
Abstract
Prenatal alcohol exposure is a major cause of neurobehavioral disabilities. MRI studies in humans have shown that alcohol is associated with white matter microstructural anomalies but these studies focused on myelin abnormalities only after birth. Only one of these studies evaluated oligodendrocyte lineage, but only for a short period during human foetal life. As data are lacking in humans and alcohol is known to impair oligodendrocyte differentiation in rodents, the present study aimed to compare by immunohistochemistry the oligodendrocyte precursor cells expressing PDGFR-α and immature premyelinating/mature oligodendrocytes expressing Olig2 in the ganglionic eminences and the frontal cortex of 14 human foetuses exposed to alcohol from 15 to 37 weeks’ gestation with age-matched controls. The human brains used in this study were obtained at the time of foetal autopsies for medical termination of pregnancy, in utero or post-natal early death. Before birth, PDGFR-α expression was strongly increased in the ganglionic eminences and the cortex of all foetuses exposed to alcohol except at the earliest stage. No massive generation of Olig2 immunoreactive cells was identified in the ganglionic eminences until the end of pregnancy and the density of Olig2-positive cells within the cortex was consistently lower in foetuses exposed to alcohol than in controls. These antenatal data from humans provides further evidence of major oligodendrocyte lineage impairment at specific and key stages of brain development upon prenatal alcohol exposure including defective or delayed generation and maturation of oligodendrocyte precursors.
Similar content being viewed by others


Introduction
Prenatal alcohol exposure (PAE) is a major non genetic cause of central nervous system (CNS) abnormalities resulting in irreversible life-long consequences which associate mental retardation or more specific neurocognitive disorders and behavioural disabilities [43]. Foetal Alcohol Syndrome (FAS) is a part of foetal alcohol spectrum disorders (FASD) which represents its most severe form. The diagnosis of FAS is based on three characteristics including in utero growth retardation (IUGR), craniofacial dysmorphism and CNS dysfunction. Children with FASD often suffer from motor delays, deficits in attention, learning and memory as well as in executive functioning and language with other less overt consequences of PAE that make up the constellation of CNS adverse outcomes. Nevertheless, children with FASD do not present the characteristic craniofacial dysmorphism and growth retardation [41].
In the mature brain, heavy chronic or binge drinking is responsible for a variety of brain injuries, notably a disproportionate loss of cerebral white matter which accounts for white matter atrophy, as glial cells are major targets of alcohol [48]. Alcohol has not only teratogenic properties but also devastating neurotoxic effects on the developing brain. Over the last decade, imaging studies with diffusion tensor imaging (DTI) have revealed changes in the organization and microstructure of callosal white matter with diffusion abnormalities extending beyond the corpus callosum in rodents and in humans with FASD, suggesting that several specific white matter regions, especially commissural, cingular and temporal connections along with deep grey matter areas are sensitive to PAE [25, 26, 31, 32, 34]. Microstructural abnormalities assessed by fractional anisotropy occur throughout the corpus callosum and have been correlated with impaired cognition in children with prenatal alcohol exposure [49]. A few years ago, it has been suggested in one human MRI study that PAE is associated with reduced white matter microstructural integrity from the neonatal period [10]. Whereas all these studies focused on newborns, children and adolescents with FASD, no human antenatal data concerning the generation of oligodendrocytes (OLs) in FAS or FASD have been reported so far, except for one human study concerning cases restricted to a short foetal period (12.2–21.4 weeks of gestation) [9]. As imaging studies indicate widespread white matter fibre tract anomalies in children and adults with FASD, it has been emphasized that PAE likely impacts on the programming of oligodendrocyte precursor cells (OPCs) [18, 48]. In the CNS, myelin is formed by processes emanating from OLs which are engaged or are preparing to engage in myelination [7, 39]. Myelination is one of the final stages of brain development, and apart from the brainstem and cerebellum which are progressively myelinated from the second half of pregnancy, this process takes place mainly during the first 30 years of postnatal life in humans [4, 25, 50].
OLs originate from neural stem cell-derived OPCs which express platelet-derived growth factor receptor-α (PDGFR-α), then differentiate into immature premyelinating OLs (pre-OLs) expressing oligodendrocyte lineage factor 4 and eventually into mature OLs expressing myelin oligodendrocyte glycoprotein, phospholipid protein and myelin basic protein (MBP) which contact axons and begin to produce myelin [7, 13]. Proliferating neural stem cells commit to the OL lineage under the influence of the transcription factors Olig1, Olig2, Nkx2.2, and Sox10 [13, 47]. In vitro studies have also revealed a primary role of Sonic Hedgehog (Shh) signalling by promoting expansion and specification of pluripotent progenitors into Olig2-positive late OPCs and immature OLs [35].
In rodents, the first wave of OPCs originates in the anterior entopeduncular area followed by two other waves arising from the lateral and caudal ganglionic eminences (LGE and CGE) [24]. OPCs require vessels as a physical substrate for migration and human OPCs have been shown to emerge from progenitor domains and to associate with the ab-luminal endothelial surface of blood vessels via Wnt-Cxcr4 (chemokine receptor 4) allowing them to “crawl along and jump between vessels” [45].
