
Abstract
Over the last few decades, chemical and physical studies on bowed string musical instruments have provided a better understanding of their wooden finished surface. Nevertheless, until now only a few of them investigated the effects of the chemical pre-treatments in the traditional making procedures. Those treatments are believed to affect wood properties, its interaction with the following treatments (i.e. varnish application) and its vibro-mechanical behaviour (that may contribute to the acoustical properties of musical instruments). In this study, two traditional alkaline treatments were applied to reference samples of spruce wood, the wood commonly used to make violins’ soundboards. An integrated analytical strategy, which combines infrared spectroscopy, analytical pyrolysis coupled to gas chromatography-mass spectrometry, and gel permeation chromatography, was employed to investigate the chemical alterations of lignocellulosic polymers (cellulose, hemicellulose, and lignin). Results have shown that the selected alkaline treatments induce the partial hydrolysis of the hemicellulose chains and a slight decrease in the crystallinity of cellulose. We could also prove: (i) the cleavage of lignin-carbohydrate complexes formed by the covalent bonds between hemicellulose and lignin in spruce wood, and (ii) the partial breaking of the hydrogen bonds network in cellulose. According to the literature, the alteration of the lignin-carbohydrate complexes is responsible for changes in wood mechanical behaviour. Hence, future perspectives of this research could outline new knowledge on the vibro-mechanical behaviour of the violin soundboard and the consequent acoustics.
Similar content being viewed by others


Introduction
The manufacturing practice of a bowed string instrument was developed during the 16th century and reached the highest grade during the 17th century in Cremona (Italy). Antonio Stradivari is the most famous proponent of this Cremonese tradition, and his instruments still represent the acme of the quality in the bowed string instruments, for their unique and unreached aesthetical and acoustical properties. These features still influence contemporary violin makers that try to reproduce and imitate the masterpieces of ancient makers. The absence of written sources on the manufacturing practice of bowed string instruments has stimulated the chemical and physical study of historical musical instruments with the aid of analytical methods, to understand the manufacturing techniques used by the old masters. Nowadays, the results of various researches allow us to have a clear view of the varnishing, the ground layer and the sizing treatments used by old makers to make the wood durable and to enhance the aesthetic and acoustic appeal of their instruments [1,2,3,4,5]. In addition, it has always been assumed that wood pre-treatments also contribute to the mechanical and hence acoustical properties of the instruments [6, 7]. As reported by Tai et al. in a recent paper [7], the author supposed that Antonio Stradivari would deliberately manipulate spruce wood. This timber was commonly used by historical violin makers for the soundboard, which is the part that most affects the acoustical properties of the instrument.
In general, wood is a very complex material, in which cellulose, hemicellulose, and lignin are packed in a composite matrix. The molecular composition and the way the components are arranged within the wood cells walls are known as ultrastructure and it can vary depending on the wood kind, the condition of growth (i.e., climate, geographical area) and, all other factors unchanged, its state of preservation or treatments done on purpose in the making processes. It determines the chemical, physical and mechanical properties of a wood species [8]. According to the most accredited wood model, the hemicellulose represents an amorphous area of transition between cellulose and lignin, thus being a highly reactive site for chemical and physical agents. The features of hemicellulose and its interface with lignin and cellulose, control the diffusion of the polar substances in the wood and its hydrothermal and viscoelastic properties [8, 9].
In the cultural heritage field, studies on the wood ultrastructure have been done mainly to investigate the state of preservation, the ongoing alterations, and the ageing processes of archaeological wooden objects [10,11,12,13,14]. On the contrary, less effort has been dedicated to studying the treatments developed by artists and craftsmen, and their characterization would represent an outstanding achievement for a deep knowledge of the making practice of wooden artefacts, in particular historical violins.
From the literature available for wooden objects, mainly furniture, physical-chemical treatments make the wood suitable for instruments. Among the physical, exposing the wood to solar radiation or high temperature for a certain period was recommended [6, 15]. Also, the practice of boiling or soaking wood in seawater was proposed by Nagyvary on the scientific evidence [16]. Nonetheless, chemical pre-treatments were widespread and could be acid or alkaline. We do not know so much about the first one [17], and the most common were the latter. Among them, the fumigation with ammonia vapour and the use of a lye-based solution [15,16,17,18,19]. Ammonia fuming is still used to stain the wood through its entire thickness, allowing the vapours to reach even the innermost structure of wood [20, 21]. The effects of this method are a wood darkening, a slight increase in anisotropy, which can then affect the wood shrinkage and swelling [21], and a significant increase in the equilibrium moisture content [22]. On the contrary, the treatment is not expected to induce any change in the mechanical properties of wood. The intensity of the wood darkening depends on the ammonia concentration in the atmosphere, the exposure time, and the wood species [23]. Concerning the lye-based solution, it is considered a strong alkali. Among the strong alkali effects, a brownish discoloration is mainly expected [21, 24].
To the best of our knowledge, the fact that the wood treatments employed by the violin makers in the traditional Cremonese practice may affect the aesthetical and acoustical properties of an instrument is supposed and specific studies dedicated to the chromatic, chemical, and mechanical changes assessment have not been carried out yet. This study aims to fill the lack of knowledge about the effects induced by chemical alkaline pre-treatments. To this purpose, reference wood samples were treated with the fuming ammonia and the potassium hydroxide pre-treatments. A multi-analytical strategy was then used to assess the aesthetic and ultrastructural transformations of the treated wood. The analytical strategy includes the application of FTIR spectroscopy in ATR mode (FTIR-ATR), analytical pyrolysis coupled to gas chromatography-mass spectrometry with in situ hexamethyldisilazane derivatization (Py(HMDS)-GC/MS), gel permeation chromatography (GPC) and X-ray diffractometry (XRD).
