
Abstract
Microbial communities in tomb soil change during archaeological excavation, and these changes can accelerate the deterioration of buried heritage. In this study, a high-throughput sequencing method was used to analyze the soil microbial diversity of tomb M88 from the Sujialong Cultural Property after careful excavation in the laboratory. The phylum of Proteobacteria, Acidobacteria and Ascomycota predominated in the tomb soil, and the dominant genera, Pseudarthrobacter, Penicillium, and Cladosporium, showed the potential to degrade residual relics in the tomb soil. These findings will help to explore the process of microbial degradation in buried heritage during long-term archaeological excavation and improve careful excavation procedures in the laboratory for further conservation. Additionally, numerous plant species were identified in the tomb soil, and most of the plants belonged to the native vegetation species. However, the identified dominant species, Glycine, Angelica, and Hippophae, should be from residual heritage rather than native species, which may provide clues for the study of funeral customs and cultural exchange in the Zeng State.
Similar content being viewed by others

Introduction
Numerous archaeological and cultural heritage is preserved in the soil, particularly in the tombs. It is known that burial conditions, especially microorganisms in the soil, play a crucial role in the deterioration of archaeological heritage [1,2,3,4,5,6]. Research on the degradation of buried heritage caused by microorganisms has been reported [3,4,5,6], but most of these studies focus on excavated heritage, such as textiles and woods. Only a few studies focused on the microorganisms in tomb soil [1, 2], and samples in these studies were collected prior to the excavation. This is because the tombs generally maintain an environment cut off from the external environment [1]. During excavation, the burial environment changes, and microorganisms from the external environment can infiltrate and alter the microbial communities in the tomb soil [1, 6]. These studies overlook the fact that excavation usually lasts for a long time, which means that the buried heritage cannot be excavated and preserved immediately. The original microbial community in the soil will be contaminated by introduced environmental microorganisms, and the growth of the invading microorganisms may accelerate the deterioration of cultural heritage [7], but few studies have investigated this process.
The identification of microbial communities is now commonly carried out by high-throughput sequencing, and this method is also used to recognize archaeological plant remains [8,9,10,11,12,13]. Kistler et al. [8] reported that high-throughput sequencing could retrieve ancient plant communities from small amounts of bulk sediment samples spanning a long-time period, and that the species of desiccated, waterlogged, or charred archaeological plant remains could also be identified with this method [11,12,13].
Careful excavation in the laboratory, in which the soil containing the tomb is extracted and excavated in the laboratory, is becoming popular in archaeological excavation because of its advantages in recovering more data on the funerary rituals of the time and preserving fragile buried cultural heritage. However, careful excavation usually takes longer than traditional excavation, which means that the cultural heritage may suffer more deterioration caused by soil microorganisms. The tomb M88, which was also called Mi Ke tomb based on the tomb owner, used in this study is an example of careful excavation in the laboratory. It was excavated from the Sujialong Cultural Property, Jingshan county, Hubei province, China in 2016 and belonged to the Zeng State (sixth century BC). Numerous tiny jades were found in the coffin area, and there were some bronzewares found between the coffin and the outer coffin. In order to collect more information about the tomb and protect excavated relics, the soil mass of the coffin area was extracted from the tomb and transferred to the laboratory of the Institute of Cultural Heritage, Shandong University in 2017. Frequent water spraying to prevent soil mass from drying out and a relatively stable environment encourage the invasion and growth of exogenous microorganisms. The high humidity of soil usually promotes the growth of microorganisms, which can accelerate the degradation of buried cultural heritage [14]. It is not yet known which specific species are supported by these conditions or whether they pose a risk to residual relics in the soil.
Within this research, we identified the microbial communities in the tomb M88 soil using high-throughput sequencing. The aim of this study was to clarify the distribution and diversity of the microbial community in tomb soil after long-term excavation, and then to explore the influence of microorganisms on the degradation of residual heritage. In addition, various plants and residual heritage of plant origin are present in the tomb, we also wanted to identify theses items from the data of high-throughput sequencing. These findings would inform future improvements in laboratory excavation, and provide some new clues to the study of the funerary system of the Zeng State.