PDGF signalling is essential for the control of OPCs’ proliferation and differentiation [5]. These mitotic progenitors/precursors express a characteristic set of markers including PDGFR-ɑ. The first PDGFR-α expressing precursors have been identified in the forebrain of human foetuses 10 weeks of gestation (WG) onwards, and are predominantly produced in higher numbers in the ganglionic eminences (GE) around 15 WG [22]. In monkeys and humans, OPCs are produced in the outer subventricular zone just when the upper layer neurons are generated, allowing for a rapid expansion and folding of the cortical surface [37]. PDGFR-ɑ is rapidly downregulated when OPCs differentiate into non-proliferating OLs, contrary to oligodendrocyte lineage transcription factor 2 (Olig2) which is expressed in OPCs, pre-OLs and OLs, knowing that the increase in OL number during development depends on OPC proliferative capacities [11, 42]. OL lineage progression in cerebral white matter occurs similarly in humans and in rodents and is mostly composed of pre-OLs around 18–27 weeks of gestation in humans and of increasingly abundant MBP-positive OLs in full-term infants [3].
Human antenatal data concerning the generation and differentiation of OLs upon PAE are lacking even though alcohol is known to impair OL differentiation in other species. Therefore, we hypothesised that during the foetal period alcohol prevents the differentiation of PDGFR-α+ OPCs into Olig2+ pre-OL or mature OL, which could explain, at least partially, the myelination defects observed on imaging studies. To test this hypothesis, we compared using immunohistochemical techniques PDGFR-α and Olig2 expression in the GE and cortical plate (CP) of foetuses antenatally exposed to alcohol with age-matched controls. The objective was to confirm the pre-existing results in humans and to provide information regarding oligodendroglial lineage development over an extended neurodevelopmental period.
Patients and methods
Patients
As previously reported [28], the brains used in this study belong to the collection which has been declared to the French Ministry of Health (collection number DC-2015-2468, cession number AC-2015-2467, located in A. Laquerrière’s Pathology Laboratory, Rouen University hospital). In each case, the parents gave their informed written consent for neuropathological studies. Autopsies were carried out in accordance with our local ethic committee and the French law.
Ten foetal control brains ranging from 16 to 36 WG were selected. Main clinical and morphological characteristics are presented in Table 1. In 6 out of the 10 cases, a medical termination of pregnancy (TOP) was carried out for pathologies other than cerebral. Two out of the 10 cases were in utero foetal death (IUFD), 2 cases were perpartum or immediate postpartum death with a cause other than cerebral or with no found cause after careful examination of the placenta and foetal organs. In all cases, the brain was macroscopically and microscopically free of detectable abnormalities. Despite the absence of lesions, patients who had been antenatally suspected of central nervous system anomalies or who had been clinically suspected of dying of neurological causes were systematically excluded.
Fourteen foetal FAS or FASD brains ranging from 15 to 37 WG whose clinical and morphological characteristics are presented in Table 2 were also studied. Causes of death were IUFD in 6 cases, TOP for foetal malformations in 7 cases and post-natal early death in the remaining case.
Methods
Autopsies had been performed according to standardized protocols [20]. Developmental age was evaluated by means of organ weights [17], skeletal measurements and by the histological maturational stages of the different viscera.
Neuropathological studies
Brain growth was evaluated according to the biometric data of Guihard-Costa and Larroche [16]. Macroscopic analysis of gyration was assessed by means of the atlas of Feess-Higgins and Larroche [14]. After fixation into a zinc-10% formalin buffer solution for at least one month, brain sections were obtained from all cortical areas and deep subcortical structures. Seven-micrometer paraffin embedded sections were stained using Haematoxylin–Eosin. The morphology of the different brain structures analysed was consistent with the foetal age.
Immunohistochemistry
Immunohistochemical analyses of OPCs and pre-OLs were carried out on six-micrometer sections obtained from paraffin-embedded material according to standardized protocols using antisera directed against Olig2 (Rabbit polyclonal, 1/200; Clinisciences, Nanterre, France) and anti-PDGFR-α (Rabbit polyclonal, 1/100; Thermofisher Scientific F67403 Illkirch Cedex, France). Noteworthy, some markers of mature oligodendrocytes such as myelin basic protein (MBP), myelin oligodendrocyte glycoprotein (MOG), phospholipid protein (PLP), adenomatous polyposis coli complex (APC) and cyclic nucleotide phosphodiesterase (CNPase) could not be used as they work on frozen tissues.