Experimentals
Wood laboratory models
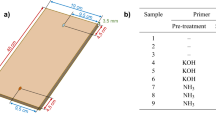
Three laboratory models (listed in Additional file 1: Table SM1), 10 cm (longitudinal direction) × 5 cm (radial direction) × 0.7 cm (transversal direction), were prepared from the same spruce (Picea abies (L.) H.Karst., 1881) wood board. They were obtained by cutting the log along the longitudinal-radial direction. According to the traditional making procedures, seasoned wood (more than 12 years—cut in 2005) was selected from Val di Fiemme (north of Italy). Before the alkaline treatments, the boards underwent three months of curing under monitored conditions (RH = 50%, T = 20 °C) to allow the wood to get in equilibrium with the new environment. The moisture content (MC) was measured by the electrical conductivity method [25]. The non-treated wood is referred to as m.0 (control sample or non-treated wood). The radial section of the mock-up named m.1 K was treated with a 1 M aqueous solution at pH = 13 of potassium hydroxide (pellets, Carlo Erba, Italy) applied by brush (to simulate the lye solution alkaline treatment). Instead, the mock-up labelled with m.1 N was kept in a sealed box saturated with ammonia vapour (NH3) (Ammonia 30% v/v, Bresciani s.r.l. Italy) for 96 h. After the treatments, and to reach the equilibrium condition with the environment, the specimens were cured under monitored conditions (RH = 50%, T = 20 °C) in a storeroom for one month to eliminate the water absorbed during the KOH treatment and the entrapped ammonia.
Analytical methods
The used techniques are complementary and can disclose specific aspects of the wood composition when combined. The analytical protocol is shown in Fig. 1 and involves the following steps:
-
1.
Aesthetical feature assessment through colorimetric measurements.
-
2.
Non-destructive qualitative evaluation of the overall wood composition by FTIR-ATR spectroscopy.
-
3.
Micro-destructive study and semi-quantitative determination of the holocellulose and lignin fraction composition by Py(HMDS)-GC/MS analysis.
-
4.
Study of the LCCs and holocellulose modification by destructive GPC analysis.
-
5.
Evaluation of the cellulose crystallinity degree by XRD analysis.

Scheme of the analytical protocol adopted in this work to evaluate the chemical modifications induced by the alkaline treatments performed on the wood samples
Steps 1 and 2 do not require any sampling or any sample pre-treatment, whereas the other steps do. The sampling was performed on the treated radial section surface of the models, in a variable quantity according to the requirement of the analytical technique (for the details see Additional file 1: Table SM1). Before the examinations, samples were dried and milled, and a peculiar preparation (described in Sect. 2.2.4) was followed before the GPC analysis. The protocol can evidence changes in the wood ultrastructure and gives information on the reactions occurring in the wood polymers under alkaline treatments.
Colorimetry
Chromatic variations of the modified wood were measured using a Konica Minolta CM-2600d portable spectrophotometer (Chiyoda, Tokyo, Japan). Data were collected in the wavelength range 400–740 nm (spectral resolution of 10 nm) using a 0.8 mm round target mask. L*, a* and b* coordinates were calculated in SCE mode (Specular Component Excluded) with a 10 standard observer and D65 illuminant according to CIELAB 1976 colour space. Fifteen measurements were performed on the treated radial section. The measurement points were identified in the centre of the squares of a grid yielding a total of 5 × 3 = 15 data before and after the treatments (t0 and t1). The mean value and the standard deviation of the mean (SDOM) were calculated for each sample. According to the literature, the colour difference ΔE*ab was calculated to obtain the magnitude of the overall colour variation [26, 27].
FTIR spectroscopy in ATR mode (FTIR-ATR)
Attenuated total reflectance (ATR) measurements were performed using a NICOLET iS5 spectrometer (Thermo Scientific, Waltham, Massachusetts, USA) equipped with iD7 ATR accessory and diamond crystal. All infrared spectra were recorded within the range of 4000–50 cm− 1 with 4 cm− 1 resolution and 32 scans. The sampling area corresponded to 1.8 mm. Three measurements were performed in the middle of the radial section of each small board, on the left, centre, and right areas, respectively. The edges of the boards were not considered to avoid the effect of the overrated sorption of the treating aqueous solution along the longitudinal direction of the fibres. A first evaluation of the spectra was carried out qualitatively by comparing the FTIR spectra of the treated samples (m.1 K and m.1 N) with the reference spectra of non-treated spruce wood (m.0). OMNIC 7.2 software package was employed for the study of bands produced by the effect of the treatments on ATR non-corrected spectra.
Pyrolysis coupled to Gas Chromatography-Mass Spectrometry with in situ hexamethyldisilazane derivatization (py(HMDS)-GC/MS)
Before analysis, the samples were previously oven-dried for 24 h at 40–50 °C and homogenised through a ball mill made of zirconium oxide (Pulverisette 23, Fritsch GmbH, Germany). For the in situ thermally assisted derivatization of pyrolysis products a silylating agent 1,1,1,3,3,3-hexamethyldisilazane (HMDS, chemical purity 99.9%, Sigma Aldrich Inc., USA) was used. The sampling was performed on the upper radial section of the models. Samples were analysed in triplicate, and the relative standard deviations associated with the calculated values were estimated below 10%. The H/L (holocellulose/lignin) ratio was then determined to assess the loss of polysaccharides or lignin as commonly used in the case of aging or treatments [28,29,30,31].
As described in previous works [11, 13, 32,33,34], an Agilent 5973 Mass Selective Detector operating in electron impact mode (EI) at 70 eV was directly combined with a GC equipment (gas chromatograph 6890, Agilent Technologies, USA) equipped with an HP- 5MS fused silica capillary column (stationary phase 5% diphenyl- 95% dimethyl-polysiloxane, 30 m × 0.25 mm i.d., Hewlett Packard, USA) and with a deactivated silica pre-column (2 m × 0.32 mm i.d., Agilent J & W, USA) able to perform the chromatographic separation and mass spectrometry determination of pyrolysis products formed in a micro-furnace of Multi-Shot Pyrolyzer EGA/Py-3030D (Frontier Laboratory, Japan). The pyrolysis and the interface temperatures were 550 °C and 280 °C, respectively. Each wood sample (about 100 µg) was put with 2 µl of HMDS into a stainless-steel cup, coated with the quartz wool, and placed in the micro-furnace. The GC oven parameters were initial temperature 50 °C, 1 min isothermal, 10 °C min− 1 to 100 °C, 2 min isothermal, 4 °C min− 1 to 190 °C, 1 min isothermal, 30 °C min− 1 to 280 °C, 30 min isothermal. The carrier gas was helium (purity 99.995%) used with a constant flow of 1.0 mL min− 1.
