
Abstract
Objectives
The aim of the present study was to develop a robust and easy to use high performance liquid chromatography (HPLC) to analyze 25(OH)D3 in human serum.
Background
Vitamin D is a fat-soluble steroid hormone precursor that is mainly produced in the skin by exposure to sunlight. It is also supplied in the diet and plays a pivotal role in calcium homeostasis and skeletal metabolism throughout life.
Methods
To assess its analytical performance, we used the RECIPE HPLC Complete Kit and an HPLC-UV instrument. Our HPLC results were compared with a validated electrochemiluminescence method.
Results
The method was linear for the lower limit of quantification from 3 ng/l up to at least 200 ng/l for 25(OH)D3, with the following equation for the regression line: y = 0.172 X + 2.45 (R2 = 0.989). Intra-assay precision was determined by extracting and quantifying 10 serum replicates from one patient. The mean was 37.875 ng/ml, the standard deviation was 0.22, and the coefficient of variation was 0.58%. Comparisons of results demonstrated good agreement between HPLC and ECL methods (R2 = 0.883).
Conclusion
The HPLC assay demonstrates excellent linearity, acceptable accuracy and precision, and good agreement with a validated ECL method. The simple sample preparation and ease of use make it practical for the routine clinical laboratory.
Similar content being viewed by others


Background
Vitamin D, a fat-soluble steroid hormone produced in the skin by exposure to sunlight or supplied via dietary sources, plays a vital role in skeletal metabolism and calcium homeostasis throughout life (Prentice et al. 2008). Vitamin D is important for the function of the cardiovascular, immune, and reproductive systems (Norman 2008; Dusso et al. 2005). Vitamin D deficiency has been implicated in various cancers (Buttigliero et al. 2011) and cardio-vascular (McGreevy and Williams 2011) and autoimmune diseases (Ascherio et al. 2010). In recent years, clinical findings showed that low vitamin D levels are associated with various non-skeletal diseases. This condition led physicians to assess vitamin D concentrations in their patients (Holick 2007; Wang 2009). Also, vitamin D deficiency is a common cause of secondary hyperparathyroidism. Elevated parathyroid (PTH) levels, especially in elderly vitamin D-deficient adults, can result in osteomalacia, increased bone turnover, reduced bone mass, and risk of bone fractures. Low 25-OH vitamin D [25(OH)D] concentrations are also associated with decreased bone mineral density. Factors contributing to low vitamin D levels include low dietary vitamin D intake, lack of exposure to sunlight, and other variables (El-Khoury et al. 2011). Clinical trials have shown that oral vitamin D supplements reduce fractures (Bischoff-Ferrari et al. 2009).
Vitamin D is biologically inert and must undergo two successive hydroxylations in the liver and kidney to become biologically active 25(OH)D. Then, active 25(OH)D is released into the circulation and connected to vitamin D binding protein (DBP) (Ascherio et al. 2010). The activity of this enzyme is regulated by numerous factors including parathyroid hormone levels and serum calcium and phosphorus (Holick 2007). One form of vitamin D, produced in skin via a photochemical reaction with 7-dehydrocholesterol, is cholecalciferol (vitamin D3) (Prentice et al. 2008). Another form is ergocalciferol (vitamin D2), which is derived from plants and used as a supplement.
The most useful marker for vitamin D status in plasma is 25-hydroxyvitamin D3 [25(OH)D3], which is formed in the liver from its precursor, vitamin D3 (Bruce et al. 2013). In the past two decades, analytical methods developed to identify vitamin D3 include capillary electrophoresis (Shi et al. 1995; Delgado-Zamarreno et al. 2002), spectrophotometry (Tütem et al. 1997), fluorimetry (Pérez-Ruiz et al. 1999), colorimetry (Amin 2001), and immunochemical and chromatographic methods (Heudi et al. 2004). Due to their simplicity and expediency, immunochemical methods are most often used to measure vitamin D concentrations (Wallace et al. 2010). The limitation of these methods is the cross-reactivity of the antibodies and their inability to differentiate between vitamins D2 and D3 with 25(OH)D metabolites (Stepman et al. 2011). The chromatographic methods require highly skilled laboratory personnel and are more accurate and reliable than the immunochemical methods. These methods show excellent analytical performance including precision data, stability, limit of quantization (LOQ), linearity, and average recovery and selectivity.