Materials and methods
Site description and soil sampling
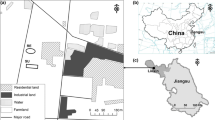
The tomb M88 is located in the south of the Sujialong Cultural Property, which contains many Zeng State tombs. All the tombs in the area are at a shallow depth from the surface, the maximum depth from the surface to the bottom of the tomb is about 200 cm. After a year of archaeological excavation, the coffin area of tomb M88 was excavated. Then, the soil mass of the coffin area, with a volume of 220 × 101 × 40 cm (Additional file 1: Fig. S1), was removed and transferred to the laboratory of the Institute of Cultural Heritage, Shandong University for careful excavation over a period of 3 years (2017–2020). During this period, the soil mass was maintained in a humid environment through spraying water to prevent cracking. Nine soil samples were collected in July 2020. The sampling sites are shown in Fig. 1. Soil samples L1, L2, L3, R1, R2, and R3 were symmetrically shaped and contained no visible residual sediments related to the tomb. The six samples were collected at a depth of 3 cm to avoid contamination with environmental microorganisms. Samples were also collected from residual relics, as follows: sample H was collected from the predicted location of the occupant’s headwear (the head area of the tomb owner), sample S was collected from a relic with an obvious fabric imprint (Fig. 2), and sample P contained material with paper-like consistency and appearing as stripes (Fig. 3).

The planform of the soil mass of the tomb M88. a The real condition of the soil; b the position of the sampling sites

The microstructure of the sample S. The red bar is 200 μm

The microstructure of the sample P. The red bar is 250 μm
Molecular analyses
DNA extraction was carried out with 0.5 g of soil sample using a HiPure Soil DNA Kit (Magen, Guangdong, China) according to the manufacturer’s instructions. High-throughput sequencing was conducted by GENEWIZ (Jiangsu, China). The V3 and V4 hypervariable regions of prokaryotic 16S rDNA were selected for the generation of amplicons, which were subjected to subsequent taxonomic analysis, with amplicon regions amplified using the forward primer “5′-CCTACGGRRBGCASCAGKVRVGAAT-3′” and the reverse primer “5′-GGACTACNVGGGTWTCTAATCC-3”. The ITS2 hypervariable region of eukaryotic ITS rDNA was targeted to generate amplicons for subsequent taxonomic analysis, with the ITS2 region amplified using a forward primer containing the sequence “5′-GTGAATCATCGARTC-3” and a reverse primer containing the sequence “5′-TCCTCCGCTTATTGAT-3”. PCR reactions were performed in triplicate in a 25 µL mixture containing 2.5 µL of TransStart Buffer, 2 µL of dNTPs, 1 µL of each primer and 20 ng of template DNA. DNA library concentrations were validated and quantified using a Qubit 3.0 Fluorometer (Thermo Fisher, USA), and each was then adjusted to a concentration of 10 nM. DNA libraries were multiplexed and loaded on an Illumina MiSeq according to the manufacturer’s instructions (Illumina, San Diego, CA, USA). Paired-end sequencing was performed followed by image analysis and base calling, which were conducted using the Control Software provided with the instrument.
The QIIME data analysis package was used for 16S and ITS rRNA data analysis. The forward and reverse reads were joined and assigned to samples based on barcodes and truncated by cutting off the barcode and primer sequence. Quality filtering of joined sequences was performed and sequences that did not fulfill the following criteria were discarded: sequence length > 200 bp, no ambiguous bases, mean quality score ≥ 20. The sequences were then compared to sequences in the reference database (RDP Gold database) using the UCHIME algorithm to detect chimeric sequences, followed by their exclusion from further analysis.
The remaining sequences were grouped into operational taxonomic units (OTUs) using the clustering program VSEARCH (1.9.6) against the Silva database for 16 S sequences and the UNITE ITS database (https://unite.ut.ee/) for ITS sequences based on a 97% sequence identity cut-off threshold. The Ribosomal Database Program (RDP) classifier, which predicts the taxonomic category down to the species level, was used to assign taxonomic categories to all OTUs using a confidence threshold of 0.8. Unclassified sequences identified using the Silva and UNITE databases were also aligned against the NCBI database. The sequences with more than 97% sequence identity were downloaded, and a phylogenetic tree was constructed using MEGA-X for further identification. Alpha diversity indices, such as the ACE, Shannon, Chao1 and Simpson indices, were calculated to assess the community diversity and enrichment of microorganisms. Beta diversity was calculated using weighted and unweighted UniFrac analysis. Based on Bray-Curtis distances and results of principal coordinates analysis (PCoA), unweighted pair group method with arithmetic mean (UPGMA) clustering trees were constructed for the different samples. Raw sequences were deposited in the NCBI Sequence Read Archive (SRA) database (accession numbers: SAMN20587851, SAMN20587867, SAMN20587868, SAMN20587885, SAMN20587886, SAMN20587899, and SAMN20587922 - SAMN20587924).
