
Abstract
Traumatic brain injury (TBI) constitutes one of the strongest environmental risk factors for several progressive neurodegenerative disorders of cognitive impairment and dementia that are characterized by the pathological accumulation of hyperphosphorylated tau (p-Tau). It has been questioned whether mouse closed-head TBI models can replicate human TBI-associated tauopathy. We conducted longitudinal histopathological characterization of a mouse closed head TBI model, with a focus on pathological features reported in human TBI-associated tauopathy. Male C57BL/6 J mice were subjected to once daily TBI for 5 consecutive days using a weight drop paradigm. Histological analyses (AT8, TDP-43, pTDP-43, NeuN, GFAP, Iba-1, MBP, SMI-312, Prussian blue, IgG, βAPP, alpha-synuclein) were conducted at 1 week, 4 weeks, and 24 weeks after rTBI and compared to sham operated controls. We conducted a systematic review of the literature for mouse models of closed-head injury focusing on studies referencing tau protein assessment. At 1-week post rTBI, p-Tau accumulation was restricted to the corpus callosum and perivascular spaces adjacent to the superior longitudinal fissure. Progressive p-Tau accumulation was observed in the superficial layers of the cerebral cortex, as well as in mammillary bodies and cortical perivascular, subpial, and periventricular locations at 4 to 24 weeks after rTBI. Associated cortical histopathologies included microvascular injury, neuroaxonal rarefaction, astroglial and microglial activation, and cytoplasmatic localization of TDP-43 and pTDP-43. In our systematic review, less than 1% of mouse studies (25/3756) reported p-Tau using immunostaining, of which only 3 (0.08%) reported perivascular p-Tau, which is considered a defining feature of chronic traumatic encephalopathy. Commonly reported associated pathologies included neuronal loss (23%), axonal loss (43%), microglial activation and astrogliosis (50%, each), and beta amyloid deposition (29%). Our novel model, supported by systematic review of the literature, indicates progressive tau pathology after closed head murine TBI, highlighting the suitability of mouse models to replicate pertinent human histopathology.
Similar content being viewed by others

Introduction
Traumatic brain injury (TBI) represents a major public health problem affecting more than 10 million people worldwide each year [36]. TBI is a leading cause of adult death and disability worldwide. It has been estimated that annually 150 to 200/1,000,000 people become disabled as a result of brain trauma, and TBI was declared a major public health problem by the National Institutes of Health in 1999 [4, 20, 53].
There is a long history of epidemiological evidence that TBI represents one of the strongest environmental risk factors for several progressive neurodegenerative disorders of cognitive impairment and dementia that are characterized by the pathological accumulation of hyperphosphorylated tau (p-Tau). In particular, TBI has been linked to Alzheimer’s disease (AD) and a unique clinicopathological entity termed chronic traumatic encephalopathy (CTE) [8, 17, 29, 46, 66, 67]. Yet, the exact mechanism(s) driving pathological tau accumulation and spread, cognitive impairment, and dementia after TBI are poorly understood. Defining the mechanisms that explain the link between TBI and dementia at the cellular level is a public health priority [61].
Mouse models of TBI-associated pathology are of great value because of the ability to conduct detailed, longitudinal histopathological characterization in a temporally accelerated fashion, as well as the unique possibility to explore underlying molecular mechanisms through genetic manipulation without confounding by comorbid conditions. This appears particularly important in light of a current lack of a uniformly accepted definition for TBI-specific tauopathy. Although pathological consensus criteria have been developed for CTE [8, 46], which is considered the prototypical TBI-associated tauopathy, challenges in applying these criteria relate to the fact that many pathologies are also present in the normally aging brain and other neurodegenerative conditions [27, 38]. Hence, distinguishing the direct effects of TBI from a sporadic progressive neurodegenerative process in humans is difficult. Moreover, animal models are needed to dissect the specific neuropathology of TBI and how pathological tau deposition relates to progressive neurodegeneration. However, concerns have been raised that murine closed head injury models may not be suitable to replicate pertinent histopathological features of human TBI-associated tauopathy [18]. Depending on the model used, variability in post-TBI histopathology may be considerable [9, 10]. Further, there are distinct differences in the cerebral anatomy as well as expression of tau isoforms in the adult brains of mice and humans [32], which could conceivably contribute to disparate pathology [41]. Therefore, it is critical to understand to what degree murine closed-head TBI can replicate human TBI-associated tau pathology and related histopathological features.