The aim of the present study was to develop a robust and easy-to-use HPLC method to analyze 25(OH)D3 in human serum. Additionally, we compared the HPLC results with those determined by electrochemiluminescence (ECL). This information is useful for physicians, clinical laboratory scientists, and producers of vitamin D reagents.
Methods
HPLC method
Reagents and standard
The reagents and standards used were HPLC grade. Acetonitrile (CH3CN) and methanol were obtained from Sigma. Serum calibrators (cholecalciferol) and controls (levels I, II, and III) for 25(OH)D3 were purchased from RECIPE (ClinChek). The commercial lyophilized serum controls and calibrators (90 μg/ml) were reconstituted in water according to the manufacturer’s instructions, aliquoted, and stored at − 20 °C. The vitamin D concentrations in the serum samples were determined using RECIPE’s HPLC complete kit. All procedures followed were in accordance with the ethical standards of the Committee on Human Experimentation Mashhad University of Medical Science (MUMS). Informed consent was obtained from all patients in the study.
Instrument and procedure
An Agilent 1100 series HPLC system equipped with a UV detector was used for the chromatographic analysis. The analytes were separated on a Hector-M C18 4.6 × 150 mm analytical column with 5.0 μm particle size. The mobile phase consisted of 90% acetonitrile and 10% methanol used in isocratic elution mode with a flow rate of 1.0 ml/min. Samples were injected using a six-port injection valve equipped with a 20 μl loop. Results were read at a wavelength of 264 nm. Data were collected and quantitated using HPLC 1100 software.
To prepare samples, we dispensed 400 μl of serum (calibrator, control, or patient sample) into 2 ml test tubes; then added 400 μl each of the precipitation and extraction reagents to the tube. The tubes were vortex-mixed for 10 s to obtain a flocculent precipitate and centrifuged at 10,000 RCF for 5 min. The clear supernatant was decanted into a 10 ml glass vial, capped, and placed in the autosampler unit of the HPLC apparatus. The extract was stable for at least 2 days at room temperature. The software calculated retention time for peak identification and peak height ratio for quantification.
Electrochemiluminescence method
Reagents and standard
The Elecsys Vitamin D3 kit (Roche) and Cobas analyzer were used to measure of 25(OH) D. The kit contains streptavidin-coated microparticles (0.72 mg/ml), reaction buffer (acetate buffer 220 mmol/l, pH 3.9, albumin 2 g/l), polyclonal anti-25(OH)D3 antibody labeled with ruthenium complex 1.5 mg/l, biotinylated 25(OH)D 0.15 mg/l, and phosphate buffer 20 mmol/l, pH 6.5. The Elecsys Vitamin D3 reagent kit was stored upright to ensure complete mixing of the microparticles before use. After opening, the kit reagents are stable for at least 1 week at 2–8 °C.
Instrument and procedure
A Hitachi Cobas e 411 electrochemiluminescence system was used to measure 25(OH)D3. The assay utilizes a 2-step 18-min incubation process. In step 1, 25(OH)D3 in a 35 μl sample competes with the biotin-labeled vitamin D in the complex contained in biotin-vitamin D/polyclonal 25(OH)D-specific ruthenium-labeled antibody. In step 2, streptavidin-coated microparticles are added and the complex attaches to the solid phase via the biotin-streptavidin interaction. The reaction mixture is aliquoted into the measuring cell and then unbound material is removed with Procell. Finally, chemiluminescent emission is induced and measured with a photomultiplier. Results are determined via an instrument-generated calibration curve by 2-point calibration and a master curve via the standard reagent.