Characterization analysis
Based on the close distance of the samples L2 and R2 from the visible residual sediments, only L1, L3, R1, and R3 were selected to analyze the tomb soil pH to avoid the influence of the residual sediments. The four samples were dried to constant weight and ground with a mortar and pestle. The pH measurement was performed with reference to the international standard [15]. However, the weight of each dried sample could not meet the minimum requirements of the pH measurement, so, L1 and R1, which were relatively similar, were mixed, as well as L3 and R3. One gram of mixed soil and 5 g of H2O were stirred for 60 min and allowed to stand for 90 min. Next, pH electrodes (Mettler Toledo S210-K, Swiss Confederation) were inserted into the supernatant, and the pH value was recorded. The pH of each sample was measured three times, and the soil used to cover the soil mass during transport was selected as the control.
We observed all samples visually, but only the samples S and P showed the imprint of residual heritage, so the two samples were analyzed by microscopic techniques to determine the micromorphological characteristics of the residual heritage. The micro-morphological characteristics of samples S and P were observed using an Ultra-Depth Three-Dimensional Microscope, DVM6 (Leica, Germany). They were then analyzed with a Renishaw inVia Confocal Raman Microscope (UK) at 532 and 785 nm excitation wavelength with an exposure time of 10 s/spectrum over five accumulations. Sample P was also examined by scanning electron microscopy (SEM) using a Quattro S device (Thermo Fisher, USA) equipped with a backscattered electron detector (BSED) and an energy-dispersive spectroscopic (EDS) probe. Each specimen was analyzed in low-vacuum mode using an accelerating voltage of 5 kV for SEM and 15 kV for EDS.
Results
Characteristics of tomb M88 soil
The pH values of the tomb soil were close to neutral, with mean pH values of 7.21 (mixed sample of L1 and R1), 6.78 (mixed sample of L3 and R3), and 7.28 (control sample).
Sample S exhibited an obvious plain weave-like structure of variable density (Fig. 2), and our previous study confirmed it was consisting of silk, which indicating that it was derived from fabric [16]. Sample P also exhibited surface imprinting, but no silk fibroin was detected in this sample, and the imprint pattern resembled the natural texture of plants rather than the structure of fabric (Fig. 3). The backscatter electron image (Additional file 1: Fig. S2) and the distribution of elements (Additional file 1: Fig. S3) both indicated the consistency of the structural components throughout the sample. High C and O content in the sample suggested that the sample was mainly composed of organic compounds. These results of microscopy techniques suggested that the residues in sample P were of plant origin. The enrichment of Hg and S in the red spots and the Raman characteristic peaks at 250 cm−1 and 342 cm−1 indicated that the red material consisted of cinnabar, while the white areas were composed of Al and Si, which indicated that these areas could be kaolin (Additional file 1: Fig. S4).
General analyses of microbial community diversity
A total of 1,320,926 valid sequence reads remaining after chimera removal were obtained from the nine samples, yielding a total of 438,358 OTUs from 16 S rDNA sequences and 718,573 OTUs from ITS rDNA sequences. Within individual samples from the total set of nine samples, the numbers of OTUs within 16 S rDNA sequences ranged from 23,248 to 71,743 per sample, while the OTU numbers for ITS rDNA sequences ranged from 42,998 to 118,627 per sample. Good’s coverage threshold was more than 97% for all samples, indicating that most of the diversity was captured. Community richness and diversity were evaluated for the nine samples using alpha diversity index-based analysis based on the Chao1, ACE, Shannon and Simpson indices (Additional file 1: Table S1). The results showed that the number of eukaryote species was lower than that of prokaryote species in each sample. Notably, sample H, P and S exhibited the higher degree of prokaryotic richness than other samples, which suggested that the residual relics promoted the growth of different bacteria. However, the structure of eukaryotic in the samples was not similar with that of prokaryotic, which may be caused by the different types and amounts of residues in different samples.
Next, community similarity was evaluated for the different samples using beta diversity indices such as PCoA (Additional file 1: Fig. S5) and a UPGMA clustering tree (Additional file 1: Fig. S6). L2 had a specific diversity compared with other samples in prokaryotic community, while R1, R3, L1, and L3 and R2, S, H, and P could be clustered into two groups based on the prokaryotic community similarity. The eukaryotic community composition of sample H was the most different from the nine samples, samples L2 and R3 had similar eukaryotic communities, and all other samples shared a similar eukaryotic group.
Distribution and diversity of organisms in the tomb soil mass
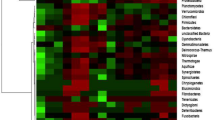
Prokaryote sequences identified in the nine samples and their relative abundances are shown in Fig. 4. Most sequences of prokaryotic populations comprising the communities of all samples were derived from bacteria of the following phyla (Fig. 4a): Proteobacteria, Acidobacteria, Actinobacteria, Bacteroidetes, Nitrospirae, Chloroflexi, and Firmicutes. Proteobacteria was the most abundant phylum in all samples (except L2), with percentages of all phyla per sample of 56.79% (L1), 26.21% (L2), 75.99% (L3), 82.73% (R1), 73.19% (R2), 75.86% (R3), 63.64% (H), 74.76% (P), and 78.41% (S). Acidobacteria was the major phylum in L2 (58.02%) and the second major phylum in six other samples (L1, L3, R1, R2, P, and S) where it ranged in prevalence from 5.77 to 20.44%. Meanwhile, Actinobacteria, Bacteroidetes, and Firmicutes also comprised a large proportion of community phyla, with averages of 3.96%, 5.02%, and 3.01% in the nine samples, respectively.

