
Abstract
Signaling by insulin-like growth factor-1 (IGF-1) is essential for the development of the central nervous system (CNS) and regulates neuronal survival and myelination in the adult CNS. In neuroinflammatory conditions including multiple sclerosis (MS) and its animal model experimental autoimmune encephalomyelitis (EAE), IGF-1 can regulate cellular survival and activation in a context-dependent and cell-specific manner. Notwithstanding its importance, the functional outcome of IGF-1 signaling in microglia/macrophages, which maintain CNS homeostasis and regulate neuroinflammation, remains undefined. As a result, contradictory reports on the disease-ameliorating efficacy of IGF-1 are difficult to interpret, together precluding its potential use as a therapeutic agent. To fill this gap, we here investigated the role of IGF-1 signaling in CNS-resident microglia and border associated macrophages (BAMs) by conditional genetic deletion of the receptor Igf1r in these cell types. Using a series of techniques including histology, bulk RNA sequencing, flow cytometry and intravital imaging, we show that absence of IGF-1R significantly impacted the morphology of both BAMs and microglia. RNA analysis revealed minor changes in microglia. In BAMs however, we detected an upregulation of functional pathways associated with cellular activation and a decreased expression of adhesion molecules. Notably, genetic deletion of Igf1r from CNS-resident macrophages led to a significant weight gain in mice, suggesting that absence of IGF-1R from CNS-resident myeloid cells indirectly impacts the somatotropic axis. Lastly, we observed a more severe EAE disease course upon Igf1r genetic ablation, thus highlighting an important immunomodulatory role of this signaling pathway in BAMs/microglia. Taken together, our work shows that IGF-1R signaling in CNS-resident macrophages regulates the morphology and transcriptome of these cells while significantly decreasing the severity of autoimmune CNS inflammation.
Similar content being viewed by others


Introduction
Microglia and border-associated macrophages (BAMs) compose the phagocytic immune resident compartment of the central nervous system (CNS) and inhabit the tissue parenchyma as well as the barrier-containing interfaces such as the choroid plexus, the leptomeninges and the perivascular spaces [25]. While the physiological and the pathological role of microglia has been extensively studied [31, 74, 77, 90], the contribution of BAMs to CNS homeostasis has only recently been investigated, thanks to technical advancements allowing to study these cells separately from short-lived bone marrow-derived myeloid cells [35, 95, 105]. These studies reveal a surprising heterogeneity of these cells within their distinct environments [95] and suggest a main role in immune surveillance, waste clearance, nutrient uptake and interaction with other patrolling cells [34]. Furthermore, experimental evidence indicates an important function of BAMs during pathological CNS conditions ranging from neurodegenerative diseases to ischemia [34] and including autoimmune neuroinflammation, in which early activation, local proliferation, augmented antigen presenting and immunomodulatory activities of BAMs have been described in different phases of disease development [45]. Nonetheless, the study of the complex role of these cells remains in its infancy [43].
Among their different pro- or anti-inflammatory actions [13], CNS-resident myeloid cells act as context-dependent immune-modulators by secreting cytokines and growth factors that support inflammation or tissue regeneration [64, 72, 98]. One of these molecules is Insulin-like growth factor 1 (IGF-1) [92] acting as a pro-remyelination [4, 61, 67, 104] and neuroprotective factor within the CNS [51] while being systemically produced by the liver as a hormone regulating the somatotropic axis. While an anti-inflammatory environment rich in IL-4 or cyclic adenosine monophosphate promotes the release of IGF-1 [8, 9, 92], a pro-inflammatory milieu (i.e. rich in IFN-γ) inhibits microglial IGF-1 production [92] and their ability to support oligodendrogenesis [7]. Beside its local production, IGF-1 levels in the CNS and in the cerebrospinal fluid (CSF) are also regulated by the import of peripheral IGF-1 across the CNS vasculature [102], a mechanism partially regulated by neuronal activity [69].
IGF-1 binds with high affinity to the IGF-1 receptor (IGF-1R) (M M Rechler and Nissley, 1985;[76], a tyrosine-kinase receptor mediating cellular growth, survival and sensing of external nutrient sources [14]. The molecule and its receptor are typically expressed in close proximity, indicating paracrine and autocrine signalling [70, 99]. In the CNS, IGF-1R is expressed by virtually all resident and invading inflammatory cells, with IGF-1 in turn being able to modulate the function of different inflammatory players [86]. In peripheral organs, signalling through IGF-1R showed an anti-inflammatory modulation of myeloid cell phenotypes in different disease models [32, 41, 50, 88]. Nonetheless, the specific actions controlled by IGF-1R signalling in microglia and BAMs remain unknown, together hindering our understanding of this pathway in CNS homeostasis and inflammation.
In this work, by employing tamoxifen-inducible genetic recombination in CX3CR1-CreERT2 × IGF-1Rfl/fl mice, we abrogated the expression of IGF-1R in CNS-resident macrophages. IGF-1R deletion did not affect the density of BAMs and microglia but led to altered cellular morphology in the brain and spinal cord. Bulk RNA sequencing of IGF-1RKO and control CNS-resident myeloid cells suggested a higher basal activation in microglia and BAMs and showed a downregulation of genes and pathways involved in BAM migration in IGF-1RKO mice. To assess whether these changes impacted the development of autoimmune CNS inflammation, we induced the neuroinflammatory model experimental autoimmune encephalomyelitis (EAE) and observed a consistently worsened clinical disease in mice carrying Igf1r deletion compared to controls.
In summary, our study describes for the first time the role of IGF-1 signalling in CNS-resident macrophages and shows that IGF-1R regulates the homeostatic phenotype and functions of these cells in turn influencing the development of autoimmune CNS inflammation.
Material and methods
Animals
Tamoxifen inducible B6.129P2(C)-Cx3cr1tm2.1(Cre(ERT2)Jung/J (Cx3Cr1-creERT2) mice were provided by Prof. Marco Prinz and crossed with C57BL/6j IGF-1R floxed mice [49] provided by Prof. Thorsten Buch and with B6.Cg-Gt(ROSA)26Sortm14(CAG−tdTomato)Hze/J (Ai14) reporter mice [58] kindly provided by Dr. Urban Deutsch. Mice were kept under specific pathogen free conditions in individually ventilated cages and fed ad libitum. Animal experiments utilized female mice between the age of 6 and 12 weeks of age and were approved by the veterinary office of the Canton of Bern, Switzerland and were performed following the Swiss legislation on animal protection.
Tamoxifen treatment
To induce Cre recombinase in six to ten weeks old IGF-1RKO−tdTomato and IGF-1RWT−tdTomato mice, we subcutaneously injected mice in four different places on the dorsal body area with a total of 200 μl of corn oil (50 μl/ spot) (Sigma-Aldrich, Cat.C8267) containing 4 mg Tamoxifen (Sigma-Aldrich, Cat. T5648) at day 0 and day 2. Tamoxifen was freshly dissolved prior to injection by thorough shaking for 2–3 h at room temperature.
DNA isolation
Mice were sacrificed by isoflurane overdose and intracardially perfused with DPBS. Following CNS harvest, 20–30 mm thick brain and spinal cord pieces were incubated in 500 μl of lysis buffer (containing 1 M Tris hydrochloride (pH 7.5) (Sigma-Aldrich), 0.5 M Ethylenediaminetetraacetic acid (EDTA) (Sigma-Aldrich, Cat. 03,685), 20% Sodium dodecyl sulfate (SDS) (Sigma-Aldrich, Cat.436143), 5 M Sodium Chloride (NaCl) (Sigma-Aldrich, S9625), and distilled water) for 2–3 h or overnight at 55 °C under shaking conditions. Following 10 min centrifugation at 13′000 rpm, 4 °C, the supernatant was transferred to tubes filled with 500 μl isopropanol and centrifugated again for 10 min, at 13′000 rpm, 4 °C. Next, the supernatant was discarded, and the DNA pellet was washed with 70% ethanol. Following centrifugation (10 min, 13′000 rpm, 4 °C), the supernatant was discarded, and the remaining ethanol in the samples was allowed to evaporate under a sterile hood. The DNA pellet was dissolved in distilled water and the concentration and purity (ratio of absorbance at 260 and 280 nm) was assessed using a NanoDrop measuring system (Thermo Fischer Scientific). The samples were frozen at −20° C until further usage.
Polymerase Chain Reaction (PCR)
DNA was amplified through polymerase chain reaction (PCR). Specifically, 1 μl of sample DNA was mixed with 12.54 μl distilled water, 4 μl of 5 × Green GoTaq flexi PCR buffer (Promega), 1.2 μl of 25 mM Magnesium Chloride (MgCl), 0.8 μl of 5 mM dNTPs, 0.13 μl Igf1r forward primer (CCC AAA CAG ACC ACC ACC A) (100 pmol/ μl), 0.13 μl Igf1r reverse primers (CTT CAG CTT TGC AGG TGC ACG) (100 pmol/ μl) and 0.2 μl GoTaq Flexi DNA Polymerase (Promega). The PCR reaction occurred under the following cycling conditions: initial denaturation (98 °C, 30 s), followed by 35 cycling times involving denaturation (98 °C, 30 s), annealing (70 °C, 30 s) and elongation (72 °C, 60 s). The reaction ended with a final elongation step at 72 °C, for 5 min. 10 μl of each sample were loaded on a 1% agarose gel and run for 50 min at 140 V. The expected band size for the Igf1r floxed is 1′200 bp, for Igf1r WT 1′100 bp and 600 bp for the Igf1r locus following genetic recombination.
EAE induction
Mice were induced with active EAE six to ten weeks post Tamoxifen treatment (after replenishment of IGF-1R+ blood monocytes by natural turnover), as previously described [44]. Briefly, at day 0, mice were immunized with an emulsion of 200 μg MOG peptide (MOG35-55, Genscript, USA) and complete Freund´s adjuvant (Santa Cruz Biotechnology) containing Mycobacterium tuberculosis (Difco) (100 μl/mouse subcutaneously, in the left and right flank and at the tail base). Furthermore, 400 ng pertussis toxin (List Biological Laboratories, Campbell, CA, USA) diluted in sterile DPBS was administered intraperitoneally at day 0 and day 2.
The weight of the mice was recorded daily and the clinical disease course was assessed using a 0–3 score scale as described in [93]. Mice displaying a 3–5% weight loss but no other motor impairments received a score of 0 (weight-loss stage), animals showing a limp tail were scored as 0.5 (day of clinical onset), mice displaying a partial weakening of hind limbs received a score of 1 and animals presenting hind limb paraparesis/paraplegia were scored with 2 (symptomatic disease peak). Mice displaying hindlimb paralysis and forelimbs paraparesis were scored with 3 and met termination criteria. For each EAE group, the disease incidence as well as the area under the curve was calculated to assess the clinical disease evolution.
