
Abstract
Background
The profiles of modified nucleosides could act as useful biomarkers for cancer and cellular stress-induced diseases. However, there are no reports of high throughput and simultaneous quantitative methods for using biomarker evaluation and discovery at the bedside.
Methods
Modified nucleosides were separated on two CAPCELL PAK ADME S3 (100 mm × 2.1 mm i.d.; 3-μm particle size) analytical columns coupled with a CAPCELL PAK ADME cartridge (10 mm × 2 mm i.d.; 3-μm particle size) guard column. Both columns were used in tandem during multi-column LC analysis to reduce analysis time. Two mobile phases were used, including 20 mM ammonium acetate adjusted to pH 5.3 using acetic acid and 1.0 M ammonium acetate/acetonitrile/water/acetic acid (1/95/5/0.03, v/v/v/v), with the post-column addition of methanol to enhance ionization efficiency. Tandem mass spectrometry detection was performed using a triple quadrupole mass spectrometer equipped with a heated electrospray ionization source in selected reaction monitoring mode.
Results
Four major nucleosides and 11 modified nucleosides, including guanosine, adenosine, uridine (U), cytidine, inosine, 1-methyladenosine, 5-methylcytidine, 2′-O-methylcytidine, 3-methylcytidine, 7-methylguanosine (m7G), 5-methyluridine (m5U), pseudouridine, 2-thiocytidine, N2-methylguanosine (m2G), N2,N2-dimethylguanosine, 2-fluoro-2′-deoxyadenosine as an internal standard, and its isotopic isomers were separated within 7 min and analyzed within 10 min. This resulted in limits of quantitation of 0.50–5.00 ng mL−1, except for m2G (10.0 ng mL−1), m7G (12.5 ng mL−1), U (12.5 ng mL−1), and m5U (50.0 ng mL−1). This method provides a wide range of linearity, with correlation coefficients greater than 0.99 for all nucleosides. Both the accuracy and precision of this method satisfied criteria of <15% for higher concentrations and <20% for the lowest concentrations.
Conclusions
In this study, we describe a rapid and selective method that uses multi-column liquid chromatography with tandem mass spectrometry (LC-MS/MS) to simultaneously quantify modified nucleosides. This global analysis will be useful for evaluating modifications in RNA.
Similar content being viewed by others

Background
Modified nucleosides are mainly included in transfer RNA (tRNA) (Agris 2004), where tRNA modifications increase conformational stability (Anderson et al. 1998), identification (Shin et al. 2011), and translational fidelity (Crick 1966; Gerber and Keller 1999; Agris 2015). For example, 1-methyadenosine (m1A) is necessary for conformational stability (Anderson et al. 1998), where the 2-thiocytidine (s2C) occurring in the anticodon loop interferes with the inosine–adenosine interaction (Agris 2015). As such, tRNA modification has been considered essential for tRNA maturation and stability. Modified nucleosides contained in most tRNA have been associated with turnover (Topp et al. 1993; Nakano et al. 1993; Sander et al. 1986); therefore, modified nucleosides, such as m1A, 2-methylguanosine (m2G), and pseudouridine (Ψ), have been reported to be cancer biomarkers (Itoh et al. 1992; Jeng et al. 2009; Djukovic et al. 2010; Cho et al. 2009). Recent studies have shown that altering the tRNA modification profile plays a major role in the cellular stress response (Chan et al. 2010, 2012, 2015; Endres et al. 2015), where 5-methylcytidine (m5C), 2′-O-methylcytidine (Cm), and N2,N2-dimethylguanosine (m2 2G) reportedly increase under oxidative conditions created by H2O2; other oxidative stress reagents do not influence these modified nucleosides (Chan et al. 2010, 2012). Alkylating stress affects 3-methylcytidine (m3C), where increasing m3C promoted the translation of ACC- and ACT-rich messenger RNA (Chan et al. 2015). In addition, inosine (I), 5-methyluridine (m5U), m5C, Cm, 7-methylguanosine (m7G), and m1A were altered by replication stress (Endres et al. 2015). These cellular stress response markers may be candidate biomarkers of cellular stress-induced diseases. The profile of modified nucleosides could act as sensitive biomarkers for cancer and be useful as potential biomarkers for investigating cellular stress-induced diseases (Table 1).