Distribution patterns of prokaryotic phyla (a) and genera (b) in the samples
The prokaryotic populations at the genus level are shown in Fig. 4b. Unclassified bacteria comprised the majority of species, accounting for 17.36- 59.92% of species in the nine communities. Cupriavidus was detected in all samples and was the most abundant genus in L1, L3, R2, P, and S, accounting for 13.72%, 20.75%, 19.15%, 16.25%, and 11.21% of genera, respectively, but only 0.03% in H. Rhodanobacter was the major genus in R1 (20.6%) and was also widely distributed among the other samples (except for L2 and H). Escherichia-Shigella was the dominant genus in sample H (13.16%) and the second most predominant group in P (7.37%) and S (7.22%). Candidatus-Koribacter and Candidatus-Solibacter were the major genera in L2 (11.44% and 7.35%, respectively), while Pseudarthrobacter, Bradyrhizobium, Variovorax and Aeromonas were the main genera in single samples such as L1 (13.04%), R3 (16.97%), R1 (10.63%), and R2 (18.5%), but were present in low abundance in the other samples.
The identified eukaryote sequences and their relative abundance levels in samples are shown in Fig. 5. Ascomycota, Streptophyta, and Ochrophyta were the dominant phyla, which together had an average abundance of 92.73%. Ascomycota was the most common fungal phylum detected in L2 (76%), R3 (87.79%), and H (56.17%), but in the other samples, the plant phylum Streptophyta predominated, accounting for 78.85% (L1), 78.75% (L3), 57.06% (R1), 79.13% (R2), 77.09% (P), and 55.63% (S) of phyla detected in the samples. Ochrophyta was detected in six samples, accounting for 11.49% (L1), 11.88% (L3), 19.61% (R1), 12.14% (R2), 12.62% (P), and 47.76% (S) of phyla detected in samples. The fungal phylum Basidiomycota was detected in seven samples, but it was only abundant in L2 (6.57%) and H (6.76%).

Distribution patterns of eukaryotic phyla (a) and genera (b) in the samples
Additionally, the eukaryotic population profile at the genus level is shown in Fig. 5b. Unclassified genera were enriched in samples L2, R3 and H, with an average abundance of 17.94% as compared to 5.77% in the other samples, while no eukaryotic genus was common to all samples. Lecanicillium was the most dominant fungal genus in L2, R3 and R1, accounting for 18.56%, 75.73%, and 8.21%, respectively. Penicillium was the second major genera in L2 (13.35%) and R3 (7.05%), and it was the predominant genera in H (9.68%). The most representative eukaryotic groups detected in the other samples were those of plants, especially Bistorta and Potentilla, which were enriched in six samples as follows: L1 (27.12% and 13.03%, respectively), L3 (23.21% and 11.88%, respectively), R1 (9,73% and 19.62%, respectively), R2 (20.36% and 12.14%, respectively), P (26.45% and 12.86%, respectively) and S (8.43% and 37.98, respectively). In addition, Geranium, Saussurea, Hippophae, Sibbaldianthe and Hedysarum were also detected in the six samples. Notably, Glycine (19.07%), and Angelica (12.66%) were detected in H but comprised only a small proportion of genera in the other samples.
Discussion
Environmental factors related to the deterioration of the buried heritage in tomb M88
Tombs in the Zeng State generally contain the body of the owner and various funerary materials, such as the commonly encountered mineral cinnabar, textiles derived from the tomb owner’s clothing (including silk and hemp fabric) and coffin cover material (wood brushed with raw lacquer), and other related items [17, 18]. The deterioration of the buried heritage depends on factors associated with the burial environment, such as the soil pH, moisture and microorganisms present at the grave site [2, 3, 19,20,21,22]. The tomb M88 was found in Hubei province, where the soil is thought to be acidic, but we found that the pH of the tomb soil that was transferred to the laboratory was neutral, as reported previously for soil from cultivated fields in Hubei province [23]. Thus, our result indicated that soil pH played a minor role in residual relic deterioration in the present case. In order to prevent cracking of the tomb soil mass, we frequently sprayed water on the surface to moisten the burial soil. It is known that water affects soil decomposition by influencing the activity of soil microorganisms [14]. A moist environment would promote the growth of microorganisms in tomb M88. In addition, the water spray introduced the environmental microbes from air and water into the tomb soil. This drove changes to the microbial community in the tomb soil and the development of new dominant strains would cause further damage to the residual heritage [7]. This implied that microorganisms might be the main factor to accelerate the degradation of buried heritage in tomb M88 and high humidity conditions may indirectly drive relic deterioration by supporting microbial growth.