Immunohistochemical procedures included a microwave pre-treatment protocol to aid antigen retrieval (pre-treatment CC1 kit, Ventana Medical Systems Inc, Tucson AZ). Incubations were performed for 32 min at room temperature using the Ventana Benchmark XT system. After incubation, slides were processed by means of the Ultraview Universal DAB detection kit (Ventana). Semi-quantitative analyses of the density in Olig2 and PDGFR-α positive cells in the GE as well as in the different layers of the frontal CP were evaluated and scored blindly by two neuropathologists (FM and AL), and together reanalyzed in case of discrepant results. Immunolabellings were scored as follows: 0: no cell labelled; +: less than 10% of cells labelled; ++: between 10 and 25% of cells labelled; +++: between 25 and 50% of cells labelled and ++++: more than 50% of the cells labelled.
Confocal analyses
In order to identify more precisely the different Olig2-positive populations, double immunolabelling was performed using Olig2 and either PDGFR-α, GABA, MAP2 or GFAP within the GE at 15–16 WG, a developmental stage in which some Olig2 positive cells could belong to other lineages than OL. The antibodies used for confocal analyses are described in Table 3. Brain sections were incubated overnight at 4 °C with various primary antibodies diluted in a buffer solution (PBS containing 1% BSA and 3% Triton X-100). Fluorescent-conjugated antibodies Alexafluor-488 and -592 were obtained from Molecular Probes (Eurogene, Or, USA). Sections were then rinsed three times with PBS for 10 min and incubated with the same incubation buffer containing the appropriate secondary antibody. Coverslips were mounted in DAPI-containing Vectashield (Vector laboratories, Cambridgeshire, UK). Non-specific binding of the secondary antibody was evaluated by omitting the primary antibodies. Images were acquired with the Leica laser scanning confocal microscope TCS SP2 AOBS (Leica Microsystems, Wetzlar, Germany). Analyses were carried out using the FIJI Is Just Image J (FIJI) software.
Results
FAS Patient’s clinical and morphological characteristics
Among the 14 cases exposed to alcohol, 3 (21%) had intra uterine growth retardation (IUGR). All but 2 cases (86%) had cranio-facial dysmorphism which was characteristic of FAS in half of these cases, associating short palpebral fissures, smooth philtrum and thin vermillion border [40]. Five cases (36%) had microcephaly with a brain weight ≤ 3rd percentile. Nine cases (57%) had other CNS anomalies already described in FAS and FASD patients, such as myelomeningocele, arhinencephaly, polymicrogyria, neuronal heterotopias and cerebellar anomalies (see Table 2). Clastic lesions were observed in 5 cases (36%) and five other cases had associated visceral anomalies (anterior coelosomia, hydronephrosis, unilateral pelvic dilatation and tetralogy of Fallot), which are known to occur in case of PAE.
Daily chronic alcohol intake throughout the pregnancy was self-reported by 7 mothers/ 14 (50%). Three out of the 7 mothers also consumed episodic high doses of alcohol named «binge drinking». In the other 7 cases, maternal alcohol intake was suspected on the basis of reports by their relatives (family and/or friends) or clinically suspected at the time of foetal autopsy according to the criteria established by Riley et al., i.e., craniofacial dysmorphism characteristic of FAS (6/7 suspected cases), microcephaly (3/7 suspected cases), other CNS lesions (5/7 suspected cases) and IUGR (2/7 suspected cases) [40]. Ten mothers (71%) had co-morbidities notably multidrug addiction, antiepileptic drugs and psychotic traits.
Semi-quantitative analysis of PDGFR-α and Olig2 immunohistochemistry in the cortical plate and ganglionic eminences
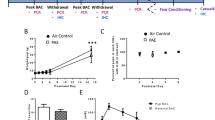
Semi-quantitative evaluation of PDGFR-α and Olig2 immunolabellings are summarised in Fig. 1 and in Table 4.

Schematic representation of PDGFR-α and Olig2 expressing cells in the GE and CP of FASD and control brains. Semi-quantitative evaluation of PDGFR-α immunoreactive OPCs in the GE displaying a delayed production at 16 WG and an increased density until the physiological disappearance of GE by comparison with control brains (a), as well as in the cortical plate of all FASD brains compared to control brains from 20 WG which persisted until 37 WG (b). Semi-quantitative evaluation of Olig2-expressing OPCs and pre-OLs in the GE in which the density of Olig2 immunoreactive cells was drastically reduced up to 24 WG in FASD brains, followed by an increasing trend to the production/differentiation between 24 and 30 WG (arrows) in FASD brains though the number of OPCs and pre-OLs remained low until regression of GE by 34 WG by comparison with control brains (c). In the cortical plate a lower density of Olig2-expressing cells in all FASD brains was observed regardless of the developmental stage compared with control brains (d). Dotted blue line: control brains; dotted red line: PAE exposed brains; black triangles: second FASD case available at a given stage
PDGFR-α expression was strongly increased in the GE and in the CP of all FASD brains in comparison with controls whatever the developmental stage with the exception of the earliest stage (Fig. 1a, b). Indeed, at 15–16 WG, PDGFR-α expression remained lower in the medial ganglionic eminences (MGE), LGE and CP of FASD brains contrary to controls (Fig. 2a–d) in which more than 60% of cells were observed. At 20 WG and at all later stages, PDGFR-α cell numbers were higher than in controls in all structures studied (Fig. 2e–l).