In this study, we characterized the histopathological features of a mouse closed head repetitive TBI (rTBI) model with specific focus on the presence and evolution of p-Tau accumulation and its association with pertinent histopathological features of human TBI-associated tauopathy according to reported consensus criteria for human CTE [8, 46]. In addition, we conducted a systematic literature review of reported tau pathology in mouse closed-head TBI and its relationship to pertinent histological features of human CTE to provide a contemporary overview of the state of the field.
Material and methods
Mouse study
Mice were randomly allocated to sham surgery versus rTBI except for 4 mice allowed to survive for 24-weeks after rTBI, which were added to gain insight into the longer-term evolution of p-Tau and associated pathology. Histological analyses were conducted by an investigator masked to the experimental groups.
Animals
Male C57BL/6 J mice (Jackson Laboratories) were socially housed with same-sex mice (n = 4 per cage) on 12-h light/dark cycle with food and water ad libitum. Spontaneously breathing mice (n = 27) weighing 26.8 ± 2.6 g (age 8–12 weeks) were subjected to closed head injury (n = 20) or sham surgery (n = 11). Brains were removed 1 week after rTBI (n = 8), 4 weeks (sham n = 8, rTBI n = 8), and 24 weeks (rTBI n = 4) for histological analyses. Additionally, we conducted AT8-staining in 3 sham operated mice that survived for 12 months to rule out any potential age-related effects on p-Tau pathology.
Anesthesia, analgesia, and traumatic brain injury induction
Animals were anesthetized with isoflurane (5% for induction, 2% for surgery, 1.5% for maintenance) in room air. Anesthesia was discontinued immediately prior to TBI and sham injury. Body temperature was monitored continuously with a rectal probe and maintained at 37.0 ± 0.5 °C. To alleviate pain, animals received 0.05 mg/kg subcutaneous buprenorphine (Med-Vet International, Mettawa, Il, USA) 30 min before anesthesia and every 6 h afterwards for 24 h. Additionally, each animal received 5 mg/kg subcutaneous carprofen (Patterson Veterinary, Devens, MA, USA) at the end of the anesthesia.
TBI was produced using a weight drop device as previously described in detail [11, 31] with the modification that animals were subjected to TBI or sham injury once daily for 5 consecutive days. Briefly, a 50 g weight was freely dropped 15 cm to strike a cylindrical polyacetal transducer rod (Delrin©, tip-diameter 2 mm, 17.4 g) that was placed with its tip directly on the exposed skull (target 2.5 mm posterior and 2.5 mm lateral from Bregma). Following TBI, the wound closed with interrupted sutures. Sham animals were anesthetized, surgically prepared (including skin incision), and placed under the impact device with the impactor touching the skull, but were not subjected to head impact. One mouse with a skull fracture was excluded.
Immunohistochemistry staining
For histology, animals received an overdose of pentobarbital (150 mg/kg Fatal-Plus, Vortech Pharmaceuticals). Then animals were perfused under isoflurane anesthesia through the ascending aorta with 50 mL saline and then with ice cold phosphate-buffered 4% paraformaldehyde (PFA) for 10 min. Brains were removed from the cranium, postfixed overnight in the same fixative, and then stored in 0.4% PFA at 4 °C until further processing. Prior to paraffin embedding brains were pre-sectioned using a brain matrix.