Isolation of CNS resident and infiltrating myeloid cells and flow cytometry analysis/ cell sorting
Mice were sacrificed by isoflurane overdose, perfused with 20 ml ice-cold DPBS and the brain and spinal cords were harvested and processed separately for CX3CR1-creERT2× IGF-1R floxed mice. The tissue was finely cut/ homogenized in collagenase medium containing 0.4 mg/ml Collagenase VIII (Sigma, C2139) and 2 U/ml DNAse (Roche) and incubated afterwards in a 37 °C water bath, under shaking conditions, for 30 min. During this incubation time, the samples were gently resuspended every 10 min to help the homogenization of the tissue. The enzymatic reaction was stopped by sample incubation with 0.5 M EDTA (1:100) for 5 min at 37 °C. The samples were then transferred on ice and filtered through a 70 μm cell strainer (SPL Life Sciences, Cat. 93,070). Following 5 min centrifugation (1′500 rpm, 4 °C), the CNS pellets were resuspended in 13 ml of 33% Percoll (Ge Healthcare Biosciences, 17-0891-01) solution and centrifugated for 30 min at 4′000 rpm, 4 °C without breaks. This step allows the creation of a density gradient formed by a myelin ring at the surface, a middle layer containing CNS resident and infiltrating myeloid cells and a blood cell ring at the bottom of the tube. The middle layer containing the cells of interest was transferred to a new tube, filtered through 100 μm cell strainer and washed twice with DPBS. Following centrifugation (5 min, 1′500 rpm, 4 °C), and supernatant was discarded and the cells were prepared for antibody staining. For this step, the cells were initially incubated with anti-CD16/32 (homemade solution) for 15 min on ice to block the FC-receptors and after DPBS wash, the cells were incubated for 30 min at 4 °C, in dark condition, with the following antibodies diluted in FACS buffer (containing 1% FCS (SeraGlob, S11500), and DPBS): CD11b (FITC or PerCP Biolegend, Clone M1/70, Cat.101230), CD45 (LSBio, Lot 62,579), CD44 (Biolegend, Brilliant Violet 605, clone IM7, Cat. 103,047), CD206 (Biolegend, Brilliant Violet 711, Clone C068C2, Cat. 141,727), Ly6C (Biolegend, Alexa Fluor 700, clone HK1.4, Cat.128024), Ly6G (Biolegend, APC/Cy7, Clone 1A8, Cat.127624), MHC-II (Biolegend, Brilliant Violet 711, Clone C068C2, Cat.141727), Fixable viability dye eFluor 506 (Invitrogen, Rockford, IL, USA, cat. 65–0866-14). The compensations were performed using unstained cells and single antibody stainings and the samples were acquired using an Attune NxT cytometer (Thermo Fisher Scientific). For data analysis, we used the software FlowJo (version 10, Ashland, OR, USA).
The following gating strategy was used to investigate the phenotype of different myeloid cell populations: after identifying cells of interest based on forward and side scatter, we selected single cells (FSC-H vs FSC-A). We performed the analysis on two distinct live cell populations: CD45highCD11b+ and CD45intermediateCD11b+ cells. Within the CD45highCD11b+ population, we selected Tomato + cells, thus identifying CD45highCD11b+Tomato+ BAMs and CD45highCD11b+Tomatoneg cells (CNS infiltrating blood-derived myeloid cells). Finally, the CD45highCD11b+TomatonegLy6GnegLy6C+ population represented CNS infiltrating monocyte-derived macrophages. CNS resident microglial cell population was identified based on CD45intermediateCD11b+Tomato+ expression. In these populations we analyzed the expression of MHC-II, CD206 and CD44. As readout, we displayed the percentage or absolute number of tdTomato positive cells (BAMs and microglia) or Ly6C + cells (infiltrating monocyte-derived macrophages) and the relative mean fluorescence intensity (MFI) calculated by subtracting the MFI of antibody staining from the MFI of isotype control staining.
For cell sorting, cells were incubated with blocking anti-CD16/32 ab (eBioscience) for 15 min on ice and after DPBS wash, cells were incubated for 45 min at 4 °C, in dark condition, with the following antibodies diluted in FACS buffer (containing 1% FCS (SeraGlob, S11500), and DPBS): CD3 (APC-Cy7 Biolegend), CD19 (APC-Cy7 Biolegend), NK1.1 (APC-Cy7 Biolegend), Ly6C (APC-Cy7 Biolegend), cell viability dye eFluor 506), CD11b (BV605 Biolegend), CD45 (BV421 Biolegend), CD206 (FITC Biolegend), P2ry12 (Biolegend, APC). Samples were sorted into RNAprotect buffer using an MoFlow Astrios (Beckman Coulter).
CNS isolation, decalcification and cryostat sections
Mice were deeply anesthetized and transcardially perfused initially with DPBS (Gibco, Paisley, UK) and then with 4% paraformaldehyde (PFA, Merk Darmstadt, Germany). Entire skulls as well as spinal columns were isolated and post-fixed in 4% PFA overnight at 4 °C. Following PBS wash, organs were decalcified for 7 to 10 days at 4 °C in 14% EDTA solution (Sigma-Aldrich, Cat. E5135) (prepared in osmose water and adjusted to a pH of 7.8 – 8) (with a change of EDTA every second day) [42]. At the end of the decalcification procedure, the softness of the bones was tested by gentle touching with a pair of tweezers. Brain and spinal cord tissues were washed with PBS, immersed for 3 days in 30% sucrose (Sigma-Aldrich, St. Louis, MO, USA) and frozen at -80 °C in O.C.T (Tissue-Tek). 40 µm sagittal brain section and 25 µm spinal cord longitudinal sections were cut using a cryostat (HM550, Thermo Fisher).
Immunofluorescence stainings of CNS tissue
IGF-1R, GFAP and myelin basic protein (MBP) immunofluorescence stainings were performed on cryostat-cut decalcified spinal cord sections as follows: for IGF-1R and GFAP stainings, the tissue was fixed with 100% ice cold acetone at −20 °C for 10 min and dried before being reconstituted in homemade 1xTris-Buffered Saline (TBS). Alternatively, for MBP staining, sections were fixed with 100% ice cold methanol at −20 °C for 10 min and immediately washed for a total of 30 min in TBS. Unspecific antibody binding was blocked for 2 h at room temperature (RT) either with 10% goat serum containing 0.1% Triton (Sigma-Aldrich, St. Louis, MO, USA) diluted in TBS (for IGF-1R and GFAP stainings) or with 5% BSA containing 0.3% Triton diluted in TBS (for MBP staining). Sections were then incubated at 4 °C overnight with the following primary antibodies: polyclonal rabbit anti-IGF1R (phospho-Y1161, Abcam, ab39398, 1:100), polyclonal rabbit anti-GFAP (Dako, Z0334, 1:100) diluted in 2% goat serum in TBS containing 0.1% Triton and a monoclonal rat anti-MBP (aa82-87, BioRad, MCA409S, 1:100) diluted in 1% BSA in TBS containing 0.3% Triton. After rinsing 3 × 10 min with TBS, spinal cord sections were incubated for 2 h at RT with the following secondary antibodies: Alexa Fluor 647 goat anti-rabbit (Invitrogen, A32733), Alexa Fluor 488 goat anti-rabbit (Invitrogen, A11008) diluted (1:200) in 2% goat serum in TBS and Alexa Fluor 488 goat anti-rat (Invitrogen, A-11006) (1:200) diluted in 1% BSA in TBS. Following a 3 × 10 min wash with TBS, slices were incubated with DAPI (1:5000 in TBS, 1 mg/ml stock, AppliChem, Darmstadt, Germany) for 10 min at RT, after which they were mounted with Mowiol 4–88 solution (Sigma-Aldrich, St Louis, MO, USA) and left to dry.
For IGF1-R staining, Z-stack images of CNS sections were acquired using a LSM800 confocal microscope (Zeiss) with 25 × and 40 × objectives. Images of GFAP and MBP stainings were acquired using a fluorescence microscope Nikon Eclipse E600 with 10 × and 20 × objectives. All images were analyzed using the software Fiji (National Institute of Health, Bethesda, MD, USA).
Analysis of inflammatory lesions, density and morphology of microglia/BAMs
To assess the density and size of inflammatory lesions, spinal cord sections from IGF-1RKO−tdTomato (n = 5) and IGF-1RWT−tdTomato mice (n = 4) were stained with DAPI and unphysiological accumulations of DAPI + structures (as indicators for immune cell infiltration) were manually selected via Fiji. Single lesion area (μm2) was calculated and number of well-defined single lesions over the entire tissue area was assessed (cm2). To define the degree of demyelination, white matter areas of spinal cord tissue sections were selected and MBP staining intensity assessed via Fiji. To assess the density and morphology of BAMs and microglial cells in healthy IGF-1RKO−tdTomato (n = 4) and IGF-1RWT−tdTomato mice (n = 4), we used the Fiji software. From each mouse, Z-stack images were acquired from brain (cortex, midbrain and cerebellum) and spinal cord (cervical, thoracic, lumbar/sacral) regions. Per region, we acquired three representative images. To analyze the density of microglia and leptomeningeal and perivascular BAMs, we used maximum intensity projection of confocal z-stack images acquired with a 25 × objective. pvMs were defined as elongated Tomato + cells delineating blood vessels. Following image conversion to binary mode, we selected three to four regions of interest per image, used a threshold and a cell circularity range tested to cover most cells in the acquired images, and used the ´count cell` function to assess the number of cells/mm2. We further assessed the morphology of individual microglia within the brain and spinal cord of WT and KO mice (confocal microscopy) as well as the morphology of leptomeningeal BAMs (two-photon intravital imaging). For the description of leptomeningeal BAMs, two-photon movies from the intact spinal cord were favored over tissue sections to better preserve the physiological morphology of these cells in their CSF-filled microenvironment. We used three different methods: Sholl analysis [60, 66, 85], skeleton analysis and fractal—lacunarity analyses [65, 106]. Each was performed on maximum intensity projection of confocal z-stack images acquired with a 40 × objective. Prior to morphological analyses, we selected images in a blinded manner. Each image was converted to binary mode and the ´unsharp mask filter´ as well as the ´noise-despecle´ function were applied in Fiji to detect as many cell branches as possible. When needed, following image thresholding, we manually reconstructed the branches lost during this procedure.
Sholl Analysis: after manually indicating the largest Sholl radius from the center of each soma, we defined the starting radius at 5 μm and the radius step size at 2 μm and selected the Sholl `intersections` method. As outcome for cell morphological analysis, we used the following parameters: the maximum number of intersections per cell irrespective of the radius (Max Inters.), the critical value/the distance from the soma where the maximum number of intersections occurred (Max.Inters. Radius) and the maximum radius where the longest branch occurred (Ending Radius) [66]. Since Sholl analysis provides only information related to cell branches, we also manually measured the soma size (area) of each analyzed cell. Skeleton analysis: on binary images, we used the `analyze skeleton 2D/3D´ function in Fiji and chose the following parameters: number of branches/ cells, maximum branch length, number of endpoints/ cell and number of junctions/ cells. Additionally, to obtain an indication about the cell complexity, we calculated the summed branch length for each cell.