Data processing was performed by comparing the mass spectra with those reported in the NIST2.4 libraries and the literature [31, 33, 35]. Peaks derived from lignin and holocellulose products were identified and integrated by AMDIS software. The areas of the peaks of interest in the chromatogram were normalised to the sum of the peak areas of all the identified holocellulose and lignin pyrolysis products. Data were averaged and expressed as percentages for the semi-quantitative assessment of the components. The percentage areas were used to calculate the relative abundances of wood pyrolysis products divided into categories of holocellulose (cyclopentenones, pyrans, furans, hydroxybenzenes and anhydrosugars) and lignin (monomers, long-chain, short-chain demethylated, carbonyl and acid) [36, 37], Additional file 1: Table SM2. Chemical changes in a single biopolymer were also assessed by analysing categorised pyrolysis products of holocellulose and lignin [28,29,30,31]. A limitation of the analytical pyrolysis method is that it does not allow the pyrolysis products of cellulose and hemicellulose to be distinguished since they form the same products during the pyrolysis process. The method is therefore able to detect chemical changes in the overall fraction of polysaccharides (holocellulose) of the analysed wood samples.
Gel permeation chromatography (GPC)
The wood samples (500 mg), were crushed in a blender, passed through a 1 mm screen and pulverized in a planetary ball mill made of zirconium dioxide 95% (PM100, Retsch, Germany). After acetylation and benzoylation of the wood samples in ionic liquid medium (for the specific procedure see [38,39,40]) they were solubilized in THF (1 mg ml− 1) and passed through a 0.45 μm GHP Acrodisc syringe filter for GPC analyses at a flow rate of 1 ml min− 1. The analyses were performed on an HP1100 series liquid chromatographic system connected to an ultraviolet UV detector set at 240 or 280 nm for benzoylated and acetylated samples. The injection port was a Rheodyne® equipped with a 20-µl loop. The column system was composed of a sequence of an Agilent PL gel 5 μm, 500 Ǻ and a PL gel 5 μm, 104 Ǻ in series.
X-Ray diffractometry (XRD)
X-ray diffractograms were recorded with a D2-PHASER spectrometer (Bruker, USA). Spectra were recorded in the range 2θ = 4–60 degrees, with a resolution of 0.016 degrees. The CuKα emission at 1.54 Å was used as an X-ray source. Cumulative spectra were obtained by recording three diffractograms for each sample. The sample was kept under rotation during each measurement to prevent iso-orientation effects. The spectra were processed with DIFFRAC (Bruker, USA). Baseline subtraction and peak fitting were performed using PeakFit (Systat Software Inc., USA). Simulated diffraction patterns were used as a reference to establish the appropriate number of peaks [41]. The crystallinity index (CI) calculated by the Segal method was used to estimate the crystallinity degree of the cellulose in each sample [41,42,43].
Results
Colorimetry
Table 1 presents the mean values and the SDOM of L*, a*, b* coordinates for the two treated samples before (t0) and after (t1) the treatment application. The ΔE* and ΔL* total variations are also calculated and reported. The variation of a* and b* coordinates is also displayed in Additional file 1: Fig. SM1, where circles represent the KOH (red – m.1 K) and the NH3 (blue – m.1 N) treated wood before (empty) and after (filled) treatments. In both cases, the effect of treatments on the aesthetic appearance of the wood is visible to the naked eye.
For the potassium hydroxide solution treated surface (m.1 K) we assessed a total colour variation (ΔE*) of 4.1, together with a decrease in L* of 3.37; the increase in b* and decrease in a* can be explained as a shift towards the yellow-green hue. Concerning the ammonia fuming treatment (mock-up m.1 N), its effect on the wood surface is more severe (ΔE*=16.2) than the one induced by potassium hydroxide and affects all the surfaces of the mock-ups (due to the vapour state of the treating agent). The main effect is a surface darkening, described by the sharp decrease in L* of 16 and a mild increase in b* of 1.91. The reaction of the ammonia with the tannin in the wood and atmospheric oxygen is chiefly responsible for this behaviour [24, 44]. It is here noted that the darkening effect induced by the ammonia fuming treatment was appreciated by traditional violin makers [24, 45] since it gives a specific nuance to the varnish.
FTIR spectroscopy in ATR mode (FTIR-ATR)
FTIR spectra of the non-treated wood (m.0 in black solid line), and of the wood after KOH (m.1 K in dark grey dashed line) and NH3 (m.1 N in grey dotted line) treatments are shown in Fig. 2. A qualitative comparison of the spectra reveals that the hemicellulose is affected by the alkaline treatments, whereas lignin and cellulose seem to be scarcely involved. Strong absorption is recognizable in correspondence of the hydrogen-bonded stretching (–OH) and C-H stretching in the broadband peaks centred around 3300 cm− 1 (1) and 2900 cm− 1 (2) and, for the first band, constant absorption intensity before and after the treatment implying an unaltered behaviour towards the bond water. The peaks between 1800 and 800 cm− 1, commonly used to describe the wood and its degradation state [31, 46,47,48], are well defined. The assigned bands are reported in Table 2. Significant changes can be distinguished in Fig. 2, mostly for the NH3 treated wood. The most evident one is the decrease in intensity of the carbohydrate band at 1736 cm− 1 and 1230 cm− 1 assigned to the acetylated galactoglucomannan (C=O and C–O stretching respectively, in acetyl units), the major hemicellulose in softwood, which can constitute up to 20 w/w% of the dry wood [49]. These modifications are clearly related to the de-acetylation effect of alkaline treatments. Other differences are the mild decrease of the peak at 896 cm− 1 representatives of the cellulose that is better recognizable by observing the increase of the peak centred at 875 cm− 1. In addition, slight variations are visible for the other bands assigned to the holocellulose at 1375, 1244, 1154, 1048, 1024 cm− 1 and the lignin at 1597, 1505, 1462, 1425, 1313, 1263 and 1244 cm− 1. A more significant variation for the KOH treated wood can be noticed for the bands assigned to the C-C aromatic skeletal vibration in lignin at 1597 and 1505 cm− 1, the strongest and stable signal of the aromatic vibration. Furthermore, the peaks at 1313 cm− 1 and 1263 cm− 1, respectively assigned to ring breathing of the syringyl and the guaiacyl, seem to decrease in intensity according to the findings of previous works about aged wood [47].

