
Abstract
This article reports on the discovery of pararealgar and semi-amorphous pararealgar in Rembrandt's masterpiece The Night Watch. A large-scale research project named Operation Night Watch was started in 2019. A variety of non-invasive analytical imaging techniques, together with paint sample research, has provided new information about Rembrandt's pigments, materials, and techniques as well as the current condition of the painting. Macroscopic X-ray fluorescence, macroscopic X-ray powder diffraction and reflectance imaging spectroscopy identified the presence of arsenic sulfide pigments and degradation products of these pigments in the doublet sleeves and embroidered buff coat worn by Lieutenant Willem van Ruytenburch (central figure to the right of Captain Frans Banninck Cocq). Examination by light microscopy of two paint samples taken from this area shows a mixture of large sharp-edged tabular yellow and orange to red pigment particles, and scanning electron microscopy-energy dispersive X-ray analysis identified these particles as containing arsenic and sulfur. Using micro-Raman spectroscopy, the yellow particles were identified as pararealgar, and the orange to red particles as semi-amorphous pararealgar. Synchrotron-based X-ray diffraction allowed visualization of the presence of multiple degradation products associated with arsenic sulfides throughout the paint layer. The discovery of pararealgar and semi-amorphous pararealgar is a new addition to Rembrandt's pigment palette. To contextualize our findings and to hypothesize why, how, and where Rembrandt obtained the pigments, we studied related historical sources. A comprehensive review of historical sources gives insight into the types of artificial arsenic sulfides that were available and suggests that a broader range of arsenic pigments could have been available in Amsterdam in the seventeenth century than previously thought. This is supported by the use of a very similar mixture of pigments by Willem Kalf (1619–1693), a contemporary artist based in Amsterdam. Together with the condition of the particles in the paint cross sections, this brings us to the conclusion that Rembrandt intentionally used pararealgar and semi-amorphous pararealgar, together with lead–tin yellow and vermilion, to create an orange paint.
Similar content being viewed by others


Introduction
Throughout history, artists made use of both naturally occurring orpiment and realgar and artificially produced arsenic sulfide pigments to produce paint with yellow, orange or red hues. The naturally occurring minerals realgar (orange to red, As4S4) and orpiment (golden yellow, As2S3), are until now the most prevalently mentioned arsenic sulfide pigments in artworks and have been used by artists since antiquity. Although artificial arsenic sulfides are known to have been produced synthetically [1], they only rarely have been reported in artworks. By using a combination of analytical techniques such as Raman spectroscopy and X-ray powder diffraction, several types of artificial arsenic sulfides have been identified and differentiated in cultural heritage objects [2,3,4,5,6,7]. Arsenic sulfide pigments are not traditionally associated with Rembrandt van Rijn’s palette. Up to now, arsenic sulfide pigments have only been identified in two paintings from his late period, The Jewish Bride (c. 1665, Rijksmuseum) and The Man in a Red Cap (c. 1665, Museum Boijmans van Beuningen) [1, 6]. In both cases artificial orpiment was found. The arsenic sulfide pigments found in The Night Watch are, however, different. In this paper, the identification and origin of the arsenic sulfide pigments in The Night Watch will be discussed. This research is carried out in the context of Operation Night Watch, a large-scale multimodal investigation of the painting, which started in July 2019.
Within arsenic sulfides three classes can be distinguished: 1. the naturally occurring minerals, 2. artificial arsenic sulfides and 3. secondary arsenic-bearing phases. Orpiment and realgar are arsenic sulfide minerals encountered in nature (class 1). This class also includes arsenopyrite (FeAsS), duranusite (As4S), dimorphite (As4S3), bonazziite (As4S4), uzonite (As4S5), alacranite (As8S9) and wakabayashilite ((As,Sb)6As4S14). The artificial arsenic sulfides (class 2) are the man-made arsenic compounds such as artificial orpiment or arsenic sulfide glass. The secondary phases (class 3) are degradation products formed from the naturally occurring minerals or the artificial arsenic sulfides. This third class includes pararealgar (As4S4), arsenolite (As2O3, cubic), claudetite (As2O3, monoclinic) and other polymorphs such as the so-called χ-phase (As4S4) [2].
The mineral orpiment, also called ‘auripigmentum’ or ‘gold pigment’, was celebrated for its bright warm yellow tone and was recommended in several artist’s manuals for use as a pigment to imitate gold [8]. The pigment particles of natural orpiment are usually coarse and exhibit a typical foliated (layered) structure [9]. Orpiment was not only used as a pigment by painters but its use was extended in day to day practices from medicinal purposes [10, 11], hair removal creams and oils [12,13,14,15,16], wax seals [17], yellow ink [18], bookbinder green (where it is mixed with indigo [17]), as well as the treatment or coating of various metals (such as silver). Even more applications can be found in literature [10, 18,19,20,21,22]. Realgar is described in many different historical sources, dating back as early as the sixteenth to eleventh century B.C. on Egyptian offering lists [23]. Its use as a pigment for oil paintings was less common.
Artificially synthetized arsenic sulfides (class 2) have been mentioned in several artists’ treatises as early as the fifteenth century [24,25,26,27] and are listed in price lists across Europe under various terms such as ‘king's yellow’, ‘king's red’, ‘arsenico citrino’, ‘risagallo’ and ‘rauschgelb’ [8, 28,29,30,31,32,33]. The manufacture of artificial arsenic sulfides is described in German, Italian, French, Dutch, and Spanish sources from the fifteenth century onward including metallurgical, alchemical, and medicinal literature [8, 11, 13, 24, 25, 27, 34,35,36,37,38,39,40,41,42,43,44,45,46,47,48,49,50,51,52]. Based on the literature, a continuous development can be noted in the production processes of these pigments. This is undoubtedly due to the increased knowledge, invention, and improvements of specialized chemical equipment (such as furnaces, retorts and sublimation vessels), as well as of chemical procedures to improve the refining of ores and the extraction of metals by early alchemists and metallurgists.
Before the fifteenth century, primarily roasting and melting procedures of the natural ores (orpiment, realgar) are described as ways to obtain artificial arsenic sulfides [25,26,27]. From the sixteenth century onwards, various sublimation processes for the natural ores are introduced, specifically to obtain so called ‘arsenic flowers’, which consisted of pure arsenic or arsenic (tri)oxide [15, 44, 53]. Additionally, the introduction of arsenic or sulfur during the heating of natural ores is reported [39, 54, 55]. From the end of the seventeenth century and the beginning of the eighteenth century, various sources describe the fusion of these ‘arsenic flowers’, obtained from the sublimation of various arsenic-containing ores (e.g. cobaltite, orpiment, realgar, löllingite, or arsenopyrite) with solid sulfur [50, 52, 56,57,58,59]. This was done in various ratios: an As:S ratio of 10:1 to obtain a yellow product and a ratio of 5:1 to obtain an orange to red product [22, 51, 52, 60,61,62]. The above-mentioned methods are considered as the dry process methods. A variety of (end)products can be obtained from the dry process method depending on multiple factors of the production process, choice of starting materials, and post processing treatments. The chemical characterization of the possible end-products is complex and largely unexplored. Grundmann and Rötter describe in detail the classification of various artificially obtained arsenic sulfides specifically with orpiment as starting material. From the nineteenth century, also a wet process method was introduced which encompassed the precipitation of materials in solution [63,64,65].
The last class (class 3) comprises the secondary phases. Both arsenic-bearing minerals and synthetic arsenic sulfide pigments are prone to degradation. Orpiment degradation can occur upon exposure to light, which causes the formation of arsenic trioxide [66]. If orpiment is present in a medium, As(V) species are formed and can migrate throughout the medium [67,68,69]. Realgar on the other side, is known to degrade to pararealgar within days upon exposure to normal light levels [70]. Pararealgar can degrade further to arsenic trioxide. During the degradation also the metastable χ-phase is formed [71]. The As-containing components found in artworks are (therefore) often complex mixtures of several compounds and difficult to trace back to the original recipe.
In general, when arsenic sulfides pigments are present in oil paintings, these are often simply described as “orpiment and/or realgar”, even though, as described above, many other arsenic sulfide pigments were available from the fifteenth century onwards. Typically, the identification of arsenic sulfides is often based on visual appearance, light microscopy, and elemental analysis. With these methods, it is not always possible to distinguish arsenic-bearing minerals from synthetic arsenic sulfides and secondary phases. As for the latter, the degradation of realgar into pararealgar was not described in detail until 1996. Before this date, the historical sources mention that light exposed realgar forms orpiment, which led to misinterpretation and up until almost 30 years ago, the friable yellow pararealgar generally was identified as orpiment [71, 72].
It is important to realize that to differentiate between the three classes of arsenic compounds, additional analytical methods of high specificity such as Raman spectroscopy and X-ray powder diffraction are essential.
In this article, two types of arsenic sulfide pigments discovered in Rembrandt's oeuvre are presented: (regular) pararealgar, which is yellow, and a semi-amorphous variant that is orange to red. The historic use, complexity of identification and interpretation of arsenic sulfides is studied in relevant historical sources to explain their use by Rembrandt.
The presence of arsenic sulfides in The Night Watch was first detected by non-invasive imaging when the entire surface of the painting was scanned with macroscopic X-ray fluorescence imaging spectroscopy (MA-XRF) (Fig. 1c). Subsequent examination of the paint surface with stereomicroscopy revealed small areas of bright orange paint (Fig. 1d) in several areas at the surface and in the underpainting of the embroidery of Willem van Ruytenburch’s buff coat (the figure dressed in yellow in the middle of the painting) which correlate with highlights in the arsenic MA-XRF map (Fig. 1c). Two paint cross sections were taken (see Fig. 1b for the sample locations) to characterize in more detail the type of arsenic sulfide present and to investigate the presence of possible chemical degradation products related to arsenic sulfides. Below, the results of light microscopy, scanning electron microscopy combined with X-ray elemental analysis (SEM–EDX), micro-Raman spectroscopy and synchrotron-based X-ray diffraction (SR-XRD) performed on these two paint cross sections are described, as well as the production processes and origin of the arsenic sulfides found. Micro-Raman spectroscopy is a very suitable method to distinguish between different arsenic sulfide pigments [72, 73]. Furthermore, the availability of artificial arsenic sulfides in Amsterdam at Rembrandt’s time was investigated by studying historical sources and by reexamination of a paint cross section from a still life painting by Willem Kalf (1619–1693) (Figure S1-2). Kalf is a contemporary of Rembrandt and also worked in Amsterdam [74].

a Rembrandt van Rijn (1606–1669), The Night Watch, 1642, oil on canvas, h 379.5 cm x w 453.5 cm, Rijksmuseum (inv. nr SK-C-5). b detail showing Willem van Ruytenburch’s embroidered buff coat and doublet sleeves with the locations of samples SK-C-5_016 and SK-C-5_017. c MA-XRF distribution map for arsenic (As-K) of a detail in the buff coat indicated with a white rectangle in (b). d stereomicroscope image of area marked by the red rectangle with arsenic hotspot indicated with a red arrow in c corresponding to bright orange paint in the stereomicroscope image (red arrow)
Results
Microscale analysis
After the discovery of arsenic-bearing paint with MA-XRF (Fig. 1C), based on the detection of arsenic independent of cobalt and nickel (indicative of the presence of the pigment smalt), further identification of the exact nature of the arsenic-containing pigments used in these areas was required. The presence of sulfur, co-localized with arsenic could not be established with MA-XRF, due to the low sensitivity of ambient MA-XRF to measure low energy X-rays emitted from low Z elements such as sulfur. Additionally, the S-K line emission lines are very close to Pb-M lines, which can result in the weak S lines to be masked by the strong Pb lines. The microscale study was done on two paint cross sections from the arsenic sulfide-rich areas. The first sample (SK-C-5_016) was taken in the proper right sleeve of Van Ruytenburch’s costume in a warm brown stripe (see Fig. 2a). During the sampling, we noticed that the paint was very brittle and crumbly, more so than in other areas of the painting. The distribution map of arsenic in this area shows that arsenic is mainly present in the warm brown stripes (Fig. 2b). The second sample (SK-C-5_017) was taken in the embroidery area of the coat, from a bright orange paint (see Fig. 2c, d). The orange paint is present in underlying layers and used as highlights in the shadows for the build-up of the gold embroidery. In some areas this layer is now exposed due to loss of the covering paint layers, and the orange paint of the underlayer can be seen by eye (see Fig. 1d).

