical procedures. TrAC Trends in Analytical Chemistry. 2012;37:61–72.
Granja B, Mendonça Filho JRH, Oliveira WS, Santos JCC. Exploring MBTH as spectrophotometric probe for total phenolic compounds determination in beverage samples Analytical Methods. DOI. 2018. https://doi.org/10.1039/C8AY00464A.
Greig SL. Sofosbuvir/velpatasvir: a review in chronic hepatitis C. Drugs. 2016;76:1567–78.
Ibrahim FA, El-Yazbi AF, Wagih MM, Barary MA. Sensitive inexpensive spectrophotometric and spectrofluorimetric analysis of ezogabine, levetiracetam and topiramate in tablet formulations using Hantzsch condensation reaction. Spectrochimica Acta Part A: Molecular and Biomolecular Spectroscopy. 2017;184:47–60.
International Conference on Harmonization; Validation of aAnalytical pProcedures. Text and Methodology, Q2A. Geneva: R1; 2005.
Jakobsen JC, Nielsen EE, Feinberg J, Katakam KK, Fobian K, Hauser G, Poropat G, Djurisic S, Weiss KH, Bjelakovic M, Bjelakovic G. Direct acting antivirals for chronic hepatitis C. Cochrane Database of Systematic Reviews. 2017;9.
Kamal AH, Mabrouk MM, Bebawy LI, Mekky MA. Spectrophotometric and robust UPLC methods for simultaneous determination of velpatasvir and sofosbuvir in their tablet. Microchemical Journal. 2019;149:103996.
McQuaid T, Savini C, Seyedkazemi S. Sofosbuvir, a significant paradigm change in HCV treatment. J Clin Translational Hepatol. 2015;3:27.
Miyano H, Toyo’'oka T, Imai K. Further studies on the reaction of amines and proteins with 4-fluoro-7-nitrobenzo-2-oxa-1, 3-diazole. Analytica Chimica Acta. 1985;170:81–7.
Mohamed HM, Lamie NT. Analytical eco-scale for assessing the greenness of a developed RP-HPLC method used for simultaneous analysis of combined antihypertensive medications. J AOAC International. 2016;99:1260–5.
Mohamed O, Alfaki A, Elbashir AA. Utilization of 4-chloro-7-nittobenzo-2-oxa-1, 3-diazol (NBD-CL) as chromogenic reagent for determination of metformin hydrochloride (MET) in pharmaceutical formulation. Asian Journal of Pharmaceutical Research and Development. 2019;7(1):19–23.
Moustapha ME, El-Gamal RM, Belal FF. Two novel UPLC methods utilizing two different analytical columns and different detection approaches for the simultaneous analysis of velpatasvir and sofosbuvir: application to their co-formulated tablet. BMC chemistry. 2019;13:118.
Nahon P, Bourcier V, Layese R, Audureau E, Cagnot C, Marcellin P, Guyader D, Fontaine H, Larrey D, De Lédinghen V, Ouzan D. Eradication of hepatitis C virus infection in patients with cirrhosis reduces risk of liver and non-liver complications. Gastroenterology. 2017;152:142–56.
Omar MA, Abdel-Lateef MA, Ali R, Derayea SM. Study on fluorescence properties of HCV antiviral (velpatasvir) and its fluorimetric determination in presence of sofosbuvir; application to stability study and human plasma. Luminescence. 2018;33:1249–56.
Omar MA, Hammad MA, Awad M. Utility of 4-chloro-7-nitrobenzo-2-oxa-1,3-diazole for development of a highly sensitive stability indicating spectrofluorimetric method for determination of salmeterol xinafoate; application to human plasma. RSC Adv. 2017;7:44773–9.
Poynard T, Yuen MF, Ratzin V, Lai CL. Viral hepatitis C. Lancet. 2003;362:2095–100.
Ragab MA, El Yazbi FA, Hassan EM, Khamis EF, Hamdy MM. Spectrophotometric analysis of two eye preparations, vial and drops, containing ketorolac tromethamine and phenylephrine hydrochloride binary mixture and their ternary mixture with chlorphenirmaine maleate. Bulletin of Faculty of Pharmacy, Cairo University. 2018;56:91–100.
