
Abstract
Background
A modified version of the QuEChERS (Quick, Easy, Cheap, Effective, Rugged, and Safe) method has been tested for quantifying six active compounds of pharmaceuticals (PhACs), i.e., two different antibiotics, two anti-inflammatories, one antifungal, and one anti-depressant, extracted from roots, leaves and stems, pulp, pits, and oil obtained from olive trees. The different matrices have been polluted with all contaminants at 25, 50, and 250 µg L−1 and the recoveries were determined by liquid chromatography tandem–mass spectrometry. The validation of the method has been carried out by determining linearity, recovery, precision, limits of detection (LODs), and limits of quantification (LOQs) values. A matrix-matched calibration for each matrix has been adopted in order to avoid the matrix effect at the aforementioned levels of fortification.
Results
The recoveries of PhACs from the different matrices were always above 70% and the relative standard deviation (RSD) always ≤ 20%, conditions required for the validation of the method. The LOD and LOQ values were always lower than 25 µg L−1, i.e., always lower than the minimum concentration used in the experiment; therefore, the method can be validated at 25, 50, and 250 µg L−1.
Conclusions
This method can represent a valid alternative to the traditional extraction methods to quantify pharmaceuticals extracted also from fatty matrices.
Graphical Abstract

Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Background
The reuse of treated wastewater is becoming a worldwide agricultural practice for the irrigation of olive trees, especially in semi-arid regions [1]. In the Mediterranean basin, the annual irrigation volume in an olive orchard ranges approx. from 1500 to 3000 m3/ha [2,3,4,5,6]. Wastewater can supplement the irrigation water requirements and can supply nutrients for crops with a reduction of the chemical fertilization, promoting the circular economy by recovering nutrients [7, 8]. Nonetheless, recent studies demonstrated the presence of organic contaminants, such as pharmaceuticals (PhACs) in treated wastewater intended for crops irrigation [9,10,11]. As a consequence, crops irrigated with treated wastewater can uptake and translocate PhACs in their tissues from few μg kg−1 to few mg kg−1 [12] and thus present a potential health risk to humans and livestock which feed on them [13, 14]. The Water Reuse Regulation EU (2020/741) [15] lays down minimum requirements for water quality and monitoring and provisions on risk management, for the safe use of reclaimed water in the context of integrated water management. The risks concerning the release of wastewater contaminants into the environment have garnered significant public concerns in the last years.
Olive oil is a lipid source within the Mediterranean diet, and its consumption brings benefits in terms of decreasing illness and reducing cardiovascular and neurological disorders, and cancers [16]. According to the data of the oil season 2021/2022, world olive oil production was about 3 million tons and its two-thirds have been obtained in Europe [17]. Although the consumption of olive oil for food purposes is the priority, olive leaves can be used in the cosmetic and pharmaceutical fields. Some bioactive molecules present in olive leaves, such as oleuropein and hydroxytyrosol, have strong beneficial properties for human and animal welfare [18]. In particular, El and Karakaya [19] reported that olive leaves exerted antihypertensive, antiatherogenic, anti-inflammatory, hypoglycemic, and hypocholesterolemic effects, in addition to being used as an extract, an herbal tea, and a powder in the human diet. With a view to recycling waste biomass, even olive pits are finding uses. Recently, Galitsopoulou et al. [20] have highlighted the phenolic, antioxidant, nutritional and microbiological properties of olive pits, hypothesizing their future use as a functional food. Another study highlighted the potential of olive pits powder as component of composites [21].
Regarding the extraction and quantification of contaminants in matrices containing high amounts of fats, the main problem is the total removal of fats before the extract passes through the chromatographic system. For this reason, the QuEChERS method that includes the clean-up steps can solve the aforementioned problem, as reported by Cunha et al. [22]. Furthermore, the coupling of the QuEChERS extraction method with the use of LC–MS or GC–MS spectrometers allows having a rapid method of quantifying different PhACs in oil and olive samples, even at very low concentration. García-Reyes et al. [23] compared different types of extraction and quantification of pesticide residues in olives and oil and found that the QuEChERS method was effective.