Lastly, we used Fractal and Lacunarity analyses as a more in-depth method to determine the complex geometry of individual cells as well as their shape/elongation. In biology, fractal analysis measures the fractal characteristics of a data set (the degree to which a cell fills the occupied space) [47]. Complementary, lacunarity analysis assesses the gaps between cell branches and the heterogeneity of their distribution. Very ramified cells display low lacunarity whereas high lacunarity indicates poor cellular branching. From these two analyses, we extracted the following parameters: cell density, span ratio and circularity (as a measure of cell shape/elongation), fractal dimension, lacunarity and maximum span across convex hull (as measures of cell complexity). The number of individual microglia analyzed was the following: WT (4 mice: brain n = 71 cells, spinal cord n = 66 cells), KO (4 mice, brain n = 83 cells, spinal cord n = 76 cells). The number of individual leptomeningeal BAMs analyzed was the following: WT (3 mice: n = 90 cells). KO (3 mice: n = 77 cells).
Two-photon intravital microscopy
The cervical spinal cord was exposed by laminectomy as previously described [38]. Briefly, animals were intubated by insertion of a canula in the trachea providing oxygen and isoflurane through a Minivent system (Harvard apparatus). Laminectomy (C2-5) was performed and spinal column fixed at C1 and C6 in a stereotactic frame. Body temperature and heart rate were recorded and stored electronically. Imaging was performed via 2-photon (LaVision TrimScope II microscope, Spectra-Physics laser). Mouse respiration via MiniVent was synchronized to picture acquisition through a triggering device (TrigViFo) reducing imaging artifacts [97]. Through an additional toolkit (VivoFollow II) [97], this synchronization allowed real-time distortion correction. Tissue displacement was automatically corrected by objective and stage adjustment through a Python script (VivoFollow I) [96]. Single images, z-stacks and videos were acquired with ImSpector software (LaVision). Raw images were processed with Fiji [82] and saved as TIF.
RNA extraction, library preparation and sequencing
Microglia and BAMs were sorted from the brain of IGF-1RKO−tdTomato and IGF-1RWT−tdTomato mice and processed for RNA sequencing. RNA extraction, library preparation and sequencing were performed at the Genomics Core Facility “KFB—Center of Excellence for Fluorescent Bioanalytics”. Total RNA was extracted from sorted cells stabilized in RNAprotect buffer according to the RNeasy Plus Micro Kit protocol (QIAGEN, Hilden, Germany). In brief, cells were stored and shipped in buffer RNAprotect at 2–8 °C. After pelleting by centrifugation for 5 min at 5′000 × g, the RNAprotect was replaced by 350 µl buffer RLT Plus and the samples were homogenized by vortexing for 30 s. Genomic DNA contamination was removed by using gDNA Eliminator spin columns. One volume of 70% ethanol was added and the samples were applied to RNeasy MinElute spin columns followed by several wash steps. Total RNA was eluted in 12 μl of nuclease free water. Purity and integrity were assessed on the Agilent 2100 Bioanalyzer with the RNA 6000 Pico LabChip reagent set (Agilent, Palo Alto, CA, USA). The SMARTer Ultra Low Input RNA Kit for Sequencing v4 (Clontech Laboratories, Inc., Mountain View, CA, USA) was used to generate first strand cDNA from 1 ng total-RNA. Double stranded cDNA was amplified by LD PCR (12 cycles) and purified via magnetic bead clean-up. Library preparation was carried out as described in the Illumina Nextera XT Sample Preparation Guide (Illumina, Inc., San Diego, CA, USA). 150 pg of input cDNA were tagged and fragmented by the Nextera XT transposome. The products were purified and amplified via a limited-cycle PCR program to generate multiplexed sequencing libraries. For the PCR step 1:5 dilutions of the unique dual indexing (i7 and I5) adapters were used. The libraries were quantified using the KAPA Library Quantification Kit—Illumina/ABI Prism User Guide (Roche Sequencing Solutions, Inc., Pleasanton, CA, USA). Equimolar amounts of each library were sequenced on a NextSeq 500 instrument controlled by the NextSeq Control Software (NCS) v2.2.0, using two 150 Cycles High Output Kits with the dual index, single-read (SR) run parameters. Image analysis and base calling were done by the Real Time Analysis Software (RTA) v2.4.11. The resulting.bcl files were converted into.fastq files with the bcl2fastq v2.18 software.
Transcriptomics analysis
Bulk RNA-seq data was aligned with STAR v2.7.8a [23] to the mouse reference genome Gencode M26 [40]. Genes with sum count expression less than 5 were filtered out. Differential expression was performed using the limma trend method [78] on the log2 counts per million (CPM) values. Heatmaps were generated with the R packages pheatmap; enrichment analysis was performed with the enrichGO function from the R package ClusterProfiler and using the Gene Ontology database.
Statistics
Statistical analysis was performed using GraphPad Prism 9 software (La Jolla, CA, USA).
All data was assessed for normality distribution. Shapiro–Wilk test was used to assess the normality for small sample sizes (< 50) while Kolmogorov Smirnov test was used for larger sample size (> 50). Data that was not normally distributed was analyzed with a non-parametric Mann Whitney U test: body weight differences before and after tamoxifen treatment, quantification of IGF-1R deletion in CNS resident perivascular BAMs, morphological analysis of microglia and BAMs, EAE AUC and incidence. We further used unpaired t test with Welch`s correction for the following sets of data: quantification of IGF-1R deletion in meningeal BAMs and microglial cells, density of CNS-resident myeloid cells following genetic ablation of IGF-1R (brain BAMs, spinal cord microglia and BAMs), morphological analysis of brain microglia (cell density, maximum branch length), spinal cord microglia (cell density, maximum span across hull, fractal dimension, number of branches, maximum branch length), spinal cord BAMs (fractal dimension, lacunarity), integrin expression and flow cytometry characterization of cell number and phenotype during EAE, number of inflammatory lesions per area, area of individual lesions during EAE, MBP staining intensity and GFAP + cells/mm2. Two-way ANOVA with Tukey`s multiple comparisons test was used for assessing the EAE severity (score differences) between IGF-1RKO−tdTomato and IGF-1RWT−tdTomato mice. All values are presented as mean ± standard error of the mean (SEM). Asterisks indicate significant differences (∗ p < 0.05, ∗ ∗ p < 0.01 and ∗ ∗ ∗ p < 0.001, ∗ ∗ ∗ ∗ p < 0.0001).
Results
Genetic deletion of Igf1r in CNS-resident microglia and BAMs
IGF-1 signalling is a main regulator of metabolism, proliferation and survival in numerous cell types, but its function in CNS-resident macrophages remains undetermined. To shed light on this unclear role, we genetically deleted the receptor Igf1r from CX3CR1-expressing myeloid cells by crossing a tamoxifen-inducible mouse model, CX3CR1-creERT2 [105], with a transgenic mouse carrying a loxP-flanked 3rd exon in the Igf1r gene (IGF-1R floxed) [49]. In the resulting CX3CR1-creERT2 × IGF-1R floxed model, recombination is achieved by subcutaneous administration of tamoxifen [19] followed by a waiting time of 5 weeks during which short-lived bone marrow-derived CX3CR1+ monocytes are replenished through natural turnover by new IGF1R+ cells. Long-lived BAMs and microglia instead maintain their IGF-1RKO status. To distinguish in situ CNS-resident macrophages from short-lived bone marrow-derived myeloid cells, mice were further crossed to a cre-inducible tdTomato reporter Ai14 mouse [58] allowing expression of the fluorescent molecule tdTomato in BAMs/microglia. Taken together, we created the new animal model CX3CR1-creERT2× IGF-1R floxed x Ai14 (henceforth IGF-1RKO−tdTomato) and its cre+ control line CX3CR1-creERT2 × Ai14 (henceforth IGF-1RWT−tdTomato) (Fig. 1A).

Characterization of IGF-1RKO mouse model. A Diagram illustrating the genetic make-up of the CX3CR1-creERT2 x IGF-1R floxed x Ai14 mouse model. B Gating strategy illustrating the sorting of tdTomato+ CD45 + CD11b + myeloid cells from the CNS of tamoxifen-treated IGF-1RKO−tdTomato and IGF1RWT−tdTomato mice. C DNA from sorted cells was tested by PCR using specific primers identifying the WT (1′100 bp), floxed (1′200 bp) or recombined (600 bp) Igf1r locus (n = 2). D Representative confocal pictures and E quantification of IGF-1R expression in Tomato + cells following staining with IGF-1R-specific antibody in CNS sections from IGF-1RKO−tdTomato (n = 3) and IGF-1RWT−tdTomato mice (n = 4): unpaired T-test with Welch´s correction, p < 0.0001 (microglia), p < 0.001 (meningeal BAMs) and Mann Whitney U test p = 0.0048 (perivascular BAMS) F) Bar graph shows difference in weight before and after tamoxifen treatment measured in IGF-1RKO−tdTomato (n = 11) and IGF-1RWT−tdTomato mice (n = 11) from 3 different experiments (Mann Whitney U test, p = 0.031). All values are presented as mean ± SEM. Asterisks indicate significant differences (∗ p < 0.05, ∗ ∗ p < 0.01 and ∗ ∗ ∗ p < 0.001, ∗ ∗ ∗ ∗ p < 0.0001)
To assess whether genetic recombination occurred in CNS-resident macrophages of IGF-1RKO−tdTomato mice, we isolated CD45highCD11b+ (BAMs) and CD45intermediateCD11b+ (microglial) cells from the CNS of tamoxifen-treated IGF-1RKO−tdTomato and IGF-1RWT−tdTomato mice (Fig. 1b). PCR analysis of sorted cells (Fig. 1c) showed that genetic recombination occurred in IGF-1RKO−tdTomato but not in IGF-1RWT−tdTomato mice. However, since recombination appeared incomplete (Fig. 1c), we quantified the efficiency of receptor deletion at a protein level on decalcified CNS sections from IGF-1RKO−tdTomato and IGF-1RWT−tdTomato control mice. Confocal analysis of sections stained with IGF-1R-specific antibodies revealed receptor expression in 27% of microglial cells and in circa 21% of BAMs in IGF-1RKO−tdTomato mice, compared to 85% of microglia and 92% of BAMs in IGF-1RWT−tdTomato mice, collectively showing a deletion efficiency of approx. 68% for microglia and circa 75% for BAMs (Fig. 1D, E).
Considering the key systemic role of IGF-1 in promoting growth [62], we further assessed the weight of mice before tamoxifen administration and five weeks later. Surprisingly, we found that IGF-1RKO−tdTomato mice gained significantly more weight compared to IGF1RWT−tdTomato mice (Fig. 1F), thus suggesting that the induced macrophage-specific IGF-1R deletion can indirectly impact the somatotropic axis of treated mice.