A simultaneous quantitative analysis method is necessary to confirm the profile of modified nucleosides. This is because there are many structural isomers, regioisomers, and isotopic isomers in modified nucleosides, where adequate chromatographic separation is the key to simultaneous analysis of modified nucleosides. On the other hand, one of the goals of these analytical methods is their use as biomarkers for diagnoses at the bedside on a daily basis. In addition, biomarker evaluation and discovery necessitates the analysis of large sample sizes to ensure statistical reliability (Koulman et al. 2009); thus, the analytical methods applied must have a high throughput. Analytical methods for modified nucleosides include liquid chromatography (LC), ultra-performance LC (UPLC), gas chromatography, or capillary electrophoresis combined with ultraviolet or mass spectrometry (MS) (Russell and Limbach 2013; Struck et al. 2011; Lee et al. 2004; Basanta-Sanchez et al. 2016). Of these methods, LC-tandem MS (LC-MS/MS) analysis is the most frequently used for modified nucleoside determination due to its high selectivity and quantitative performance. Several studies have reported good chromatographic separation; however, these methods require long times to separate nucleosides (Djukovic et al. 2010; Cho et al. 2009; Struck et al. 2011; Lee et al. 2004). Rodríguez-Gonzalo et al. (2016) reported an analytical method that quickly and simultaneously determined nucleosides; however, this method has not been used to study significant cancer biomarkers or cellular stress markers, such as m5C, Cm, and m2 2G. Basanta-Sanchez et al. (2016) demonstrated a highly sensitive, accurate, quantitative UPLC-MS/MS nucleosides analysis method; however, this method applies to cellular RNA modifications and is thus unsuitable for human biological samples, such as blood and urine, due to its narrow calibration range. Several studies have attempted to increase LC analysis throughput, while also showing the utility of the multiple column LC method (Korfmacher et al. 1999; Cass et al. 2001; Oertel et al. 2002; Orton et al. 2013; Lee et al. 2015; Patel et al. 2016). This approach is helpful for reducing the total run time without sacrificing chromatographic separation, as using multiple columns reduces separation and wash times. In this study, we developed a rapid, selective method that simultaneously quantifies modified nucleosides through a multi-column LC-MS/MS system.
Methods
Chemicals
A nucleoside test mix, purchased from Sigma-Aldrich (St. Louis, MO, USA), containing 50 μg/mL of cytidine (C), 25 μg/mL of guanosine (G), 25 μg/mL of I, 25 μg/mL of m1A, 50 μg/mL of m5C, 20 μg/mL of Cm, 100 μg/mL of m3C methosulfate, 25 μg/mL of m7G, 100 μg/mL of m5U, 25 μg/mL of Ψ, 10 μg/mL of s2C dehydrate, and 25 μg/mL of uridine (U) was used. Adenosine (A) and 2-fluoro-2′-deoxyadenosine (f2dA) were purchased from Sigma-Aldrich. m2G was purchased from Santa Cruz Biotechnology, Inc., (Santa Cruz, CA, USA). m2 2G was purchased from Toronto Research Chemicals (Toronto, Canada). Methanol (MeOH) and acetonitrile (MeCN) of LC/MS grade were obtained from Kanto Chemical (Tokyo, Japan). LC/MS-grade ammonium acetate (CH3COONH4) and acetic acid (CH3COOH) were obtained from Wako Pure Chemical Industries. Ultrapure-grade water was prepared using a PURELAB Ultra from Organo (Tokyo, Japan).