a Detail of the 5 µm resolution photograph of the sleeve of Van Ruytenburch’s costume. The blue ‘X’ indicates the location of sample SK-C-5_016 [75]. b As-K MA-XRF distribution map of the detail in a. c Detail of the 5 µm resolution photograph of the embroidered edge of Van Ruytenburch’s buff coat. The red ‘X’ indicates the sample location of SK-C-5_017 [75]. d As-K MA-XRF distribution map of the detail in c. Stereoscopic micrographs of the sample locations for SK-C-5_016 (e) and SK-C-5_017 (f) indicated respectively with a blue and red arrow. g Light microscope image of sample SK-C-5_016 in dark field (DF) mode. h Light microscope image of sample SK-C-5_017 in DF mode. i Light microscope image of sample SK-C-5_016 under UV (365 nm) light. j Light microscope image of sample SK-C-5_017 under UV (365 nm) light. k SEM backscattered-electron (BSE) image of sample SK-C-5_016. l SEM-BSE image of sample SK-C-5_017. White arrow indicates arsenic trioxide formation
Figure 2g and i show the light microscopy images of sample SK-C-5_016 under, respectively, dark field (DF) settings and under UV (365 nm) illumination. This sample was taken in Van Ruytenburch’s sleeve (Fig. 2a, e). The images show a yellow translucent degraded paint layer with only few distinct pigment particles. This translucent layer does not only consist of (low Z) organic material, as can be deduced from the brighter grey in this area in the SEM backscattered-electron (BSE) image. The grey value is brighter than the organic embedding medium (top and bottom) and other low Z materials in the sample. The brighter grey contrast is a consequence of the presence of arsenic and lead in this translucent layer, confirming that the paint is largely degraded. The presence of both arsenic and lead were confirmed by SEM–EDX (see Figure S5d,e). This translucent layer looks amorphous and rather homogenous. One large yellow As-containing particle can be found in the bottom part of the sample (see Figure S5e). Furthermore, we find a few large particles of lead–tin yellow and lead white, some carbon-based black and organic brown pigment (possibly Cassel earth), some tiny iron containing particles (possibly earth pigment) and several red lake particles.
The images of sample SK-C-5_017 in Fig. 2h and j show a dense layer of yellow and orange to red particles, and an occasional tiny bright red particle (probably vermilion). Additionally, some lead–tin yellow and gypsum are present in this layer. Especially in the light microscopy image under UV light (Fig. 2j) the large yellow and orange to red particles can be clearly distinguished from other paint components such as the binding medium. The particles have different sizes (up to 30 µm in diameter) and shapes, but the edges are always rather angular, reminiscent of glass-like pigments such as smalt [76]. This is especially visible when the cross section is studied with SEM (see BSE-SEM images of sample SK-C-5_017 in Fig. 2l). SEM–EDX measurements confirmed the presence of arsenic and sulfur in these particles. The bulky, sharp-edged particles have a very different morphology than the arsenic sulfide particles identified in two other paintings by Rembrandt that contain arsenic sulfides. In both The Jewish Bride and The Man in a Red Cap, small (2 to 5 µm in diameter) perfectly spherical yellow amorphous arsenic sulfide particles were identified by using a combination of light microscopy, SEM–EDX, XRD and Raman spectroscopy [4, 6]. This type of amorphous arsenic sulfide was identified as purified artificial orpiment glass, belonging to class 2 [63].
Raman spectroscopy was used to determine the exact arsenic sulfide species in the samples taken from The Night Watch. Measurements were performed on several large and small particles in cross Sections 016 and 017. In Fig. 3a, four measurement locations (A-D) in sample 017 are shown. The Raman spectra collected on the yellow particles in sample 017 all show similar characteristics. Two of these Raman spectra (A-B) are shown in Fig. 3c. The spectra include all Raman bands of pararealgar (As4S4). Especially the pattern of four distinct peaks between 300 and 400 cm−1 and the two strong peaks around 230 cm−1 are very characteristic for pararealgar [72]. The peak around 275 cm−1 is also a good indicator of pararealgar, as it does not overlap with any peaks related to orpiment or realgar. Figure S3 shows several Raman spectra taken in the large yellow particle of sample 016, which can also be identified as pararealgar.

a Light microscope image in (DF) mode of sample SK-C-5_017 with the Raman measurements locations from A-D. b Light microscope image in (DF) mode of sample SK-A-199_R9/4 taken from the painting Still Life with a Silver Jug and a Porcelain Bowl by Willem Kalf with the Raman measurements locations from E–H. c Raman spectra taken in locations A-D of sample SK-C-5_017, with a reference spectrum of pararealgar (RRUFF database ID: R150123). d Raman spectra taken in locations E–H of sample SK-A-199_R9/4, with a reference spectrum of pararealgar (RRUFF database ID: R150123)
The Raman spectra of the orange to red particles (two representative spectra, collected in measurement locations C and D in Fig. 3a, are shown in Fig. 3c) are also very similar to one another. In comparison to the yellow particles, the Raman spectral range between 300 and 400 cm−1 shows much less defined peaks. The peaks around 175 and 203 are less intense in the red particles. Additionally, when looking at the ratio between the peak at 345 cm−1 and the peak at 275 cm−1, the peak at 275 cm−1 is relatively less intense in these particles. The broadening of the peaks between 300 and 400 cm−1 resembles an effect that can also be observed in the Raman spectra of amorphous arsenic sulfides [68]. The broadening of Raman bands is an indication of a more amorphous material compared to the crystalline phase and could have been caused by heating or subliming of pararealgar to obtain a more reddish hue [1, 27, 39, 41]. Because the typical peaks of pararealgar are still present and the broadening of the bands is not as severe as seen in total amorphous arsenic sulfides [65], we will refer to the orange to red particles as “semi-amorphous” pararealgar in the rest of the text.
In sample 017, the yellow (crystalline pararealgar) and orange to red particles (semi-amorphous pararealgar) are homogeneously distributed within the paint layer and no color change or gradient of color is observed within the particles. The edges of these particles are sharp and there is no disruption of the paint-pigment matrix observed both by SEM and light microscopy. In the top left (Fig. 2l, white arrow), an in-situ formed degradation product can be observed. In this particle, SEM–EDX confirmed the presence of mainly arsenic and oxygen, suggesting the presence of arsenic trioxide, a degradation product formed within the paint layer. The area around the particle appears disrupted and has more vacant space compared to the remainder of the paint layer. If the formation of pararealgar resulted from in-situ light-induced degradation, we would expect a similar effect around the yellow and orange to red particles. We would then also expect that the yellow particles (consisting of more crystalline pararealgar) would be located near the top of the paint sample and the orange to red particles (semi-amorphous pararealgar) near the bottom, where there was less exposure to light, as is the case with the formation of arsenolite from the arsenic sulfides in this study and from orpiment in a previous study [69]. However, this is not the case here. This suggests that in The Night Watch, both the pararealgar (yellow) and the semi-amorphous pararealgar (orange to red) were originally used by the artist and that neither is the result of in situ chemical alteration.
Sample from a painting by a contemporary 17th-century artist
To further explore the possibility of the use of pararealgar as a pigment, paint samples from different artists known to have used arsenic sulfides within the Rijksmuseum collection were examined with Raman spectroscopy. In one case, namely a cross section from Still Life with a Silver Jug and a Porcelain Bowl by Willem Kalf (1655 – 1660, Rijksmuseum, Amsterdam) a very similar paint mixture in comparison to the mixture in The Night Watch was found. Figure 3b shows the light microscope image of a paint cross section taken in the orange (fruit) in the decorative plate (see Supporting Information for a photo of the painting (Figure S1) and the UV light micrograph of the sample (Figure S2)). The upper paint layer contains a mixture of sharp-edged yellow and orange to red particles. SEM–EDX measurements indicate the presence of arsenic and sulfur in these particles. Two representative Raman spectra from the more yellow particles are shown in Fig. 3d, E–F. Similar to the yellow particles in The Night Watch, these particles can be identified as pararealgar. For the orange to red particles again we observe a similar Raman spectrum (two representative spectra shown in Fig. 3d, G-H), but with a broadening of the peaks between 300 and 400 cm−1 and a relative decrease of the Raman specific peak at 275 cm−1, indicative of the presence of semi-amorphous pararealgar. The painting by Willem Kalf is dated about 15 years after The Night Watch and both painters lived in Amsterdam. This seems to suggest that a variety of different arsenic sulfide pigments were available to artists working in Amsterdam at the time. The source of the arsenic sulfides and their presence in Amsterdam is further discussed in the discussion section.
Radiation damage
Pararealgar is known to be a light-induced degradation product of realgar as well as of the minerals in the alacranite (As8S9) series, both the natural and synthetic phases [2]. Light-induced degradation can also take place due to irradiation by the Raman laser. Literature shows that this mainly takes place upon irradiation with green light (506.0–544.0 nm) [77]. On the samples of The Night Watch, the measurements shown in this study were carried out with a red (785 nm) laser, but a few test measurements were done with a green (532 nm) laser. Laser intensities can be found in the Materials and Methods section. No changes in the spectra were observed after repeating the measurements. The paint sample of the painting by Willem Kalf was only measured with the 785 nm laser, and the spectra are very similar to those of The Night Watch. Again, the surface of the paint sample and the spectra did not change during the measurements.
SR-µ-XRD
A 2D SR-μ-XRD scan collected at beamline ID13 at ESRF (Grenoble, France) shows the presence of pararealgar in paint sample 017 of The Night Watch. The distribution of pararealgar shows small particles of pararealgar throughout the sample (see Fig. 4b). There is no visible correlation between the large yellow pigment particles we observed under the light microscope and the pararealgar distribution. This is likely due to the large size of the pigment particles (> 10 µm) with respect to the beam size (< 1–2 µm). Thus, the resulting distribution map of pararealgar is strongly dependent on the orientation of the large particles, instead of the presence or absence of the pigment. Additionally, the XRD results were obtained in transmission mode and the LM was performed in reflection mode.

a Light microscope image in (DF) mode of sample SK-C-5_017 with SR-μ-XRD crystalline phase-specific distribution maps of the same area of the cross section showing b pararealgar, c arsenolite, d mimetite and e schultenite. The XRD maps are tilted a few degrees in respect to a. In b–e, a darker color corresponds to a higher scaling parameter and therefor a higher diffraction intensity of the given compound
Three degradation products related to arsenic sulfides were identified in sample 017: arsenolite (Fig. 4c, As2O3), mimetite (Fig. 4d, lead arsenate, Pb5(AsO4)2Cl) and schultenite (Fig. 4e, lead arsenate, PbHAsO4). Figure S4 shows the average XRD pattern of sample 017 (collected at beamline P06, PETRA III, DESY, Hamburg, Germany) and the identified crystalline components in the sample. Mimetite and schultenite are known degradation products of arsenic sulfides that form from mobile As(V) species (i.e., arsenate ions, degradation products of arsenic sulfides) [62,63,64] and lead ions [78,79,80,81]. Mimetite is a relatively stable and insoluble lead arsenate, stable in the pH range from 3 up to 12, depending on the total activity of the arsenates. Relative low concentrations of Pb(II) and As(V) ions are necessary for the formation of mimetite to take place [82]. Arsenolite is known to be a light-induced degradation product of orpiment and pararealgar, which explains why it is mainly present close to the surface and in the crack of the paint sample, where the highest levels of light exposure can be expected. Schultenite is a lead arsenate that is stable under more acidic conditions in comparison to mimetite, and higher concentrations of dissolved Pb(II) and As(V) ions need to be present compared to mimetite [82]. Mimetite was mainly present in the bottom area of the paint sample. The formation of either lead arsenates can be an indication of a difference in the local conditions at the interface between and within the paint layer(s). MA-XRF scans of The Night Watch show that lead is present throughout the entire painting. Lead is present as the commonly used pigment lead white, the yellow pigment lead–tin yellow, a lead-containing impregnation of the canvas [83] and possibly as addition to the oil or later varnish layers [84]. Lead–tin yellow was specifically used in the costume of Van Ruytenburch and particles were found in both cross sections, 017 and 016 (see Figures S4, S6); however, we are not able to assess whether this pigment is the source of the lead ions in the lead arsenates, or that they originate by dissolution of one of the other lead-containing components of the painting. Chloride ions, needed for the formation of mimetite, are often present in oil paintings. Either the lead white used in the painting or chloride ions present in the ambient environment of the painting are the most likely sources [78].