Taken together, tamoxifen administration in IGF-1RKO−tdTomato mice resulted in IGF-1R ablation from the majority of microglia/BAMs in the mouse CNS and led to a significantly higher weight gain in IGF-1RKO−tdTomato compared to IGF-1RWT−tdTomato mice.
IGF-1R deletion leads to morphological changes in BAMs and microglia
Since IGF-1 signalling regulates cell survival and proliferation [39], we assessed whether IGF-1R deletion affected the numbers of microglia as well as perivascular and meningeal BAMs, but found no significant change in their density in either the brain or the spinal cord of IGF-1RKO−tdTomato and IGF-1RWT−tdTomato mice (Fig. 2A, B – microglia, Fig. 2C, D – perivascular BAMs, Fig. 2E, F – meningeal BAMs).

The density of CNS-resident myeloid cells following Igf-1r genetic ablation. A Representative pictures (scale bar 100 μm) and B bar graph illustrating the density of Tomato + microglia in brain and spinal cord sections from IGF-1RWT−tdTomato mice (n = 4, number of cells analysed: microglia brain n = 49, microglia spinal cord n = 28) and IGF-1RKO−tdTomato (n = 4 mice, number of cells analysed: microglia brain n = 45, Microglia spinal cord n = 42). C Representative pictures of Tomato + perivascular BAMs (pvBAMs) in the brain (scale bar zoom-out image: 100 μm, scale bar zoom-in image: 50 μm) and D bar graph illustrating the density of pvBAMs in brain and spinal cord sections (pooled p = 0.0048 together) from IGF-1RWT−tdTomato (n = 4, number of cells analysed: 31) and IGF-1RKO−tdTomato (n = 4 mice, number of cells analysed: 33). E Representative pictures and F bar graph illustrating the density of Tomato + meningeal BAMs (mBAMs) in brain and spinal cord sections from IGF-1RWT−tdTomato (n = 4, number of cells analysed: mBAMs brain n = 31, mBAMs spinal cord n = 13) and IGF-1RKO−tdTomato (n = 4 mice, number of cells analysed: BAMs brain n = 26, BAMs spinal cord n = 20). No statistically significant differences were observed between either group. Statistical analysis performed by unpaired t test with Welch’s correction: brain microglia, p = 0.833, spinal cord microglia p = 0.684, pvBAMs p = 136, brain mBAMs p = 0.828 and spinal cord mBAMs p = 0.3878
Considering that microglia functionality and activation are directly related to dynamic changes in their cell shape, size and ramifications [21, 36, 47], we analyzed the morphology of individual microglial cells on tissue sections from the brain and spinal cord of healthy IGF-1RKO−tdTomato and IGF-1RWT−tdTomato mice by Sholl analysis [60, 66, 85], skeleton analysis [2] and fractal analysis [65, 106]. Notably, brain microglia from IGF-1RKO−tdTomato mice displayed significantly increased ramifications (increased number of branches, higher total cell branch length and maximum branch length, more junctions and endpoints of contact) and a more complex morphology (defined by fractal dimension and the maximum distance between branches in a convex hull) (Fig. 3A, B, Additional file 1: Fig. 1A). In parallel, in the spinal cord of IGF-1RKO−tdTomato mice, microglia displayed lower lacunarity as well as an increased cell density and ending radius (defined by the maximum Sholl radius) (Fig. 3C, D, Additional file 1: Fig. 1B). Next, we assessed whether the morphology of BAMs would also be affected by the absence of IGF-1R. Given their location in confined perivascular spaces as well as their typical elongated shape stretching along the vessel, we refrained from analysing differences in pvMs morphology. However, we imaged by 2-photon microscopy the cervical spinal cords of anesthetized IGF-1RKO−tdTomato and IGF-1RWT−tdTomato mice and analyzed the morphology of leptomeningeal BAMs in vivo. Despite similar cell size and shape, BAMs from IGF-1RKO−tdTomato mice displayed higher lacunarity, more branches and an increased number of endpoints and branch junctions compared to control mice (Fig. 3E, F, Additional file 1: Fig. 1C).

Morphological characterization of CNS-resident myeloid cells following Igf-1r genetic ablation. To assess the morphology of CNS resident microglia (brain, spinal cord) and leptomeningeal BAMs in IGF-1RKO−tdTomato (n = 4 mice) compared to IGF-1RWT−tdTomato (n = 4 mice), we conducted three sets of analyses (Sholl analysis, skeletal analysis and fractal-lacunarity analysis – described in the method section). A Representative pictures of microglia from the brain of IGF-1RKO−tdTomato and IGF-1RWT−tdTomato mice and B quantification of number of branches per cell (p = 0.0001), fractal dimension (p = 0.0006), ending radius (p < 0.0001) of brain microglia (unpaired t test with Welch’s correction, approximative n° of cells analysed per group: IGF-1RKO−tdTomato (n = 84) IGF-1RWT−tdTomato (n = 71)). C Representative pictures of microglia morphology from the spinal cord of IGF-1RKO−tdTomato and IGF-1RWT−tdTomato mice and D quantification of number of branches per cell (p = 0.498), fractal dimension (p = 0.406), ending radius (p = 0.034) of spinal cord microglia (unpaired t test with Welch’s correction, approximative n° of cells analysed per group: IGF-1RKO−tdTomato (n = 75) IGF-1RWT−tdTomato (n = 66)). E Representative pictures of BAMs from the spinal cord leptomeninges of IGF-1RKO−tdTomato and IGF-1RWT−tdTomato mice and F quantification number of branches per cell (p = 0.034), fractal dimension (p = 0.760), ending radius (p = 0.049) of spinal cord BAMs (unpaired t test with Welch’s correction, approximative n. of cells analysed per group: IGF-1RKO−tdTomato (n = 78) IGF-1RWT−tdTomato (n = 91)). All values are presented as mean ± SEM. Asterisks indicate significant differences (∗ p < 0.05, ∗ ∗ p < 0.01 and ∗ ∗ ∗ p < 0.001, ∗ ∗ ∗ ∗ p < 0.0001)
Together, these results indicate that the lack of IGF-1R in CNS resident myeloid cells induced significant changes in the morphology of these cells, in turn suggesting that IGF-1 signalling regulates the overall phenotype of BAMs and microglia during homeostasis.
RNA analysis reveals distinct transcriptomic changes in microglia and BAMs upon IGF-1R deletion
To dissect the functional impact of IGF-1R deletion in CNS-resident phagocytes, we isolated BAMs and microglial cells from the brain and meninges of IGF-1RKO−tdTomato and IGF-1RWT−tdTomato control mice (Additional file 2: Fig. 2A) and performed bulk RNA sequencing analysis. Transcriptomic changes were observed both in IGF-1RKO−tdTomato microglia and BAMs compared to their IGF-1RWT counterparts from IGF1RWT−tdTomato mice (Additional file 2: Fig. 2B). Surprisingly, given the substantial changes in cellular morphology observed in IGF-1RKO−tdTomato mice, microglial transcriptome was only partially modified by Igf1r deletion (Fig. 4A). Within these cells we found the ATPase 2 (Atp7b) gene, which is involved in the export of excess intracellular copper [110], to be the most upregulated gene in IGF-1RKO−tdTomato microglia, together with Aspm, which is involved in the regulation of the cell cycle [108] (Fig. 4B). Gene ontology (GO) enrichment analysis further showed that multiple differentially expressed genes involved in ribosomal biogenesis and functions were also upregulated in microglia from IGF-1RKO−tdTomato mice compared to controls (Additional file 2: Fig. 2C).

Transcriptomic analysis and expression of adhesion molecules in microglia and BAMs following genetic ablation of Igf1r. A Volcano plot and B heatmap showing the differential expression analysis of microglia from IGF-1RKO−tdTomato and IGF-1RWT−tdTomato mice sorted from brain. C Volcano plot and D heatmap showing the differential expression analysis of BAMs sorted from the brain of IGF-1RKO−tdTomato and IGF-1RWT−tdTomato mice E Functional enrichment analysis of BAMs from IGF-1RKO−tdTomato and IGF-1RWT−tdTomato mice using the Biological Process pathways from the Gene Ontology database, above: pathways upregulated in IGF-1RKO−tdTomato mice, below: pathways downregulated in IGF-1RKO−tdTomato mice. F–H Flow cytometry analysis of expression of α4 (CD49d), αL (CD11a) and β2 (CD18) integrins and PSGL-1 in BAMs, microglia and splenic macrophages from IGF-1RKO−tdTomato (n = 3) and IGF-1RWT−tdTomato (n = 3) mice. Data represented as mean ± SEM. Statistical analysis was performed by using unpaired t test with Welch´s correction, *p < 0.05: BAMs (α4: p = 0.757; β2: p = 0.629, PSGL-1: p = 0.034), Microglia (α4: p = 0.591; β2: p = 0.466, PSGL-1: p = 0.907), splenic macrophages (α4: p = 0.539; αL: p = 0.281; β2: p = 0.629, PSGL-1: p = 0.402)
Conversely, RNA analysis of IGF-1RKO−tdTomato BAMs showed the upregulation of genes involved in IFN-γ signalling (Gbp10, Tgtp1), cell activation (Slamf9), myeloid cell differentiation (Hoxa7), signal transduction (Stat5, Capn5, Adcy1), plasma membrane dynamics (Mtss2) and lysosomal degradation (Hgsnat) (Fig. 4C, D). In parallel, genes involved in intercellular interactions (Mfap4, Spata13, Jam2), immunoregulation (Itgb3, Plxdc2, Siglec-h), inhibition of apoptosis (Arhgap24), disease associated genes (Cd9) and cell cycle (Ccne1) were downregulated in BAMs from IGF-1RKO−tdTomato compared to IGF-1RWT−tdTomato mice (Fig. 4C, D). GO enrichment analysis in BAMs from IGF-1RKO−tdTomato mice showed decreased RNA processing and splicing and leukocyte migration, while pathways upregulated indicated increased GTPase and Ras-mediated signalling, cellular growth and amoeboidal-like migration (Fig. 4E). Given the observed changes in genes involved in cellular movement, we isolated microglia and BAMs from the CNS of tamoxifen-treated IGF-1RWT−tdTomato and IGF-1RKO−tdTomato mice and analysed the surface expression of adhesion molecules such as integrin αL, β2, α4, αM, and selectin ligand PSGL-1 via flow cytometry. Expression of αL could not be detected in neither BAMs nor microglia (Fig. 4F, G), but the integrin was readily detected on control splenocytes (Fig. 4H). While the expression levels of integrin α4, αL and β2 were comparable between IGF-1RKO and IGF-1RWT cells, PSGL-1 appeared significantly downregulated in BAMs but not microglia from IGF1RKO−tdTomato mice (Fig. 4F, G).