Standard solutions
Stock standard solutions of 100 μg mL−1 for A, m2G, m2 2G, m3C, m5U, and f2dA; at 50.0 μg mL−1 for C and m5C; at 25.0 μg mL−1 for m1A, I, G, m7G, U, and Ψ; at 20.0 μg mL−1 for Cm; and at 10.0 μg mL−1 for s2C were prepared in 20.0 mM CH3COONH4, adjusted to a pH of 5.3 using CH3COOH. These stock solutions were stored at −30 °C in brown glass bottles. Working solutions were prepared by appropriate dilution of these stock solutions.
LC-MS/MS conditions
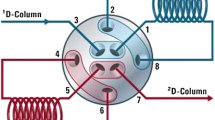
The LC system was based on a NANOSPACESI-2 LC system (Shiseido, Tokyo, Japan), with a CAPCELL PAK ADME S3 (100 mm × 2.1 mm i.d.; 3-μm particle size; Shiseido, Tokyo, Japan) analytical column, coupled with a CAPCELL PAK ADME cartridge (10 mm × 2 mm i.d.; 3-μm particle size; Shiseido, Tokyo, Japan) guard column. Our multiple column LC system is composed of one auto sampler, two six-port valves, four pumps, and two analytical columns coupled with a guard column; this system selects the column in line with the mass spectrometer. Both columns are used in parallel during multi-column LC analysis. One column is used as an analytical column, while the other is washed and prepared for the next injection. Figure 1 shows flow diagrams and valve positions of this multiple column analysis setup, where method A is separated by column A and method B is separated by column B. Valve A was activated to switch between the two columns when the sample was injected. Valve B (the MS divert valve) was switched to MS/MS after 0.5 min of injection. All systems were controlled via the autosampler trigger signal. Two mobile phases were used, including 20 mM CH3COONH4 adjusted to pH 5.3 using CH3COOH (mobile phase A) and 1.0 M CH3COONH4/MeCN/H2O/CH3COOH (1/95/5/0.03, v/v/v/v) (mobile phase B), with the post-column addition of MeOH to enhance ionization efficiency. Other LC condition parameters are summarized in Table 2.

Flow diagram outlining multi-column analysis. Method A is separated by column A, and method B is separated by column B. One column is used as the analytical column, while the other column is washed and prepared for the next injection
MS/MS detection was performed using a triple quadrupole mass spectrometer equipped with a heated electrospray ionization (HESI) source (TSQ Quantum Ultra, Thermo Fisher Scientific, San Jose, CA, USA). This system operated under the following conditions: positive mode ionization, spray voltage 4.0 kV, vaporizer temperature 450 °C, sheath gas pressure 60 psi, auxiliary pressure 40 psi, capillary temperature 300 °C, collision gas pressure 1.0 mTorr, tube lens offset 90 V, and collision energy 20 eV. Descriptions of selected reaction monitoring (SRM) transitions can be found in previous studies and are summarized in Table 3. (Lee et al. 2004; Su et al. 2014; Fan et al. 2006) The LC-MS/MS system was controlled by Xcalibur software (Thermo Fisher Scientific), which was also used to collect data.
Calibration
All peaks were automatically integrated using the Xcalibur software, and compound concentrations were calculated from calibration curves describing the relationship between peak ratios and areas, using f2dA as an internal standard (IS). Calibration curves for system A ranged from 1.00–200 ng mL−1; specifically, m1A ranged from 1.25–500 ng mL−1; I, G, C from 2.50–500 ng mL−1; m2G from 10.0–2000 ng mL−1; m5C from 0.50–500 ng mL−1; m7G, U from 12.5–500 ng mL−1; m2 2G from 1.00–2000 ng mL−1; m5U from 50.0–2000 ng mL−1; m3C from 1.39–696 ng mL−1; Cm from 1.00–400 ng mL−1; s2C from 4.39–175 ng mL−1; and Ψ from 5.00–500 ng mL−1 (including an IS of 200 ng mL−1). Calibration curve linearity was evaluated over at least six concentrations.
Precision and accuracy
To evaluate accuracy and precision, we analyzed four standard solution concentrations consisting of five samples on three separate days. Accuracy was calculated from the percentage deviation of the mean from the true value, and the precision was expressed as the relative error and coefficient of variation (CV, %).