Distribution and diversity of microorganisms in tomb M88
Importantly, the profiles of bacterial communities among the nine samples revealed relationships with sampling sites (Fig. 1b and Additional file 1: Fig. S6a). For example, microbial community profiles at the edges of the soil mass (L1, L3, R1 and R3) differed from profiles associated with the H, P, and S relic samples, which harbored obvious archaeological residues. The R2 sampling site was in close proximity to the P and S sampling sites, which may have explained why these samples yielded similar bacterial community profiles. The phylum Firmicutes was identified only in the H, P and S samples, which contained relic residues. Most of the bacteria genera (in this study Aeromonas, Escherichia-Shigella, Klebsiella, Kurthia and Raoultella) belonging to this phylum are conditional pathogens that can cause respiratory and intestinal diseases in humans [24, 25]. During careful excavation in the laboratory, excavators need to be in close contact with the soil mass, and these microorganisms may enter the human body through the skin or respiratory tract. It suggested that excavators should take greater precautions when excavating the residual heritage. Pseudarthrobacter was also enriched in the above samples (H, P, S, and R2). Pseudarthrobacter is known to excrete dextranase, an enzyme than can degrade polysaccharides such as cellulose, an important component of plant [18]. Numerous residual heritage in tomb M88 is plant origin, for example, the coffin, plant layers in sample P, and some recorded hemp fabric [17, 18]. It suggested that Pseudarthrobacter could accelerate the degradation of these heritage by disrupting the plant structure during excavation. heritageCupriavidus, which is known for its ability to resist toxic heavy metals, is normally associated with sites harboring high concentrations of heavy metals [3, 26,27,28]. Here, this genus was detected in most samples (except for H, L2 and R3), and its detection likely resulted from the diffusion of copper ions from bronzeware artifacts into the tomb soil as numerous bronzewares were excavated in the tomb.
Interestingly, although the numbers of identified fungal sequences were lower than the numbers of identified bacterial sequences in the samples, the total numbers of ITS OTU sequences greatly exceeded the numbers of 16S OTU sequences, and the profiles of eukaryotic communities indicated they were much more diverse than prokaryotic communities. Hypervariable regions of ITS sequences are often used to identify plants [29], so, the ITS OTUs can be composed of both fungi and plants. Fungal DNA was only detected in high concentrations in the L2, R3 and H samples, which was likely related to the physical heights of the sampling sites; the tomb plan depicted in Fig. 1 did not occupy a single plane, with sample H collected at a greater height than the other samples, while R3 was collected from the lowest site. L2 lay at the edge of the soil mass, and the plant matter in this sample may have been lost. Thus, some samples may have lacked plant-based material, leading to the abundance of fungi.
With regard to sequences associated with specific fungal phyla, Ascomycota phylum sequences were predominant among the nine samples, and Penicillium, Lecanicillium, Cladosporium, Pseudogymnoascus, and Blumeria genera-associated sequences were predominant in most of the samples. Organisms belonging to these genera are relevant in that they have been shown to be prevalent in the soil and air of the tomb environment [21, 22, 30]. In addition, previous studies showed that Penicillium and Cladosporium were associated with the deterioration of cultural relics, such as textiles, wood, lacquer and bone [5, 31,32,33], and thus, these fungi may pose a threat to residual heritage in tomb M88.
To sum up, Proteobacteria, Acidobacteria and Ascomycota were the dominant phylum in the soil of tomb M88, but no one genus was dominant in all samples. Pseudarthrobacter, Penicillium and Cladosporium were predominant in the samples with residual heritage, and these microorganisms could accelerate the degradation of the residual heritage through metabolites such as enzymes and acids. Isothiazolinone is a biocide with a wide antimicrobial spectrum. Previous study reported that isothiazolinone was effective against most dominant microorganisms present in tomb M88 [34], and this biocide was also added to the sprinkler system to inhibit the growth of microorganisms in the excavation site of Nanhai no. 1 Shipwreck [35]. It suggested that biocides such as isothiazolinone could be added to water during moisturizing treatment to prevent the biodeterioration of residual heritage in tomb soil.