Taken together, IGF-1R deletion from CNS-resident macrophages led to distinct transcriptomic changes in microglia and BAMs but mostly impacted BAM physiology by regulating pathways involved in RNA processing, growth, intracellular signalling and migration. Accordingly, the absence of IGF-1R also led to the decreased surface expression of the adhesion molecule PSGL-1 in BAMs. Overall, these changes during homeostasis might have a significant impact on the role of BAMs/microglia upon CNS pathology.
IGF-1R deletion in CNS resident microglia/macrophages increases EAE severity
IGF-1 is considered a potential therapeutic agent for the treatment of several CNS pathologies including multiple sclerosis (MS) [3, 12, 68, 73, 101]. However, IGF-1 administration in MS patients [29] and in the EAE model [5, 10, 18, 22, 33, 52, 54, 56, 103] showed no efficacy or contradictory outcomes. The interpretation of these results is difficult because of the expression of IGF-1R on multiple CNS and immune cell types [27].
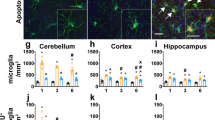
To understand the role of IGF-1R signalling in CNS-resident macrophages during autoimmune CNS inflammation, we thus induced EAE in tamoxifen-treated IGF-1RWT−tdTomato and IGF-1RKO−tdTomato mice. Notably, IGF-1RKO−tdTomato mice showed a substantially worsened clinical course compared to control mice, with an increased area under the curve and a clinical score that appeared significantly higher than in control mice as soon as 3 days after clinical disease onset and throughout the subsequent symptomatic peak and chronic EAE stages (Fig. 5A, B). Disease incidence was however comparable between experimental groups, thus suggesting that IGF-1R absence did not affect peripheral development of autoimmunity (Fig. 5C). Reflecting the worsened clinical course in IGF-1RKO−tdTomato mice, we observed significantly higher number and a trend toward an increased size of inflammatory lesions in the inflamed spinal cord of IGF-1RKO−tdTomato compared to IGF-1RWT−tdTomato controls at EAE peak (Fig. 5D, E). Furthermore, immunostaining against the myelin protein MBP and the astrocyte marker GFAP in tissue sections from IGF-1RKO−tdTomato and IGF-1RWT−tdTomato mice revealed a trend toward increased demyelination and astrogliosis in absence of IGF-1R (Fig. 5F-I).

Deletion of IGF1R from CNS-resident myeloid cells leads to worsened clinical signs and tissue inflammation in EAE. EAE was induced in tamoxifen-treated IGF-1RWT−tdTomato (n = 13) and IGF-1RKO−tdTomato mice (n = 21) Shown are: A clinical score of disease (mean ± SEM of animals pooled together from 5 independent experiments), statistical analysis: two-way ANOVA, multiple comparisons test of score differences per EAE day between IGF-1RWT−tdTomato and IGF-1RKO−tdTomato mice. B area under the curve (AUC) (Mann Whitney U test, p = 0.001) and C) disease incidence in the immunized mice (Mann Whitney U test, p = 0.999). Data is represented as mean ± SEM. D At the symptomatic peak of disease, CNS tissue was isolated from IGF-1RWT−tdTomato (n = 4) and IGF-1RKO−tdTomato mice (n = 5) and DAPI staining was performed to assess lesion density (number of inflammatory lesions/cm2, unpaired t test with Welch`s correction, p = 0.0335) and Area of individual lesions (μm2). Per mouse, four to five regions have been analysed and the mean was calculated. Each dot represents mean values obtained per mouse. Data is represented as mean ± SEM. (E) Representative image displaying spinal cord lesions in 1RWT−tdTomato and IGF-1RKO−tdTomato mice at peak, white dotted lines indicate single lesions, scale bar 100 μm. (F) Following immunostaining against MBP, the degree of demyelination was quantified, displayed is MBP relative staining intensity. Data is represented as mean ± SEM, ). Representative images displaying MBP + staining of spinal cords at EAE peak in IGF-1RWT−tdTomato and IGF-1RKO−tdTomato, scale bar 100 μm. H Similarly, after immunostaining against the astrocyte marker GFAP, we quantified the degree of astrogliosis (displayed as GFAP + cell density—cells/mm2) I Representative pictures of GFAP + staining in spinal cords at EAE peak in IGF-1RWT−tdTomato and IGF-1RKO−tdTomato, scale bar 100 μm. Asterisks indicate significant differences (∗ p < 0.05, ∗ ∗ p < 0.01 and ∗ ∗ ∗ p < 0.001, ∗ ∗ ∗ ∗ p < 0.0001)
To further characterize the functional impact of IGF-1R deletion on myeloid cells during inflammation, we quantified the total number of CNS-resident and infiltrating macrophages and assessed differences in their functional phenotype via flow cytometry in IGF-1RKO−tdTomato vs IGF-1RWT−tdTomato mice during EAE (Additional file 3: Fig. 3A). At the symptomatic peak of EAE the total number of microglial cells and the expression levels of MHC-II were comparable between experimental groups (Additional file 3: Fig. 3B). Furthermore, despite a statistically non-significant increase in the number of BAMs and infiltrating Ly6C+ monocyte-derived cells in the CNS of IGF-1RKO−tdTomato mice, no significant differences were detected in expression of the phenotypic markers MHC-II, CD206 and CD44 (Additional file 3: Fig. 3C, D). Flow cytometric analysis at the chronic stage of EAE yielded comparable results (data not shown).
In summary, IGF-1R deletion from the majority of CNS-resident macrophages resulted into a significantly stronger clinical development of anti-myelin immunity in the EAE model.
Discussion
IGF-1 is an essential component of the somatotropic axis regulating body growth [63]. While its systemic ablation leads to aberrant postnatal organ development [100], mice carrying deletion of its receptor IGF-1R die embryonically [53]. In humans, IGF1R haploinsufficiency is associated with impaired growth [71]. Notably, beside its systemic hormonal function, IGF-1 signalling acts as an organ-specific modulator of physiological cellular processes [27].
In the CNS, IGF-1 is locally secreted by microglia and BAMs [92] and in parallel is imported to the cerebrospinal fluid (CSF) from the bloodstream, with CSF levels of this growth factor regulated by the concentration of IGF-1 in the serum and by active import through neurovascular coupling [15, 69, 102]. Low levels of IGF-1 in the CSF have been associated with the development of autism-spectrum disorders [107], thus indicating a key physiological role of this molecule at CNS interfaces. Accordingly, while the parenchymal expression of IGF-1R substantially decreases following CNS development [27], IGF-1R expression in resident cells at the border areas of the CNS remains unchanged throughout adulthood [80]. However, the autocrine/paracrine action of this molecule in regulating the physiology of IGF-1-producing BAMs and microglia remains surprisingly undetermined. Together, this missing information hampers our understanding of IGF-1 functions in the CNS and the potential therapeutic use of IGF-1 upon tissue damage.
To shed light on the role of IGF-1R signalling in CNS-resident myeloid cells, we here made use of a novel mouse model (IGF-1RKO−tdTomato) in which Igf1r was conditionally deleted in BAMs and microglia. Even though IGF-1R deletion targeted only 68% of microglia and 75% for BAMs, receptor ablation resulted in significant changes at a cellular and, surprisingly, systemic level. More specifically, 5 weeks after recombination, IGF-1RKO−tdTomato mice showed a significant increase in body weight compared to IGF-1RWT−tdTomato controls. Homozygous Igf1r ablation in hypothalamic neurons or hemizygous Igf1r deficiency in neuronal and glial cells were previously reported to influence body mass, leading to a decrease in weight [1, 46], however no reports of growth changes upon myeloid-cell specific mutations in IGF-1R exist. It remains unclear whether the observed effect could be attributed to the deletion of IGF-1R from microglia and BAMs, potentially affecting IGF-1 sensing in pituitary and hypothalamic neurons participating in the somatotropic axis [63], or whether other CX3CR1+ macrophages, potentially targeted by our ablation approach in peripheral organs, contributed to the increase in body mass.
Beside these systemic changes, deletion of IGF-1R massively impacted the physiology of BAMs and microglia. Analysis of tissue sections and intravital imaging revealed that both leptomeningeal spinal cord BAMs and brain microglia in IGF-1RKO−tdTomato mice displayed more complex ramification with higher degree of branches, endpoints and junctions per cell compared to cells from IGF-1RWT−tdTomato mice. While the correlation between morphological changes and functional alterations in BAMs remains unclear, highly ramified microglia are normally associated with increased homeostatic surveillance and neuroprotective functions [81], with the complexity of microglia ramification regulated by the sensing of neuronal activity and extracellular nucleotides [59]. This is opposed to the “hypertrophic” appearance of these cells upon activation [81] or to “dystrophic” cells observed during ageing and Alzheimer’s disease [28]. Parallel analysis of cell ramifications in the spinal cord did not reveal significant differences between IGF-1RKO and IGF-1RWT microglia but indicated a higher radius of cells from IGF-1RKO−tdTomato mice – similar to brain microglia- suggestive of increased homeostatic surveillance [26, 75]. In parallel, however, spinal cord microglia from IGF-1RKO−tdTomato mice showed decreased lacunarity, i.e. rotational variation of these cells compared to their counterparts in IGF-1RWT−tdTomato animals. While the functional meaning of this parameter is context-dependent [48], decreased lacunarity in microglia was observed following IL-1β stimulation [26]. Furthermore, IGF-1RKO microglia in the spinal cord showed a higher density (i.e. process thickness) again similar to cytokine-stimulated microglia [26]. In summary, morphological analysis revealed significant differences between BAMs and microglia in IGF-1RKO−tdTomato and IGF-1RWT−tdTomato mice but could not provide univocal indications about the differential activation of these cells in situ.
In parallel, transcriptomic analysis revealed that microglia from IGF-1RKO−tdTomato mice increased expression of Aspm, a positive regulator of the cell cycle [11] and Atp7b, a gene involved in cellular activation during Alzheimer´s disease [110]. Accordingly, GO analysis indicated increased ribosomal biogenesis, a process typically characterizing microglia activation e.g. in response to LPS stimulation [87]. Overall, these changes suggested an augmented basal activation of microglia following IGF-1R deletion.
When analysing the transcriptome of BAMs, several key changes could be observed between IGF-1RKO and IGF-1RWT cells. Firstly, upregulation of genes downstream of IFN-γ signalling (Gbp10, Tgtp1) and downregulation of genes involved in immunoregulation (Itgb3, Plxdc2, Siglec-h) [37, 84, 91] implied alteration of immune functions in IGF-1RKO BAMs compared to control.
Secondly, pathways linked to metabolic activation were altered in the absence of IGF-1R, and more specifically pathways governing catabolic processes and mRNA processing were downregulated while cell growth processes were upregulated in IGF-1RKO cells from IGF-1RKO−tdTomato mice. Accordingly, IGF-1R signalling is described as regulator of cellular metabolism in the CNS [27].