Abdallat et al. [24], in a study conducted in Jordan, found diclofenac in olives, but not in twigs and leaves, suggesting a high rate of plant uptake, especially during the olive’s growth period. Oueslati et al. [1] tested the pomological characteristics and the olive oil quality after the irrigation with untreated industrial poultry wastewater, but no consideration has been made on the possible fate of organic contaminants in the plant organs and in the oil. Christou et al. [12] reported a low potential of table olives for contaminants of emerging concern uptake. To our knowledge, few studies have been carried out on the fate of PhACs in the olive tree and in the resulting oil. Instead, several studies have been conducted on the extraction and quantification of PhACs in fruits and vegetables [13, 25,26,27,28,29,30]. More studies have instead been conducted on the fate of pesticides in olives and oil. Recently, García-Vara et al. [31] validated a modified QuEChERS method for the extraction of 42 pesticides including organophosphates, phenylureas, anilines, neonicotinoids, and others from olives. Previously, Anagnostopoulos and Miliadis [32] validated a short sample preparation step based on acetonitrile extraction coupled with a gas and liquid chromatography–tandem mass spectrometry to determine the residues of 32 pesticides in olive oil and olives. Gilbert-López et al. [33] also developed a QuEChERS method coupled with liquid chromatography–electrospray tandem mass spectrometry to quantify 104 pesticides in olives.
The objective of this study was to evaluate the QuEChERS extraction approach for the identification and quantification of some active substances, more frequently reported in treated wastewater [34, 35], that can enter and translocate in olive tree with the irrigation.
We believe that this study can help researchers in the critical step of extracting PhACs from complex matrices such as oily ones. The possibility of using this method for the simultaneous extraction of some PhACs from complex matrices represents the innovation of this study. In addition, this method is eco-sustainable, because requires low quantity of solvents and water, and limited spaces with respect to other traditional extraction methods. In this way, useful and rapid indications would be obtained regarding the fate of PhACs in oil and olives, given that studies on this subject are not available in the literature. Finally, we believe that this research can provide a support tool applicable for the regulatory environmental monitoring programs in the olive sector and in all fields dealing with oily matrices.
Materials and methods
Plant material
Plant samples were collected by 3-year-old olive plants (Olea europaea (L.) cv. Ogliarola) grown in open field located in Apulia region (southern Italy). In particular, the following organs were collected: fine roots (1–5 mm in diameter), 1-year vegetative shoots from which mature leaves and stems were sampled, olive fruit from which pulp and pit were separated, and olive oil.
Selected pharmaceuticals
Acetonitrile (ACN), LC–MS grade water, magnesium sulfate anhydrous (MgSO4), sodium citrate (Na citrate), and primary secondary amine (PSA) were purchased from Sigma-Aldrich. The extraction tube (citrate buffer) contained 0.5 g of Na Citrate Dibasic Sesquihydrate, 1 g Na Citrate Tribasic Dihydrate, 1 g NaCl, and 4 g of MgSO4, while the clean-up tube contained 900 mg of MgSO4 and 150 mg of PSA for roots, and 900 mg of MgSO4, 150 mg of PSA, and 150 mg octadecyl silica (C18) for pulp, seeds, oil, leaves, and stems. The QuE-Lab® Tubes used for the extractions were bought from Lab Instruments (Italy). The analytical standards (purity > 99%) of carbamazepine, climbazole, clarithromycin, diclofenac, ketoprofen, and sulfamethoxazole were supplied from Lab Instruments (Italy). Table 1 shows the physico-chemical characteristics of selected PhACs.
Pharmaceuticals stock standard and internal standards solution
Ten mg of the pure standard of each PhAC for pharmaceuticals stock standard solution and the same quantity of isotopically labeled internal standards for internal standards solution were dissolved in 10 mL of ACN (climbazole, ketoprofen) and methanol (MeOH) (carbamazepine, diclofenac, clarithromycin, sulfamethoxazole) to obtain the single standard solutions at 1000 mg L−1. Successively, to obtain the pharmaceuticals stock standard solution at 50 mg L−1, 1 mL of each standard solution was dissolved in 14 mL of the solvent and stored at − 18 ± 3 °C in the dark. Matrix-matched calibration curves were established at three different concentration levels for each analyte (25, 50, and 250 µg L−1) by spiking blank samples of the selected matrices after the extraction process, in order to assess the linearity of the analytical method. The same final concentrations levels of the external standards were used to obtain the external standard calibration curves.