PDGFR-ɑ immunoreactivities in the GE and CP of FASD and control brains. Lower densities of PDGFR-ɑ expressing OPCs in the GE of FASD brain at 15 WG by comparison with the control aged 16 WG in which most of the cells were immunolabelled (OM X20) (a, b), with a similar pattern observed in the CP of FASD and control brain (c, d). But from 20 WG, higher densities of PDGFR-ɑ expressing OPCs in the GE of FASD brains compared with control brains (OM X20) (e, f), as observed in the CP (g, h), with the same pattern found at 30 WG in the GE (OM X20) (i, j) and in the CP (k, l). (OM: original magnification; scale bar: 0.35 mm)
At 16 WG, more than 60% Olig2-positive precursors and pre-OLs were identified in the MGE and LGE of the control brain, whereas Olig2-positive cells were less numerous in the FASD brain (Fig. 3a, b). Moreover, Olig2 immunoreactivity was essentially identified in the MGE of the FASD brain and not yet in the LGE. At 22 WG, Olig2-positive cell density was drastically reduced in the GE of FASD brains (Fig. 3c–f), and remained low until the regression of GE that normally occurs around 34 WG. No massive generation of Olig2 immunoreactive cells was identified in FASD brains until the end of the pregnancy, except for three small peaks of generation in FASD brains between 24 and 30 WG (Figs. 1c, 3g, h), contrary to controls in which a major production was observed between 16 and 24 WG.

Olig2 immunoreactivities in the GE of FASD and control brains. Lower densities of Olig2 expressing cells in the GE of FASD brain at 15 WG by comparison with the GE of control brain at 16 WG in which more than 50% of Olig2 positive cells were observed (a, b) the most striking differences between FASD and control GE being observed at 22 WG (c, d). Similar differences, though less pronounced, were also noted at 24 WG in the FASD brain (case 5) compared to the control (e, f) contrary to what was noted in the FASD brain (case 6), in which an intense immunoreactivity was observed arguing for a delayed production/differentiation starting from this term (g, h)
At all developmental stages, the density of Olig2-positive cells within the CP was consistently higher in each control brain in comparison with FASD brains (Figs. 1d, 4), with the highest density in the control cortical plate (around 80% of the cells) observed at 16 WG, contrasting with the paucity of immunolabelled cells in FASD (Fig. 4a, b).

Olig2 immunoreactivities in the CP of FASD and control brains. Significantly lower densities of Olig2-positive cells in FASD brains compared to control brains at 15–16 WG (OM X10) (a, b) with a similar pattern observed at 20 WG (OM X10) (c, d). At 30 WG, Olig2-positive cells remained scarce in the FASD brain contrary to the control, in which Olig2-positive cells were located in all layers of the cortical plate (OM X20) (e, f) with an increase in density in the superficial layers at 33 WG in the control cortical plate only (OM X20) (g, h). (OM: original magnification; scale bar: 0.35 mm)
Confocal analyses
At early stages, a diffuse and intense immunolabelling of cells with Olig2 antibody was identified. As Olig2 is known to be also expressed in immature neurons and interneurons as well as in a subpopulation of astrocytes, we performed double immunolabellings using Olig2 and either MAP2, GABA, GFAP or PDGFR-α to evaluate more precisely the percentage of Olig2-positive OPCs [22, 23, 30]. No co-expression of Olig2 with MAP2, GABA, GFAP or PDGFR-α within the GE and CP was observed (Additional file 1: Fig. S1).
Discussion
In living children with FASD aged from 10 to 17, imaging studies have revealed lower white matter volumes and sometimes a complete lack of myelination of major white matter tracts in the brain as well as white matter microstructure alterations [39, 48, 49]. However, the impact of alcohol on oligodendrocyte lineage and by extension on myelination has essentially been studied using in vitro experiments or animal models. Alcohol can disrupt myelination at any stage of OL development but targets particularly OPCs which are more vulnerable to excitotoxic damage, free radicals and pro-inflammatory cytokines than mature OLs [2, 39]. Furthermore, it has been shown in a study performed on primary mouse OL cultures that acetaldehyde, the metabolic byproduct of alcohol is lethal to OLs which are much more sensitive to acetaldehyde than to alcohol, particularly upon long-term alcohol exposure [6].