Lastly, IGF-1R absence from BAMs affected cellular adhesion and migratory properties, as shown by the increased expression of Slamf9 [24], the decreased expression of genes involved in intercellular interactions (Mfap4, Spata13, Jam2) and decreased “leukocyte migration” process in the GO enrichment analysis from IGF-1RKO−tdTomato mice. Along the same line, GTPase signalling processes were upregulated in IGF-1RKO cells from IGF-1RKO−tdTomato mice. This change might be linked to the function of IGF-1R as hybrid receptor able to activate G-protein regulated signalling [17], the latter mediating the regulatory effects of IGF-1 on cell adhesion and migration [16]. Furthermore, again suggesting altered adhesion and migratory capability of IGF-1RKO cells, BAMs from IGF-1RKO−tdTomato mice showed lower surface expression of PSGL-1, a molecule involved in leukocyte migration with immunomodulatory signalling functions [94].
Taken together, morphological and transcriptomic analysis of CNS-resident myeloid cells upon IGF-1R deletion indicated complex variations of microglia/BAM physiology affecting migration, adhesion, metabolic activation and immune response in these cells.
Not surprisingly, these extensive functional alterations profoundly impacted the development of autoimmune CNS inflammation, with IGF-1RKO−tdTomato mice showing a significant worsening of clinical EAE progression and of tissue inflammation compared to controls. CNS-resident macrophages exert both protective and detrimental roles during autoimmune neuroinflammation similar to tissue-invading monocyte-derived macrophages [44, 55, 89]. BAMs and microglia become activated even before the development of clinical signs [6, 20] and secrete inflammatory cytokines and chemokines recruiting peripheral immune cells into the CSF [79, 83]. The observed clinical worsening of EAE, also reflected by increased density of inflammatory lesions in IGF-1RKO−tdTomato mice, might thus suggest that IGF-1R signalling in BAMs/microglia exert a significant protective role limiting the development of CNS autoimmunity. While the mechanism underlying this amelioration remain unclear, the comparable number and expression of activation markers in CNS myeloid cells from IGF-1RKO−tdTomato compared to IGF-1RWT−tdTomato mice suggest that this disease-ameliorating effect is mostly mediated via an indirect modulation of the CNS environment by microglia/macrophages upon IGF-1R deletion.
Conclusions
Our study in the IGF1RKO−tdTomato model shed light for the first time on the role of IGF-1 signalling in CNS-resident macrophages. We here showed that loss of IGF-1R in these cells leads to: (I) altered cellular morphology; (II) regulation of cellular transcriptome affecting adhesive, metabolic and immune functions; (III) worsened autoimmune CNS inflammation. Furthermore, even though the observed changes might be affected by cellular heterogeneity and by the differential efficiency in receptor deletion, our work suggests differences in the role of IGF-1R between BAMs and microglia, with microglia phenotypically affected by absence of the receptor and BAMs displaying broader transcriptomic changes upon Igf1r deletion. BAMs are strategically located at CNS interfaces and extensively interact with invading immune cells during pathology [30, 45, 109]. Notably, since we observed a massive impact on the evolution of neuroinflammation upon a genetic modification affecting only 70% of BAMs, our model suggests a central role of these cells in inflammatory responses within the CNS.
Taken together, given the results obtained in our deletion model, it is tempting to speculate that IGF-1R in CNS-resident macrophages play a tonic anti-inflammatory function similar to the one described in peripheral macrophages [32, 41, 50, 88]. Further experiments are however needed to corroborate this hypothesis.
Availability of data and materials
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Abbreviations
- CNS:
-
Central nervous system
- MS:
-
Multiple sclerosis
- EAE:
-
Experimental autoimmune encephalomyelitis
- BAM:
-
Barrier-associated macrophage
- IGF-1:
-
Insulin-like growth factor 1
- IGF-1R:
-
Insulin-like growth factor 1 receptor
References
Al-Samerria S, Radovick S (2021) The role of insulin-like growth factor-1 (IGF-1) in the control of neuroendocrine regulation of growth. Cells 10.
Arganda-Carreras I, Fernández-González R, Muñoz-Barrutia A, Ortiz-De-Solorzano C (2010) 3D reconstruction of histological sections: application to mammary gland tissue. Microsc Res Tech 73:1019–1029
Arpa J, Sanz-Gallego I, Medina-Báez J, Portela LV, Jardim LB, Torres-Aleman I et al (2011) Subcutaneous insulin-like growth factor-1 treatment in spinocerebellar ataxias: an open label clinical trial. Mov Disord 26:358–359
Barres BA, Hart IK, Coles HS, Burne JF, Voyvodic JT, Richardson WD et al (1992) Cell death and control of cell survival in the oligodendrocyte lineage. Cell 70:31–46
Bilbao D, Luciani L, Johannesson B, Piszczek A, Rosenthal N (2014) Insulin-like growth factor-1 stimulates regulatory T cells and suppresses autoimmune disease. EMBO Mol Med 6:1423–1435
Borjini N, Paouri E, Tognatta R, Akassoglou K, Davalos D (2019) Imaging the dynamic interactions between immune cells and the neurovascular interface in the spinal cord. Exp Neurol 322:113046
Butovsky O, Landa G, Kunis G, Ziv Y, Avidan H, Greenberg N et al (2006) Induction and blockage of oligodendrogenesis by differently activated microglia in an animal model of multiple sclerosis. J Clin Invest 116:905–915
Butovsky O, Talpalar AE, Ben-Yaakov K, Schwartz M (2005) Activation of microglia by aggregated beta-amyloid or lipopolysaccharide impairs MHC-II expression and renders them cytotoxic whereas IFN-gamma and IL-4 render them protective. Mol Cell Neurosci 29:381–393
Butovsky O, Ziv Y, Schwartz A, Landa G, Talpalar AE, Pluchino S et al (2006) Microglia activated by IL-4 or IFN-gamma differentially induce neurogenesis and oligodendrogenesis from adult stem/progenitor cells. Mol Cell Neurosci 31:149–160
Cannella B, Pitt D, Capello E, Raine CS (2000) Insulin-like growth factor-1 fails to enhance central nervous system myelin repair during autoimmune demyelination. Am J Pathol 157:933–943
Capecchi MR, Pozner A (2015) ASPM regulates symmetric stem cell division by tuning cyclin E ubiquitination. Nat Commun 6:8763
Carro E, Trejo JL, Gomez-Isla T, Leroith D, Torres-Aleman I (2002) Serum insulin-like growth factor I regulates brain amyloid-beta levels. Nat Med 8:1390–1397
Cherry JD, Olschowka JA, O’banion, M.K. (2014) Neuroinflammation and M2 microglia: the good, the bad, and the inflamed. J Neuroinflammation 11:98
Clara MC, Rickey RR, Wei-Hua L, George J, Sonya CP, Carolyn AB (2000) Insulin-like growth factor 1 regulates developing brain glucose metabolism. Proc Natl Acad Sci 97:10236–10241
Coculescu M (1999) Blood-brain barrier for human growth hormone and insulin-like growth factor-I. J Pediatr Endocrinol Metab 12:113–124
Crudden C, Girnita A, Girnita L (2015) Targeting the IGF-1R: The Tale of the Tortoise and the Hare. Front Endocrinol (Lausanne) 6:64
Crudden C, Song D, Cismas S, Trocmé E, Pasca S, Calin GA et al. (2019) Below the surface: IGF-1R therapeutic targeting and its endocytic journey. Cells 8.
Cusick MF, Libbey JE, Trede NS, Fujinami RS (2014) Targeting insulin-like growth factor 1 leads to amelioration of inflammatory demyelinating disease. PLoS ONE 9:e94486
Danielian PS, Muccino D, Rowitch DH, Michael SK, Mcmahon AP (1998) Modification of gene activity in mouse embryos in utero by a tamoxifen-inducible form of Cre recombinase. Curr Biol 8:1323–1326
Davalos D, Ryu JK, Merlini M, Baeten KM, Le Moan N, Petersen MA et al (2012) Fibrinogen-induced perivascular microglial clustering is required for the development of axonal damage in neuroinflammation. Nat Commun 3:1227
Davis BM, Salinas-Navarro M, Cordeiro MF, Moons L, De Groef L (2017) Characterizing microglia activation: a spatial statistics approach to maximize information extraction. Sci Rep 7:1576
Ditoro D, Harbour SN, Bando JK, Benavides G, Witte S, Laufer VA et al (2020) Insulin-like growth factors are key regulators of T helper 17 regulatory T cell balance in autoimmunity. Immunity 52:650-667.e610
Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S et al (2013) STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29:15–21
Dollt C, Michel J, Kloss L, Melchers S, Schledzewski K, Becker K et al (2018) The novel immunoglobulin super family receptor SLAMF9 identified in TAM of murine and human melanoma influences pro-inflammatory cytokine secretion and migration. Cell Death Dis 9:939
Engelhardt B, Vajkoczy P, Weller RO (2017) The movers and shapers in immune privilege of the CNS. Nat Immunol 18:123–131
Fernández-Arjona MDM, Grondona JM, Fernández-Llebrez P, López-Ávalos MD (2019) Microglial morphometric parameters correlate with the expression level of IL-1β, and allow identifying different activated morphotypes. Front Cell Neurosci 13:472
Fernandez AM, Torres-Alemán I (2012) The many faces of insulin-like peptide signalling in the brain. Nat Rev Neurosci 13:225–239
Franco-Bocanegra DK, Mcauley C, Nicoll JaR, Boche D (2019) Molecular mechanisms of microglial motility: changes in ageing and alzheimer's disease. Cells 8.
Frank JA, Richert N, Lewis B, Bash C, Howard T, Civil R et al (2002) A pilot study of recombinant insulin-like growth factor-1 in seven multiple sderosis patients. Mult Scler 8:24–29
Galea I, Palin K, Newman TA, Van Rooijen N, Perry VH, Boche D (2005) Mannose receptor expression specifically reveals perivascular macrophages in normal, injured, and diseased mouse brain. Glia 49:375–384
Gao Z, Tsirka SE (2011) Animal models of MS reveal multiple roles of microglia in disease pathogenesis. Neurol Res Int 2011:383087
Ge RT, Mo LH, Wu R, Liu JQ, Zhang HP, Liu Z et al (2015) Insulin-like growth factor-1 endues monocytes with immune suppressive ability to inhibit inflammation in the intestine. Sci Rep 5:7735
Genoud S, Maricic I, Kumar V, Gage FH (2005) Targeted expression of IGF-1 in the central nervous system fails to protect mice from experimental autoimmune encephalomyelitis. J Neuroimmunol 168:40–45
Gerganova G, Riddell A, Miller AA (2022) CNS border-associated macrophages in the homeostatic and ischaemic brain. Pharmacol Ther 240:108220
Goldmann T, Wieghofer P, Jordão MJ, Prutek F, Hagemeyer N, Frenzel K et al (2016) Origin, fate and dynamics of macrophages at central nervous system interfaces. Nat Immunol 17:797–805
Gomez-Nicola D, Perry VH (2015) Microglial dynamics and role in the healthy and diseased brain: a paradigm of functional plasticity. Neuroscientist 21:169–184
Guan Y, Du Y, Wang G, Gou H, Xue Y, Xu J et al (2021) Overexpression of PLXDC2 in stromal cell-associated M2 macrophages is related to EMT and the progression of gastric cancer. Front Cell Dev Biol 9:673295
Haghayegh Jahromi N, Tardent H, Enzmann G, Deutsch U, Kawakami N, Bittner S et al. (2017). A novel cervical spinal cord window preparation allows for two-photon imaging of T-cell interactions with the cervical spinal cord microvasculature during experimental autoimmune encephalomyelitis. Front Immunol 8.