Extraction and quantification of pharmaceuticals
Fresh spiked or unspiked samples of olive roots, leaves and stems, pulp, seeds, or oil were added to 6 mL of MilliQ water into polypropylene centrifuge tubes and shaken vigorously for 1 min by using a Vortex mixer at maximum speed (Vortex Fisher Scientific FB15013 TopMix; Fig. 1). Successively, 10 mL of ACN and the relative internal standard at the middle level of calibration curve were added to the solutions. The use of isotopically labeled internal standards helps compensate for any matrix effect (signal suppression/enhancement) and further improve accuracy and precision [36]. Aliquots of a pharmaceuticals stock standard solution were added to achieve the concentrations of 25, 50, and 250 µg L−1, respectively. The tubes were hand-shaken for 1 min, and then salting-out with citrate buffer was performed. The combination between MgSO4 and NaCl allowed to reduce the amount of matrix components co-extracted and to influence the peak shapes and areas of several contaminants [37]. The MgSO4 during the clean-up process facilitates the partitioning of solvents, improves the recovery of polar analytes, and removes any water from the organic phase [36, 38]. After the addition of the salt, the tubes were immediately manually shaken for 1 min to prevent the formation of MgSO4 conglomerates [36] and centrifuged for 5 min at 3700 rpm. Clean-up step of samples was carried out as reported in Fig. 1. PSA is typically used to remove fatty acids, sugars, organic acids, lipids, and some pigments, while C18 has been used for leaves and stems, pulp, seeds, and oil to remove their higher lipid contents compared to those of roots [39]. The same procedure was applied without the addition of the pharmaceuticals stock standard solution to check the possible presence of selected contaminants in samples. Figure 1 shows the scheme of the QuEChERS method used for the extraction of PhACs from different parts of olive plants and olive oil. The different amounts of sample used for the extraction of PhACs are related to the different water contents in the starting matrices.

Workflow of the optimization of the QuEChERS process. ACN: acetonitrile; PSA: primary secondary amine; MgSO4: magnesium sulfate anhydrous; NaCl: Sodium Chloride; C18: Octadecylsilane
Data acquisition was done in full scan, positive mode, by a Thermo Scientifc™ UltiMate 3000 UHPLC equipped with a degasser, a high-pressure gradient pump, a WPS autosampler, a column oven, and a Q Exactive mass spectrometer. Ten microliter of each sample and of selected syringe standards were injected in Accucore™ aQ C18 Polar Endcapped (2.6 μm; 100 × 2.1 mm) column (Thermo Fisher Scientifc) and maintained at 40 °C. The system conditions are reported in De Mastro et al. [40]. Data acquired were processed by the Thermo Xcalibur 4.0.27.10, Chromeleon, and Trace Finder 3.3 methods, and 5-ppm mass tolerance was used for each extracted ion chromatogram. Table 2 provides UHPLC detection parameters used for the analysis.
Validation of QuEChERS method
The validation of the QuEChERS method was done according to Caldas et al. [41]. Three levels of fortification, i.e., 25, 50, and 250 µg L−1 for each contaminant replicated six times, were used to evaluate the recovery. The calibration curves were obtained by plotting the area ratio (peak area of the analyte divided by the peak area of the internal standard) of each calibration level against its corresponding concentration. Prior to calibration, blank extracts of the studied samples were measured to ensure they did not contain PhACs. Additional to the instrument calibration using the calibration solution, quality control samples were prepared with blank matrix with PhACs and internal standard enrichment at concentration of 50 µg L−1 and were injected every 5 samples during the analyses, confirmed with concentration variation lower than 20% with respect to the theoretical concentration. The coefficient of determination (r2) of each analytical curve has been used to evaluate the linearity of the calibration curve. The precision of the validation method has been evaluated through the relative standard deviation (RSD%). The limit of detection (LOD) and the limit of quantification (LOQ) have been determined according to the ISO 11843-2.
Results and discussion
The quantifiers and qualifiers were selected, considering previous studies and available mass spectrometry databases. Average recoveries of all the tested compounds were determined using six replicates at three concentration levels. The absolute recoveries of PhACs from the different matrices were always above 70% and the relative standard deviation (RSD) always ≤ 20%, conditions that met the validation requirements according to the guidelines of Sante et al. [42] (Table 3).
The average recovery, considering the matrices globally, was 85.34%. In particular, the greatest recovery occurred in the pulp (90.11%), the least in the pit (81.88%), while for oil the average recovery was 82.14% (Table 3, Fig. 2).

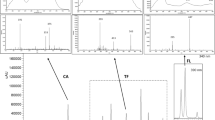
Liquid chromatography–high-resolution mass spectrometry (LC–HRMS) chromatograms of oil samples spiked with pharmaceuticals stock standard solution at concentration of 50 μg L−1
Considering the individual PhAC, the maximum recovery occurred with carbamazepine (94.92%), while the minimum recovery with sulfamethoxazole (76.58%). The differences in the recovery percentages can be presumably ascribed to the different chemical properties of PhACs studied. In fact, SMX is a poorly soluble compound, and therefore, it may have had a lower affinity with the organic solvent and be extracted in smaller quantities than the other PhACs.
Table 4 reports the linearity of the method in roots, leaves and stems, pulp, pits, and oil, respectively, evaluable through the correlation coefficients (r2).