Results and discussion
Typical HPLC chromatograms of calibrator and patient samples are shown in Fig. 1. The retention time for 25(OH)D3 was 4.6 min, and the total analysis time was 8.0 min, typical for conventional HPLC assay. The analytical performance was evaluated by the precision data, stability, limit of quantization (LOQ), linearity, and average recovery for 25 (OH) D3 using commercial calibrator serums. The analytical accuracy was estimated at 25 and 90 ng/ml and their percent relative errors were 1.004 and 1.03%, respectively. The method was linear for the lower limit of quantification (LLQ) from 3 ng/l up to at least 200 ng/l for 25(OH)D3, with the following regression line equation: y = 0.172 X + 2.45 (R2 = 0.989).

Typical HPLC chromatograms of calibrator (a) and patient samples (b). Concentrations were 90 and106 ng/ml, respectively
Analyses of standard, control, and serum samples showed that the 25(OH)D extract remained stable at room temperature for 2 days. Stability results from three patient samples are shown in Table 1.
In a precision study (n = 10) using patient sera at 38 ng/ml for 25(OH)D3, the mean concentration was 37.875 ng/ml, the SD was 0.22, and the CV was 0.58%, indicating the high precision of our method.
The analytical recoveries for 25(OH)D were performed by adding a standard to the patient samples. The vitamin D concentration in the patient sample was 58 ng/ml. This sample was selected for recovery analysis. After recovery, 100 μl of the patient sample was added to 300 μl of the 90 ng/ml vitamin D standard. The average vitamin D concentration in the serum sample was 83.25 ng/ml. The average recovery rate obtained was 99.1% (SD = 1.47%). Thus, this chromatographic system is suitable for quantitative determination of 25(OH)D3. Figure 2 shows the chromatograms before and after recovery.

HPLC chromatograms of a serum sample before (a) and after (b) recovery
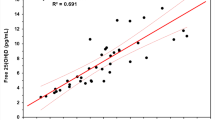
25(OH)D concentrations in 114 serum samples were analyzed by HPLC and compared with those obtained by electrochemiluminescence. The results were similar for the two methods (n = 114; R2 = 0.883) (Fig. 3).

Evaluation of 25-OH vitamin D in human serum samples with HPLC and ECL
In the last two decades high vitamin D deficiency rates have been reported in adults and children worldwide (Galunska et al. 2014). Therefore, many researchers and physicians have tried to estimate vitamin D status (Galunska et al. 2014). With the expansion of the vitamin D tests, differences increased in testing methodologies, standards, reference ranges, and interpretation of results (Carter 2009). Measurement of vitamin D metabolites in plasma is challenging because they are lipophilic, tightly bound to DBP, and at very low concentrations, i.e., in the nanomolar range for 25(OH)D2 and 25(OH)D3 (Shimada et al. 2001; Ding et al. 2010). Immunoassay methods measure total metabolite concentration and cannot distinguish between the D2 and D3 forms. In addition, 25(OH)D cross-reacts with 24,25(OH)2 D3(de Castro et al. 1999; Lind et al. 1997). Currently, however, immunochemical assays are routinely used in laboratories due to their convenience, speed, turnaround, and cost-effectiveness (Wootton 2005). Most of the disadvantages of immunochemical methods were improved by chromatographic methods. These methods measure both 25(OH)D2 and 25(OH)D3 quantitatively and selectively (Stepman et al. 2011; Guo et al. 2006). Liquid chromatograph/mass spectrometry (LC-MS/MS) is widely used as the reference method (Carter 2009) and is a reliable diagnostic tool able to distinguish 25(OH)D2 from 25(OH)D3 (Krasowski 2011; van den Ouweland et al. 2010). Although LC-MS/MS can be the premier method, most clinical laboratories do not use this technique because of the substantial cost and need for highly trained operators (Lensmeyer et al. 2006). In contrast to the LC-MS/MS, with its expensive instrumentation and requirement for considerable technical expertise, HPLC is less technically demanding and less expensive (Lensmeyer et al. 2006). With HPLC, a simple sample preparation procedure of protein precipitation followed by liquid-liquid extraction ensures effective release of 25(OH)D from the binding protein (Galunska et al. 2014).