Results and discussion
Chromatographic separation
Our MS/MS system was used to detect four major nucleosides and 11 modified nucleosides and f2dA (IS), which were separated by HPLC in <10 min using a CAPCELL PAK ADME column (Fig. 2). The resultant retention times using the two-column, parallel system were as follows: C (2.30 min), U (2.71 min), Ψ (1.56 min), m5C (2.27 min), m3C (3.63 min), Cm (3.79 min), m5U (3.96 min), s2C (3.28 min), A (5.15 min), I (3.61 min), m1A (2.99 min), G (3.75 min), m7G (4.69 min), m2G (3.39 min), m2 2G (5.41 min), and f2dA (IS) (6.16 min). The 10-min analytical time is half the previously reported time of 20 min (Djukovic et al. 2010; Cho et al. 2009; Russell and Limbach 2013; Struck et al. 2011; Lee et al. 2004; Basanta-Sanchez et al. 2016) Longer re-equilibration time allows reproducibility on column separation. However, one-third of the total analysis time was spent on re-equilibration. To circumvent this problem, we separated re-equilibration time from total analysis time using the multiple column system. It is important to note that our LC system consists of the least equipment to perform the analytical and wash gradient compared to previous reports (Korfmacher et al. 1999; Cass et al. 2001; Oertel et al. 2002; Orton et al. 2013; Lee et al. 2015; Patel et al. 2016). This is simple and easy-to-use for most LC/MS users. We also note that the peak at 2.30 min in the chromatogram of U corresponds to the C + 1 isotope peak, where our method can differentiate between the U and C + 1 isotope peak. Additionally, this method can differentiate between m5U for m5C + 1 and m3C + 1, m5U for m5C + 2 and m3C + 2, and I for A + 1. Typical chromatograms for the standard solutions are shown in Fig. 2 and demonstrate sufficient separation using an ADME column. Simultaneous quantitative analysis of modified nucleosides was previously performed using the ODS column (Djukovic et al. 2010; Russell and Limbach 2013; Lee et al. 2004; Basanta-Sanchez et al. 2016; Rodríguez-Gonzalo et al. 2016). Chromatographic separation on the ODS column depends on the hydrophobic interaction, which enables separation by the number of methyl groups (Núñez et al. 2007). However, the ODS column is generally unsuitable for the separation of hydrophilic compounds, such as nucleosides, because of the weak interaction due to low surface polarity. An extended time is required for sufficient separation of nucleosides on the ODS column. An ADME column is composed of an adamantyl functional group with polymer-coated silica. While retaining the hydrophobic interaction similarly to the ODS column, it also has higher surface polarity. Therefore, the ADME column provides good separation of hydrophilic compounds. In fact, separation of hydrophilic compounds, such as amino acids, carboxylic acids, and other hydrophilic metabolites on ADME columns has been reported (Mochizuki et al. 2015; Uno et al. 2015; Song et al. 2016). Using this unique surface property of the ADME column, we achieved good chromatographic separation of nucleosides and rapid analysis. Tubercidin was previously used as an internal standard (Mishima et al. 2014). However, we used f2dA because it was not separated from other nucleosides under this chromatographic condition.

SRM product ion chromatograms of modified nucleoside standards in HESI positive ionization mode. SRM selected reaction monitoring, HESI heated electrospray ionization
Comparison of peak area ratio using multiple column LC mode
As this method uses multiple LC columns to reduce runtime, we evaluated column identity by investigating the ratio between target peak areas and IS area using the two columns. No differences in area ratios for the nucleosides using either column were detected (Fig. 3), proving that our method can be used in multi-column LC mode.

Mean ± SE area ratio/mean area ratio (%) obtained via multiple column LC mode. The mean area ratio was calculated from an average of the column A and column B area ratios for each nucleoside. SE standard error; LC: liquid chromatography
Limit of quantification (LOQ) and linearity
The LOQ corresponds to the lowest concentration that is quantifiable given accuracy criteria of < ±15% and a signal-to-noise ratio >10 AU. The LOQ for our system was measured as 1.00–5.00 ng mL−1, except for m2G (10.0 ng mL−1), m7G (12.5 ng mL−1), U (12.5 ng mL−1), and m5U (50.0 ng mL−1).