To our knowledge, no study concerning the effects of alcohol during human brain development has focused on OPC production from the GE. Nevertheless, using in vivo and in vitro mouse models, it has been demonstrated that alcohol hinders basal progenitor proliferation in the ventricular zone/subventricular zone (VZ/SVZ) by interfering with the cell cycle at G1-S transition from early development [38]. It might therefore be suggested that a similar mechanism occurs in the LGE and CGE, which could explain defective or delayed production of OPCs during foetal life. Finally, only one study by Darbinian et al. has been published concerning oligodendroglial lineage generation and differentiation in FAS human brains over the period covering the second trimester of pregnancy, from 12.2 to 21.4 WG [9]. By means of mRNA and flow cytometry analyses, these authors showed that ethanol (EtOH) exposure was associated with an increased proportion of cells that express protein markers for early OL progenitors and with a reduced proportion of cells expressing mature OL markers. Nevertheless, the use of brain homogenates in this study did not make it possible to determine in which specific regions oligodendrocyte production was affected, i.e., CP and/or GE. The present study shows not only that oligodendrocyte production is delayed in the GE but also that the proportion of cells expressing maturing OL markers is reduced in FAS brains later in development, supporting the hypothesis that this defect in differentiation persists at least until birth. Such alterations could be partly explained by an enhanced apoptosis as caspase-3 activation has been shown to be substantially increased in EtOH exposed human foetuses [9]. During rodent brain development, alcohol has also been shown to impair astrocyte and oligodendrocyte differentiation and to increase apoptosis [51]. Furthermore, a study performed in foetal macaque brains exposed to alcohol has shown that the decrease in OLs observed in comparison with control brains was linked to massive apoptosis, a single in utero alcohol exposure triggering widespread acute apoptotic death of OLs throughout white matter regions at a rate higher than 12 times compared to the physiological OL apoptosis rate [7]. This study also highlighted the fact that OLs become sensitive to the apoptogenic effect of alcohol at the time they are beginning to generate myelin constituents in their cytoplasm, i.e., when they become positive for MBP and negative for PDGFR-α, a fact we could not confirm in our human cohort, as myelination starts from birth only [7]. From this study, it could be suggested that in addition to a defective and/or delayed generation of OPCs, apoptosis also likely contributes to the decrease of Olig2 positive cells observed in human FASD brains, which was also observed by Darbinian et al. [9]. Epigenetic mechanisms could play an additional role in cell fate specification of brain precursor cells as alcohol is known to induce oxidative stress that alters gene expression, in particular Shh, which promotes expansion and specification of multipotent progenitors into OPCs and immature OLs [12, 35]. Upon alcohol exposure, several other signalling pathways through which alcohol may directly disrupt OPC differentiation and survival have also been implicated, such as PDGFRα, Wnt3a and Wnt5a, which regulate OL cell fate specification, differentiation and proliferation [21, 27, 46]. The increase in PDGFR-α positive OPCs observed in our study could therefore be related to a deregulation of Wnt signalling, preventing OPCs from progressing towards OL differentiation.
Whereas changes in OL morphology, maturation, differentiation and survival have been reported in third trimester-equivalent preclinical models of FASD [7, 8, 52], very little is known about the deleterious effects of alcohol on OL lineage derived from distinct telencephalic germinal zones [15, 18]. In 2017, Newville et al. found a drastic decrease in the number of mature OLs and proliferating OPCs within the corpus callosum of alcohol-exposed mice at postnatal day 16, but neither mature OLs nor OPCs derived from the postnatal SVZ were numerically affected, indicating ontogenetic heterogeneity in susceptibility to alcohol [33]. Several studies performed in rodents have demonstrated that myelination is delayed upon PAE, consisting in a weak expression of MBP, reduced myelin thickness and myelin alterations at the ultrastructural level, which impair the formation of neuronal circuits and conduction of neuronal signals [18, 32, 36].
Another mechanism which regulates the formation of myelin around axons consists in interactions between OPCs which receive excitatory and inhibitory inputs mediated by glutamate and GABA, and developing axons. Recent studies have demonstrated that a significant proportion of grey matter myelin in the cortex forms on the axons of local inhibitory interneurons in both rodents and humans [29, 44]. During development, GABA likely acts as a local environmental cue to control myelination and thus influences the conduction velocity of action potentials in the CNS. Nevertheless, data remain controversial since endogenous GABA by interacting with GABAA receptors has been shown either to increase or to decrease proliferation, apoptosis and consequently oligodendrocyte numbers, as well as shortening internode length, which allows for faster saltatory conduction velocities. Similar discrepancies have also been observed regarding glutamate which blocks proliferation and progression of OPCs but also promotes myelin formation [19, 53].
It has long been acknowledged that in rodents, Olig2 expressing progenitors in the MGE give rise to GABAergic interneurons at early developmental stages and oligodendrocytes thereafter [30]. In humans, Olig2 has been mainly detected in the proliferative zones of the ganglionic eminences between 5–15 post-conceptional weeks prior to the expression of oligodendrocyte precursor markers. By 20 WG, these cells spread throughout the cortex, and co-express markers for immature neurons, neurogenic radial glia and intermediate progenitors [22, 23]. Using immunohistochemistry on 8–12 post-conceptional week human sections, Olig2 immunoreactivity has been shown to be expressed in GABAergic cells of the proliferative zones of the MGE and septum [1]. In the present study, the detection of more than 60% of Olig2 immunoreactive cells at the earliest stage could correspond to OPCs admixed with other nerve cell populations. Confocal studies together with quantitative analyses allowed us to show that Olig2 was specifically expressed in maturing OL from an early developmental stage and was not co-expressed with MAP2, GABA, GFAP or PDGFR-α.