In sample 016, the paint looks much more degraded than the orange paint in 017. Mimetite was the main arsenic containing component identified by SR-μ-XRD. No recognizable XRD pattern for pararealgar was collected in the large pigment particle in the sample (large particle in Fig. 2g). Mimetite was present throughout the entire paint sample, as can be seen in Figure S5. This shows that mobile arsenate and lead ions were present within the entire paint layer, which was also confirmed with SEM–EDX (see Figure S5d and e).These arsenate ions initially derive from arsenic sulfides that have partly dissolved and then start migrating through the paint system [66,67,68,69, 85, 86]. Whether the presence of mimetite has changed the color of this area (now a warm brown) is difficult to assess as the number of pigment particles in this paint sample is limited. However, the formation of this degradation product might have an effect on the translucency of the paint layer [80]. The lead ions necessary for this formation might have derived from the lead white or lead–tin yellow pigment particles present in this paint layer (see Figure S5c for the lead–tin yellow distribution), but, as mentioned before, it is difficult to identify the exact source of the lead ions. The average XRD pattern of sample 016 can be found in Figure S6.
Historical sources
Next to the microscale technical analysis of the paint samples, we studied a large body of historical sources to investigate the production and availability of natural and artificial arsenic sulfides along with their trade routes, prices and how they were sold in seventeenth century Amsterdam. Based on the history of the production of artificial arsenic sulfides found in the historical sources and the creation date of The Night Watch, 1642, it is expected that the available artificial arsenic sulfides are an end-product of a roasting, melting or sublimation process with natural ores as a starting product with possibly the addition of arsenic or sulfur. We therefore not only looked for the artificial type, but also for arsenic and the natural ores in general. Based on this information, we were able to place our findings in historical context and hypothesize how pararealgar and semi-amorphous pararealgar ended up in The Night Watch.
Amsterdam
Based on historical sources, it is evident that a variety of natural and artificial arsenic sulfides were manufactured and traded during Rembrandt’s time and were available in Amsterdam. Due to the geological position of the Dutch Republic, which is not considered a significant mining country in terms of mineral resources, we know that the arsenic sulfides were imported. However, it remains uncertain whether the arsenic sulfides could have also been artificially treated or processed in the Netherlands and Amsterdam specifically for the purpose of selling them as artists pigments. No direct accounts have been found yet regarding a specialized (pigment) manufacturer of artificial arsenic sulfides in the seventeenth century. Nevertheless, the knowledge on subliming metals, including the sublimation of orpiment and arsenic have been reported in some of the Dutch (al)chemical, medicinal and metallurgical literature. Johan Rudolf Glauber, a German apothecary and chemist in Amsterdam describes the sublimation of various minerals in his description of new philosophical furnaces (1651) including a product made from arsenic and orpiment, which he notes are poisonous but useful for painters [87]. In Chimische medici (1647), Johannes Poppen includes a recipe on how to make arsenic fully red like a ruby by subliming orpiment with arsenolite, while Danish scientist Ole Worm, in his work Museum Wormianum, published in Amsterdam, describes the sublimation process of orpiment to obtain artificial realgar [45, 46]. The production is also described by painters Willem Beurs at the end of the seventeenth century and later in 1758 by Robert Dossie [8, 32]. The former describes ‘king's yellow’ as a product of roasting natural orpiment in the sun, while Robert Dossie provides detailed descriptions of two sublimation processes: one involving the combination of arsenic and sulfur, and the other involving the sublimation of natural orpiment.
The discussion on artificial arsenic sulfides in Dutch literature however are rather sporadic and later in time compared to the more extensive recipes in German and Italian literature that date back from the early fifteenth century. Although the Dutch painter Willem Beurs describes the manufacturing of roasting orpiment in the sun, it is unlikely that Rembrandt made the pigments himself but rather bought them readymade, similar to the other pigments in his palette. Painters were aware that arsenic sulfide pigments were highly toxic, and the roasting or subliming of arsenic sulfides required specific equipment. This was also the case for the production of other synthetically produced pigments such as dry-process vermilion, for which Amsterdam was famous in the seventeenth century and was done for instance by the manufacturer Pekstok in Amsterdam [17].
Production and import
In the seventeenth century, Amsterdam was an important center of commerce and with its lucrative position and easy access to southern Europe, England, Scandinavia and the Baltics, the city benefited from important overseas trade networks [88, 89]. Both the natural and artificial forms of arsenic sulfide found their way to Amsterdam, most likely through imports from Italy or Germany. Venice and Germany played pivotal roles as prominent trade centers and production sites in this context. Jacques Le Moine de L'Espine, in his work Le negoce d’Amsterdam (1694), and Jacques Savary des Brûlons, in his Dictionnaire Universel de commerce, highlight Venice as a significant city from where orpiment was shipped [90,91,92]. Similarly, Brûlons, Nicolas Lémery (1743), and Robert de Farvacques (1741) specifically mention Germany as the source of artificial arsenic sulfides, which were produced and distributed throughout Europe [60, 61, 93]. The production of artificial arsenic sulfides appear to have been closely intertwined with the mining of arsenic-containing ores such as cobaltite and arsenopyrite [22, 53, 93,94,95,96,97]. Specialized arsenic smelters in Germany employed roasting techniques to eliminate the arsenic content from cobalt ores, for instance with the aim of producing zaffre, the intermediate product that is used to make the pigment smalt, as illustrated (Fig. 5a, b) and described in Johann Kunckel’s Ars Vitraria Experimentalis (1679) [47, 98]. Cronstedt in 1788 notes that the majority of the commercially traded arsenic derived from the cobalt works in Saxony, where zaffre was produced [10]. Grundmann et al. noted the presence of those smelters in for instance Ehrenfreidersdorf, Freiberg and Reichenstein in East Germany, Joachimsthal and Strassegg, Rotgülden and Walchen in Austria [73, 99]. From the sixteenth century there was also an intensive trade in white arsenic oxide and in yellow and red artificial arsenic sulfides (rauschgelbe, rauschrot) from arsenic smelters in Austria to Venice [73].

a, b Pages from Johann Kunckel’s Ars Vitraria Experimentalis (Franckfurt und Leipzig, 1679)[47] showing a A furnace for roasting cobalt ores, with the long horizontal chimney duct for the recovery of arsenic, b a furnace for the sublimation of arsenic recovered from the long horizontal duct of the master chimney shown in (a). In this furnace, the arsenic powder is heated, and the sublimate is made into solid pieces. The source describes that both yellow and red arsenic end products are produced here. c A page from the Weimar taxa of 1674 which includes the prices for white, yellow and red arsenic (‘Arsenici albi, citrini, rubri’) and both common and ‘the best’ orpiment (‘auripigmenti comm., optimi’) (SUB Göttingen library, 8 MED FOR 38_11) [102]
Both the natural and the artificial arsenic sulfides are listed already at the end of the fourteenth century in an inventory of Pietro Fasolis, a merchant/dealer in pigments in Piemonte, as ‘risalgardi’: pigments with an intense orange color, consisting of arsenic and sulfur, similar to natural orpiment but also available in synthetic form (“pigmento di colore arancio intenso constituito da solfuro di arsenico, come l’orpimento, noto sia in forma naturale che di sintesi”) [33]. The inventory of the Venetian merchant Jacopo de Benedetti (1594) lists various qualities and types (at 860 kg) (“oropimento comun [sic]m[asenda]do; oropimento intt cernido; oropimento c[hiaro]; Oropimento mezan”) and indicates that they represent an important weight fraction of his supply [100, 101]. Amsterdam had an important trade route with Venice and orpiment is specifically listed as one of the items imported to Amsterdam, which might have easily encompassed the artificial arsenic sulfides [90].
Price and availability
The artificial arsenic sulfides were probably sold in Amsterdam by apothecaries or local merchants together with the natural minerals. Local merchants cooperated with traders from other towns, and products were also sold via public sales events in Amsterdam. The inn ‘De Bracke Gront’, for example, which was only a short distance from Rembrandt's home in the Sint Antoniesbreestraat, hosted public sales on “drogeries waren”, chemical or pharmaceutical goods, among which dyes, dried fruits, and probably pigments [90, 103]. Given their toxicity, the permission to trade and sell arsenic goods were likely more regulated than that of other chemicals. A Copenhagen pharmacopeia from 1672 for example states that only pharmacists were allowed to buy and sell ‘arsenicum’ [104]. A permission from the Utrecht city council on February 12, 1669 is granted to two painters to trade in red ‘arsenicum’, perchance the artificial kind. The document furthermore specifies that aside from pharmacies only pigment merchants with granted authorization were allowed to trade this pigment and that they have to keep a record of the name, place of residence, quantity and time of purchase of the buyers [105]. Around fifteen artificial arsenic sulfides with different physical properties were also found in a miniature apothecary of a collector’s cabinet from 1730, likely originating from Delft and now in the collection of the Rijksmuseum (BK-1956–44)[106]. Although the cabinet does not have a historical inventory list of the content, the minerals and artificial arsenic sulfide match closely to the detailed description of some artificial arsenic sulfides described by Abraham Gottlob Werner (1791) of a similar mineral cabinet [107]. This demonstrates that various and different types of artificial arsenic sulfides were available and collected.
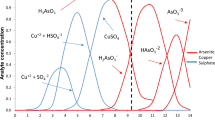
To investigate the pricing and availability of various types of arsenic sulfides, we examined seventeenth century pharmacopeias and price lists (see Table S1 in Supporting Information). Unfortunately, Dutch pharmacopeias provide less comprehensive information compared to their German and Danish counterparts. They lack price lists and offer less detailed descriptions of the products. While the Dutch sources frequently mention orpiment and the general term "arsenic," the German sources from the early sixteenth and seventeenth centuries provide specific references to several artificial arsenic sulfides, along with their corresponding prices. This is likely due to the regional proximity of the cities or known trade routes to the manufacturing source. The artificial arsenic sulfides (described as ‘arsenic citrin’, ‘gelber arsenic’, ‘arsenic rubr. or rubei’, ‘Rauschgelbe’) in the initial years of the seventeenth century generally have the same price as natural orpiment while later in the seventeenth century they can become twice as expensive (Fig. 5c). The difference in price is probably linked to the manufacturing: while in the sixteenth century, the natural ores were used as a starting product, during the seventeenth century the process refined, and products were obtained from the sublimation of white arsenic and sulfur. In comparison to other pigments, both the natural and artificial arsenic sulfide pigments were priced averagely. Based on the pharmacy taxae, they are generally twice as expensive as lead white, comparable to or a bit more expensive than lead–tin yellow and consistently cheaper than vermilion.
Discussion
The results from the analytical analyses with light microscopy, SEM–EDX, micro-Raman spectroscopy, and SR-μ-XRD of samples SK-C-5_017 and SK-C-5_016 suggest that Rembrandt used a mixture of yellow pararealgar and an orange to red semi-amorphous pararealgar to obtain the bright orange paint in Van Ruytenburch’s buff coat.
Three explanations can be given for the presence of (either crystalline or semi-amorphous) pararealgar in The Night Watch:
-
1)
Pararealgar has formed in the paint layer due to degradation of the originally used arsenic sulfides.
-
2)
Pararealgar was present as a contaminant in the original pigments which the painter used, the result of degradation during mining, processing, transport, trade and/or sale of the arsenic sulfide pigments. In this case the pararealgar was unintentionally used by the artist.
-
3)
Pararealgar and semi-amorphous pararealgar were intentionally used as pigment. Either the two pigments were mixed by the artist, or the mixture of pigments was sold in Amsterdam. Although Rembrandt was known for his complex pigment mixtures, the very similar pigment mixture observed in the painting by Willem Kalf suggests the second scenario.
To date, the presence of pararealgar in European paintings and art objects has only been confirmed in a few cases. In 1995, the first finding of pararealgar in an Italian painting was done by Corbeil and Helwig, where they used XRD to identify pararealgar in Holy Family and Saint (tentatively attributed to school of Titian). Around that same time, Trentelman et al. identified pararealgar in Tintoretto’s The Dreams of Men [72]. This work presents the first Raman spectrum of pararealgar recorded. Both these studies already mention that it is difficult to determine if the pararealgar was used as a pigment or whether it has degraded from realgar. Vermeulen et al. identified pararealgar in a seventeenth century still life painting by Abraham Mignon Festoon of flowers and insects and a polychrome sculpture The holy family by Walter Pompe, dated ca. 1760 [5]. Pararealgar was also identified in Domenico Tintoretto’s Entry of Philip II into Mantua by Grundmann et al. [73]. Vermeulen et al. consider the pararealgar to be a possible intended painting material or at least the starting material for producing artificial arsenic sulfides, while Grundmann et al. consider the pararealgar detected in Tintoretto as a degradation of an ‘originally’ employed artificial realgar. Also in other artworks and heritage objects, pararealgar has been identified and multiple authors suggest that the pararealgar could have been used as a pigment [108,109,110,111,112,113]. Zuena et al. suggest the use of pararealgar as starting product to make an amorphous arsenic sulfide [114]. In two cases where pararealgar was identified in paintings it was found together with remnants of realgar and the paint was very degraded. In this case, the authors assume that the pararealgar has formed in-situ [115, 116]. These remnants could also be contaminants picked up during the collection of the pararealgar from an ore.