The response of the detector was linear for each PhAC in the range considered with r2 between 0.9986 and 0.9997 for roots, between 0.9965 and 0.9998 for leaves and stems, between 0.9994 and 0.9999 for pulp, between 0.9976 and 0.9996 for pits, and between 0.9995 and 0.9999 for oil. Bragança et al. [43] also reported a good linearity for PhACs isolated from soil in the range 1.5–500 μg kg−1.
The analysis of three blank samples without PhACs has been conducted to assess the selectivity of the method and to ensure they did not contain the studied compounds. The matrix effect has been compensated by a matrix-matched calibration, as reported also in other studies [40, 44,45,46]. The average LOD value ranged from 4.24 to 11.27 μg kg−1 for oil and pits, respectively, while the average LOQ values from 8.49 to 18.05 μg kg−1 for oil and stems and leaves, respectively (Table 4). Since LOD and LOQ values appeared always lower than the minimum concentration used in the experiments, the method can be validated in the range 25–250 μg kg−1. The results of this study confirmed that the QuEChERS method with its clean-up step makes it possible to obtain excellent recoveries by purifying also the fat matrix to the maximum.
The average recovery of each PhAC was decreasing in the order CBZ > CLZ > CLR > DCF > KTP > SMX. This result can be ascribed to a different interaction of the single molecules with the solvent (ACN and water) in the presence of the citrate buffer. Although ACN is considered an excellent solvent for the extraction of many compounds with the least interference [45, 47, 48], an important parameter that influences the extraction percentage of a compound is its pKa [49]. When the pH of the extraction solution is lower than the pKa of the compound, the non-ionized form of a compound prevails, and it is more easily extracted. Since the pH of the extraction solution was 6.8, CLR and CBZ were in their undissociated form and, therefore, showed higher percentages of recovery. Regarding the CLZ, its pKa was similar to the pH of the extraction solution and it was also probably easily extracted. The recoveries of DCF, KTP, and SMZ were directly related to their pKa values, in the sense of lower pKa corresponded to lower recoveries, due to the dissociated forms prevailing over the undissociated ones. Other authors tested the acidification of ACN or water with acetic acid [43, 50] to increase the recoveries of compounds with low pKa. In contrast, to increase the recoveries of compounds with high pKa, Kvicalova et al. [51] proposed the addition of ammonium to ACN.
To our knowledge, no other study has been conducted on the fate of PhACs in oil and olives, probably due to the complexity of these matrices. In fact, fatty matrices can damage the chromatographic system. About this, the clean-up step provided by the QuEChERS method with MgSO4-PSA-C18 can trap fatty acids, without to lose the planar structure of pesticides that remain in ACN [52]. Some studies have been conducted on the extraction of pesticides from these matrices with the QuEChERS method with satisfactory results, confirming the effectiveness of the clean-up step [22, 23, 53]. López-Blanco et al. [54] tested different clean-up sorbents for the analysis of different kind of pesticides in olives and olive oil and found good results in terms of recoveries using PSA + C18 as sorbent material. Finally, previous studies suggested to increase the solvent/sample ratio to improve the recovery of very lipophilic molecules with QuEChERS method for high fat samples [53, 55, 56].
Conclusions
The proposed modified QuEChERS method, as well as being valid for roots, stems, and leaves, can be considered suitable for the extraction of PhACs from fat-rich matrices such as oil and olives. The clean-up step foreseen by the method allows to limit the damages to the chromatographic system deriving from the co-extraction of fats, consuming small amount of organic solvent. The QuEChERS method also allows to extract many samples simultaneously, reducing the time of the analysis and the costs. To our knowledge, no other QuEChERS method has been validated for the quantification of pharmaceuticals extracted from roots, stems, leaves, oil, and olives, and results of this study may be useful for further studies on the fate of these contaminants along the food chain. Additional studies are needed to optimize the method for acid-sensitive or base-sensitive compounds, by correcting the pH of the extractive solution. Finally, this study can be extended to the study of other widely consumed fatty matrices of vegetable origin.
Availability of data and materials
The dataset used and/or analyzed during the current study is available from the corresponding author on reasonable request.
Abbreviations
- QuEChERS:
-
Quick, easy, cheap, effective, rugged, and safe
- LOD:
-
Limit of detection
- LOQ:
-
Limit of quantification
- PhACs:
-
Pharmaceuticals
- GC:
-
Gas chromatography
- LC:
-
Liquid chromatography
- MS:
-
Mass spectrometry
- MS/MS:
-
Tandem mass spectrometry
- HPLC:
-
High-performance liquid chromatography
- ACN:
-
Acetonitrile
- MeOH:
-
Methanol
- PSA:
-
Primary secondary amine
- UHPLC:
-
Ultra high-performance liquid chromatography
- R 2 :
-
Coefficient of determination
- RSD:
-
Relative standard deviation
- LC–HRMS:
-
Liquid chromatography–high-resolution mass spectrometry
References
Oueslati A, Dabbou S, Methneni N, Montevecchi G, Nava V, Rando R, Bartolomeo G, Antonelli A, Di Bella G, Ben Mansour H. Pomological and olive oil quality characteristics evaluation under short time irrigation of olive trees cv. chemlali with untreated industrial poultry wastewater. Sustainability. 2023;15(5):4198. https://doi.org/10.3390/su15054198.