FTIR spectra of the control (m.0 in black solid line), the KOH (m.1 K in dark grey dashed line) and NH3 (m.1 N in grey dotted line) treated wood. Vertical lines correspond to the peaks of interest listed in Table 2. Holocellulose and lignin bands are indicated in green and orange, respectively
Pyrolysis coupled to Gas Chromatography-Mass Spectrometry with in situ hexamethyldisilazane derivatization (py(HMDS)-GC/MS)
Analytical pyrolysis allowed us to determine 37 compounds belonging to holocellulose and lignin. The spruce is a conifer; therefore, the pyrolysis products of the lignin were mainly of the guaiacyl type. Pyrolytic profiles of all analysed samples are qualitatively similar and differ in the relative abundances of some compounds.
The percentage composition of pyrolysis compounds obtained for reference (m.0) and treated woods (m.1 K and m.1 N) are reported in TAdditional file 1: able SM3. The KOH treated wood (m.1 K) shows a very similar percentage composition in respect to the non-treated wood (m.0) − 55.1% and 56.5% for the Holocellulose and 44.9% and 43.5% for the lignin—suggesting that no significant changes occurred. On the contrary, the ammonia treatment (m.1 N) produces a higher relative amount of pyrolysis products derived from the holocellulose (70.2%), which can indicate the alteration of polysaccharide polymeric structure but also cross-linking between hemicelluloses and lignin. The pyrolytic holocellulose/lignin ratio (H/L) calculated for the three replicas of each analysed sample also again confirms that the ammonia treatment was more effective than the potassium hydroxide one: H/L ratio is constant for m.1 K (H/L 1,2 ± 0,2) compared to m.0 (H/L 1,3 ± 0,2) while increases up to 10% in the m.1 N specimen (H/L 2.4 ± 0,2). More detailed information on chemical modifications of the wooden structure at the molecular level can be obtained evaluating holocellulose and lignin pyrolysis products categorized into groups, as listed in Additional file 1: Table SM2. Figure 3 (panel a and b) shows the percentage abundances of each category of holocellulose and lignin pyrolytic products. Concerning the holocellulose products (Fig. 3a) in the reference wood (m.0), the most abundant is the cyclopentenones group; the anhydrosugars, pyrans, hydroxybenzenes, and furans follow. The poor effectiveness of the KOH treatment (m.1 K) in the modification of the polysaccharide fraction of the wood is confirmed by the unchanged holocellulose profile compared to the untreated wood; differently, the holocellulose in m.1 N specimen undergoes modification. The alteration of the polymeric structure is proven by the decrease of the relative abundance of the cyclopentenones (about 40%) accompanied by the strong increase of the anhydrosugars (about 60%) and by a reduction in the hydroxybenzenes formation. A decrease in cyclopentenones and at the same time an increase in the relative abundances of anhydrosugars is related to the depolymerisation of polysaccharides. Therefore, we may suggest the reduction of cross-linking in the structure involving the polysaccharides caused by ammonia treatment (m.1 M) in agreement with literature data [31, 32, 36, 50].

Percentage abundances of categories of the holocellulose (a) and the lignin (b) pyrolysis products in the analysed samples
The percentage areas of the six categories of lignin pyrolysis products (monomers, short-chain, long-chain, carbonyl, carboxyl, and demethylated/demethoxylated) are shown in Fig. 3b. In the reference wood (m.0), monomers are the most abundant, followed by short- and long-chains. On the other hand, the treated samples provided a lower percentage area of lignin monomers and a higher percentage area of short and long-chain pyrolysis products. These variations were more evident for m.1 N than for m.1 K, again confirming that ammonia fuming was a more effective treatment than the potassium hydroxide solution. The decrease in the abundance of monomers and the increase of short-chain pyrolysis products in archaeological wood is often used as a degradation/alteration index for this material. The increased yield of secondary pyrolysis products such as short and long-chain compounds can be related to a less cross-linked structure, not only of the lignin polymer but also of the lignin-hemicellulose network. It is known, in fact, that lignin with a reduced cross-linking degree is less thermally stable and can undergo further degradation upon pyrolysis [32].
Gel permeation chromatography (GPC)
GPC analyses at 240 nm after benzoylation and at 280 nm after acetylation provided information about the whole cell wall components (cellulose, hemicelluloses, lignin, and their connections) and the lignocellulosic fractions only comprising aromatic moieties (mainly LCCs, lignin carbohydrates complexes), respectively [40, 51]. By comparing the results of the reference wood (m.0) and the treated samples (m.1 K and m.1 N), any variation ascribable to the molecular weight distribution can represent a kind of transformation or degradation of the specific components.
By observing the GPC profiles (Fig. 4) the treatments seem to affect the molecular weight distributions of the lignocellulosic components. The profiles after benzoylation (Fig. 4a), related to the entire wood components, are almost superimposable, but a partial shift of the profiles at higher elution times could be detected. This shift can be ascribed to partial hydrolysis or other degradation phenomena involving the whole lignocellulosic matrix. These results are confirmed after acetylation (Fig. 4b) where a shift of the profiles and a little enrichment in the low molecular weight area is observed for the acetylated m.1 K and m.1 N (KOH solution and NH3 vapour, respectively). Since these chromatograms account for the LCCs, we can consider the possibility that the degradation started from those components. It seems that the carbohydrate’s chains were partially hydrolysed releasing low molecular weight LCCs, clearly detected at 22 min (Fig. 4b) [49]. These observations confirm and integrate the FTIR characterization of milled wood where the band at 1730 cm− 1 (acetyl) disappeared after NH3 and KOH treatments.