In the case of The Night Watch, the in-situ formation of (artificial) realgar into pararealgar seems unlikely since there is no indication of the presence or remnants of realgar, crystalline or amorphous, in the paint cross sections. As described earlier, both the yellow and orange to red particles are sharp edged and the particles and the surroundings look unaltered, unlike in light-degraded areas. Additionally, in the case of formation of pararealgar due to light degradation, we would expect to see a gradient of in-situ formed pararealgar from the top towards the bottom of the paint cross section. This leads us to the conclusion that a mixture of pararealgar (yellow) and the semi-amorphous pararealgar (orange to red) were originally used by the artist, probably for its specific hue, making the third hypothesis cited above the most likely scenario.
The morphology of the arsenic sulfide particles closely corresponds to the pararealgar particles found in Domenico Tintoretto’s Entry of Philip II into Mantua (part of the Gonzaga cycle, 1579/80, Alte Pinakothek, Munich). Grundmann et al. hypothesize that all pararealgar particles have converted from artificial realgar or alacranite to pararealgar in the paint layer [73]. As their Raman results do not show presence of any other arsenic sulfides apart from pararealgar, or any gradient of degradation in the paint layer, we expect that also in this case pararealgar could have been used by the artist.
The use of pararealgar as a pigment or as a starting material to obtain an artificial arsenic sulfide pigment, however, is not entirely surprising. The degradation rate of realgar into pararealgar is relatively fast. A short exposure to sunlight, possibly aided by some mechanical grinding of the realgar particles while preparing the paint, already starts the process. As mentioned above, before the discovery of pararealgar in 1980 [117], this light-induced polymorph of realgar was generally assumed to be orpiment based on its yellow color [72]. The preparation of artificial arsenic sulfides from orpiment is described in numerous sources from the early fifteenth century onwards. It is therefore possible that between the mining, processing, transport, grinding, the selling and the use by the artist, realgar has already altered to pararealgar. We have to assume that some of the material sold in seventeenth century Amsterdam as orpiment, contained or consisted of pararealgar, or was sold as a different hue or grade of orpiment as listed in the inventory of the Venetian merchant Jacopo de Benedetti. It is therefore plausible that pararealgar was employed as a pigment, as we have seen here in The Night Watch and in Still Life with a Silver Jug and a Porcelain Bowl by Willem Kalf, and was mentioned in previous studies finding pararealgar in paintings and other cultural heritage objects.
The literature describing the manufacturing of artificial arsenic sulfides, largely focusses on the transmutation or ability to change the colors of minerals to obtain various shades from bright yellow, golden, warm yellow, orange, red to deep red. In the Paduan manuscript from 1585 we read: “to make a most beautiful orange yellow color” out of orpiment; in the bnf Ms fr. 640 manuscript sublimation of orpiment is suggested with a little arsenic to obtain orange [27, 118]. Raffaello Borghini describes that burning orpiment “makes another strong color” [41]. And an anonymous Spanish treatise describes roasting and burning the natural orpiment to obtain half-tints [1]. Brûlons also notes that the artificial red arsenic sulfide, the product of heating orpiment, is only used by painters and chosen in big pieces, heavy, brilliant and deep in color [92]. A recent publication by Cruz et al. also describes the use and the variety of artificial arsenic sulfide pigments in Europe as understudied and probably underestimated in current literature[3].
Rembrandt’s choice
As vermilion and lead–tin yellow were also found in the paint mixture, Rembrandt clearly aimed for a bright orange tone with a high color strength that allowed to create an illusion of the gold thread embroidery in Van Ruytenburch’s costume. The artificial orange to red arsenic sulfide might have offered different optical and rheological paint properties as compared to the mineral form of orpiment and realgar. However, as far as we know no reconstruction-based research has been conducted to compare the paint properties between the natural and artificially produced arsenic sulfides. Van Loon et al. proposed, with regard to Rembrandt’s The Jewish Bride (Rijksmuseum), that the bright yellow particles (2 to 5 µm in diameter), a purified form of artificial orpiment glass obtained by a sublimation reaction (dry process), found in a complex mixture with lead–tin yellow, vermilion, a little earth pigment and significant amounts of lake, were thought to have been added to give brightness and intensity to the brown shadow tones of Isaac’s sleeve [6]. In the same way, Rembrandt often added a few tiny bright red particles of vermilion to ‘lift and brighten’ the color of the paint [119].
Conclusion
Microscopic research using light microscopy, SEM–EDX, micro-Raman spectroscopy and SR-µ-XRD has identified the presence of two types of arsenic sulfide pigments in The Night Watch: the yellow pararealgar and an orange to red semi-amorphous pararealgar. These arsenic sulfides were not found before in the oeuvre of Rembrandt. Although pararealgar has so far mainly been thought of as a degradation product of realgar and other arsenic sulfides, we propose that pararealgar was available in seventeenth century Amsterdam as a pigment and that Rembrandt used it to paint with. There is no evidence to suggest it was formed in-situ due to chemical alteration of the paint. The orange to red semi-amorphous particles also exhibit some characteristics of pararealgar. This might be due to partial degradation of a fully amorphous arsenic sulfide, but we would rather propose that these semi-amorphous orange to red particles were obtained by intentionally heating pararealgar particles to obtain a pigment with a more pronounced orange to red hue.
Although most heritage-related literature still refer to orpiment and realgar in relation to arsenic sulfide pigments, this research as well as other studies, propose that there was a broader spectrum of arsenic sulfide pigments available to (Dutch) artists in the seventeenth century. This is in accordance with historical sources as well as catalogues of collection cabinets that describe long lists of different arsenic sulfides, both natural and artificial and with different colors ranging from yellow, through orange, to red. The use of yellow pararealgar and an orange to red semi-amorphous pararealgar by Rembrandt in Amsterdam in the seventeenth century is supported by the identification of a very similar paint mixture in a painting by Willem Kalf that was painted in the same city some 15 years later. Extensive literature research of historical sources also shows the contemporary knowledge of manipulating both natural or artificial arsenic sulfides to obtain different shades of yellow to red. These findings confirm that identifying arsenic sulfides pigments in cultural heritage objects should be done by molecule specific techniques such as Raman spectroscopy and/or XRD to identify the exact species. It highlights that the collection of arsenic sulfide pigments used by artists in the seventeenth century was much larger than commonly perceived. Also in the case of Dutch seventeenth century oil paintings, pararealgar should not only be considered as an in-situ formed degradation product, but as a separate pigment as well.
Materials and methods
Macro-scale analysis
Macro X-ray fluorescence (MA-XRF) imaging
Elemental maps of The Night Watch were acquired with the commercially available MA-XRF scanner M6 Jetstream from Bruker Nano GmbH (Berlin, Germany). The M6 Jetstream consists of a measuring head equipped with a 30 W Rh-target microfocus X-ray tube, a polycapillary lens, and a dual 60 mm2 X-Flash silicon drift detector (SDD) with a beryllium window (energy resolution < 145 eV at Mn-Kα) that is moved over the surface of the painting by means of an X, Y-motorized stage, enabling a scan area of 80 × 60 cm2. The entire painting was scanned at 50 kV, a current of 200 µA, with a 500 µm step size, and a dwell time of 35 ms.
A total of 56 scans, 8 rows and 7 columns, with an overlap of approximately 20% (horizontal and vertical) were needed to scan the whole painting. The M6 Jetstream was securely fastened with a custom-made rotating platform (Bronnenberg) on the scissor lifts. To retain a constant instrument-to- painting distance (ca. 10 mm from the x-ray snout and the painting), maintaining an approximate 240 µm spot size and reducing fluctuation of the attenuation of the x-ray fluorescence signals in ambient air, the scanner was carefully positioned and aligned for every scan.
The resulting spectral data cubes of the MA-XRF scanning, shown in this paper, were processed using data analysis software packages PyMCA (Python Multichannel Analyzer) and Datamuncher gamma 1.4 and 1.5 [120, 121]. PyMCA was used to perform spectral fitting and deconvolution, in order to resolve overlapping peaks of different elements using the MCA Hypermet function, in this case relevant for the overlapping K lines of arsenic with the lead L lines. After spectral deconvolution, the separate elemental distribution images were compiled with the Datamuncher software. In the ensuing 2D distribution maps, each pixel carries information on the calculated net peak intensities of the emission lines of the element, with a grey scale linear to the detected intensities.
Micro-scale analysis
Light microscopy
In 2020, two paint cross sections were taken from the arsenic containing areas of the costume of Van Ruytenburch. Sample SK-C-5_016 was taken in the proper right sleeve of Van Ruytenburch’s costume in a warm brown stripe. Sample SK-C-5_017 was taken from the embroidered border of the sleeveless buff coat in an orange highlight on top of a brown underlayer. Sample SK-A-199_R9/4 was taken by Arie Wallert in 1997, from a location in the orange color in the middle orange (fruit). The paint samples were embedded in— Poly-pol PS230: a two-component polyester mounting resin (Poly-Service Amsterdam, the Netherlands) and polished using a sample holder and Micromesh sheets up to grade 12,000 (Micro-Surface Finishing Products Inc., Wilton, Iowa, USA) [122].
The cross sections were photographed in bright field (BF) and dark field (DF) mode, and under ultraviolet radiation (UV365 nm) at spatial resolutions of 0.27 µm/pixel (200 ×) and 0.11 µm/pixel (500 ×) with a Zeiss Axio Imager.A2m microscope (Carl Zeiss Microscopy, LLC, United States) equipped with a Zeiss AxioCam 506 color digital camera. White light was provided by a LED lamp and a Colibri 2 controller for UV-fluorescence microscopy (LED light source at 365 nm, using a filter cube composed of a 365 nm excitation filter (EX G 365), a beam splitter at 395 nm (BS FT 395), and an emission long-pass filter at 420 nm (EM LP 420)). All images were obtained and processed in the image-acquisition software Zen 2 pro (blue edition) with extended depth of focus (MEDF) facilities and observed on a calibrated Eizo Color Edge CG277 BK computer screen.
SEM–EDX
Scanning electron microscopy studies in combination with energy dispersive X-ray spectroscopy (SEM–EDX) were performed on a FEI NovaNano SEM 450 variable pressure electron microscope equipped with a ThermoFisher NSS EDX system. The uncoated samples were examined under low vacuum and BSE images were obtained with an acceleration voltage of 15 kV, spot 3, GAD detector, 90 Pa and a 5 mm eucentric working distance. The samples were analyzed in high vacuum with a 4 nm thick tungsten coating to improve surface conductivity, using a Leica EM ACE600 Coater (8E−3 mbar). BSE-images were obtained with an accelerating voltage of 15 kV, spot 3, 6 mm working distance. The EDX maps were also collected under high vacuum conditions, with an accelerating voltage of 20 kV, spot 5 and a map resolution of 512 by 340 pixels and a magnification of 650x.
Synchrotron µ-XRPD
ID13, ESRF
2D SR-µ-XRD imaging was carried out on cross section SK-C-5_017 at ID13 beamline of the ESRF storage ring (ESRF, The European Synchrotron, 71 Avenue des Martyrs, CS40220, 38043 Grenoble Cedex 9, France). This research was done via the “Historical Materials BAG”, which is a community proposal that gives 10 European institutes the opportunity for guaranteed beamtime at two X-ray powder diffraction (XRPD) beamlines ID13 and ID22 at the ESRF [123, 124]. ID13 is an ESRF undulator beamline dedicated to high-lateral resolution diffraction and scattering experiments using focused monochromatic X-ray beams [124]. Samples were mounted vertically, perpendicular to the 13 keV x-ray beam and the beam was focused to achieve a size of 2 × 2 μm2 (h × v). An optical microscope looking upstream towards the samples allowed for positioning of the sample. Diffraction signals were recorded in transmission geometry using an EIGER-X 4 M single photon counting detector (Dectris Ltd., Switzerland) and calibration of the diffraction setup was performed by means of a corundum (Al2O3) reference sample. The embedding medium of the cross section was thinned down to ca. 1–2 mm before the measurements.
An area of 70 × 125 µm2 was mapped using a step size of 2 µm in horizontal direction and 2 µm in vertical direction. A dwell time of 10 ms was used to acquire the diffraction patterns.