Linearity
Calibration curves for all nucleosides are summarized in Table 3. Correlation coefficients were greater than 0.99 for all nucleosides, such that the calibration curves showed good linearity within the quantification range. In comparison with previous studies (Djukovic et al. 2010; Cho et al. 2009; Lee et al. 2004; Rodríguez-Gonzalo et al. 2016), these results suggest that our method is applicable to concentrations of modified nucleosides found within biological samples.
Accuracy and precision
The accuracy ranged from 85.05–114.7% for the highest three concentrations and 80.03–119.6% for the lowest concentrations across all nucleosides (Table 4). Precision ranged from 1.073–14.84% for the highest concentrations of all compounds and 1.731–19.92% for the lowest concentrations of nucleosides (Table 4). These results demonstrate that the method presented has both good reliability and repeatability, where data satisfied criteria of <15% within higher concentrations and <20% within the lowest concentrations.
Conclusions
This study describes a selective, high-throughput method to simultaneously quantify modified nucleosides using a novel multi-column LC-MS/MS analysis. This method selectivity separated modified nucleosides and their isotopic isomers quickly (within 7 min), analyzing them within 10 min using a unique multiple-column setup, whose linearity spans a wide concentration range. Both values of accuracy and precision satisfied criteria of <15% for higher concentrations and <20% for the lowest concentration. This unique method provides a fast, reliable means of analyzing the profile of modified nucleosides, which could be used to diagnose at bedside on a daily basis.
References
Agris PF. Decoding the genome: a modified view. Nucleic Acids Res. 2004;32(1):223–38.
Agris PF. The importance of being modified: an unrealized code to RNA structure and function. RNA. 2015;21(4):552–4.
Anderson J, Phan L, Cuesta R, Carlson BA, Pak M, Asano K, Björk GR, Tamame M, Hinnebusch AG. The essential Gcd10p-Gcd14p nuclear complex is required for 1-methyladenosine modification and maturation of initiator methionyl-tRNA. Genes Dev. 1998;12(23):3650–62.
Basanta-Sanchez M, Temple S, Ansari SA, D'Amico A, Agris PF. Attomole quantification and global profile of RNA modifications: epitranscriptome of human neural stem cells. Nucleic Acids Res. 2016;44(3):e26.
Cass RT, Villa JS, Karr DE, Schmidt Jr DE. Rapid bioanalysis of vancomycin in serum and urine by high-performance liquid chromatography tandem mass spectrometry using on-line sample extraction and parallel analytical columns. Rapid Commun Mass Spectrom. 2001;15(6):406–12.
Chan CT, Dyavaiah M, DeMott MS, Taghizadeh K, Dedon PC, Begley TJ. A quantitative systems approach reveals dynamic control of tRNA modifications during cellular stress. PLoS Genet. 2010;6(12):e1001247.
Chan CT, Pang YL, Deng W, Babu IR, Dyavaiah M, Begley TJ, Dedon PC. Reprogramming of tRNA modifications controls the oxidative stress response by codon-biased translation of proteins. Nat Commun. 2012;3:937.
Chan CT, Deng W, Li F, DeMott MS, Babu IR, Begley TJ, Dedon PC. Highly predictive reprogramming of tRNA modifications is linked to selective expression of codon-biased genes. Chem Res Toxicol. 2015;28(5):978–88.
Cho SH, Choi MH, Lee WY, Chung BC. Evaluation of urinary nucleosides in breast cancer patients before and after tumor removal. Clin Biochem. 2009;42(6):540–3.
Crick FH. Codon—anticodon pairing: the wobble hypothesis. J Mol Biol. 1966;19(2):548–55.
Djukovic D, Baniasadi HR, Kc R, Hammoud Z, Raftery D. Targeted serum metabolite profiling of nucleosides in esophageal adenocarcinoma. Rapid Commun Mass Spectrom. 2010;24(20):3057–62.