Rodríguez-Sousa AA, Barandica JM, Aguilera PA, Rescia AJ. Examining potential environmental consequences of climate change and other driving forces on the sustainability of spanish olive groves under a socio-ecological approach. Agriculture. 2020;10(11):509. https://doi.org/10.3390/agriculture10110509.
Gucci R, Fereres E, Goldhamer DA. Olive. In: Steduto P, Hsiao TC Fereres E, Raes D, editors. Crop yield response to water. Irrigation and drainage paper, n 66. 2nd edition. Rome: FAO 2012; p. 300–313. ISBN 978-92-5-107274-5.
Gucci R, Caruso G, Gennai C, Esposto S, Urbani S, Servili M. Fruit growth, yield and oil quality changes induced by deficit irrigation at different stages of olive fruit development. Agric Water Manag. 2019;212:88–98. https://doi.org/10.1016/j.agwat.2018.08.022.
Palese AM, Pasquale V, Celano G, Figliuolo G, Masi S, Xiloyannis C. Irrigation of olive groves in Southern Italy with treated municipal wastewater: effects on microbiological quality of soil and fruits. Agric Ecosyst Environ. 2009;129(1–3):43–51. https://doi.org/10.1016/j.agee.2008.07.003.
Lavee S, Hanoch E, Wodner M, Abramowitch H. The effect of predetermined deficit irrigation on the performance of cv. Muhasan olives (Olea europaea L.) in the eastern coastal plain of Israel. Sci Hortic. 2007;112:156–63. https://doi.org/10.1016/j.scienta.2006.12.017.
Aghtape AA, Ghanbari A, Sirousmehr A, Siahsar B. Effect of irrigation with wastewater and foliar fertilizer application on some forage characteristics of foxtail millet (Setaria italica). Plant Physiol Biochem. 2011;3:34–42.
Sioud O, Beltifa A, Ayeb N, Mansour HB. Characterization of industrial dairy wastewater and contribution to reuse in cereals culture: Study of phytotoxic effect. Austin J Environ Toxicol. 2016;2:1013.
Frascaroli G, Reid D, Hunter C, Roberts J, Helwig K, Spencer J, Escudero A. Pharmaceuticals in wastewater treatment plants: a systematic review on the substances of greatest concern responsible for the development of antimicrobial resistance. Appl Sci. 2021;11:6670. https://doi.org/10.3390/app11156670.
Gorovits R, Sobol I, Akama K, Chefetz B, Czosnek H. Pharmaceuticals in treated wastewater induce a stress response in tomato plants. Sci Rep. 2020;10:1856. https://doi.org/10.1038/s41598-020-58776-z.
Semerjian L, Shanableh A, Semreen MH, Samarai M. Human health risk assessment of pharmaceuticals in treated wastewater reused for non-potable applications in Sharjah, United Arab Emirates. Environ Int. 2018;121:325–31. https://doi.org/10.1016/j.envint.2018.08.048.
Christou A, Papadavid G, Dalias P, Fotopoulos V, Michael C, Bayona JM, Piña B, Fatta-Kassinos D. Ranking of crop plants according to their potential to uptake and accumulate contaminants of emerging concern. Environ Res. 2019;170:422–32. https://doi.org/10.1016/j.envres.2018.12.048.
Christou A, Karaolia P, Hapeshi E, Michael C, Fatta-Kassinos D. Long-term wastewater irrigation of vegetables in real agricultural systems: concentration of pharmaceuticals in soil, uptake and bioaccumulation in tomato fruits and human health risk assessment. Water Res. 2017;109:24–34. https://doi.org/10.1016/j.watres.2016.11.033.
Helmecke M, Fries E, Schulte C. Regulating water reuse for agricultural irrigation: risks related to organic micro-contaminants. Environ Sci Eur. 2020;32:1–4. https://doi.org/10.1186/s12302-019-0283-0.
Regulation (EU) 2020/741 of the European Parliament and of the Council of 25 May 2020 on minimum requirements for water reuse (Text with EEA relevance).