Azimuthal integration was performed using a dedicated Jupyter Notebook [123]. The software package XRDUA was employed to perform compound identification and execute quantitative analysis by Rietveld refinement [125]. The obtained 2D distributions shown in this article are based on the global scaling factor obtained from the fitting procedure.
P06 beamline, PETRA III, DESY
Samples SK-C-5_016 was analyzed with SR-μ-XRD at beamline P06, PETRA III, DESY (Hamburg, Germany). This hard X-ray micro- and nano-probe beamline is suited for X-ray powder diffraction imaging experiments at the (sub-)micrometric scale. A Kirkpatrick-Baez optical system was used to focus the 21 keV beam to a diameter of 0.5 μm and a flux of ca. 1010 photons/s. The sample was mounted on a plastic frame that could be moved in the XYZ directions. An EIGER-X 4 M detector (Dectris Ltd., Switzerland) was used to collect the diffraction signals. The sample was placed 18 cm in front of the detector to achieve a sufficiently wide angular range.
An area of 150 by 270 µm2 was mapped using a step size of 1 µm in both directions. A dwell time of 0.25 s was used.
Micro Raman spectroscopy
Raman spectra shown in this study were acquired with a Renishaw inVia confocal micro-Raman spectrometer with a Peltier-cooled (-60°) CCD detector (1020 × 256 px), using a Renishaw HPNIR 785 nm diode laser as excitation source, in combination with a 1200 l/mm grating. A silicon reference sample was used to calibrate the instrument. To avoid laser induced damage, a laser power of less than 0.0001% of the maximum intensity was used for the first measurements, and then the power was carefully increased until a satisfactory spectrum was collected. The spectra shown in this study were collected with a laser power of 2.1 mW with neutral density filters (0.5% of the maximum laser intensity). Spectra were collected using a 100 × objective and an exposure time of 1 s and 3 accumulations. WiRE 4 Raman software was used for the data collection and to remove the baseline. An in-house written MATLAB script was used to normalize the spectra. Measurements were acquired on several big and small particles in samples 016 and 017 with variations in laser power, acquisition time and accumulations. Not all spectra are shown in this study. Additionally, some spectra were collected using a Renishaw 532 nm (RL532C50) solid state laser as excitation source, using a 50 × objective and a laser power of approximately 0.03–0.13 mW.
Availability of data and materials
All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
References
Richter M, Grundmann G, van Loon A, Keune K, Boersma A, Rapp K. The occurrence of artificial orpiment (dry process) in northern European painting and polychromy and evidence in historical sources. 2007. p. 167–92.
Gliozzo E, Burgio L. Pigments—Arsenic-based yellows and reds. Archaeol Anthropol Sci. 2022;14:4.
Cruz AJ, Melo HP, Valadas S, Miguel C, Candeias A. The matter from which an orange colour is made: on the arsenic pigment used in a portuguese mannerist painting. Heritage. 2022;5:2646–60.
Burgio L, Manca R, Browne C, Button V, Horsfall Turner O, Rutherston J. Orange for gold? Arsenic sulfide glass on the V&A Leman Album. J Raman Spectrosc. 2019;50:1169–76.
Vermeulen M, Saverwyns S, Coudray A, Janssens K, Sanyova J. Identification by Raman spectroscopy of pararealgar as a starting material in the synthesis of amorphous arsenic sulfide pigments. Dyes Pigm. 2018;149:290–7.
van Loon A, Noble P, Krekeler A, Van der Snickt G, Janssens K, Abe Y, et al. Artificial orpiment, a new pigment in Rembrandt’s palette. Heritage Science. 2017;5:26.
Grundmann G, Richter M. Types of dry-process artificial arsenic sulphide pigments in cultural heritage. 2012. p. 119–44.
Beurs, Wilhelmus. De groote waereld in’t kleen geschildert, of Schilderagtig tafereel van’s weerelds schilderyen, kortelijk vervat in ses boeken: verklarende de hooftverwen, haare verscheide mengelingen in oly, en der zelver gebruik. Omtrent de meeste vertoningen van de zigtbare natuire. Leersaamelijk den liefhebbers en leerlingen der ed. schilderkonst medegedeelt [The Big World Painted Small]. T’Amsterdam: By Johannes en Gillis Janssonius van Waesberge; 1692.
West FE. Orpiment and Realgar. In: West Fitzhugh E, editor. Artists’ pigments: a handbook of their history and characteristics, vol. 3. Washington: Publishing Office, National Gallery of Art; 1997. p. 47–80.
Cronstedt AF. An Essay Towards a System of Mineralogy: In Two Volumes. Charles Dilly, in the Poultry; 1788.
Glauber JR, French J. A description of new philosophical furnaces, or, A new art of distilling, divided into five parts: whereunto is added a description of the tincture of gold, or the true aurum potabile: also the first part of the mineral work: set forth and published for the sakes of them that are studious of the truth [Internet]. London: Printed by Richard Coats, for Tho. Williams ...; 1651 [cited 2022 Aug 12]. https://catalog.hathitrust.org/Record/101821734. Accessed 12 Aug 2022.
Braekman (ed.) WL. Medische en technische Middelnederlandse recepten [Medical and technical Middle Dutch recipes]. Koninklijke Vlaamse Academie voor Taal- en Letterkunde [Internet]. Gent: DBNL; 1975. https://www.dbnl.org/tekst/_med002medi01_01/colofon.php. Accessed 12 Aug 2022.
Biringuccio V. De la pirotechnia. Libri. 10 doue ampiamente si tratta non solo di ogni sorte & diuersita di miniere, ma anchora quanto si ricerca intorno à la prattica di quelle cose di quel che si appartiene a l’arte de la fusione ouer gitto de metalli come d’ogni altra cosa simile a questa. Composti per il s. Vanoccio Biringuccio sennese .. [On Pyrotechnics. Books. 10 where extensively treated not only of every kind & diversity of mines, but also of what is sought around the practice of those things concerning the art of casting or casting of metals as of any other similar thing to this. Composed by Mr. Vanoccio Biringuccio from Siena]. per Curtio Navò & fratelli al segno del Lion; 1540.
Birelli GB, Uffenbach P. Alchimia nova, das ist, Die güldene Kunst selbst, oder, Aller Künsten Mutter: sampt dero heimlichen Secreten, vnzehlichen verborgenen Kindern vnd Früchten .../ auss dem Italianischen dess edlen vnd vesten Hanss Baptiste Birelli von Senis ; auff das fleissigst verteutscht durch Petrum Vffenbachium [Alchimia nova, that is, The Golden Art itself, or, Mother of All Arts: along with its secret secrets, numerous hidden children, and fruits ... / translated from Italian by the noble and steadfast Hans Baptiste Birelli von Senis; diligently translated by Petrus Vffenbachium] [Internet]. Getruckt zu Franckfurt am Mayn: Bey Niclass Hoffman; 1603. http://catalog.hathitrust.org/api/volumes/oclc/83289700.html. Accessed 12 Aug 2022.
Birelli G. Opere di Giouambatista Birelli. Tomo Primo. Nel qual si tratta dell’alchimia, suoi membri, vtili, curiosi, & diletteuoli. Con la vita d’Hermete, con due tauole, l’una de’ capitoli, & l’altra delle cose notabili [Works of Giouambatista Birelli. Volume One. In which the alchemy is discussed, its useful, curious, and delightful members. With the life of Hermes, with two tables, one of the chapters, and the other of notable things]. 1601.
Arnaud de Villeneuve. Le Tresor des pouvres : selon maistre Arnoult de Villenove, maistre Gérard de Solo, et plusieurs aultres Docteurs en medicine de Montpellier. Nouvellement imprimé et corrigé [The Treasure of the Poor: according to Master Arnoult de Villenove, Master Gérard de Solo, and several other Doctors in medicine from Montpellier. Newly printed and corrected.] [Internet]. Lyon: Nourry, Claude (1470?-1533); 09. http://gallica.bnf.fr/ark:/12148/bpt6k10980071. Accessed 12 Aug 2022.
Pekstok P. De Pekstok-notities [The Pekstok notes] [Internet]. Amsterdam; 1691. https://archief.amsterdam/inventarissen/file/07b3a3fe95ef936bcf763c039c93c322
Witgeest S. Het verbetert en vermeerdert natuurlyk toover-boek. Of ’t nieuw speel-toneel der konsten (onder ps. Simon Witgeest) [The Improved and Enlarged Natural Magic Book. Or the new play stage of the arts (under the pseudonym Simon Witgeest)]. Amsterdam: Jan ten Hoorn; 1698.
Morley CL. Collectanea chymica Leydensia. Apud Henricum Drummond; 1684.
Blankaart S. Collectanea medico-physica, oft, Hollands jaar-register der genees- en natuur-kundige aanmerkingen van gantsch Europa &c. : beginnende met het jaar MDCLXXX : door eigen ondervinding en gemeen-making van verscheide heeren en liefhebbers [Collectanea Medico-Physica, or, Dutch Yearbook of Medical and Physical Observations from the Whole of Europe, etc.: Starting from the Year 1680: Through Personal Experience and Contributions from Various Gentlemen and Enthusiasts] [Internet]. t’Amsterdam : By Johan ten Hoorn, boekverkooper ...; 1680. http://archive.org/details/collectaneamedic00blan. Accessed 21 Jul 2022.
L’Emery N. Het Philosoophische Laboratorium, Of der Chymisten Stook-Huis (etc.) [The Philosophical Laboratory, or the Chemists’ Distillery]. Hoorn; 1683.
Houttuyn M (Martinus), Linné C von, Philips JC, Bayer FM, H. de Vries (Firm) former owner D. Natuurlyke historie : of, Uitvoerige beschryving der dieren, planten, en mineraalen [Natural History: or, Detailed Description of Animals, Plants, and Minerals] [Internet]. Te Amsterdam : By F. Houttuyn [and others]; 1761. http://archive.org/details/natuurlykehistor34hout. Accessed 12 Aug 2022.
Iversen E. Some ancient Egyptian paints and pigments: a lexicographical study. [København: I kommission hos Munksgaard; 1955.
Cennini C, Thompson DV. The craftsman’s handbook. New York: Dover Publications; 1960.
Ms. 1975/1491 Trierer Malerbuch [Internet]. Trier; https://artechne.hum.uu.nl/node/87697
MS 1243. Bibliotheca Riccardiana Firenze; 15th century.
Merrifield MP (Mary P. Original treatises, dating from the XIIth to the XVIIIth centuries, [o]n the arts of painting : in oil, miniature, mosaic, and on glass ; of gilding, dyeing, and the preparation of colours and artificial gems ; preceded by a general introduction ; with translations, prefaces, and notes. By Mrs. Merrifield ... In two volumes [Internet]. London : John Murray, Albemarle Street; 1849. http://archive.org/details/originaltreatis00merrgoog. Accessed 10 Aug 2022.
Apotecken Tax vnd ordnung aller Ertzneien so der Apotecken der Furstlichen Stad Lignitz im Jar nach Christi vnsers lieben HErrn geburt 1567. durch die verordente Visitatorn der billigkeit nach gestelt welche in diesem jtzt lauffenden 1568. jare auff befelh eines Erbarn Wolveisen Raths der Stad Lignitz von dem Doctore Baldasar Summer jtziger zeit alda Fuerstlichen bestelten Leib vnd Stad Artzt wider vbersehen in das werck gebracht vnd in den druck verfertiget. [Pharmacy Tax and Regulation of all Medicines for the Pharmacies of the Princely City of Lignitz in the Year after the Birth of our dear Lord 1567. arranged by the appointed inspectors of fairness, which in this now running year 1568, on the command of a respected Wolweisen Council of the City of Lignitz by Doctor Baldasar Summer currently appointed as the Prince’s personal and city physician, was reviewed and completed, and prepared for print] [Internet]. Wittenberg: Johann Schwertel; 1568. http://resolver.staatsbibliothek-berlin.de/SBB0001796500000000
Burmester A, Haller U, Krekel C. Pigmenta et Colores: The Artist’s Palette in Pharmacy Price Lists from Liegnitz (Silesia). Trade in Artists’ Materials: Markets and Commerce in Europe to. 2010;1700:314–24.