GPC profile of the wood samples from m.0, m.1 K and m.1 N after benzoylation (a) and acetylation (b)
X-Ray diffractometry (XRD)
The XRD spectra obtained for m.0, m.1 K and m.1 N are reported in Fig. 5. The diffraction pattern corresponds to cellulose Iβ phase, with diffraction peaks at 2θ = 20.5°, 22.4° and 23.9°, corresponding to the (201), (002) and (040) diffraction planes, respectively. Cellulose Iβ is the most common cellulose allomorph in nature [41, 42] and it is also expected to show a double diffraction peak around 2θ = 16°, corresponding to the (101) and (10 − 1) diffraction planes. These peaks are not clearly visible in XRD spectra. This could be due to an iso-orientation effect of the crystalline phase of cellulose, likely derived from an insufficient pulverization of the samples. However, as all samples have been treated in the same way, a comparison of their diffraction patterns can provide reliable information on the changes induced by the two pre-treatments. The intensity of the reflection signals decreased after treatments, and the effect was more evident after ammonia fuming (m.1 N) than KOH solution treatment (m.1 K). The calculated Crystallinity Indices (CI) of cellulose shown in Fig. 5 (right upper corner) confirm this behaviour. Comparing the obtained values for reference wood (m.0–0.63) and the samples, the index decreases for m.1 K to 0.57 and further to 0.41 for m.1 N. This evidence suggests that the treatments tend to reduce the cellulose crystallinity, most significantly after NH3 fuming than KOH application.

Diffractograms for samples of spruce wood (m.0) treated with KOH solution (m.1 K) and ammonia (m.1 N). The Crystallinity Indices (CI) value for each sample is shown
Discussion
Our findings highlighted how the effect of alkaline treatments are different from those observed in other similar contexts and believed to produce a milder but comparable outcome (e.g., delignification and degradation of archaeological wood) [8, 23, 46, 47, 52, 53]. While the effects of the alkaline medium on the aesthetical feature are well described and distinguished—by the darkening and the mild yellowing of the surface for the ammonia fuming and KOH solution treatments, respectively—the impact of both considered treatments on the wood composition is analogous. The FTIR-ATR experiments showed that most of the changes induced by the alkaline treatments affected hemicellulose and lignin, while the signals of cellulose were less affected. In addition, the Py(HMDS)-GC/MS experiments showed that the pyrolytic behaviour of both holocellulose and lignin was influenced by the pre-treatments, confirming the FTIR results. The degradation of holocellulose consisted in the deacetylation of the O-acetyl-galactoglucomannan moieties, and in partial hydrolysis of the glycosidic bonds, which led to an increase in the pyrolytic yields of secondary products such as anhydrosugars. The most extensive degradation of hemicellulose compared to cellulose is most likely ascribable to the less organized structure, which makes hemicellulose more reactive towards the alkaline reactants [8]. The most likely degradation mechanism for hemicellulose is partial hydrolysis of the glycosidic bonds [55].
The results of the GPC analyses provide further insight into the degradation mechanisms. The profiles obtained after benzoylation suggest that the overall effect of the alkaline treatments on the degree of polymerization of the wood components is small. This confirms that the hydrolysis of hemicellulose was only partial. On the other hand, significant changes were observed in the GPC profiles obtained after acetylation, suggesting a more significant cleavage of the bonds in lignin and of the LCCs. LCCs are covalent connections between hemicellulose and lignin, and most of the LCCs in softwoods involve the arabinoglucuronoxylan units in hemicellulose [56]. This leads us to conclude that the alkaline treatments prompt the cleavage of the LCCs and that the hydrolysis of hemicellulose mainly interests the monomers directly bound to lignin.
Regarding cellulose, very little information can be obtained from the adopted techniques, except for the XRD analysis that specifically estimates the crystallinity degree. The crystallinity indices of treated samples were only slightly lower than the untreated one, probably not enough to hypothesise the hydrolysis of the cellulose fibres, but suitable to propose a more feasible dissolution process. The effect of the alkaline treatment on the cellulose can be assumed to be similar to the swelling of the cellulose fibres network that is obtained by mercerization [57,58,59,60,61]. The swelling of cellulose fibres leads to a partial cleavage of the hydrogen bonds network in cellulose and it could be the reason why a moderate change in crystallinity was observed for the treated samples [58, 62,63,64,65].
As an overall consideration, the alkaline treatments seem to mostly affect hemicellulose, which is the most reactive component of wood towards chemical treatments. Due to its key role in the LCCs that act as a bridge between cellulose and lignin, even a small degradation of hemicellulose influences their interactions and therefore the ultrastructure of the cell wall [66]. The modification of the wood matrix—based on the loss of LCCs integrity—is expected to change the mechanical properties of wood [9, 65, 67,68,69]. Precisely in these circumstances, properties like hydrophilicity and the stiffness of pre-treated wood can play a pivotal role in the interaction with the following preparation layer in the violin making process (e.g., the hydrophilic sizing and ground coat) likewise the acoustic properties of the finished instrument.
Conclusions
Understanding the chemical processes occurring during the pre-treatment of a violin making procedure is fundamental to achieving awareness for conservation. We proved that pre-treatments in alkaline media affect the wood composition, and thus its chemical and physical properties. Particularly, we evidenced LCCs modifications, responsible for wood mechanical behaviour. Though further investigations—based on vibro-mechanical tests to measure the variation of the vibro-mechanical properties of wood—are in progress, we may state that the observed chemical modifications affect the acoustical properties of modern and historical instruments. The results herein reported, shed light on the pre-treatments of wood—the step of the lutherie procedure which is often cited by the literature but completely unknown—and its effect on the wood chemistry which has been only supposed until now. The most recent investigations on historical bowed string instruments seem to confirm that the construction procedures and the presence of specific materials in the coating system (e.g. oil-resinous varnish) could not alone determine the quality of a violin, and suggest that the artificial manipulation of the wood also could be a significative responsible [70]. We strongly believe that our findings could represent an important achievement in the literature landscape, able to provide new useful insights for future research and concretely point at a new experimental direction to investigate the acoustic properties of the Cremonese string instruments. Moreover, the use of reference wood specimens enabled us to follow the chemical processes which cannot be evidenced by studying only historical violins. Such new knowledge could help modern luthiers in choosing the best making and restoration procedures.