Hakuno F, Takahashi SI (2018) IGF1 receptor signaling pathways. J Mol Endocrinol 61:T69-t86
Harrow J, Denoeud F, Frankish A, Reymond A, Chen CK, Chrast J et al. (2006) GENCODE: producing a reference annotation for ENCODE. Genome Biol 7 Suppl 1, S4.1–9.
Higashi Y, Sukhanov S, Shai SY, Danchuk S, Tang R, Snarski P et al (2016) Insulin-like growth factor-1 receptor deficiency in macrophages accelerates atherosclerosis and induces an unstable plaque phenotype in apolipoprotein E-deficient mice. Circulation 133:2263–2278
Hsu M, Rayasam A, Kijak JA, Choi YH, Harding JS, Marcus SA et al (2019) Neuroinflammation-induced lymphangiogenesis near the cribriform plate contributes to drainage of CNS-derived antigens and immune cells. Nat Commun 10:229
Ivan DC, Walthert S, Berve K, Steudler J, Locatelli G (2020) Dwellers and trespassers: mononuclear phagocytes at the borders of the Central nervous system. Front Immunol 11:609921
Ivan DC, Walthert S, Locatelli G (2021) Central nervous system barriers impact distribution and expression of iNOS and arginase-1 in infiltrating macrophages during neuroinflammation. Front Immunol 12:666961
Jordão MJC, Sankowski R, Brendecke SM, Sagar Locatelli G, Tai YH et al. (2019) Single-cell profiling identifies myeloid cell subsets with distinct fates during neuroinflammation. Science 363.
Kappeler L, De Magalhaes Filho C, Dupont J, Leneuve P, Cervera P, Périn L et al (2008) Brain IGF-1 receptors control mammalian growth and lifespan through a neuroendocrine mechanism. PLoS Biol 6:e254
Karperien A, Ahammer H, Jelinek HF (2013) Quantitating the subtleties of microglial morphology with fractal analysis. Front Cell Neurosci 7:3
Karperien AL, Jelinek HF (2015) Fractal, multifractal, and lacunarity analysis of microglia in tissue engineering. Front Bioeng Biotechnol 3:51
Klöting N, Koch L, Wunderlich T, Kern M, Ruschke K, Krone W et al (2008) Autocrine IGF-1 action in adipocytes controls systemic IGF-1 concentrations and growth. Diabetes 57:2074–2082
Knuever J, Willenborg S, Ding X, Akyüz MD, Partridge L, Niessen CM et al (2015) Myeloid cell-restricted insulin/IGF-1 receptor deficiency protects against skin inflammation. J Immunol 195:5296–5308
Lewitt MS, Boyd GW (2019) The role of insulin-like growth factors and insulin-like growth factor-binding proteins in the nervous system. Biochem Insights 12:1178626419842176
Li W, Quigley L, Yao DL, Hudson LD, Brenner M, Zhang BJ et al (1998) Chronic relapsing experimental autoimmune encephalomyelitis: effects of insulin-like growth factor-I treatment on clinical deficits, lesion severity, glial responses, and blood brain barrier defects. J Neuropathol Exp Neurol 57:426–438
Liu JP, Baker J, Perkins AS, Robertson EJ, Efstratiadis A (1993) Mice carrying null mutations of the genes encoding insulin-like growth factor I (Igf-1) and type 1 IGF receptor (Igf1r). Cell 75:59–72
Liu X, Yao DL, Webster H (1995) Insulin-like growth factor I treatment reduces clinical deficits and lesion severity in acute demyelinating experimental autoimmune encephalomyelitis. Mult Scler 1:2–9
Locatelli G, Theodorou D, Kendirli A, Jordão MJC, Staszewski O, Phulphagar K et al (2018) Mononuclear phagocytes locally specify and adapt their phenotype in a multiple sclerosis model. Nat Neurosci 21:1196–1208
Lovett-Racke AE, Bittner P, Cross AH, Carlino JA, Racke MK (1998) Regulation of experimental autoimmune encephalomyelitis with insulin-like growth factor (IGF-1) and IGF-1/IGF-binding protein-3 complex (IGF-1/IGFBP3). J Clin Invest 101:1797–1804
Rechler A MM, Nissley SP (1985) The nature and regulation of the receptors for insulin-like growth factors. Annu Rev Physiol 47:425–442
Madisen L, Zwingman TA, Sunkin SM, Oh SW, Zariwala HA, Gu H et al (2010) A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nat Neurosci 13:133–140
Madry C, Attwell D (2015) Receptors, ion channels, and signaling mechanisms underlying microglial dynamics. J Biol Chem 290:12443–12450
Mcgill BE, Barve RA, Maloney SE, Strickland A, Rensing N, Wang PL et al (2018) Abnormal microglia and enhanced inflammation-related gene transcription in mice with conditional deletion of Ctcf in Camk2a-Cre-expressing neurons. J Neurosci 38:200–219
Mcmorris FA, Smith TM, Desalvo S, Furlanetto RW (1986) Insulin-like growth factor I/somatomedin C: a potent inducer of oligodendrocyte development. Proc Natl Acad Sci U S A 83:822–826
Milman S, Atzmon G, Huffman DM, Wan J, Crandall JP, Cohen P et al (2014) Low insulin-like growth factor-1 level predicts survival in humans with exceptional longevity. Aging Cell 13:769–771
Milman S, Huffman DM, Barzilai N (2016) The somatotropic axis in human aging: framework for the current state of knowledge and future research. Cell Metab 23:980–989
Miron VE, Boyd A, Zhao JW, Yuen TJ, Ruckh JM, Shadrach JL et al (2013) M2 microglia and macrophages drive oligodendrocyte differentiation during CNS remyelination. Nat Neurosci 16:1211–1218
Morrison H, Young K, Qureshi M, Rowe RK, Lifshitz J (2017) Quantitative microglia analyses reveal diverse morphologic responses in the rat cortex after diffuse brain injury. Sci Rep 7:13211
Morrison HW, Filosa JA (2013) A quantitative spatiotemporal analysis of microglia morphology during ischemic stroke and reperfusion. J Neuroinflammation 10:4
Mozell RL, Mcmorris FA (1991) Insulin-like growth factor I stimulates oligodendrocyte development and myelination in rat brain aggregate cultures. J Neurosci Res 30:382–390
Nagano I, Ilieva H, Shiote M, Murakami T, Yokoyama M, Shoji M et al (2005) Therapeutic benefit of intrathecal injection of insulin-like growth factor-1 in a mouse model of Amyotrophic Lateral Sclerosis. J Neurol Sci 235:61–68
Nishijima T, Piriz J, Duflot S, Fernandez AM, Gaitan G, Gomez-Pinedo U et al (2010) Neuronal activity drives localized blood-brain-barrier transport of serum insulin-like growth factor-I into the CNS. Neuron 67:834–846
O’donnellFrederickKradyVannucciWood SLTJJKSJTL (2002) IGF-I and microglia/macrophage proliferation in the ischemic mouse brain. Glia 39:85–97
Ocaranza P, Golekoh MC, Andrew SF, Guo MH, Kaplowitz P, Saal H et al (2017) Expanding genetic and functional diagnoses of IGF1R Haploinsufficiencies. Horm Res Paediatr 87:412–422
Pérez-Martín M, Cifuentes M, Grondona JM, López-Avalos MD, Gómez-Pinedo U, García-Verdugo JM et al (2010) IGF-I stimulates neurogenesis in the hypothalamus of adult rats. Eur J Neurosci 31:1533–1548
Pini G, Scusa MF, Congiu L, Benincasa A, Morescalchi P, Bottiglioni I et al (2012) IGF1 as a potential treatment for Rett syndrome: safety assessment in six rett patients. Autism Res Treat 2012:679801
Plastini MJ, Desu HL, Brambilla R (2020) Dynamic responses of microglia in animal models of multiple sclerosis. Front Cell Neurosci 14:269
Pontes B, Ayala Y, Fonseca AC, Romão LF, Amaral RF, Salgado LT et al (2013) Membrane elastic properties and cell function. PLoS ONE 8:e67708
Prager D, Melmed S (1993) Insulin and insulin-like growth factor I receptors: are there functional distinctions? Endocrinology 132:1419–1420
Prinz M, Jung S, Priller J (2019) Microglia biology: one century of evolving concepts. Cell 179:292–311
Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W et al (2015) limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res 43:e47
Russi AE, Brown MA (2015) The meninges: new therapeutic targets for multiple sclerosis. Transl Res 165:255–269
Russo VC, Gluckman PD, Feldman EL, Werther GA (2005) The insulin-like growth factor system and its pleiotropic functions in brain. Endocr Rev 26:916–943
Savage JC, Carrier M, Tremblay M (2019) Morphology of microglia across contexts of health and disease. Methods Mol Biol 2034:13–26
Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T et al (2012) Fiji: an open-source platform for biological-image analysis. Nat Methods 9:676–682
Schläger C, Körner H, Krueger M, Vidoli S, Haberl M, Mielke D et al (2016) Effector T-cell trafficking between the leptomeninges and the cerebrospinal fluid. Nature 530:349–353
Schmitt H, Sell S, Koch J, Seefried M, Sonnewald S, Daniel C et al (2016) Siglec-H protects from virus-triggered severe systemic autoimmunity. J Exp Med 213:1627–1644
Sholl DA (1956) The measurable parameters of the cerebral cortex and their significance in its organization. Prog Neurobiol 1956:324–333
Smith TJ (2010) Insulin-like growth factor-I regulation of immune function: a potential therapeutic target in autoimmune diseases? Pharmacol Rev 62:199–236
Sousa C, Golebiewska A, Poovathingal SK, Kaoma T, Pires-Afonso Y, Martina S et al. (2018). Single-cell transcriptomics reveals distinct inflammation-induced microglia signatures. EMBO Rep 19.