Designatio Et Valor, Omnium Materialium, Et Medicamentorum, tam Simplicium, quam Compositorum, quæ in Officinis Gedanensibus reperiuntur & venduntur = Verzeichnüß und Taxa Aller Marterialien und Artzneyen, so wol der Einfachen als zusammen gesetzen, welche in den Dantziger Apotheken zu finden seyn, und verkauffet werden. [List and Price of All Materials and Medicines, both Simple and Compound, Found and Sold in the Pharmacies of Danzig] [Internet]. Gdańsk: Dawid Fryderyk Rhete ( -1694); 1668. https://pbc.gda.pl/publication/12022
Apothecken-Ordnung und Taxa Derer in denen Apothecken der Churfl. Sächs. alten freyen Berg-Stadt Freybergk in Meissen/ befindlichen Medicamenten und Materialien : durch E.E. Rath daselbsten auffgerichtet und publiciret Anno 1673. [Pharmacy Regulation and Price List of the Medicines and Materials Found in the Pharmacies of the Electoral Saxon Ancient Free Mining Town of Freiberg in Meissen: Established and Published by the Noble Council there in the Year 1673] [Internet]. Freyberg: Zacharias Becker; 1673. http://digitale.bibliothek.uni-halle.de/vd17/470090. Accessed 4 Sep 2023.
Dossie R. The handmaid to the arts [by R. Dossie.]. Nourse; 1758.
Gabrieli BO. L’inventario della spezieria di Pietro Fasolis e il commercio dei materiali per la pittura nei documenti piemontesi (1332–1453). Parte prima [The Inventory of Pietro Fasolis’ Apothecary and the Trade of Painting Materials in Piedmontese Documents (1332–1453). Part One]. Bollettino della società storica pinerolese. 2012;29:7–43.
Bartl A. Der “Liber illuministarum” aus Kloster Tegernsee: Edition, Übersetzung und Kommentar der kunsttechnologischen Rezepte [The ‘Liber illuministarum’ from Tegernsee Monastery: Edition, Translation, and Commentary on Art Technological Recipes]. Franz Steiner Verlag; 2005.
Beuther D. David Beuthers, Gewesenen Thurfürstl. Sächsischen Probation-Meisters zu Dreßden, und Philosophi Adepti, Zwey rare Chymische Tractate: Darinnen Nicht nur alle Geheimnisse der Probier-Kunst, Derer Ertze und Schmeltzung derselben, Sondern auch Die Mögligkeit der Verwandelung, Der geringen Metallen in bessere, gar deutlich gezeiget werden. Aus einem alten raren, von Anno 1514. biß 1582. geschriebenen Buche zum ersten mahl in Druck gegeben. Deme Beygefüget dieses Autoris Universal, oder Vollkommener Bericht von der wahren Alchymie [David Beuthers, Former Electoral Saxon Assayer in Dresden, and Adept Philosopher, Two Rare Chemical Tracts: Wherein Not Only All the Secrets of the Assaying Art, of Ores and Their Melting, But Also the Possibility of Transformation, of Base Metals into Better Ones, Are Clearly Demonstrated. Published in Print for the First Time from an Old Rare Book Written from 1514 to 1582. Accompanied by this Author’s Universal, or Complete Account of True Alchemy]. Leipzig: Martini; 1717.
Agricola G. De Re Metallica translated from the First Latin Edition of 1556 [Internet]. New York: Dover Publications; 1950 [cited 2023 Sep 5]. https://www.gutenberg.org/files/38015/38015-h/38015-h.htm. Accessed 5 Sep 2023.
Dioscoride. Les six livres de Pedacion Dioscoride d’Anazarbe de la matière médicinale , translatez de latin en francois [The six books of Pedanius Dioscorides of Anazarbus on medical materials, translated from Latin into French] [Internet]. Lyon: Payan; 1559 [cited 2023 Sep 6]. https://gallica.bnf.fr/ark:/12148/bpt6k96067706. Accessed 6 Sep 2023.
Mattioli PA. De I Discorsi Di M. Pietro Andrea Matthioli Sanese, Medico Cesareo, Et Del Serenissimo Principe Ferdinando Arcidvca D’Avstria, Etc. Nelli sei libri Di Pedacio Dioscoride Anazarbeo Della Materia Medicinale ... Con due Tauole copiosissime spettanti l’vna à ciò, che in tutta l’opera si contiene; & l’altra alla cura di tutte l’infirmità del corpo humano: La quale contiene il III. IIII. V. VI. & vltimo libro [The Discourses of Mr. Pietro Andrea Matthioli of Siena, Imperial Physician, and of the Most Serene Prince Ferdinand Archduke of Austria, Etc. In the six books of Pedanius Dioscorides of Anazarbus on medical materials... With two very extensive tables, one concerning everything contained in the entire work, and the other concerning the treatment of all the ailments of the human body: Which includes the III. IV. V. VI. & last book]. Venice: Appresso Bartolomeo de gli Alberti; 1604.
Making and Knowing Project, Smith P, Rosenkranz N, Uchacz T, Taape T, Goodbarge C, et al., editors. Secrets of Craft and Nature in Renaissance France: A Digital Critical Edition and English Translation of BnF Ms. Fr. 640 [Internet]. New York: Making and Knowing Project; 2020. https://edition640.makingandknowing.org.
Lomazzo GP. Trattato dell’arte de la pintura, Libro Terzo: Del Colore, Cap VI [Treatise on the Art of Painting, Book Three: On Color, Chapter VI]. Milan; 1584.
Borghini R. Il riposo, in cui della pittura e della scultura si favella, de’ piu illustri Pittori, e scultori, e delle piu famose opere loro si fa mentione; e le cose principali appartenenti a dette arti s’insegnano [Il riposo, in which painting and sculpture are discussed, mention is made of the most illustrious painters and sculptors, and their most famous works are mentioned; and the principal things belonging to these arts are taught]. Florence: Giorgio Maresotti; 1584.
de Medici A. Apparato della Fonderia Dell’illustrissimo et eccellentiss. Sig. D. Antonio Medici. Nella quale si contiene tutta l’arte Spagirica di Teoprasto Paracelso, & sue medicine. Et altri segreti bellissimi. Vol.2 [Apparatus of the Foundry of the Most Illustrious and Most Excellent Mr. D. Antonio Medici. In which is contained all the Spagyric art of Theophrastus Paracelsus, and his medicines. And other beautiful secrets. vol.2] [Internet]. 1604. https://artechne.hum.uu.nl/node/91933. Accessed 6 Sep 2023.
Leão DN do. Descripcao do reino de Portugal. Per Duarte Nunez do Leao, desembragador da casa da supplicao. Dirigida ao illustissimo & muito excellente snor Dom Diogo da Sylva .. [Description of the Kingdom of Portugal. By Duarte Nunez do Leao, Judge of the Casa da Supplicacao. Addressed to the Most Illustrious and Most Excellent Lord Dom Diogo da Silva]. Lisboa: Iorge Rodriguez; 1610.
Castelli P. Chalcanthinum dodecaphorion siue Duodecim dubitationes in vsu olei vitrioli et defensio antiquorum in arsenici, atque sandarachae potu, Ad Raymundum Mindererum Medicum Germanum Eloquentissimum. auctore Petro Castello philosopho, ac medico Romano [Chalcanthinum dodecaphorion, or Twelve Doubts on the Use of Vitriol Oil and Defense of the Ancients in the Drinking of Arsenic and Sandarach. To Raymond Minderer, a Most Eloquent German Physician. By Peter Castello, Philosopher and Roman Physician] [Internet]. Rome: Iacobi Mascardi; 1619. http://archive.org/details/ita-bnc-mag-00001015-001. Accessed 6 Sep 2023.
Poppen J. Chimische medici: Het tweede deel. Van der bereydinghe der metalen/mineralen/ende kruyden/etc. Elck in hare quinta essentie te brengen/etc. [Chemical Medicines: The Second Part. Concerning the Preparation of Metals/Minerals/and Herbs/etc. Each to be brought to its Quintessence/etc]. Amsterdam: gedruckt by Thuenis Jacobsz, Boeck-verkooper op het Water/inde Loots-Man; 1647.
Worm O. Museum Wormianum, seu historia rerum rariorum, tam naturalium, quam artificialium, tam domesticarum, quam exoticarum, quae Hafniae Danorum in aedibus authoris servantur [Museum Wormianum, or History of Rare Things, both Natural and Artificial, both Domestic and Exotic, which are preserved in the House of the Author in Copenhagen, Denmark]. Elzevirius; 1655.
Kunckel J. Ars Vitraria Experimentalis, Oder Vollkommene Glasmacher-Kunst [The Experimental Glass Art, or Perfect Glassmaker’s Craft] [Internet]. 1. Auflage. Frankfurt (Main); Leipzig; Jena: Selbstverlag; 1679. Deutches Textarchiv: http://www.deutschestextarchiv.de/kunckel_glasmacher_1679. Accessed 7 Mar 2024.
Lemery N. Cours de chymie contenant la manière de faire les opérations qui sont en usage dans la médecine, par une méthode facile ... Par Nicolas Lemery [Chemistry Course containing the Method of Carrying out Operations Used in Medicine, by an Easy Method ... By Nicolas Lemery]. 5th ed. Paris: Michallet; 1683.
Kunckel J. Wieder neu aufgerichtete und vergrösserte in zwey Theilen angewiesene curieuse Kunst- und Werck-Schul: darinnen jedes Theils oder Buches Innhalt auf folgendem Blat zu ersehen. Der Neu-aufgerichteten und Vergrösserten Jn Sechs Bücher oder Theilen verfasten curieusen Kunst- und Werk-Schul, sehr verlangter nunmehr erfolgter Anderer Theil. 2 [Newly Re-established and Enlarged Curious Art and Craft School, Divided into Two Parts: Wherein the Contents of Each Part or Book are to be Seen on the Following Page. The Newly Re-established and Enlarged Curious Art and Craft School, Composed in Six Books or Parts, much Desired Now Completed Second Part.2]. Nurnberg: JohannZieger; 1707.
Henkel JF. “Pyritologia, Oder: Kieß-Historie : Als des vornehmsten Minerals, Nach dessen Nahmen, Arten, Lagerstätten, Ursprung, Eisen, Kupffer, unmetallischer Erde, Schwefel, Arsenic, Silber, Gold, einfachen Theilgen, Vitriol und Schmeltz-Nutzung” [Pyritologia, or: Pyrite History: As the Most Important Mineral, According to its Name, Types, Deposits, Origin, Iron, Copper, Non-Metallic Earth, Sulfur, Arsenic, Silver, Gold, Simple Elements, Vitriol, and Smelting Use] [Internet]. Leipzig: Johann Christian Martini; 1725. https://www.digitale-sammlungen.de/en/details/bsb10283779. Accessed 5 Sep 2023.
Boerhaave H. Elementa chemiae: quae anniversario labore docuit in publicis, privatisque, scholis Hermannus Boerhaave [Elements of Chemistry: Taught annually in public and private schools by Hermann Boerhaave]. Leiden: Severinus, Isaak; 1732.
Sarmento J de C. Materia medica physico-historico-mechanica. Reyno mineral. london; 1735.
Beuther D. Davids Beuthers, der Medicin Doctoris Universal, und Vollkommener Bericht, Von der hochberümbten Kunst der Alchymj und seinen in solcher erlangten, und erkundigten Secreten. und Kunststücklein: Daraus die Gewißheit und Perfection dieser vortrefflichen Kunst zuerkennen. Sampt beygefügtem Gespräch, von Betrug und Irrweg, etlicher unerfahrnen Laboranten, so sich betrieglich vor Alchymisten dargeben [David Beuthers, the Doctor of Medicine’s Universal, and Complete Account, On the Highly Renowned Art of Alchemy and his Attained and Discovered Secrets. and Artifices: From Which the Certainty and Perfection of this Excellent Art Can be Recognized. Along with an Added Conversation, on Deception and Misdirection, of Some Inexperienced Laborers, Who Deceitfully Present Themselves as Alchemists]. Fitzer; 1631.
Collegio medico di Firenze. Ricettario fiorentino di nuovo illustrato [Florentine Recipe Book Newly Illustrated] [Internet]. Marescotti, Giorgio, fl.; 1597. http://archive.org/details/hin-wel-all-00000667-001. Accessed 10 Jul 2023.
Libavius A. Alchymia Andreæ Libavii : Recognita, Emendata, Et aucta, tum dogmatibus & experimentis nonnullis : Tvm Commentario Medico Physico Chymico, Qvi Exornatvs Est Variis Instrumentorum Chymicorum picturis : partim aliunde translatis, partim plane nouis ... [Andreas Libavius’ Alchemy: Recognized, Corrected, and Augmented, with Some Doctrines and Experiments: Also with a Medical-Physical-Chemical Commentary, Embellished with Various Pictures of Chemical Instruments: Some Translated from Elsewhere, Some Entirely New] [Internet]. Frankfurt: Excudebat Joannes Saurius; 1606. http://sammlungen.ub.uni-frankfurt.de/drucke/11852207. Accessed 6 Sep 2023.