Availability of data and materials
The datasets used and/or analysed during the current study are available from the corresponding author on reasonable request.
Abbreviations
- FTIR-ATR:
-
FTIR Spectroscopy in ATR mode
- GPC:
-
Gel permeation chromatography
- LCCs:
-
Lignin carbohydrates complexes
- MC:
-
Moisture content
- Py(HMDS)-GC/MS:
-
Analytical pyrolysis coupled to gas chromatography-mass spectrometry with in situ hexamethyldisilazane derivatization
- RH:
-
Relative humidity
- SDOM:
-
Standard deviation of the mean value
- XRD:
-
X-Ray diffractometry
References
Tai BH. Stradivari’s varnish a review of scientific findings−part I. J Violin Soc Am VSA. 2007;XXI:119–44.
Tai BH. Stradivari’s varnish: A review of scientific findings, part II. J Violin Soc Am. 2009;22:60–90.
Echard JP, Lavédrine B. Review on the characterisation of ancient stringed musical instruments varnishes and implementation of an analytical strategy. J Cult Herit. 2008;9:420–9.
Fiocco G, Rovetta T, Gulmini M, Piccirillo A, Licchelli M, Malagodi M. Spectroscopic analysis to characterize finishing treatments of ancient bowed string instruments. Appl Spectrosc. 2017;71:2477–87.
Invernizzi C, Fiocco G, Iwanicka M, Targowski P, Piccirillo A, Vagnini M, Licchelli M, Malagodi M, Bersani D. Surface and interface treatments on wooden artefacts: Potentialities and limits of a non-invasive multi-technique study. Coatings. 2021;11:29.
Obataya E. Effects of natural and artificial ageing on the physical and acoustic properties of wood in musical instruments. J Cult Herit. 2017;27:S63–9.
Tai H-C, Chen P-L, Xu J-W, Chen S-Y. Two-photon fluorescence and second harmonic generation hyperspectral imaging of old and modern spruce woods. Opt Express. 2020;28:38831–41.
Fengel D, Wegener G. Wood: chemistry, ultrastructure, reactions. Berlin: Walter De Gruyter & Co.; 2011.
Tarasov D, Leitch M, Fatehi P. Lignin-carbohydrate complexes: properties, applications, analyses, and methods of extraction: a review. Biotechnol Biofuels. 2018;11:269.
Colombini MP, Orlandi M, Modugno F, Tolppa EL, Sardelli M, Zoia L, Crestini C. Archaeological wood characterisation by PY/GC/MS, GC/MS, NMR and GPC techniques. Microchem J. 2007;85:164–73.
Colombini MP, Lucejko JJ, Modugno F, Orlandi M, Tolppa EL, Zoia L. A multi-analytical study of degradation of lignin in archaeological waterlogged wood. Talanta. 2009;80:61–70.
Zoia L, Tamburini D, Orlandi M, Łucejko JJ, Salanti A, Tolppa EL, Modugno F, Colombini MP. Chemical characterisation of the whole plant cell wall of archaeological wood: an integrated approach. Anal Bioanal Chem. 2017;409:4233–45.
Degano I, Modugno F, Bonaduce I, Ribechini E, Colombini MP. Recent advances in analytical pyrolysis to investigate organic materials in heritage science. Angew Chem Int Ed. 2018;57:7313–23.
Lucejko JJ, Tamburini D, Modugno F, Ribechini E, Colombini MP. Analytical pyrolysis and mass spectrometry to characterise lignin in archaeological wood. Appl. Sci. 2021;11:240.
Broecke L. Cennino Cennini’s Il Libro dell’Arte. A new English translation and commentary with Italian transcription. London: Archetype Publications; 2015.
Nagyvary J. The chemistry of a Stradivarius. Chem Eng News. 1988;66:24–31.
Lemery N. Recueil Des Curiositez Rares & Nouvelles des plus Admirables Effets de la Nature & de l’Art: Composé de quantité de beaux Secrets gallans & autres… Leide: Chez Pierre vander Aa; Paris: Merchand Libraire; 1684.
Ruscelli G. Secreti di don Alessio piemontese nuouamente stampati. Con una bellissima aggiunta de’ Secreti hauti da un religioso pratichissimo et eccellente et esperimentati. Lucca; 1557.
Fioravanti LB. Compendio de i Secreti Rationali, di m. Leonardo Fiorauanti Bolognese, Medico, & Cirurgico. Diuiso in Cinque Libri. Operanon Meno Diletteuole, che Utile, alla Medicina, & Cirurgia, ad Alchimisti, a Quelle Donne, che si Dilettano di Belletti, & a Molte Alt. Torino; 1592.
Weigl M, Pöckl J, Müller U, Pretzl H, Grabner M. UV-resistance of ammonia treated wood. In: 3rd European conference on wood modification. 15th-16th October 2007.
Kropat M, Hubbe MA, Laleicke F. Natural, accelerated, and simulated weathering of wood: a Review. BioResources. 2020;15:9998–10062.
Weigl M, Pöckl J, Grabner M. Selected properties of gas phase ammonia treated wood. Eur J Wood Wood Prod. 2009;67:103–9.
Miklečić J, Španić N, Jirouš-Rajković V. Wood color changes by ammonia fuming. BioResources. 2012;7.3:3767–78.
Behnood A, Van Tittelboom K, De Belie N. Methods for measuring pH in concrete: a review. Constr Build Mater. 2016;105:176–88.
Dietsch P, Franke S, Franke B, Gamper A, Winter S. Methods to determine wood moisture content and their applicability in monitoring concepts. J Civ Struct Heal Monit. 2015;5:115–27.
Luo MR, Cui G, Rigg B. The development of the CIE 2000 colour-difference formula: CIEDE2000. Color Res Appl. 2001;26:340–50.
Sharma G, Wu W, Dalal EN. The CIEDE2000 color-difference formula: Implementation notes, supplementary test data, and mathematical observations. Col Res Appl, 2005;30:21–30.