Rageh AH, El-Shaboury SR, Saleh GA, Mohamed FA. Spectophotometric method for determination of certain cephalosporins using 4-chloro-7-nitrobenzo-2-oxa-1, 3-diazole (NBD-Cl). Natural Science. 2010;2(08):828–40.
Rani JS, Devanna N. A new RP-HPLC method development and validation for simultaneous estimation of sofosbuvir and velpatasvir in pharmaceutical dosage form. Int. J. Eng. Technol. Sci. Res. 2017;4:145–52.
Rezk MR, Basalious EB, Badr KA. Novel determination of sofosbuvir and velpatasvir in human plasma by UPLC–MS/MS method: application to a bioequivalence study. Biomedical Chromatography. 2018;32:e4347.
Rezk MR, Monir HH, Marzouk HM. Novel determination of a new antiviral combination; sofosbuvir and velpatasvir by high performance thin layer chromatographic method; application to real human samples. Microchemical Journal. 2019a;146:828–34.
Rezk MR, Monir HH, Marzouk HM. Spectrophotometric assessment of the brand new antiviral combination: sofosbuvir and velpatasvir in their pure forms and pharmaceutical formulation. Spectrochimica Acta Part A: Molecular and Biomolecular Spectroscopy. 2019b;213:159–66.
Ribeiro DS, Prior JA, Santos JL, Lopes JA, Lima JL. Exploiting the oxidative coupling reaction of MBTH for indapamide determination. Talanta. 2009;79:1161–8.
Saraya RE, Elhenawee M, Saleh H. Development of a high-performance thin-layer chromatographic method for the simultaneous determination of newly co-formulated antiviral drugs sofosbuvir and velpatasvir in their pure forms and tablet dosage form. J Planar Chromatography-Modern TLC. 2019;32:141–7.
Sowjanya K, Thejaswini JC, Gurupadayya BM, Indupriya M. Spectrophotometric determination of pregabalin using Gibb’s and MBTH reagent in pharmaceutical dosage form. Der Pharma Chemica. 2011;3(1):112–22.
Tahir MS, Ahmed M, Issam AM, Qadir MA, Adnan A, Rashid A, Quratulain SYED, Hussain R, Hin LK, Ikram R. Determination of sofosbuvir and velpatasvir in solid pharmaceutical dosage form by hplc-pda and its application to in vitro dissolution studies. Latin American Journal of Pharmacy. 2018;37:1067–73.
Van Aken K, Strekowski L, Patiny L. EcoScale, a semi-quantitative tool to select an organic preparation based on economical and ecological parameters. Beilstein journal of organic chemistry. 2006;2(1):3.
Vanaja B, Vageesh NM, Kistayya C, Urukundu V. RP-HPLC method development and validation for simultaneous estimation of sofosbuvir and velpatasvir in pure and pharmaceutical dosage form. Innovat International Journal Of Medical & Pharmaceutical. Sciences. 2018;3.
Wahbi AA, El-Yazbi FA, Barary MH, Sabri SM. Derivative spectrophotometric analysis of two-component mixtures using a compensation technique. Analyst. 1992;117:785–9.
Walash MI, Belal F, El-Enany N, Elmansi H. Development and validation of stability indicating method for determination of sertraline following ICH guidelines and its determination in pharmaceuticals and biological fluids. Chem Cent J. 2011;5:61.
Williams R. Global challenges in liver disease. Hepatology. 2006;44:521–6.
Acknowledgements
Not applicable
Funding
The authors declare that this work was not funded by any agencies.
Author information
Authors and Affiliations
Contributions
Amira F. El-Yazbi: Supervision, Conceptualization, Methodology, Investigation and Writing- Original draft preparation, Reviewing and Editing. Nourhan E. Elashkar: Methodology, Data analysis, Data Curation, Data Validation and Writing- Original draft preparation. Karim M. Abdel-Hay, Hytham M. Ahmed and Wael Talaat: Supervision, Reviewing and Editing. The authors read and approved the final manuscript
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable
Consent for publication
Not applicable
Competing interests
The authors declare that they have no conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
El-Yazbi, A.F., Elashkar, N.E., Abdel-Hay, K.M. et al. Eco-friendly analytical methods for the determination of compounds with disparate spectral overlapping: application to antiviral formulation of sofosbuvir and velpatasvir. J Anal Sci Technol 12, 7 (2021). https://doi.org/10.1186/s40543-021-00257-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40543-021-00257-7