Rodriguez-Lopez P, Lozano-Sanchez J, Borras-Linares I, Emanuelli T, Menendez JA, Segura-Carretero A. Structure-biological activity relationships of extra-virgin olive oil phenolic compounds: health properties and bioavailability. Antioxidants. 2020;9(8):685. https://doi.org/10.3390/antiox9080685.
International Olive Oil Council (IOC)—The world of Olive Oil (2022). https://www.internationaloliveoil.org/the-world-of-olive-oil/?lang=it. Accessed 12th June 2022.
Di Meo CM, Izzo F, Rocco M, Zarrelli A, Mercurio M, Varricchio E. Mid-infrared spectroscopic characterization: new insightson bioactive molecules of Olea europea L. leaves from selected italian cultivars. Infrared Phys Technol. 2022;127:104439. https://doi.org/10.1016/j.infrared.2022.104439.
El SN, Karakaya S. Olive tree (Olea europaea) leaves: potential beneficial effects on human health. Nutr Rev. 2009;67:632–8. https://doi.org/10.1111/j.1753-4887.2009.00248.x.
Galitsopoulou A, Salepi C, Karagianni F. Transforming olive pits into functional foods: evaluation of phenolic, antioxidant, nutritional and microbiological properties. Funct Foods Health Dis. 2022;12(11):614–26. https://doi.org/10.31989/ffhd.v12i10.1011.
Koutsomitopoulou AF, Bénézet JC, Bergeret A, Papanicolaou GC. Preparation and characterization of olive pit powder as a filler to PLA-matrix bio-composites. Powder Technol. 2014;255:10–6. https://doi.org/10.1016/j.powtec.2013.10.047.
Cunha SC, Lehotay SJ, Mastovska K, Fernandes JO, Beatriz M, Oliveira PP. Evaluation of the QuECHERS sample preparation approach for the analysis of pesticide residues in olives. J Sep Sci. 2007;30:620–32. https://doi.org/10.1002/jssc.200600410.
García-Reyes JF, Ferrer C, Gómez-Ramos MJ, Fernández-Alba AR, García-Reyes JF, Molina-Díaz A. Determination of pesticide residues in olive oil and olives. Trends Anal Chem. 2007;26:239–51. https://doi.org/10.1016/j.trac.2007.01.004.
Abdallat GA, Salameh E, Shteiwi M, Bardaweel S. Pharmaceuticals as emerging pollutants in the reclaimed wastewater used in irrigation and their effects on plants, soils, and groundwater. Water. 2022;14:1560. https://doi.org/10.3390/w14101560.
Mordechay EB, Mordehay V, Tarchitzky J, Chefetz B. Pharmaceuticals in edible crops irrigated with reclaimed wastewater: Evidence from a large survey in Israel. J Hazard Mater. 2021;416:126184. https://doi.org/10.1016/j.jhazmat.2021.126184.
Wu X, Dodgen LK, Conkle JL, Gan J. Plant uptake of pharmaceutical and personal care products from recycled water and biosolids: a review. Sci Total Environ. 2015;536:655–66. https://doi.org/10.1016/j.scitotenv.2015.07.129.
Malchi T, Maor Y, Tadmor G, Shenker M, Chefetz B. Irrigation of root vegetables with treated wastewater: evaluating uptake of pharmaceuticals and the associated human health risks. Environ Sci Technol. 2014;48:9325–33. https://doi.org/10.1021/es5017894.
Riemenschneider C, Al-Raggad M, Moeder M, Seiwert B, Salameh E, Reemtsma T. Pharmaceuticals, their metabolites, and other polar pollutants in field-grown vegetables irrigated with treated municipal wastewater. J Agric Food Chem. 2016;64:5784–92. https://doi.org/10.1021/acs.jafc.6b01696.
Margenat A, Matamoros V, Díez S, Cañameras N, Comas J, Bayona JM. Occurrence and human health implications of chemical contaminants in vegetables grown in peri-urban agriculture. Environ Int. 2019;124:49–57. https://doi.org/10.1016/j.envint.2018.12.013.
de Santiago-Martín A, Meffe R, Teijón G, Martínez Hernández V, López-Heras I, Alonso Alonso C, Arenas Romasanta M, de Bustamante I. Pharmaceuticals and trace metals in the surface water used for crop irrigation: Risk to health or natural attenuation? Sci Total Environ. 2020;705:135825. https://doi.org/10.1016/j.scitotenv.2019.135825.
García-Vara M, Postigo C, Palma P, Bleda MJ, LópezdeAlda M. QuEChERS-based analytical methods developed for LC–MS/MS multiresidue determination of pesticides in representative crop fatty matrices: olives and sunflower seeds. Food Chem. 2022;386:132558. https://doi.org/10.1016/j.foodchem.2022.132558.