Łucejko JJ, Zborowska M, Modugno F, Colombini MP, Praogonekdzyński W. Analytical pyrolysis vs. classical wet chemical analysis to assess the decay of archaeological waterlogged wood. Anal Chim Acta. 2012;745:70–7.
Lucejko JJ, Modugno F, Ribechini E, Tamburini D, Colombini MP. Analytical instrumental techniques to study archaeologicalwood degradation. Appl Spectrosc Rev. 2015;50:584–625.
Pizzo B, Macchioni N, Capretti C, Pecoraro E, Sozzi L, Fiorentino L. Assessing the wood compressive strength in pile foundations in relation to diagnostic analysis: the example of the Church of Santa Maria Maggiore, Venice. Constr Build Mater. 2016;114:470–80.
Tamburini D, Łucejko JJ, Pizzo B, Mohammed MY, Sloggett R, Colombini MP. A critical evaluation of the degradation state of dry archaeological wood from Egypt by SEM, ATR-FTIR, wet chemical analysis and Py(HMDS)-GC-MS. Polym Degrad Stab. 2017;146:140–54.
Tamburini D, Łucejko JJ, Zborowska M, Modugno F, Prądzyński W, Colombini MP. Archaeological wood degradation at the site of Biskupin (Poland): Wet chemical analysis and evaluation of specific Py-GC/MS profiles. J Anal Appl Pyrolysis. 2015;115:7–15.
Tamburini D, Łucejko JJ, Ribechini E, Colombini MP. New markers of natural and anthropogenic chemical alteration of archaeological lignin revealed by in situ pyrolysis/silylation-gas chromatography-mass spectrometry. J Anal Appl Pyrolysis. 2016;118:249–58.
Braovac S, Tamburini D, Łucejko JJ, McQueen C, Kutzke H, Colombini MP. Chemical analyses of extremely degraded wood using analytical pyrolysis and inductively coupled plasma atomic emission spectroscopy. Microchem J. 2016;124:368–79.
Mattonai M, Tamburini D, Colombini MP, Ribechini E. Timing in analytical pyrolysis: Py(HMDS)-GC/MS of glucose and cellulose using online micro reaction sampler. Anal Chem. 2016;88:9318–25.
Moldoveanu S. Analytical pyrolysis in polymeric carbohydrates. In: Analytical pyrolysis of natural organic polymers. Amsterdam: Elsevier Science B.V; 1998. p. 217–316.
Tamburini D, Łucejko J-J, Ribechini E, Colombini M-P. Snapshots of lignin oxidation and depolymerization in archaeological wood: an EGA-MS study. J Mass Spectrom. 2015;50:1103–1113.
King AWT, Zoia L, Filpponen I, Olszewska A, Haibo XIE, Kilpeläinen I, Argyropoulos DS. In situ determination of lignin phenolics and wood solubility in imidazolium chlorides using31P NMR. J Agric Food Chem. 2009;57:8236–43.
Zoia L, King AWT, Argyropoulos DS. Molecular weight distributions and linkages in lignocellulosic materials derivatized from ionic liquid media. J Agric Food Chem. 2011;59:829–38.
Salanti A, Zoia L, Tolppa EL, Orlandi M. Chromatographic detection of lignin-carbohydrate complexes in annual plants by derivatization in ionic liquid. Biomacromolecules. 2012;13:445–54.
French AD. Idealized powder diffraction patterns for cellulose polymorphs. Cellulose. 2014;21:885–96.
French AD, Santiago Cintrón M. Cellulose polymorphy, crystallite size, and the Segal Crystallinity Index. Cellulose. 2013;20:583–8.
Segal L, Creely JJ, Martin AE, Conrad CM. An empirical method for estimating the degree of crystallinity of native cellulose using the X-ray diffractometer. Text Res J. 1959;29:786–94.
Tinkler CK. “Fumed” oak and natural brown oak. Biochem J. 1921;15:477–86.
Čermák P, Dejmal A. The effect of heat and ammonia treatment on colour response of oak wood (Quercus robur) and comparison of some physical and mechanical properties. Maderas Cienc y Tecnol. 2013;15:3.
Pandey KK, Pitman AJ. FTIR studies of the changes in wood chemistry following decay by brown-rot and white-rot fungi. Int Biodeterior Biodegrad. 2003;52:151–60.
Gupta BS, Jelle BP, Gao T. Wood facade materials ageing analysis by FTIR spectroscopy. Proc Inst Civ Eng Constr Mater. 2015;168:219–31.
Cavallaro G, Agliolo Gallitto A, Lisuzzo L, Lazzara G. Comparative study of historical woods from XIX century by thermogravimetry coupled with FTIR spectroscopy. Cellulose. 2019;26:8853–65.
Lundqvist J, Teleman A, Junel L, Zacchi G, Dahlman O, Tjerneld F, Stålbrand H. Isolation and characterization of galactoglucomannan from spruce (Picea abies). Carbohydr Polym. 2002;48:29–39.
Lucejko JJ, Tamburini D, Zborowska M, Babiński L, Modugno F, Colombini MP. Oak wood degradation processes induced by the burial environment in the archaeological site of Biskupin (Poland). Herit Sci. 2020;8:44.
Zoia L, Salanti A, Orlandi M. Chemical characterization of archaeological wood: Softwood Vasa and hardwood Riksapplet case studies. J Cult Herit. 2015;16:428–37.
Balaban M, Uçar G. The effect of the duration of alkali treatment on the solubility of polyoses. Turkish J Agric For. 1999;23:667–71.
Tamburini D, Łucejko JJ, Zborowska M, Modugno F, Cantisani E, Mamoňová M, Colombini MP. The short-term degradation of cellulosic pulp in lake water and peat soil: a multi-analytical study from the micro to the molecular level. Int Biodeterior Biodegrad. 2017;116:243–59.
Bay MS, Karimi K, Nasr Esfahany M, Kumar R. Structural modification of pine and poplar wood by alkali pretreatment to improve ethanol production. Ind Crops Prod. 2020;152:112506.