Distribution and diversity of plants in tomb M88
Most M88 tomb soil samples were enriched with plant-derived content (> 75%), except for samples H (33.72), L2 (13.97%), and R3 (0.27%). Potentilla, Bistorta, Saussurea, Sibbaldianthe, Poa, Euphrasia and Euphorbia were the main plant genera identified in this study. Li et al. reported that these species were also widely deposited in the soil from the Qujialing archaeological site (6000–300 a. BP), which was located less than 70 km away from the M88 tomb [36]. And these plants are common in the area today. The tomb was exposed for a long period of time during excavation, and the distance between the tomb and the surface was small, modern plants, especially roots or pollen, may have infiltrated. So it is difficult to distinguish whether these identified plants belonged to ancient or modern plants.
In addition, some plants not belonging to this region were identified in the tome soil, and there was also no possibility of human contamination during the excavation. Most samples were enriched with Hippophae, especially the sample S which contained visible traces of residual silk. This plant species usually lives in relatively dry areas, including areas within north, northwest and southwest China [37]. However, Hubei is a region in central China that has always had abundant rainfall (even in ancient times), implying that Hippophae was brought to the tomb site from other regions. Currently, Hippophae is an ingredient used to make yellow dye [38], a fact that aligns with our findings that Hippophae was the most abundant DNA detected in silk residues of tomb M88. This suggested that Hippophae was used to dye the clothes of the tomb owner. Nevertheless, there is no existing documented record describing the use of Hippophae to make dye in ancient China, warranting additional research to investigate this possibility. Next, Geranium sequence representation in P exceeded that of the other samples. When considered together with SEM results showing that sample P had a paper-like structure composed of plant leaves, this suggested that Geranium leaves were the major constituent of sample P. We also found that Glycine and Angelica were only dominant in the head area of the grave owner. Some plants or artifacts based on these genera were usually used to cover on the grave owner or the coffin based on the funerary system of the Zeng State [17, 18]. These findings may provide some new archaeological information for the study of the tomb.
Conclusion
Here, we analyzed the microbiome profiles of the soil of tomb M88, which was carefully excavated in the laboratory. Proteobacteria, Acidobacteria and Ascomycota were the most dominant microorganisms in the tomb soil, and Pseudarthrobacter, Penicillium and Cladosporium, the dominant genera in the microbial community, have the ability to accelerate the biodeterioration of residual heritage. The humid environment created by the moisturizing treatment promoted the growth of these microorganisms, which suggested that some biocides should be added to the spray water during the future carful excavation in the laboratory. In addition, the identification of plants might provide valuable archeological clues to the funerary system and cultural exchange.
Availability of data and materials
The raw sequences of high-throughput sequencing were deposited in the NCBI Sequence Read Archive (SRA) database (accession numbers: SAMN20587851, SAMN20587867, SAMN20587868, SAMN20587885, SAMN20587886, SAMN20587899, and SAMN20587922–SAMN20587924).
References
Xu J, Wei Y, Jia H, Xiao L, Gong D. A new perspective on studying burial environment before archaeological excavation: analyzing bacterial community distribution by high-throughput sequencing. Sci Rep. 2017;7:41691. https://doi.org/10.1038/srep41691.
Kazarina A, Gerhards G, Petersone-Gordina E, Kimsis J, Pole I, Zole E, et al. Analysis of the bacterial communities in ancient human bones and burial soil samples: tracing the impact of environmental bacteria. J Archaeol Sci. 2019;109:104989. https://doi.org/10.1016/j.jas.2019.104989.
Margariti C. The effects of micro-organisms in simulated soil burial on cellulosic and proteinaceous textiles and the morphology of the fibres. Stud Conserv. 2021;66(5):282–97. https://doi.org/10.1080/00393630.2020.1812245.
Pangallo D, Kraková L, Chovanová K, Bučková M, Puškarová A, Šimonovičová A. Disclosing a crypt: microbial diversity and degradation activity of the microflora isolated from funeral clothes of Cardinal Peter Pázmány. Microbiol Res. 2013;168:289–99. https://doi.org/10.1016/j.micres.2012.12.001.
Blanchette RA. A review of microbial deterioration found in archaeological wood from different environments. Int Biodeterior Biodegrad. 2000;46:189–204. https://doi.org/10.1016/S0964-8305(00)00077-9.
Caneva G, Isola D, Lee HJ, Chung YJ. Biological risk for hypogea: shared data from etruscan tombs in italy and ancient tombs of the Baekje Dynasty in Republic of Korea. Appl Sci. 2020;10:6104. https://doi.org/10.3390/app10176104.
Sterflinger K, Piñar G. Microbial deterioration of cultural heritage and works of art—tilting at windmills? Appl Microbiol Biotechnol. 2013;97:9637–46. https://doi.org/10.1007/s00253-013-5283-1.