Conclusions
HPLC enables reliable quantification of both 25(OH)D3 and 25(OH)D2. The relatively short analysis time makes it useful for routine clinical laboratory practice. The HPLC procedure is the preferred technique for measuring these two metabolites due to its high selectivity and efficient analytical performance. Due to its rapid separation, high sensitivity, and accurate quantification, HPLC has become popular for the detection of vitamins in various matrices.
Abbreviations
- CV:
-
Coefficient of variation
- ECL:
-
Electrochemiluminescence
- HPLC:
-
High-performance liquid chromatography
- LLQ:
-
Lower limit of quantification
- PTH:
-
Parathyroid hormone
- SD:
-
Standard deviation
References
Amin AS. Colorimetric determination of tocopheryl acetate (vitamin E) in pure form and in multi-vitamin capsules. Eur J Pharm Biopharm. 2001;51(3):267–72.
Ascherio A, Munger KL, Simon KC. Vitamin D and multiple sclerosis. Lancet Neurol. 2010;9(6):599–612.
Bischoff-Ferrari HA, Willett WC, Wong JB, Stuck AE, Staehelin HB, Orav EJ, et al. Prevention of nonvertebral fractures with oral vitamin D and dose dependency: a meta-analysis of randomized controlled trials. Arch Intern Med. 2009;169(6):551–61.
Bruce SJ, Rochat B, Béguin A, Pesse B, Guessous I, Boulat O, et al. Analysis and quantification of vitamin D metabolites in serum by ultra-performance liquid chromatography coupled to tandem mass spectrometry and high-resolution mass spectrometry–a method comparison and validation. Rapid Commun Mass Spectrom. 2013;27(1):200–6.
Buttigliero C, Monagheddu C, Petroni P, Saini A, Dogliotti L, Ciccone G, et al. Prognostic role of vitamin d status and efficacy of vitamin D supplementation in cancer patients: a systematic review. Oncologist. 2011;16(9):1215–27.
Carter GD. 25-Hydroxyvitamin D assays: the quest for accuracy. Clin Chem. 2009;55(7):1300–2.
de Castro ML, Fernández-Romero JM, Ortiz-Boyer F, Quesada J. Determination of vitamin D3 metabolites: state-of-the-art and trends. J Pharm Biomed Anal. 1999;20(1–2):1–17.
Delgado-Zamarreno M, Gonzalez-Maza I, Sanchez-Perez A, Carabias-Martinez R. Separation and simultaneous determination of water-soluble and fat-soluble vitamins by electrokinetic capillary chromatography. J Chromatogr A. 2002;953(1–2):257–62.
Ding S, Schoenmakers I, Jones K, Koulman A, Prentice A, Volmer DA. Quantitative determination of vitamin D metabolites in plasma using UHPLC-MS/MS. Anal Bioanal Chem. 2010;398(2):779–89.
Dusso AS, Brown A, Slatopolsky E. Vitamin D. Am J Physiol Ren Physiol. 2005;289:F8–28.
El-Khoury JM, Reineks EZ, Wang S. Progress of liquid chromatography-mass spectrometry in measurement of vitamin D metabolites and analogues. Clin Biochem. 2011;44(1):66–76.
Galunska BT, Gerova D, Boncheva M, Svinarov D. HPLC method for measuring the circulating levels of 25-hydroxy vitamin D: validation and comparison with ID LC/MS/MS and immunoassay. Integr Food Nutr Metab. 2014;1(2):119–23.
Guo T, Taylor RL, Singh RJ, Soldin SJ. Simultaneous determination of 12 steroids by isotope dilution liquid chromatography-photospray ionization tandem mass spectrometry. Clin Chim Acta. 2006;372(1–2):76–82.
Heudi O, Trisconi M-J, Blake C-J. Simultaneous quantification of vitamins A, D3 and E in fortified infant formulae by liquid chromatography–mass spectrometry. J Chromatogr A. 2004;1022(1–2):115–23.
Holick MF. Vitamin D deficiency. N Engl J Med. 2007;357(3):266–81.
Krasowski MD. Pathology consultation on vitamin D testing. Am J Clin Pathol. 2011;136(4):507–14.