Conclusion
The present study provides further evidence that there is major oligodendrocyte lineage impairment at all stages of brain development upon PAE, consisting in defective/delayed generation, migration and maturation of oligodendrocyte precursors. Since oligodendrocyte development and myelin are a target of alcohol, the disruption of oligodendrocyte differentiation and of myelination process is very likely responsible for inadequate establishment of neuronal networks and inefficient conduction of neuronal signals. Disruption of oligodendrocyte generation and differentiation, together with GABA interneuronopathy that we previously identified [28], most likely contribute to developmental encephalopathy and subsequent life-long neuro-behavioural disabilities.
Availability of data and materials
Most data generated or analyzed during this study are included in this article. Additional datasets are available from the corresponding author on request.
Abbreviations
- CGE:
-
Caudal Ganglionic Eminence
- CNS:
-
Central Nervous System
- CP:
-
Cortical Plate
- DTI:
-
Diffusion Tensor Imaging
- FAS:
-
Foetal Alcohol Syndrome
- FASD:
-
Foetal Alcohol Spectrum Disorder
- GE:
-
Ganglionic Eminences
- IUFD:
-
In Utero Foetal Death
- IUGR:
-
Intra Uterine Growth Retardation
- LGE:
-
Lateral Ganglionic Eminences
- MBP:
-
Myelin Basic Protein
- MGE:
-
Medial Ganglionic Eminence
- OLs:
-
Oligodendrocytes
- Olig2:
-
Oligodendrocyte lineage transcription factor 2
- OPCs:
-
Oligodendrocyte Precursor Cells
- PAE:
-
Prenatal Alcohol Exposure
- PDGFRα:
-
Platelet-Derived Growth Factor Receptor alpha
- Pre-OL:
-
Pre-Oligodendrocyte
- Shh:
-
Sonic Hedgehog
- SVZ:
-
Subventricular Zone
- TOP:
-
Termination of Pregnancy
- VZ:
-
Ventricular Zone
- WG:
-
Weeks Gestation
References
Alzubi A, Lindsay S, Kerwin J, Looi SJ, Khalil F, Clowry GJ (2017) Distinct cortical and sub-cortical neurogenic domains for GABAergic interneuron precursor transcription factors NKX21, OLIG2 and COUP-TFII in early Foetal human telencephalon. Brain Struct Funct 222(5):2309–2328
Back SA, Gan X, Li Y, Rosenberg PA, Volpe JJ (1998) Maturation-dependent vulnerability of oligodendrocytes to oxidative stress-induced death caused by glutathione depletion. J Neurosci 18(16):6241–6253
Barateiro A (1843) Fernandes A (2014) Temporal oligodendrocyte lineage progression: in vitro models of proliferation, differentiation and myelination. Biochim Biophys Acta 9:1917–1929
Bartzokis G, Lu PH, Heydari P, Couvrette A, Lee GJ, Kalashyan G et al (2012) Multimodal magnetic resonance imaging assessment of white matter aging trajectories over the lifespan of healthy individuals. Biol Psychiatry 72(12):1026–1034
Collarini EJ, Pringle N, Mudhar H, Stevens G, Kuhn R, Monuki ES et al (1991) Growth factors and transcription factors in oligodendrocyte development. J Cell Sci Suppl 15:117–123
Coutts DJ, Harrison NL (2015) Acetaldehyde, not ethanol, impairs myelin formation and viability in primary mouse oligodendrocytes. Alcohol Clin Exp Res 39(3):455–462
Creeley CE, Dikranian KT, Johnson SA, Farber NB, Olney JW (2013) Alcohol-induced apoptosis of oligodendrocytes in the Foetal macaque brain. Acta Neuropathol Commun 1:23
Dalitz P, Cock M, Harding R, Rees S (2008) Injurious effects of acute ethanol exposure during late gestation on developing white matter in Foetal sheep. Int J Dev Neurosci 26(5):391–399
Darbinian N, Darbinyan A, Merabova N, Bajwa A, Tatevosian G, Martirosyan D et al (2021) Ethanol-mediated alterations in oligodendrocyte differentiation in the developing brain. Neurobiol Dis 148:105181
Donald KA, Roos A, Fouche JP, Koen N, Howells FM, Woods RP et al (2015) A study of the effects of prenatal alcohol exposure on white matter microstructural integrity at birth. Acta Neuropsychiatr 27(4):197–205
Egawa N, Shindo A, Hikawa R, Kinoshita H, Liang AC, Itoh K et al (2019) Differential roles of epigenetic regulators in the survival and differentiation of oligodendrocyte precursor cells. Glia 67(4):718–728
Ehrhart F, Roozen S, Verbeek J, Koek G, Kok G, van Kranen H et al (2019) Review and gap analysis: molecular pathways leading to Foetal alcohol spectrum disorders. Mol Psychiatry 24(1):10–17
Emery B (2010) Regulation of oligodendrocyte differentiation and myelination. Science 330(6005):779–782
- Feess-Higgins A, Larroche JC (1987) Development of the human Foetal brain. In: Masson (ed) Paris, INSERM CNRS