Kistler L, Bieker VC, Martin MD, Pedersen MW, Ramos Madrigal J, Wales N. Ancient plant genomics in archaeology, herbaria, and the environment. Annu Rev Plant Biol. 2020;71:605–29. https://doi.org/10.1146/annurev-arplant-081519-035837.
Brown TA, Cappellini E, Kistler L, Lister DL, Oliveira HR, Wales N, et al. Recent advances in ancient DNA research and their implications for archaeobotany. Veg Hist Archaeobot. 2015;24:207–14. https://doi.org/10.1007/s00334-014-0489-4.
Przelomska NAS, Armstrong CG, Kistler L. Ancient plant DNA as a window into the cultural heritage and biodiversity of our food system. Front Ecol Evol. 2020;8:74. https://doi.org/10.3389/fevo.2020.00074.
Tanaka K, Zhao CF, Wang NY, Kubota S, Kanehara M, Kamijo N, et al. Classification of archaic rice grains excavated at the Mojiaoshan site within the Liangzhu site complex reveals an Indica and Japonica chloroplast complex. Food Prod Process Nutr. 2020;2:15. https://doi.org/10.1186/s43014-020-00028-8.
Schworer C, Leunda M, Alvarez N, Gugerli F, Sperisen C. The untapped potential of macrofossils in ancient plant DNA research. New Phytol. 2022;235:391–401. https://doi.org/10.1111/nph.18108.
Crump SE, Frechette B, Power M, Cutler S, de Wet G, Raynolds MK. Ancient plant DNA reveals high arctic greening during the last interglacial. Proc Natl Acad Sci USA. 2021;118:e2019069118. https://doi.org/10.1073/pnas.2019069118.
Carter DO, Yellowlees D, Tibbett M. Moisture can be the dominant environmental parameter governing cadaver decomposition in soil. Forensic Sci Int. 2010;200:60–6. https://doi.org/10.1016/j.forsciint.2010.03.031.
ISO 10390:2021. Soil, treated biowaste and sludge: determination of pH. Geneva: International Organization for Standardization; 2021.
Zheng H, Yang H, Zhou Y, Li T, Ma Q, Wang B, et al. Rapid enrichment and detection of silk residues from tombs by double-antibody sandwich ELISA based on immunomagnetic beads. Anal Chem. 2021;93:14440–7. https://doi.org/10.1021/acs.analchem.1c02556.
Wang B. A study on graves of zeng state period. 2009. kns.cnki.net/KCMS/detail/detail.aspx?dbname=CMFD2009&filename=2200915854.nh. (in Chinese).
Zhu W. On the Funeral System Reflected in Shisangli and Jixili in Yili. 2008. kns.cnki.net/KCMS/detail/detail.aspx?dbname=CMFD2009&filename=2009081038.nh. (in Chinese).
Zhou C, Byard RW. Factors and processes causing accelerated decomposition in human cadavers—an overview. J Forensic Leg Med. 2011;18:6–9. https://doi.org/10.1016/j.jflm.2010.10.003.
Lowe AC, Beresford DV, Carter DO, Gaspari F, O’Brien RC, Stuart BH, et al. The effect of soil texture on the degradation of textiles associated with buried bodies. Forensic Sci Int. 2013;231:331–9. https://doi.org/10.1016/j.forsciint.2013.05.037.
Li Y, Huang Z, Petropoulos E, Ma Y, Shen Y. Humidity governs the wall-inhabiting fungal community composition in a 1600-year tomb of Emperor Yang. Sci Rep. 2020;10:8421. https://doi.org/10.1038/s41598-020-65478-z.
Ma W, Wu F, Tian T, He D, Zhang Q, Gu JD, et al. Fungal diversity and its contribution to the biodeterioration of mural paintings in two 1700-year-old tombs of China. Int Biodeterior Biodegrad. 2020;152:104972. https://doi.org/10.1016/j.ibiod.2020.104972.
Zhang YP, Wu Y, Zheng XW. Trend analysis and influencing factors of soil acidity and alkalinity in Hubei. Resour Environ Eng. 2018;32:30–4. https://doi.org/10.16536/j.cnki.issn.1671-1211.2018.S1.005.
Janda JM, Abbott SL. The genus aeromonas: taxonomy, pathogenicity, and infection. Clin Microbiol Rev. 2010;23:35–73. https://doi.org/10.1128/CMR.00039-09.
Barson WJ, Marcon MJ. Klebsiella and Raoultella species. In: Principles and practice of pediatric infectious diseases. 4th ed. Edinburgh: Elsevier Inc.; 2012. https://doi.org/10.1016/B978-1-4377-2702-9.00140-9.