Anagnostopoulos C, Miliadis GE. Development and validation of an easy multiresidue method for the determination of multiclass pesticide residues using GC–MS/MS and LC–MS/MS in olive oil and olives. Talanta. 2013;112:1–10. https://doi.org/10.1016/j.talanta.2013.03.051.
Gilbert-López B, García-Reyes JF, Lozano A, Fernández-Alba AR, Molina-Díaz A. Large-scale pesticide testing in olives by liquid chromatography–electrospray tandem mass spectrometry using two sample preparation methods based on matrix solid-phase dispersion and QuEChERS. J Chromatogr A. 2010;1217:6022–35. https://doi.org/10.1016/j.chroma.2010.07.062.
Adeleye AS, Xue J, Zhao Y, Taylor AA, Zenobio JE, Sun Y, Han Z, Salawu OA, Zhu Y. Abundance, fate, and effects of pharmaceuticals and personal care products in aquatic environments. J Hazard Mater. 2022;424:127284. https://doi.org/10.1016/j.jhazmat.2021.127284.
Rodriguez-Mozaz S, Vaz-Moreira I, Varela Dell Giustina S, Llorca M, Barcelo D, Schubert S, Berendonk TU, MichaelKordatou I, Fatta-Kassinos D, Martinez JL, et al. Antibiotic residues in final effluents of European wastewater treatment plants and their impact on the aquatic environment. Environ Int. 2020;140:105733. https://doi.org/10.1016/j.envint.2020.105733.
Kumari A, Bhattacharya B, Agarwal T, Paul V, Kumar Maurya V, Chakkaravarthi S, Simal-Gandara J. Method development and validation for acrylamide in potato cutlet by UHPLC-MS/MS. Food Control. 2023;151:109817. https://doi.org/10.1016/j.foodcont.2023.109817.
Gonzalez-Curbelo MA, Socas-Rodriguez B, Herrera-Herrera AV, Gonzalez-Salamo J, Hernandez-Borges J, Rodriguez-Delgado MA. Evolution and applications of the QuEChERS method. Trends Anal Chem. 2015;71:169–85. https://doi.org/10.1016/j.trac.2015.04.012.
Bonerba E, Ceci E, Montemurro N, Tantillo G, Di Pinto A, Celano GV, Bozzo G. Rapid modified QuEChERS method for pesticides detection in honey by high-performance liquid chromatography UV-visible. Ital J Food Saf. 2014;3(2):1647. https://doi.org/10.4081/ijfs.2014.
Perestrelo R, Silva P, Porto-Figueira P, Pereira JAM, Silva C, Medina S, Câmara JS. QuEChERS—fundamentals, relevant improvements, applications and future trends. Anal Chim Acta. 2019;1070:1–28. https://doi.org/10.1016/j.aca.2019.02.036.
De Mastro F, Cocozza C, Traversa A, Cacace C, Mottola F, Mezzina A, Brunetti G. Validation of a modified QuEChERS method for the extraction of multiple classes of pharmaceuticals from soils. Chem Biol Technol Agric. 2022;9:49. https://doi.org/10.1186/s40538-022-00305-3.
Caldas SS, Bolzan CM, Cerqueira MB, Tomasini D, Furlong EB, Fagundes C, Primel EG. Evaluation of a modified QuEChERS extraction of multiple classes of pesticides from a rice paddy soil by LC-APCI-MS/MS. J Agric Food Chem. 2011;59:11918–26. https://doi.org/10.1021/jf202878s.
Guidance SANTE 11312/2021—Analytical quality control and method validation procedures for pesticide residues analysis in food and feed. European Commission Directorate-General for Health and Food Safety. 2021. pp. 1–46.
Bragança I, Plácido A, Paíga P, Domingues VF, Delerue-Matos C. QuEChERS: a new sample preparation approach for the determination of ibuprofen and its metabolites in soils. Sci Total Environ. 2012;433:281–9. https://doi.org/10.1016/j.scitotenv.2012.06.035.
Acosta-Dacal A, Rial-Berriel C, Diaz-Diaz R, del Mar B-S, Luzardo OP. Optimization and validation of a QuEChERS-based method for the simultaneous environmental monitoring of 218 pesticide residues in clay loam soil. Sci Tot Environ. 2021;753:142015. https://doi.org/10.1016/j.scitotenv.2020.142015.
Correia-Sa L, Fernandes VC, Carvalho M, Calhau C, Domingues VF, Delerue-Matos C. Optimization of QuEChERS method for the analysis of organochlorine pesticides in soils with diverse organic matter. J Sep Sci. 2012;35:1521–30. https://doi.org/10.1002/jssc.201200087.