Pott JH. Exercitationes chymicae, de sulpphuribus metallorum, de auripigmento, de solutione corporum particulari, de terra foliata tartari, de acido vitrioli vinoso et de acido nitri vinoso; sparsim hactenus editae, jam vero collectae restitutae a mendis repurgatae ... illustratae [Chemical Exercises, on the Sulfides of Metals, on Orpiment, on the Dissolution of Specific Bodies, on Tartar Foliata Earth, on Vinous Vitriolic Acid and on Vinous Nitric Acid; Scattered thus far, now Collected, Restored from Errors, Purged... and Illustrated]. Berlin: Rüdigerus; 1738.
Charas M. Pharmacopée royale galénique et chymique [Royal Galenic and Chemical Pharmacopoeia]. L’Auteur; 1676.
Académie des sciences. Mémoires de mathématique et de physique, Présentés à l’Académie Royale des Sciences, par divers Sçavans, et lûs dans Ses Assemblées: Tome Premier [Memoirs of Mathematics and Physics, Presented to the Royal Academy of Sciences by Various Scholars, and Read at its Meetings: Volume One] [Internet]. Paris: De L’imprimerie Royale; 1750. https://gallica.bnf.fr/ark:/12148/bpt6k3478z
Lemery N (1645–1715) A du texte. Dictionnaire universel des drogues simples ... ouvrage dépendant de la “Pharmacopée universelle”, par feu M. Lemery,... 3e édition... [Universal Dictionary of Simple Drugs ... a work related to the ‘Universal Pharmacopoeia’, by the late Mr. Lemery,... 3rd edition...] [Internet]. Paris: impr. de la Vve d’Houry; 1733. https://gallica.bnf.fr/ark:/12148/bpt6k9667536f. Accessed 10 Jul 2023.
Farvacques R de. Medicina pharmaceutica, of groote algemeene schatkamer der drôgbereidende geneeskonst [Medicina pharmaceutica, or the Great General Treasury of Drug-Preparing Medicine] [Internet]. Leiden: I. Severinus; 1741. http://archive.org/details/b30411154. Accessed 30 Jun 2023.
Lémery N, Putten C van, Witt I de. Woordenboek of algemeene verhandeling der enkele droogeryen ... : in ’t Fransch beschreven door den heer Nicolaes Lemery [Dictionary or General Treatise on Simple Drugs ... : Described in French by Mr. Nicolaes Lemery] [Internet]. Rotterdam: Jan Daniel Beman; 1743. http://archive.org/details/woordenboekofalg00leme. Accessed 30 Jun 2023.
Wallerius JG. Systema mineralogicum, quo corpora mineralia in classes, ordines, genera et species, suis cum varietatibus divisa, describuntur, atque observationibus, experimentis et figuris aeneis illustrantur A Johan. Gotsch. Wallerio [Systema Mineralogicum, in which Mineral Bodies are Divided into Classes, Orders, Genera, and Species, with their Varieties, Described, and Illustrated with Observations, Experiments, and Copper Engravings by Johan. Gotsch. Wallerio]. Vienna: Ex officina Krausiana; 1778.
Rötter C, editor. Auripigment, Orpiment: Studien zu dem Mineral und den künstlichen Produkten = Studies on the mineral and the artifical products. München: Siegl; 2007.
Vermeulen M. Natural and amorphous arsenic sulfide pigments : characterization, degradation and influence of the binding medium. [Antwerpen]: Universiteit Antwerpen; 2017.
Vermeulen M, Palka K, Vlček M, Sanyova J. Study of dry- and wet-process amorphous arsenic sulfides: Synthesis, Raman reference spectra, and identification in historical art materials. J Raman Spectrosc. 2019;50:396–406.
Keune K, Mass J, Mehta A, Church J, Meirer F. Analytical imaging studies of the migration of degraded orpiment, realgar, and emerald green pigments in historic paintings and related conservation issues. Heritage Science. 2016;4:10.
Keune K, Mass J, Meirer F, Pottasch C, van Loon A, Hull A, et al. Tracking the transformation and transport of arsenic sulfide pigments in paints: synchrotron-based X-ray micro-analyses. J Anal At Spectrom. 2015;30:813–27.
Vermeulen M, Nuyts G, Sanyova J, Vila A, Buti D, Suuronen J-P, et al. Visualization of As( iii ) and As( v ) distributions in degraded paint micro-samples from Baroque- and Rococo-era paintings. J Anal At Spectrom. 2016;31:1913–21.
Broers FTH, Janssens K, Nelson Weker J, Webb SM, Mehta A, Meirer F, et al. Two pathways for the degradation of orpiment pigment (As2S3) found in paintings. J Am Chem Soc. 2023;145:8847–59.
Jovanovski G, Makreski P. Intriguing minerals: photoinduced solid-state transition of realgar to pararealgar—direct atomic scale observation and visualization. ChemTexts. 2020;6:5.
Pratesi G, Zoppi M. An insight into the inverse transformation of realgar altered by light. Am Miner. 2015;100:1222–9.
Trentelman K, Stodulski L, Pavlosky M. Characterization of pararealgar and other light-induced transformation products from realgar by raman microspectroscopy. Anal Chem. 1996;68:1755–61.
Grundmann G, Ivleva N, Richter M, Stege H, Haisch C. The rediscovery of sublimed arsenic sulphide pigments in painting and polychromy: applications of Raman microspectroscopy. The National Gallery Technical Bulletin 30th Anniversary Conference, London, UK, 2009. 2011;269–76.
Wallert A, Berg A van den. Still lifes: techniques and style: an examination of paintings from the Rijksmuseum. Amsterdam, Zwolle: Rijksmuseum Waanders; 1999.
Erdmann RG. 717 gigapixel visible light composite image (5 µm resolution) for Rembrandt’s Night Watch [Internet]. 2021. https://images.erdmann.io/curtain.html?i=Rijksmuseum/SK-C-5/SK-C-5_VIS_5-um_2020-09-08[l%3DVIS.5.µm], published online 2021-09-23
Roy A, editor. Artists’ pigments: a handbook of their history and characteristics, Volume 2. Reprinted. National Gallery of Art, Washington, Archetype Publications Ltd., London; 2012.
Wu F, Zhang Y, Li Y, Wang Y, Ma J, Wang F. Photoinduced effects of monochromatic visible light with different wavelengths on realgar. J Raman Spectroscopy. 2022;53:1533–9.
Simoen J, De Meyer S, Vanmeert F, de Keyser N, Avranovich E, Van der Snickt G, et al. Combined Micro- and Macro scale X-ray powder diffraction mapping of degraded Orpiment paint in a 17th century still life painting by Martinus Nellius. Heritage Science. 2019;7:83.
Vanmeert F, de Keyser N, van Loon A, Klaassen L, Noble P, Janssens K. Transmission and reflection mode macroscopic x-ray powder diffraction imaging for the noninvasive visualization of paint degradation in still life paintings by Jan Davidsz. de Heem. Anal Chem. 2019;91:7153–61.
De Keyser N, Broers F, Vanmeert F, De Meyer S, Gabrieli F, Hermens E, et al. Reviving degraded colors of yellow flowers in 17th century still life paintings with macro- and microscale chemical imaging. Sci Adv. 2022;8:eabn6344.
Monico L, Prati S, Sciutto G, Catelli E, Romani A, Quintero Balbas D, et al. Development of a multi-method analytical approach based on the combination of synchrotron radiation X-ray micro-analytical techniques and vibrational micro-spectroscopy methods to unveil the causes and mechanism of darkening of “fake-gilded” decorations in a Cimabue painting. J Anal At Spectrom. 2022;37:114–29.
Magalhães MCF, Silva MCM. Stability of Lead(II) arsenates. Monatsh Chem. 2003;134:735–43.
Broers FTH, Verslype I, Bossers KW, Vanmeert F, Gonzalez V, Garrevoet J, et al. Correlated x-ray fluorescence and ptychographic nano-tomography on Rembrandt’s The Night Watch reveals unknown lead “layer.” Sci Adv. 2023;9:9394.
Gonzalez V, Fazlic I, Cotte M, Vanmeert F, Gestels A, De Meyer S, et al. Lead(II) formate in Rembrandt’s night watch: detection and distribution from the macro- to the micro-scale. Angew Chem Int Edn. 2023. https://doi.org/10.1002/anie.202216478.
Mirazimi M, Mohammadi M, Liu W. Kinetics and mechanisms of arsenic and sulfur release from crystalline orpiment. Miner Eng. 2021;170: 107032.
Mirazimi M, Fan J, Liu W. Kinetics of arsenic and sulfur release from amorphous arsenic trisulfide. Hydrometallurgy. 2021;200: 105555.
Glauber JR, French J. A description of new philosophical furnaces, or, A new art of distilling, divided into five parts : whereunto is added a description of the tincture of gold, or the true aurum potabile : also the first part of the mineral work : set forth and published for the sakes of them that are studious of the truth [Internet]. London : Printed by Richard Coats, for Tho. Williams ...; 1651. http://archive.org/details/descriptionofnew00glau. Accessed 12 Aug 2022.
Winkelman PH, editor. Bronnen tot de geschiedenis van den Oostzeehandel [Sources for the History of the Baltic Sea Trade] [Internet]. Huygens institute; 1971. https://resources.huygens.knaw.nl/retroboeken/oostzeehandel/#page=65&accessor=toc&source=4. Accessed 30 Jun 2023.
Prak M. The Dutch Republic in the Seventeenth Century: The Golden Age. Cambridge University Press; 2005. https://doi.org/10.1017/CBO9780511817311
L’Espine JLM de. Le negoce d’Amsterdam, ou Traité de sa banque, de ses changes, des Compagnies orientales, & occidentales, des marchandises que l’on en tire, & que l’on y apporte des plus considérables villes de l’Europe, & des autres parties du monde, & de leurs poids & mesures [The Commerce of Amsterdam, or Treatise on its Bank, its Exchanges, the East and West India Companies, the Goods traded there, and brought from the most significant cities of Europe and other parts of the world, and their weights and measures]. chez Pierre Brunel; 1694.
Le Moine de l’Espine J, Le Long I. De koophandel van Amsterdam, naar alle gewesten des werelds. Bestaande, in een verhandeling, van de waaren en koopmenschappen, die men daar heenen sendt en wederom ontfangt: benevens vergelykingen der munten, maaten en gewigten, en op wat wyse men over en weër wisselt .. [The Trade of Amsterdam, to all Regions of the World. Consisting of a Treatise on the Goods and Merchandise sent there and received back, along with comparisons of currencies, measures, and weights, and how exchange is conducted] [Internet]. Rotterdam, Ph. Losel; 1753 http://archive.org/details/dekoophandelvan01lemo. Accessed 10 Aug 2022.
Savary Des Bruslons J. Dictionnaire universel de commerce, contenant tout ce qui concerne le commerce qui se fait dans les quatre parties du monde. Tome 2 / ... Ouvrage posthume du Sr Jacques Savary Des Bruslons... continué... et donné au public, par Philémon-Louis Savary,... [Universal Dictionary of Commerce, containing everything concerning the commerce conducted in the four parts of the world. Volume 2 / ... Posthumous work of Mr. Jacques Savary Des Bruslons... continued... and published, by Philémon-Louis Savary] [Internet]. 4 vol.; in-fol. les héritiers Cramer et les frères Philibert, editor. Genève: les héritiers Cramer et les frères Philibert; 1744. https://gallica.bnf.fr/ark:/12148/bpt6k5656941q. Accessed 14 Apr 2023.
Buys E. Nieuw en volkomen woordenboek van konsten en weetenschappen; bevattende alle de takken der nuttige kennis, met naaukeurige beschryvingen, zo van de onderscheidene machines, werktuigen, gereedschappen, figuuren, en ontwerpen dienende om dezelve op te helderen; als meede van de klassen, soorten, toebereidselen, en het gebruik van de voortbrenzels der natuur, het zy dieren, planten, mineraalen, aardgewassen, of vochten; mitsgaders de koningryken, provintien, steden, dorpen, en andere merkwaardige plaatzen door de geheele waereld [New and complete dictionary of arts and sciences; containing all branches of useful knowledge, with accurate descriptions of various machines, tools, instruments, figures, and designs used to illustrate them; as well as of the classes, types, preparations, and uses of natural products, whether animals, plants, minerals, vegetables, or fluids; as well as the kingdoms, provinces, cities, villages, and other remarkable places throughout the world]. By S. J. Baalde; 1777.
Alston C. Lectures On The Materia Medica: Containing The Natural History of Drugs, Their Virtues And Doses .... In Two Volumes. Edward Dilly; 1770.