Endres L, Dedon PC, Begley TJ. Codon-biased translation can be regulated by wobble-base tRNA modification systems during cellular stress responses. RNA Biol. 2015;12(6):603–14.
Fan H, Li SP, Xiang JJ, Lai CM, Yang FQ, Gao JL, Wang YT. Qualitative and quantitative determination of nucleosides, bases and their analogues in natural and cultured Cordyceps by pressurized liquid extraction and high performance liquid chromatography–electrospray ionization tandem mass spectrometry (HPLC–ESI–MS/MS). Anal Chim Acta. 2006;567(2):218–28.
Gerber AP, Keller W. An adenosine deaminase that generates inosine at the wobble position of tRNAs. Science. 1999;286(5442):1146–9.
Itoh K, Konno T, Sasaki T, Ishiwata S, Ishida N, Misugaki M. Relationship of urinary pseudouridine and 1-methyladenosine to activity of leukemia and lymphoma. Clin Chim Acta. 1992;206(3):181–9.
Jeng LB, Lo WY, Hsu WY, Lin WD, Lin CT, Lai CC, Tsai FJ. Analysis of urinary nucleosides as helper tumor markers in hepatocellular carcinoma diagnosis. Rapid Commun Mass Spectrom. 2009;23(11):1543–9.
Korfmacher WA, Veals J, Dunn-Meynell K, Zhang X, Tucker G, Cox KA, Lin CC. Demonstration of the capabilities of a parallel high performance liquid chromatography tandem mass spectrometry system for use in the analysis of drug discovery plasma samples. Rapid Commun Mass Spectrom. 1999;13(20):1991–8.
Koulman A, Lane GA, Harrison SJ, Volmer DA. From differentiating metabolites to biomarkers. Anal Bioanal Chem. 2009;394(3):663–70.
Lee SH, Jung BH, Kim SY, Chung BC. A rapid and sensitive method for quantitation of nucleosides in human urine using liquid chromatography/mass spectrometry with direct urine injection. Rapid Commun Mass Spectrom. 2004;18(9):973–7.
Lee H, Mun DG, Bae J, Kim H, Oh SY, Park YS, Lee JH, Lee SW. A simple dual online ultra-high pressure liquid chromatography system (sDO-UHPLC) for high throughput proteome analysis. Analyst. 2015;140(16):5700–6.
Mishima E, Inoue C, Saigusa D, Inoue R, Ito K, Suzuki Y, Jinno D, Tsukui Y, Akamatsu Y, Araki M, Araki K, Shimizu R, Shinke H, Suzuki T, Takeuchi Y, Shima H, Akiyama Y, Toyohara T, Suzuki C, Saiki Y, Tominaga T, Miyagi S, Kawagisihi N, Soga T, Ohkubo T, Yamamura K, Imai Y, Masuda S, Sabbisetti V, Ichimura T, Mount DB, Bonventre JV, Ito S, Tomioka Y, Itoh K, Abe T. Conformational change in transfer RNA is an early indicator of acute cellular damage. J Am Soc Nephrol. 2014;25(10):2316–26.
Mochizuki T, Takayama T, Todoroki K, Inoue K, Min JZ, Toyo'oka T. Towards the chiral metabolomics: liquid chromatography-mass spectrometry based DL-amino acid analysis after labeling with a new chiral reagent, (S)-2,5-dioxopyrrolidin-1-yl-1-(4,6-dimethoxy-1,3,5-triazin-2-yl)pyrrolidine-2-carboxylate, and the application to saliva of healthy volunteers. Anal Chim Acta. 2015;875:73–82.
Nakano K, Nakao T, Schram KH, Hammargren WM, McClure TD, Katz M, Petersen E. Urinary excretion of modified nucleosides as biological marker of RNA turnover in patients with cancer and AIDS. Clin Chim Acta. 1993;218(2):169–83.