Rashid A, Nawaz S, Barker H, Ahmad I, Ashraf M. Development of a simple extraction and clean-up procedure for determination of organochlorine pesticides in soil using gas chromatography–tandem mass spectrometry. J Chromatogr A. 2010;1217:2933–9. https://doi.org/10.1016/j.chroma.2010.02.060.
Fernandes VC, Domingues VF, Mateus N, Delerue-Matos C. Organochlorine pesticide residues in strawberries from integrated pest management and organic farming. J Agric Food Chem. 2011;59:7582–91. https://doi.org/10.1021/jf103899r.
Ramalhosa MJ, Paiga P, Morais S, Delerue-Matos C, Prior Pinto Oliveira MB. Analysis of polycyclic aromatic hydrocarbons in fish: evaluation of a quick, easy, cheap, effective, rugged, and safe extraction method. J Sep Sci. 2009;32:3529–38. https://doi.org/10.1002/jssc.200900351.
Vera J, Correia-Sa L, Paiga P, Braganca I, Fernandes VC, Domingues VF, Delerue-Matos C. QuEChERS and soil analysis. An overview. Sample Prep. 2013. https://doi.org/10.2478/sampre-2013-0006.
Wang YH, Du LW, Zhou XM, Tan HH, Bai LY, Zeng DQ, Tian H. QuEChERS extraction for high performance liquid chromatographic determination of pyrazosulfuron-ethyl in soils. J Chem Soc Pak. 2012;34:28–32.
Kvicalova M, Doubravova P, Jobanek R, Jokesova M, Ocenaskova V, Sussenbekova H, Svobodova A. Application of different extraction methods for the determination of selected pesticide residues in sediments. Bull Environ Contam Toxicol. 2012;89:21–6. https://doi.org/10.1007/s00128-012-0622-y.
Wong J, Zhang K, Tech K, Hayward DG, Makovi CM, Krynistky AJ, Schenck F, Banerjee KK, Dasgupta S, Brown D. Multiresidue pesticide analysis in fresh produce by capillary gas chromatography-mass spectrometry/selective ion monitoring (gc-ms/sim) and -tandem mass spectrometry (gc-ms/ms). J Agric Food Chem. 2010;58:5868–83.
Chamkasema N, Harmon T. Analysis of pesticides in olive oil using a modified QuEChERS method with LC-MS/MS and GC-MS/MS. J Reg Sci. 2015;01:16–35.
López-Blanco R, Nortes-Méndez R, Robles-Molina J, Moreno-González D, Gilbert-López B, García-Reyes JF, Molina-Díaz A. Evaluation of different cleanup sorbents for multiresidue pesticide analysis in fatty vegetable matrices by liquid chromatography tandem mass spectrometry. J Chromatogr A. 2016;22(1456):89–104. https://doi.org/10.1016/j.chroma.2016.06.019.
Lehotay S, Mastovska K, Yun S. Evaluation of two fast and easy methods for pesticide residue analysis in fatty food matrixes. J AOAC Int. 2005;88:630–8.
Koesukwiwat U, Lehotay SJ, MaÅtovsk K, Dorweiler KJ, Leepipatpiboon NN. Extension of the quechers method for pesticide residues in cereals to flaxseeds, peanuts, and doughs. J Agric Food Chem. 2010;58:5950–8. https://doi.org/10.1021/jf902988b.
Acknowledgements
Not applicable.
Funding
This research was funded by MINISTERO DELL’ISTRUZIONE DELL’UNIVERSITÀ E DELLA RICERCA, Italy, and Grant number 2017C5CLFB.
Author information
Authors and Affiliations
Contributions
GB, CC, FDM, AT, BD, and ANM contributed to conceptualization; GB, CC, FDM, AT, and FM contributed to methodology; FDM, PN, and FM did formal analysis; GB, CC, FDM, FM, AT, BD, and ANM performed investigation; GB provided resources; FDM, AT, PN, ANM, and FM were responsible for software; FDM, AT, and FM performed data curation; FDM, AT, and ANM were involved in writing-original draft preparation; GB, CC, and BD were involved in writing-review and editing; GB, FDM, and FM did validation; GB, CC, FDM, FM, AT, BD, and ANM contributed to visualization; GB and BD were involved in funding acquisition; GB, CC, and BD did supervision. All the authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This manuscript is an original research and has not been published or submitted in other journals.
Consent for publication
All the authors agreed to publish in this journal.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Brunetti, G., Traversa, A., De Mastro, F. et al. Evaluation of the QuEChERS extraction approach for the analysis of active compounds of pharmaceuticals in olive tree portions. Chem. Biol. Technol. Agric. 10, 80 (2023). https://doi.org/10.1186/s40538-023-00454-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40538-023-00454-z