Lensmeyer GL, Wiebe DA, Binkley N, Drezner MK. HPLC method for 25-hydroxyvitamin D measurement: comparison with contemporary assays. Clin Chem. 2006;52(6):1120–6.
Lind C, Chen J, Byrjalsen I. Enzyme immunoassay for measuring 25-hydroxyvitamin D3 in serum. Clin Chem. 1997;43(6):943–9.
McGreevy C, Williams D. New insights about vitamin D and cardiovascular disease: a narrative review. Ann Intern Med. 2011;155(12):820–6.
Norman AW. From vitamin D to hormone D: fundamentals of the vitamin D endocrine system essential for good health. Am J Clin Nutr. 2008;88(2):491–9.
Pérez-Ruiz T, Martínez-Lozano C, Tomás V, Martín J. Flow-injection fluorimetric determination of vitamin K1 based on a photochemical reaction. Talanta. 1999;50(1):49–56.
Prentice A, Goldberg GR, Schoenmakers I. Vitamin D across the lifecycle: physiology and biomarkers. Am J Clin Nutr. 2008;88(2):500–6.
Shi H, Ma Y, Humphrey JH, Craft NE. Determination of vitamin A in dried human blood spots by high-performance capillary electrophoresis with laser-excited fluorescence detection. J Chromatogr B Biomed Appl. 1995;665(1):89–96.
Shimada K, Mitamura K, Higashi T. Gas chromatography and high-performance liquid chromatography of natural steroids. J Chromatogr A. 2001;935(1–2):141–72.
Stepman HC, Vanderroost A, Van Uytfanghe K, Thienpont LM. Candidate reference measurement procedures for serum 25-hydroxyvitamin D3 and 25-hydroxyvitamin D2 by using isotope-dilution liquid chromatography–tandem mass spectrometry. Clin Chem. 2011;57(3):441–8.
Tütem E, Apak R, Günaydı E, Sözgen K. Spectrophotometric determination of vitamin E (α-tocopherol) using copper (II)-neocuproine reagent. Talanta. 1997;44(2):249–55.
van den Ouweland JM, Beijers AM, Demacker PN, van Daal H. Measurement of 25-OH-vitamin D in human serum using liquid chromatography tandem-mass spectrometry with comparison to radioimmunoassay and automated immunoassay. J Chromatogr B Anal Technol Biomed Life Sci. 2010;878(15–16):1163–8.
Wallace A, Gibson S, De La Hunty A, Lamberg-Allardt C, Ashwell M. Measurement of 25-hydroxyvitamin D in the clinical laboratory: current procedures, performance characteristics and limitations. Steroids. 2010;75(7):477–88.
Wang S. Epidemiology of vitamin D in health and disease. Nutr Res Rev. 2009;22(2):188–203.
Wootton AM. Improving the measurement of 25-hydroxyvitamin D. Clin Biochem Rev. 2005;26(1):33–6.
Acknowledgements
This research was supported and funded by Pardis Clinical and Genetic Laboratory. We thank referral cases of Pardis for contributing in this project and koromabzarteb company.
Availability of data and materials
This data will be shared to clinical laboratories to measure vitamin D at a lower cost using HPLC instead of ECL.
Author information
Authors and Affiliations
Contributions
FK contributed to the conception and design, analysis and interception of data, drafting of the article, and final approval of the version to be published. SNayerabadi, SNahid, and AA helped in the conception and design, and acquisition of data. AM helped in the conception and design, acquisition of data, and drafting of the article. AV contributed to the conception and design, revising of the article, and final approval of the version to be published. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
All procedures followed were in accordance with the ethical standards of the committee on human experimentation MUMS (Mashhad University of Medical Science). Informed consent was obtained from all patients for being included in the study.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Keyfi, F., Nahid, S., Mokhtariye, A. et al. Evaluation of 25-OH vitamin D by high performance liquid chromatography: validation and comparison with electrochemiluminescence. J Anal Sci Technol 9, 25 (2018). https://doi.org/10.1186/s40543-018-0155-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40543-018-0155-z