Karl Cäsar Leonhard CC “von ” L. Hütten-erzeugnisse und andere auf künstlichem Wege gebildete Mineralien als Stützpuncte ... [Metal Products and Other Minerals Formed Artificially as Reference Points] [Internet]. Schweizerbart; 1858. http://archive.org/details/httenerzeugniss00leongoog. Accessed 30 Jun 2023.
Buonanni F. Neuer Tractat von Firniß- Laquir und Mahler-Künsten [New Treatise on Varnish, Lacquer, and Painting Arts]. Breßlau und Leipzig: Pietsch; 1746.
Seccaroni C, Haldi J-P. Cobalto, zaffera, smalto dall’antichità al XVIII secolo [Cobalt, Zaffre, Enamel from Antiquity to the XVIII Century]. Rome: Enea; 2016.
Berrie BH. Mining for color: new blues, yellows, and translucent paint. Early Sci Med. 2015;20:308–34.
Grundmann G, Modl D, Bojar H-P, Gilg H. Die Arsenhütte Zuckenhut/Strassegg (Steiermark Österreich) Archäologischer Erstnachweis der Produktion künstlicher Arsensulfid-Pigmente [The Arsenic Works in Zuckenhut/Strassegg (Styria, Austria): Archaeological First Evidence of the Production of Artificial Arsenic Sulfide Pigments]. Metalla. 2009;240–5.
Krischel R. The Inventory of the Venetian ‘Vendecolori’Jacopo de’Benedetti: The Non-Pigment Materials. Kirby et al, Trade in Artists’ Materials. 2010;253–66.
Batur K, Radić RI. Archaeological evidence of Venetian trade in colouring materials: the case of the Gnalić shipwreck. Trading paintings and painters’ materials. 2019;1550–800.
Erneuerte und verbesserte Medicinal- und Apotheker Ordnung, Des Durchleuchtigsten Fürsten ... Johann-Ernstens, Hertzogs zu Sachsen, Jülich, Cleve und Berg ... Samt beygefügter Taxa, derer, in der privilegirten Apotheken zu Weimar befindlichen Wahren u. Artzeneyen zu Jedermans Wissenschaft und Nachricht in Druck gegeben, den 19. Novembris 1673 [Renewed and Improved Medicinal and Apothecary Regulations, of the Most Serene Prince Johann-Ernst, Duke of Saxony, Jülich, Cleve, and Berg ... Together with the Attached Price List of Goods and Medicines in the Privileged Pharmacies in Weimar, Printed for Everyone’s Knowledge and Information, on November 19, 1673] [Internet]. Weimar: Müller; 1674. http://resolver.sub.uni-goettingen.de/purl?PPN396385885
Advertentie. Amsterdamse courant [Internet]. Dag. 1745 Jun 3. https://resolver.kb.nl/resolve?urn=ddd:010714662:mpeg21:a0009. Accessed 4 Jul 2023.
Københavns universitet Lægevidenskabelige fakultet. Catalogus&valor medicamentorum simplicium&compositorum in officinis Hafniensibus prostantium. Apotecker Taxt paa alt hvis i Kiøbenhaffn hos de fire privilegerede Apotheckere til Kiøbs findis, etc. [Edited by T. Bartholinus.] Lat., Dan.&Ger [Catalog and Value of Simple and Compound Medicines available in the Pharmacies of Copenhagen. Price list for everything found in the four privileged pharmacies in Copenhagen]. P. Haubold; 1672.
Hallema A. Dokters en apothekers te Utrecht in de 17e eeuw. Twee merkwaardige ordonnanties van 1655 betreffende doktershonoraria [Doctors and Pharmacists in Utrecht in the 17th Century. Two Remarkable Ordinances from 1655 Regarding Doctors’ Fees]. Ned Tijdschr Geneeskd. 1956;100:2914–9.
Baarsen RJ, Cadée G, van Gelder R, van Duin P, Bierman AI, van der Brugge-Mulder JJ. Collector’s Cabinet with Miniature Apothecary’s Shop. van Duin P, editor. Amsterdam: Rijksmuseum; 2017.
Werner AG [Hrsg. Ausführliches und systematisches Verzeichnis des Mineralien-Kabinets des weiland kurfürstlich sächsischen Berghauptmans Herrn Karl Eugen Pabst von Ohain, der Leipziger und St. Petersburger ökonomischen Gesellschaft Mitgliede, und der königlich sardinischen Gesellschaft der Wissenschaften zu Tur [Detailed and Systematic Catalog of the Mineral Cabinet of the late Electoral Saxon Chief Mining Officer Mr. Karl Eugen Pabst von Ohain, Member of the Leipzig and St. Petersburg Economic Societies, and of the Royal Sardinian Society of Sciences in Turin] [Internet]. Freiberg und Annaberg: Craz; 1791. https://doi.org/10.11588/diglit.2527
Clark RJH, Gibbs PJ. Identification of lead(II) sulfide and pararealgar on a 13th century manuscript by Raman microscopy. Chem Commun. 1997. https://doi.org/10.1039/a701837a
Burgio L, Clark RJH. Comparative pigment analysis of six modern Egyptian papyri and an authentic one of the 13th centuryBC by Raman microscopy and other techniques. J Raman Spectrosc. 2000;31:395–401.
Rosalie David A, Edwards HGM, Farwell DW, De Faria DLA. Raman spectroscopic analysis of ancient egyptian pigments. Archaeometry. 2001;43:461–73.
Burgio L, Clark RJH, Theodoraki K. Raman microscopy of Greek icons: identification of unusual pigments. Spectrochim Acta Part A Mol Biomol Spectrosc. 2003;59:2371–89.
Burgio L, Clark RJH, Muralha VSF, Stanley T. Pigment analysis by Raman microscopy of the non-figurative illumination in 16th- to 18th-century Islamic manuscripts. J Raman Spectroscopy. 2008;39:1482–93.
Wu M, Zhang Y, Zhang B, Li L. Study of colored lacquerwares from Zenghou Yi Tomb in Early Warring States. New J Chem. 2021;45:9434–42.
Zuena M, Fondi V, Adembri B, Della Ventura G, Sodo A. A multidisciplinary approach to investigate the peculiar artistic technique of Scagliola: modern age decorations in Hadrian’s Villa (Tivoli, Italy). Vib Spectrosc. 2022;123: 103465.
Mahon D, Centeno SA, Wypyski MT, Salomon XF, Bayer A. Technical Study of Three Allegorical Paintings by Paolo Veronese: “The Choice between Virtue and Vice, Wisdom and Strength,” and “Mars and Venus United by Love.” Metropolitan Museum studies in art, science and technology. Metropolitan Museum of Art; 2010.
Dunkerton J, Spring M, Billinge R, Kalinina K, Morrison R, Macaro G, et al. Titian’s painting technique before 1540. London: National Gallery; 2013.
Roberts AC, Ansell HG, Bonardi M. Pararealgar, a new polymorph of AsS, from British Columbia. Can Mineral. 1980;18:525–7.
Smith P, Rosenkranz N, Uchacz T, Taape T, Goodbarge C, Pitman S, et al. Secrets of craft and nature in renaissance France: a digital critical edition and english translation of BnF Ms. Fr. 640. 2020;
Bomford D, Kirby J, Roy A, Rüger A, White R. Art in the making: Rembrandt. New Haven: Yale University Press; 2006.
Alfeld M, Janssens K. Strategies for processing mega-pixel X-ray fluorescence hyperspectral data: a case study on a version of Caravaggio’s painting Supper at Emmaus. J Anal At Spectrom. 2015;30:777–89.
Solé VA, Papillon E, Cotte M, Walter Ph, Susini J. A multiplatform code for the analysis of energy-dispersive X-ray fluorescence spectra. Spectrochim Acta Part B. 2007;62:63–8.
van Loon A, Keune K, Boon JJ. Improving the Surface Quality of Paint Cross-sections for Imaging Analytical Studies with Specular Reflection FTIR and Static-SIMS. art’05—8th International Conference on “Non Destructive Investigations and Micronalysis for the Diagnostics and Conservation of the Cultural and Environmental Heritage.” Lecce (Italy); 2005.
Cotte M, Gonzalez V, Vanmeert F, Monico L, Dejoie C, Burghammer M, et al. The “Historical Materials BAG”: a new facilitated access to synchrotron x-ray diffraction analyses for cultural heritage materials at the european synchrotron radiation facility. Molecules. 2022;27:1997.
Riekel C, Burghammer M, Davies R. Progress in micro- and nano-diffraction at the ESRF ID13 beamline. IOP Conf Ser: Mater Sci Eng. 2010;14: 012013.
De Nolf W, Vanmeert F, Janssens K. XRDUA: crystalline phase distribution maps by two-dimensional scanning and tomographic (micro) X-ray powder diffraction. J Appl Cryst. 2014;47:1107–17.
Acknowledgements
Susan Smelt and Anna Krekeler are acknowledged for their help in the collection of MA-XRF scans. Robert Erdmann is acknowledged for the execution, processing, stitching and registration of the 5-μm photography of The Night Watch[75]. Leila Sauvage is acknowledged for her help with the execution of the 5-μm photography. Arie Wallert is acknowledged for the sampling and embedding of paint sample SK-A-199_R9/4. Marine Cotte is acknowledged for help during the BAG beamtime at ESRF. Ken Vidar Falch and Jan Garrevoet are acknowledged for their help during the beamtime at DESY. We would like to thank Günter Grundmann for an insightful discussion regarding pararealgar and amorphous arsenic sulfides. Team Operation Night Watch is thanked for their collaboration and input during this research.
Funding
The ESRF beamtime was granted through the peer-reviewed BAG proposal HG-172 at ID13. The Historical materials BAG has been implemented with support from the European Union's Horizon 2020 research and innovation programme under grant agreement No 870313, Streamline. We acknowledge DESY (Hamburg, Germany), a member of the Helmholtz Association HGF, for the provision of experimental facilities. Parts of this research were carried out at PETRA III. Beamtime was allocated for proposal I-20190926. This research was supported in part through the Maxwell computational resources operated at the Deutsches Elektronen-Synchrotron DESY, Hamburg, Germany. The research leading to this result has been supported by the project CALIPSOplus under the Grant Agreement 730872 from the EU Framework Programme for Research and Innovation HORIZON 2020. This project was supported by the Netherlands Institute for Conservation+Art+Science+(NICAS) and the Dutch Research Council (NWO) (3D2P project, project number 628.007.031). The main partner of Operation Night Watch is AkzoNobel. Operation Night Watch is made possible by The Bennink Foundation, C.L. de Carvalho-Heineken, PACCAR Foundation, Piet van der Slikke & Sandra Swelheim, American Express Foundation, Familie De Rooij, Het AutoBinck Fonds, TBRM Engineering Solutions, Dina & Kjell Johnsen, Familie D. Ermia, Familie M. van Poecke, Bruker Nano Analytics, Henry M. Holterman Fonds, Irma Theodora Fonds, Luca Fonds, Piek-den Hartog Fonds, Stichting Zabawas, Cevat Fonds, Johanna Kast-Michel Fonds, Marjorie & Jeffrey A. Rosen, Stichting Thurkowfonds, Familie Van Ogtrop Fonds, FedEx Express, Airbnb, NICAS, the Night Watch Fund, the City of Amsterdam and the Amsterdam Museum. The University of Antwerp acknowledges support through Interreg project Smart*Light 2.0.
Author information
Authors and Affiliations
Contributions
The initial manuscript was written by NDK & FB; Conceptualization: NDK, FB, KK; data acquisition/processing: Sample taking (AvL, PN), Sample preparation and light microscopy (AvL), SEM–EDX (NDK, AvL, FB), Raman spectroscopy (NDK, FB, AvL), SR-micro-XPRD acquisition and processing at P06 (FB, VG, AG, SDM) at ESRF for SK-C-5_017 (FV, FB, NDK), research into the historical sources (NDK). NDK, FB, FV, AvL, FG, SDM, AG, VG, EH, PN, FM, KJ and KK have read, revised and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Competing interests
Authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
40494_2024_1350_MOESM1_ESM.docx
Supplementary Material 1. Supporting Information PDF including additional images, Raman spectroscopy data, XRD data and an overview of historical sources.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
De Keyser, N., Broers, F.T.H., Vanmeert, F. et al. Discovery of pararealgar and semi-amorphous pararealgar in Rembrandt's The Night Watch: analytical study and historical contextualization. Herit Sci 12, 237 (2024). https://doi.org/10.1186/s40494-024-01350-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40494-024-01350-x