Guerri C, Pascual M, Renau-Piqueras J (2001) Glia and Foetal alcohol syndrome. Neurotoxicology 22:593–599
Guihard-Costa AM, Larroche JC (1990) Differential growth between the Foetal brain and its infratentorial part. Early Hum Dev 23:27–40
Guihard-Costa AM, Ménez F, Delezoide AL (2002) Organ weights in human foetuses after formalin fixation: standards by gestational age and body weight. Pediatr Dev Pathol 5:559–578
Guizzetti M, Zhang X, Goeke C, Gavin DP (2014) Glia and neurodevelopment: focus on Foetal alcohol spectrum disorders. Front Pediatr 2:123
Hamilton NB, Clarke LE, Arancibia-Carcamo IL, Kougioumtzidou E, Matthey M, Káradóttir R et al (2017) Endogenous GABA controls oligodendrocyte lineage cell number, myelination, and CNS internode length. Glia 65(2):309–321
Haute Autorité de santé. Standard protocol for Foetal or perinatal autopsy (2014) Ann Pathol 34(6):415–33
Ille F, Sommer L (2005) Wnt signaling: multiple functions in neural development. Cell Mol Life Sci 62(10):1100–1108
Jakovcevski I, Filipovic R, Mo Z, Rakic S, Zecevic N (2009) Oligodendrocyte development and the onset of myelination in the human Foetal brain. Front Neuroanat 1(3):5
Jakovceski I, Zecevic N (2005) Olig transcription factors are expressed in oligodendrocytes and neuronal cells in the human Foetal CNS. J Neurosci 25:10064–10073
Kessaris N, Fogarty M, Iannarelli P, Grist M, Wegner M, Richardson WD (2006) Competing waves of oligodendrocytes in the forebrain and postnatal elimination of an embryonic lineage. Nat Neurosci 9(2):173–179
Lebel C, Rasmussen C, Wyper K, Walker L, Andrew G, Yager J et al (2008) Brain diffusion abnormalities in children with Foetal alcohol spectrum disorder. Alcohol Clin Exp Res 32(10):1732–1740
Lebel C, Roussotte F, Sowell ER (2011) Imaging the impact of prenatal alcohol exposure on the structure of the developing human brain. Neuropsychol Rev 21:102–118
Luo J, Miller MW (1998) Growth factor-mediated neural proliferation: target of ethanol toxicity. Brain Res Rev 27(2):157–167
Marguet F, Friocourt G, Brosolo M, Sauvestre F, Marcorelles P, Lesueur C et al (2020) Prenatal alcohol exposure is a leading cause of interneuronopathy in humans. Acta Neuropathol Commun 8(1):208
Micheva KD, Wolman D, Mensh BD, Pax E, Buchanan J, Smith SJ et al (2016) A large fraction of neocortical myelin ensheathes axons of local inhibitory neurons. Elife 5:e15784
Miyoshi G, Butt SJ, Takebayashi H, Fishell G (2007) Physiologically distinct temporal cohorts of cortical interneurons arise from telencephalic Olig2-expressing precursors. J Neurosci 27(29):7786–7798
Moore EM, Migliorini R, Infante MA, Riley EP (2014) Foetal alcohol spectrum disorders: recent neuroimaging findings. Curr Dev Disord Rep 1(3):161–172
Newville J, Howard TA, Chavez GJ, Valenzuela CF, Cunningham LA (2021) Persistent myelin abnormalities in a third trimester-equivalent mouse model of fetal alcohol spectrum disorder. Alcohol Clin Exp Res. https://doi.org/10.1111/acer.14752
Newville J, Valenzuela CF, Li L, Jantzie LL, Cunningham LA (2017) Acute oligodendrocyte loss with persistent white matter injury in a third trimester equivalent mouse model of Foetal alcohol spectrum disorder. Glia 65(8):1317–1332
Norman AL, Crocker N, Mattson SN, Riley EP (2009) Neuroimaging and Foetal alcohol spectrum disorders. Dev Disabil Res Rev 15(3):209–217
Ortega JA, Radonjić NV, Zecevic N (2013) Sonic hedgehog promotes generation and maintenance of human forebrain Olig2 progenitors. Front Cell Neurosci 7:254
Ozer E, Sarioglu S, Güre A (2000) Effects of prenatal ethanol exposure on neuronal migration, neuronogenesis and brain myelination in the mice brain. Clin Neuropathol 19(1):21–25
Rash BG, Duque A, Morozov YM, Arellano JI, Micali N, Rakic P (2019) Gliogenesis in the outer subventricular zone promotes enlargement and gyrification of the primate cerebrum. Proc Natl Acad Sci 116(14):7089–7094
Riar AK, Narasimhan M, Rathinam ML, Henderson GI, Mahimainathan L (2016) Ethanol induces cytostasis of cortical basal progenitors. J Biomed Sci 23:6
Rice J, Gu C (2019) Function and mechanism of myelin regulation in alcohol abuse and alcoholism. BioEssays 41(7):e1800255
Riley EP, Infante MA, Warren KR (2011) Foetal alcohol spectrum disorders: an overview. Neuropsychol Rev 21(2):73–80
Riley EP, McGee CL (2005) Foetal alcohol spectrum disorders: an overview with emphasis on changes in brain and behavior. Exp Biol Med 230(6):357–365