Spadaro O, Camell CD, Bosurgi L, Nguyen KY, Youm YH, Rothlin CV et al (2017) IGF1 shapes macrophage activation in response to immunometabolic challenge. Cell Rep 19:225–234
Spiteri AG, Wishart CL, Pamphlett R, Locatelli G, King NJC (2022) Microglia and monocytes in inflammatory CNS disease: integrating phenotype and function. Acta Neuropathol 143:179–224
Stratoulias V, Venero JL, Tremblay M, Joseph B (2019) Microglial subtypes: diversity within the microglial community. Embo j 38:e101997
Su X, Esser AK, Amend SR, Xiang J, Xu Y, Ross MH et al (2016) Antagonizing integrin β3 increases immunosuppression in cancer. Cancer Res 76:3484–3495
Suh HS, Zhao ML, Derico L, Choi N, Lee SC (2013) Insulin-like growth factor 1 and 2 (IGF1, IGF2) expression in human microglia: differential regulation by inflammatory mediators. J Neuroinflammation 10:37
Tietz SM, Zwahlen M, Haghayegh Jahromi N, Baden P, Lazarevic I, Enzmann G et al (2016) Refined clinical scoring in comparative EAE studies does not enhance the chance to observe statistically significant differences. Eur J Immunol 46:2481–2483
Tinoco R, Otero DC, Takahashi AA, Bradley LM (2017) PSGL-1: A new player in the immune checkpoint landscape. Trends Immunol 38:323–335
Van Hove H, Martens L, Scheyltjens I, De Vlaminck K, Pombo Antunes AR, De Prijck S et al (2019) A single-cell atlas of mouse brain macrophages reveals unique transcriptional identities shaped by ontogeny and tissue environment. Nat Neurosci 22:1021–1035
Vladymyrov M, Abe J, Moalli F, Stein JV, Ariga A (2016) Real-time tissue offset correction system for intravital multiphoton microscopy. J Immunol Methods 438:35–41
Vladymyrov M, Haghayegh Jahromi N, Kaba E, Engelhardt B, and Ariga A (2020) VivoFollow 2: Distortion-Free Multiphoton Intravital Imaging. Front Phys 7.
Voss EV, Škuljec J, Gudi V, Skripuletz T, Pul R, Trebst C et al (2012) Characterisation of microglia during de- and remyelination: can they create a repair promoting environment? Neurobiol Dis 45:519–528
Walter HJ, Berry M, Hill DJ, Logan A (1997) Spatial and temporal changes in the insulin-like growth factor (IGF) axis indicate autocrine/paracrine actions of IGF-I within wounds of the rat brain. Endocrinology 138:3024–3034
Wang J, Zhou J, Powell-Braxton L, Bondy C (1999) Effects of Igf1 gene deletion on postnatal growth patterns. Endocrinology 140:3391–3394
Wilczak N, Chesik D, Hoekstra D, De Keyser J (2008) IGF binding protein alterations on periplaque oligodendrocytes in multiple sclerosis: implications for remyelination. Neurochem Int 52:1431–1435
Yan H, Mitschelen M, Bixler GV, Brucklacher RM, Farley JA, Han S et al (2011) Circulating IGF1 regulates hippocampal IGF1 levels and brain gene expression during adolescence. J Endocrinol 211:27
Yao DL, Liu X, Hudson LD, Webster HD (1995) Insulin-like growth factor I treatment reduces demyelination and up-regulates gene expression of myelin-related proteins in experimental autoimmune encephalomyelitis. Proc Natl Acad Sci U S A 92:6190–6194
Ye P, Carson J, D’ercole AJ (1995) In vivo actions of insulin-like growth factor-I (IGF-I) on brain myelination: studies of IGF-I and IGF binding protein-1 (IGFBP-1) transgenic mice. J Neurosci 15:7344–7356
Yona S, Kim KW, Wolf Y, Mildner A, Varol D, Breker M et al (2013) Fate mapping reveals origins and dynamics of monocytes and tissue macrophages under homeostasis. Immunity 38:79–91
Young K, Morrison H (2018). Quantifying microglia morphology from photomicrographs of immunohistochemistry prepared tissue using imagej. J Vis Exp.
Yuan ZF, Mao SS, Shen J, Jiang LH, Xu L, Xu JL et al (2020) Insulin-like growth factor-1 down-regulates the phosphorylation of FXYD1 and rescues behavioral deficits in a mouse model of rett syndrome. Front Neurosci 14:20
Zeng WJ, Cheng Q, Wen ZP, Wang JY, Chen YH, Zhao J et al (2020) Aberrant ASPM expression mediated by transcriptional regulation of FoxM1 promotes the progression of gliomas. J Cell Mol Med 24:9613–9626
Zhang Z, Zhang ZY, Wu Y, Schluesener HJ (2012) Lesional accumulation of CD163+ macrophages/microglia in rat traumatic brain injury. Brain Res 1461:102–110
Zheng Z, White C, Lee J, Peterson TS, Bush AI, Sun GY et al (2010) Altered microglial copper homeostasis in a mouse model of Alzheimer’s disease. J Neurochem 114:1630–1638
Acknowledgements
We thank Dr. Urban Deutsch, Mark Liebi, Albert Witt and animal caretakers at the Theodor Kocher Institute for precious help with genotyping and mouse colony maintenance; Prof. Jens Brüning for donating the IGF1R floxed line. We also thank the Microscopy Imaging Centre Bern for usage of shared equipment.
Funding
This work was supported by a Scherbarth Foundation grant, by a Research Grant funded by the Swiss Multiple Sclerosis Society and by FISM (Fondazione Italiana Sclerosi Multipla – cod. 2019/R-Single/001 and financed or co-financed with the ‘5 per mille’ public funding) awarded to GL. Furthermore, BE and GL received financial support from the ITN consortium ENTRAIN.
Author information
Authors and Affiliations
Contributions
DCI, KCB, SW, GM, KB, EB, FF, HL, JS and GL performed and analysed experiments; TB offered help with DNA analysis and kindly provided the IGF1R floxed model; MP supervised the transcriptome analysis and provided the CX3CR1-creERT2 model, GL designed the experiments and wrote the manuscript; BE edited the manuscript, BE and GL supervised the study.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Animal work was performed under the license BE138/20 approved by the local veterinary cantonal authority.
Competing interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1.
Morphological characterization of CNS-resident myeloid cells following ablation of IGF-1R.To assess the morphology of CNS resident microglia (brain and spinal cord) and leptomeningeal BAMs in IGF-1RKO-tdTomato (n=4 mice) compared to IGF-1RWT-tdTomato (n=4 mice), we performed Sholl analysis, skeletal analysis and fractal-lacunarity analysis (see methods for more details). The parameters obtained from these 3 morphological analyses were grouped into 4 categories indicating cell size (comprising quantification of soma size, cell density, critical value defined as distance from soma where maximum number of branches occurred), cell shape/elongation (characterized by span ratio and circularity), cell complexity (quantification of cell lacunarity and maximum cell span across a convex hull) and cell ramification (illustrated through summed branch length/cell, maximum branch length/ cell, maximum intersections/cell, number of endpoints/cell (measurement of cell contact with environment), number of junctions/ cell). Number of cells analysed: Brain Microglia: IGF-1RKO-tdTomato (n=84), IGF-1RWT-tdTomato (n=71); Spinal Cord Microglia: IGF-1RKO-tdTomato (n=75) IGF-1RWT-tdTomato (n=66); Leptomeningeal BAMs: IGF-1RKO-tdTomato (n=78) IGF-1RWT-tdTomato (n=91). All values are presented as mean ± SEM. Statistical analysis was performed by using unpaired t test with Welch’s correction for normally distributed data or Mann-Whitney U test for non-normally distributed data. Asterisks indicate significant differences (∗p < 0.05, ∗∗p < 0.01 and ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001).
Additional file 2.
Transcriptomic changes in microglia and BAMs upon IGF-1R deletion. A) Sorting strategy of microglia (CD45+CD11b+P2ry12+tdTomato+) and BAMs (CD45+CD11b+P2ry12-CD206+tdTomato+). Cells were sorted from the brain of IGF-1RKO-tdTomato and IGF-1RWT-tdTomato mice directly into RNA protect buffer. B) Venn diagram showing the concordance of gene expression differences in microglia and BAMs from IGF-1RKO-tdTomato and IGF-1RWT-tdTomato mice C) Functional enrichment analysis of the microglia from IGF-1RKO-tdTomato and IGF-1RWT-tdTomato mice using all pathways from the Gene Ontology database. Left, pathways upregulated in IGF-1RKO-tdTomato mice; right, pathways downregulated in IGF-1RKO-tdTomato mice.
Additional file 3.
Flow cytometry characterization of CNS-myeloid cell number and phenotype at the symptomatic peak of EAE. A) Gating Strategy for analysis of CNS resident myeloid cells isolated from brain and spinal cords including leptomeninges of IGF-1RKO-tdTomato and IGF-1RWT-tdTomato mice at EAE peak (clinical manifestation: hind leg paralysis). Cells were identified based on their size and granularity, using the forward versus side scatter gating (FSC vs SSC). Following single cell gating (FSC-H vs FSC-A) and selection of alive cells (live-dead staining), we gated for CD45 vs CD11b expression and analyzed two distinct cell populations: CD45highCD11b+ cells and CD45intermediateCD11b+ cells. Within the CD45highCD11b+ cell population, we further gated on Tomato positivity, thus identifying CNS-resident CD45highCD11b+Tomato+ BAMs (C) and CD45highCD11b+Tomatoneg cells illustrating CNS infiltrating blood-derived myeloid cells. The latter population was further gated for Ly6G vs Ly6C positivity and CD45highCD11b+TomatonegLy6GnegLy6C+ population representing CNS infiltrating monocyte-derived macrophages (D) was selected for the final analysis. CNS resident microglial cell population was identified based on CD45intermediateCD11b+Tomato+ expression (B). Representative histograms of marker expression profiles of MHCII, CD44, CD206 on respective myeloid cell populations analysed. We display the absolute number of CNS resident microglial cells (D), BAMs (E) and CNS-infiltrating monocyte-derived macrophages (C) in IGF-1RKO-tdTomato (n=4) and IGF-1RWT-tdTomato (n=4) mice at EAE peak and relative expression (mean fluorescence intensity of sample relative to isotype control staining) or percentage of positive cells of MHC-II, CD206, CD44 molecules. No statistically significant differences were observed in either of the parameters checked within the three populations of cells investigated between in IGF-1RKO-tdTomato and IGF-1RWT-tdTomato mice. All values are presented as mean ± SEM. Statistical analysis was performed by using unpaired t test with Welch`s correction.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Ivan, D.C., Berve, K.C., Walthert, S. et al. Insulin-like growth factor-1 receptor controls the function of CNS-resident macrophages and their contribution to neuroinflammation. acta neuropathol commun 11, 35 (2023). https://doi.org/10.1186/s40478-023-01535-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40478-023-01535-8