Ghavidel A, Scheglov A, Karius V, Mai C, Tarmian A, Vioel W, Vasilache V, Sandu I. In-depth studies on the modifying effects of natural ageing on the chemical structure of European spruce (Picea abies) and silver fir (Abies alba) woods. J Wood Sci. 2020;66:77.
Lawoko M, Henriksson G, Gellerstedt G. Characterisation of lignin-carbohydrate complexes (LCCs) of spruce wood (Picea abies L.) isolated with two methods. Holzforschung. 2006;60:156–61.
Medronho B, Lindman B. Brief overview on cellulose dissolution/regeneration interactions and mechanisms. Adv Colloid Interface Sci. 2015;222:502–8.
Song Y, Zhang J, Zhang X, Tan T. The correlation between cellulose allomorphs (I and II) and conversion after removal of hemicellulose and lignin of lignocellulose. Bioresour Technol. 2015;193:164–70.
Budtova T, Navard P. Cellulose in NaOH–water based solvents: a review. Cellulose. 2016;23:5–55.
Peng H, Dai G, Wang S, Xu H. The evolution behavior and dissolution mechanism of cellulose in aqueous solvent. J Mol Liq. 2017;49:11121–30.
Swensson B, Larsson A, Hasani M. Dissolution of cellulose using a combination of hydroxide bases in aqueous solution. Cellulose. 2020;27:101–12.
Mittal A, Katahira R, Himmel ME, Johnson DK. Effects of alkaline or liquid-ammonia treatment on crystalline cellulose: changes in crystalline structure and effects on enzymatic digestibility. Biotechnol Biofuels. 2011;4:41.
Lionetto F, Del Sole R, Cannoletta D, Vasapollo G, Maffezzoli A. Monitoring wood degradation during weathering by cellulose crystallinity. Mater. 2012;5:1910–22.
Mukarakate C, Mittal A, Ciesielski PN, Budhi S, Thompson L, Iisa K, Nimlos MR, Donohoe BS. Influence of crystal allomorph and crystallinity on the products and behavior of cellulose during fast pyrolysis. ACS Sustain Chem Eng. 2016;4:4662–74.
Xu E, Wang D, Lin L. Chemical structure and mechanical properties of wood cellwalls treated with acid and alkali solution. Forests. 2020;11:87.
Zimmermann T, Richter K, Bordeanu N, Sell J. Arrangement of cell-wall constituents in chemically treated Norway spruce tracheids. Wood Fiber Sci. 2007;39:221–31.
Ishikura Y, Abe K, Yano H. Bending properties and cell wall structure of alkali-treated wood. Cellulose. 2010;17:47–55.
Moon RJ, Wells J, Kretschmann DE, Evans J, Wiedenhoeft AC, Frihart CR. Influence of chemical treatments on moisture-induced dimensional change and elastic modulus of earlywood and latewood. Holzforschung. 2010;64:683–92.
You TT, Zhang LM, Zhou SK, Xu F. Structural elucidation of lignin-carbohydrate complex (LCC) preparations and lignin from Arundo donax Linn. Ind Crops Prod. 2015;71:65–74.
Su CK, Chen SY, Chung JH, Li GC, Brandmair B, Huthwelker T, Huthwelker T, Fulton JL, Borca CM, Huang SJ, Nagyvary J, Tseng HH, Chang CH, Chung DT, Vescovi R, Tsai YS, Cai W, Lu BJ, Xu JW, Hsu CS, Wu JJ, Li HZ, Jheng YK, Lo SF, Chen HM, Hsieh YT, Chung PW, Chen CS, Sun YC, Chan JCC, Tai HC. Materials engineering of violin soundboards by Stradivari and Guarneri. Angew Chemie Int Ed. 2021;60:19144–54.
Acknowledgements
The work is part of a wide project ANIMA—ANalysis and Investigation of Materials and Acoustics which involved Comune di Cremona city and the Cultural District of Violin Making of Cremona. The authors would like to thank Comune di Cremona city for funding and sponsoring the ANIMA project; the master makers M° Arrè Pino, M° Luca Baratto, M° Bardella Elena, M° Borchardt Gaspar, M° Cavagnoli Roberto, M° Cavanna Marcello, M° Piccinotti Barbara, M° Shotaro Nishimura and M° Voltini Alessandro which are part of the Cultural District of Violin Making for providing and explaining their knowledge about traditional practices; RIVOLTA SNC di M. e S. RIVOLTA (Desio (MI), Italy; Scuola Internazionale di Liuteria of Cremona; prof. Claudio Canevari of the University of Pavia which supported us with expertise, places and tools for the laboratory models preparation and prof. Curzio Merlo of the Laboratorio di Diagnostica applicata a.i. Beni Culturali di Cr.Forma.
Funding
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Author information
Authors and Affiliations
Contributions
MA designed the experimentation, prepared the laboratory samples, carried out the acquisition and the processing of the data, wrote and edited the original draft; DC supervised and validate, and cared the review and editing; GF edited and reviewed the manuscript and the figures; MM prepared sampling, carried out the analysis and the data processing, validated and edited the results; JJL carried out the acquisition and the processing of the data, supervised the methodology and the analysis, validated the results; LZ prepared the samples, supervised the methodology, carried out the acquisition and the processing of the data, validated the results, reviewed the manuscript. MPC supervised the project and the methodology, validated, and reviewed the manuscript. MM supervised and validated the project, the methodology and reviewed and editing the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Table SM1
. Laboratory models preparation and sampling strategy for the destructive tests. Table SM2. Lignin and Holocellulose pyrolysis products identified in spruce wood and classified into categories. Table SM3. Semi-quantitative results obtained by Py(HMDS)-GC/MS. H/L ratio for the studied samples. Fig. SM1. a* and b* coordinates measured before (empty circle) and after (filled circle) the KOH (red) and the NH3 (blue) treatments, respectively.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Albano, M., Comelli, D., Fiocco, G. et al. Chemical modification of wood induced by the traditional making procedures of bowed string musical instruments: the effect of alkaline treatments. Herit Sci 10, 76 (2022). https://doi.org/10.1186/s40494-022-00718-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40494-022-00718-1