Rivers LE, Young KM, Rizzi M, Jamen F, Psachoulia K, Wade A et al (2008) PDGFRA/NG2 glia generate myelinating oligodendrocytes and piriform projection neurons in adult mice. Nat Neurosci 11(12):1392–1401
Roozen S, Peters GY, Kok G, Townend D, Nijhuis J, Koek G et al (2018) Systematic literature review on which maternal alcohol behaviours are related to Foetal alcohol spectrum disorders (FASD). BMJ Open 8(12):e022578
Stedehouder J, Couey JJ, Brizee D, Hosseini B, Slotman JA, Dirven CMF et al (2017) Fast-spiking parvalbumin interneurons are frequently myelinated in the cerebral cortex of mice and humans. Cereb Cortex 27(10):5001–5013
Tsai HH, Niu J, Munji R, Davalos D, Chang J, Zhang H et al (2016) Oligodendrocyte precursors migrate along vasculature in the developing nervous system. Science 351(6271):379–384
Vangipuram SD, Lyman WD (2012) Ethanol affects differentiation-related pathways and suppresses Wnt signaling protein expression in human neural stem cells. Alcohol Clin Exp Res 36(5):788–797
van Tilborg E, de Theije CGM, van Hal M, Wagenaar N, de Vries LS, Benders MJ et al (2018) Origin and dynamics of oligodendrocytes in the developing brain: Implications for perinatal white matter injury. Glia 66(2):221–238
Wilhelm CJ, Guizzetti M (2016) Foetal alcohol spectrum disorders: an overview from the glia perspective. Front Integr Neurosci 9:65
Wozniak JR, Muetzel RL, Mueller BA, McGee CL, Freerks MA, Ward EE et al (2009) Microstructural corpus callosum anomalies in children with prenatal alcohol exposure: an extension of previous diffusion tensor imaging findings. Alcohol Clin Exp Res 33(10):1825–1835
Young KM, Psachoulia K, Tripathi RB, Dunn SJ, Cossell L, Attwell D et al (2013) Oligodendrocyte dynamics in the healthy adult CNS: evidence for myelin remodeling. Neuron 77(5):873–885
Young C, Roth KA, Klocke BJ, West T, Holtzman DM, Labruyere J et al (2005) Role of caspase-3 in ethanol-induced developmental neurodegeneration. Neurobiol Dis 20(2):608–614
Zoeller RT, Butnariu OV, Fletcher DL, Riley EP (1994) Limited postnatal ethanol exposure permanently alters the expression of mRNAS encoding myelin basic protein and myelin-associated glycoprotein in cerebellum. Alcohol Clin Exp Res 18(4):909–916
Zonouzi M, Scafidi J, Li P, McEllin B, Edwards J, Dupree JL et al (2015) GABAergic regulation of cerebellar NG2 cell development is altered in perinatal white matter injury. Nat Neurosci 18(5):674–682
Acknowledgements
The authors are grateful to Nikki Sabourin-Gibbs, Rouen University Hospital, for her help in editing the manuscript.
Funding
This work was supported by Rouen University Hospital, Normandy University, the Institut National de la Santé et de la Recherche Médicale (INSERM; UMR1245), the Fondation de France, the Fondation de l’Avenir and the Agence Nationale de la Recherche (ANR). MB is a recipient of a fellowship from the French Research Ministry.
Author information
Authors and Affiliations
Contributions
FM, BG and AL designed the study. FS, PM and SM collected the clinical data. FM and AL performed and interpreted the pathological analyses. CL and MB performed immunohistochemical and confocal analyses and FM and AL analyzed the results. FM, GF and AL wrote the manuscript. BG, GF and SM critically reviewed the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participants
Autopsy and neuropathological analyses were performed after appropriate informed consent was obtained from the parents.
Competing interests
The authors declare no competing interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information

Additional file 1: Figure S1
. Confocal analyses in the GE of a normal brain at 16 WG Double immunolabellings using Olig2 (red) and MAP2 (green) (a), Olig2 (red) and GFAP (green) (b), Olig2 (red) and GABA (green) (c) and using Olig2 (red) and PDGFR-α (green) (d) did not reveal any co-expression with neurons, interneurons, astrocytes and oligodendroglial precursor markers
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

{kind=link}
Cite this article
Marguet, F., Brosolo, M., Friocourt, G. et al. Oligodendrocyte lineage is severely affected in human alcohol-exposed foetuses. acta neuropathol commun 10, 74 (2022). https://doi.org/10.1186/s40478-022-01378-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40478-022-01378-9