Mergeay M, Monchy S, Vallaeys T, Auquier V, Benotmane A, Bertin P, et al. Ralstonia metallidurans, a bacterium specifically adapted to toxic metals: towards a catalogue of metal-responsive genes. FEMS Microbiol Rev. 2003;27:385–410. https://doi.org/10.1016/S0168-6445(03)00045-7.
Mergeay M, Van Houdt R. Cupriavidus metallidurans CH34, a historical perspective on its discovery, characterization and metal resistance. FEMS Microbiol Ecol. 2021;97:fiaa247. https://doi.org/10.1093/femsec/fiaa247.
Shi Z, Zhang Z, Yuan M, Wang S, Yang M, Yao Q, et al. Characterization of a high cadmium accumulating soil bacterium, Cupriavidus sp. WS2. Chemosphere. 2020;247:125834. https://doi.org/10.1016/j.chemosphere.2020.125834.
Zhao H, Wu W, Zheng YL, Pan HM, Zhai JY. Application of rDNA-ITS sequence analysis method in medicinal plants research. Lishizhen Med Materia Med Res. 2009;20:959–62.
Vasanthakumar A, DeAraujo A, Mazurek J, Schilling M, Mitchell R. Microbiological survey for analysis of the brown spots on the walls of the tomb of King Tutankhamun. Int Biodeterior Biodegrad. 2013;79:56–63. https://doi.org/10.1016/j.ibiod.2013.01.014.
Turner-walker G. Part 1 analytical approaches in the chemical and microbial degradation of bones and teeth. 2008.
Abdel-Azeem AM, Held BW, Richards JE, Davis SL, Blanchette RA. Assessment of biodegradation in ancient archaeological wood from the Middle Cemetery at Abydos, Egypt. PLoS ONE. 2019;14:e0213753. https://doi.org/10.1371/journal.pone.0213753.
Pyzik A, Ciuchcinski K, Dziurzynski M, Dziewit L. The bad and the good-microorganisms in cultural heritage environments—an update on biodeterioration and biotreatment approaches. Mater (Basel). 2021;14:177. https://doi.org/10.3390/ma14010177.
Mattea R, Thomas W, Lionel N, Lionel M, Patrick DM, Marcelino TS, et al. Current and future chemical treatments to fight biodeterioration of outdoor building materials and associated biofilms: moving away from ecotoxic and towards efficient, sustainable solutions. Sci Total Environ. 2022;802:149846. https://doi.org/10.1016/j.scitotenv.2021.149846.
Li NS, Chen Y, Shen DW. “Nanhai no. 1” study on the conservation of shipwreck excavation site (2014–2016). Beijing: Science Press; 2017. p. 85–92.
Li YG, Hou SF, Mo DW. Records for pollen and charcoal from Qujialing archaeological site of Hubei and ancient civilization development. J Palaeogeogr. 2009;11:702–10.
Pundir S, Garg P, Dviwedi A, Ali A, Kapoor VK, Kapoor D, et al. Ethnomedicinal uses, phytochemistry and dermatological effects of Hippophae rhamnoides L.: a review. J Ethnopharmacol. 2021;266:113434. https://doi.org/10.1016/j.jep.2020.113434.
Ciesarová Z, Murkovic M, Cejpek K, Kreps F, Tobolková B, Koplík R, et al. Why is sea buckthorn (Hippophae rhamnoides L.) so exceptional? A review. Food Res Int. 2020;133:109170. https://doi.org/10.1016/j.foodres.2020.109170.
Acknowledgements
We thank LetPub (www.letpub.com) for its linguistic assistance during the preparation of this manuscript. We thank Qinglin Ma, Institute of Cultural Heritage, Shandong University for his help and continuous support.
Funding
This work was supported by the China Postdoctoral Science Foundation (2019M662390), the Shandong Provincial Natural Science Foundation, China (ZR2020QC045).
Author information
Authors and Affiliations
Contributions
LZ and QF contributed to the collection of the samples, prepared figures and reviewed drafts of the paper. TL contributed to study design and paper preparation, data analysis and wrote the main manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additionalfile 1: Table S1.
Alpha diversity of the nine samples. Figure S1. The location of thesoil mass cut from the tomb M88. Figure S2.The SEM-EDS results of the sample P. FigureS3. The distribution of elements in the sample P. Figure S4. The EDS results of the sample P in different sites. Figure S5. Principal co-ordinates analysis of the (a)prokaryotic and (b) eukaryotic communities in the samples. Figure S6. UPGMA Tree of the (a) prokaryotic and (b) eukaryoticcommunities in the samples.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Zhu, L., Fang, Q. & Li, T. Distribution and diversity of organisms in tomb soil excavated in the laboratory: a case study of tomb M88 from Sujialong Cultural Property, China. Herit Sci 10, 166 (2022). https://doi.org/10.1186/s40494-022-00803-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40494-022-00803-5