Núñez O, Ikegami T, Miyamoto K, Tanaka N. Study of a monolithic silica capillary column coated with poly(octadecyl methacrylate) for the reversed-phase liquid chromatographic separation of some polar and non-polar compounds. J Chromatogr A. 2007;1175:7–15.
Oertel R, Richter K, Fauler J, Kirch W. Increasing sample throughput in pharmacological studies by using dual-column liquid chromatography with tandem mass spectrometry. J Chromatogr A. 2002;948(1-2):187–92.
Orton DJ, Wall MJ, Doucette AA. Dual LC − MS platform for high-throughput proteome analysis. J Proteome Res. 2013;12(12):5963–70.
Patel DC, Wahab MF, Armstrong DW, Breitbach ZS. Advances in high-throughput and high-efficiency chiral liquid chromatographic separations. J Chromatogr A. 2016;1467:2–18.
Rodríguez-Gonzalo E, Herrero-Herrero L, García-Gómez D. Development, validation and application of a fast analytical methodology for the simultaneous determination of DNA- and RNA-derived urinary nucleosides by liquid chromatography coupled to tandem mass spectrometry. J Chromatogr B Analyt Technol Biomed Life Sci. 2016;1019:132–9.
Russell SP, Limbach PA. Evaluating the reproducibility of quantifying modified nucleosides from ribonucleic acids by LC-UV-MS. J Chromatogr B Analyt Technol Biomed Life Sci. 2013;923–924:74–82.
Sander G, Topp H, Heller-Schöch G, Wieland J, Schöch G. Ribonucleic acid turnover in man: RNA catabolites in urine as measure for the metabolism of each of the three major species of RNA. Clin Sci. 1986;71(4):367–74.
Shin BS, Kim JR, Walker SE, Dong J, Lorsch JR, Dever TE. Initiation factor eIF2γ promotes eIF2-GTP-Met-tRNAi(Met) ternary complex binding to the 40S ribosome. Nat Struct Mol Biol. 2011;18(11):1227–34.
Song Y, Song Q, Li J, Zhang N, Zhao Y, Liu X, Jiang Y, Tu P. An integrated strategy to quantitatively differentiate chemome between Cistanche deserticola and C. tubulosa using high performance liquid chromatography-hybrid triple quadrupole-linear ion trap mass spectrometry. J Chromatogr A. 2016;1429:238–47.
Struck W, Waszczuk-Jankowska M, Kaliszan R, Markuszewski MJ. The state-of-the-art determination of urinary nucleosides using chromatographic techniques “hyphenated” with advanced bioinformatic methods. Anal Bioanal Chem. 2011;401(7):2039–50.
Su D, Chan CT, Gu C, Lim KS, Chionh YH, McBee ME, Russell BS, Babu IR, Begley TJ, Dedon PC. Quantitative analysis of ribonucleoside modifications in tRNA by HPLC-coupled mass spectrometry. Nat Protoc. 2014;9(4):828–41.
Topp H, Duden R, Schöch G. 5,6-Dihydrouridine: a marker ribonucleoside for determining whole body degradation rates of transfer RNA in man and rats. Clin Chim Acta. 1993;218(1):73–82.
Uno K, Takayama T, Todoroki K, Inoue K, Min JZ, Toyo’oka T. Evaluation of a novel positively-charged pyrrolidine-based chiral derivatization reagent for the enantioseparation of carboxylic acids by LC-ESI-MS/MS. Chromatography. 2015;36(2):57–60.
Acknowledgements
This work was supported in part by JSPS KAKENHI grant number 16H04704 (YT, YM, HT).
Authors’ contributions
DJ performed experimental and analytical work and prepared an initial draft of the article. KS and SN assisted method development and analysis. YK, HT, YM, TA, and YT reviewed the manuscript. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Jinno, D., Kanemitsu, Y., Saitoh, K. et al. Rapid and selective simultaneous quantitative analysis of modified nucleosides using multi-column liquid chromatography-tandem mass spectrometry. J Anal Sci Technol 8, 1 (2017). https://doi.org/10.1186/s40543-017-0110-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40543-017-0110-4