Immunohistochemistry was performed against p-TauSer−202/Thr205, (AT8, 1:250, Thermo Fisher Scientific, Cat# MN1020, RRID: AB_223647), p-TauThr231 (AT180, 1:250, Thermo Fisher Scientific, Cat# MN1040, RRID: AB_223649), TAR DNA-binding protein 43 (TDP-43, 1:250, Proteintech, Cat# 10,782–2-AP, RRID: AB_615042), pTDP-43Ser−409/410 (1:250, Proteintech, Cat# 22,309–1-AP, RRID: AB_11182943), neuronal nuclei (NeuN, 1:200, Proteintech, Cat# 26,975–1-AP, RRID: AB_2880708), glial fibrillary acidic protein (GFAP, 1:250, Agilent, Cat# Z0334, RRID: AB_10013382), ionized calcium binding adaptor molecule 1 (1:250, Iba-1, Wako, Cat# 019–19,741, RRID: AB_839504), myelin basic protein (MBP, 1:200, Santa Cruz Biotechnology, Cat# M3821, RRID: AB_1841021), SMI-312 (1:200, BioLegend Cat# 837,904, RRID: AB_2566782), beta amyloid precursor protein (βAPP, 1:200, Zymed, CT695, Cat# 51–2700, RRID: AB_2533902), α-synuclein (1:250, Biolegend, Cat# 824,301, RRID: AB_2564879), and immunoglobulin G (IgG, 1:100, Abcam, Cat# ab6708, RRID: AB_956005). For chromogenic staining, tissue sections labeled with the primary antibodies (AT8, AT180, NeuN, βAPP, α-synuclein, IgG) were incubated with appropriate biotin-conjugated secondary antibodies followed by avidin–biotin complex (Vector Laboratories) incubation and treatment with diaminobenzidine as directed by the manufacturer. Hematoxylin counterstaining was used for AT8, AT180, βAPP, and α-synuclein labeled tissues. For immunofluorescence staining tissue sections labeled with the primary antibodies (TDP-43, pTDP-43, NeuN, GFAP, Iba-1, MBP, SMI-312, βAPP, alpha-synuclein) were incubated in appropriate secondary antibodies conjugated with Alexa Fluor 488 (1:250, Abcam, Cat# ab150113, RRID: AB_2576208 and Cat# ab150077, RRID: AB_2630356), Alexa Fluor 555 (1:250, Abcam, Cat# ab150106, RRID: AB_2857373), and Alexa Fluor 647 (1:250, Abcam, Cat# ab150075, RRID: AB_2752244 and Cat# ab150115, RRID: AB_2687948). Omitting the primary antibody in a subset of slides served as negative controls.
Prussian blue staining
To assess for microhemorrhages, sections were stained for Prussian blue reaction using an Iron Stain Kit (# HT20, Sigma-Aldrich), following the manufacturer’s instructions.
Microscopy
Paraffin sections, 10-µm thick coronal, were obtained at approximately Bregma -3.07 mm (impact center; s3), −1.67 mm (adjacent to the impact center; s2), and + 1.21 mm (remote to the impact center; s1) for histological assessment (Fig. 1a, b). For quantitative and qualitative analyses of AT8, NeuN, GFAP, Iba-1, MBP, and SMI-312 staining one coronal section each from these coordinates (s1-s3) was used. For qualitative analyses of βAPP, alpha-synuclein, IgG, and Prussian blue staining we reviewed one coronal section each from s1-s3. For quantitative and qualitative analyses of TDP-43 and pTDP-43 one coronal section from s2 was used. All histological analyses were performed by an investigator masked to the animal groups.

Patterns and evolution of p-Tau accumulation after closed head repetitive traumatic brain injury (rTBI). (a) Approximate location of the impact center over the intact mouse skull (blue circle) relative to the brain sections sampled for histological analysis (s1-s3; dashed lines). (b) Approximate location of p-Tau positive cells at 4 weeks after rTBI (composite of 8 mice; each red dot represents 8 p-Tau positive cells, blue ellipses indicate the spatial relation between the impactor and brain surface). Black boxes indicate the location of photomicrographs shown in panel c-e (box 1) and f–h (box 2). (c) Intact cerebral cortex without p-Tau accumulation at 1 week after rTBI. Progressive accumulation of p-Tau in the superficial layers of the cerebral cortex at (d) 4 weeks and (e) 24 weeks after rTBI. P-Tau accumulation in the corpus callosum at (f) 1 week, (g) 4 weeks, and (h) 24 weeks after rTBI (red asterisks). (f) In contrast to later time points, AT8-immunoreactivity at 1 week was restricted to dot-like staining in a subset of cells (arrowheads). Examples of p-Tau accumulation in (i) perivascular, (j) subpial, (k, l) periventricular, and (l) mammillary body locations as well as (m) at the depth of the superficial longitudinal fissure (white arrowheads) in perivascular (black arrowheads) and subpial (red arrowheads) locations at 4 weeks after rTBI. Scale bars are 30 µm (in c–j), 1 mm (k), and 300 µm (in l–m)
Image acquisition and quantification
To acquire images of all stained sections for subsequent offline analysis we used a Leica DM6 B microscopy system equipped with a brightfield DMC5400 color CMOS camera and an immunofluorescent DFC9000 sCMOS camera.
To determine the spatial distribution of p-Tau accumulation after rTBI, sections were imaged at 63 × magnification and the approximate location of all AT8-positive profiles systematically recorded within each of the sampled sections and transferred to a standard atlas [52] to provide a visual representation of the observed tau pathology relative to the impact location. Given reported p-Tau accumulation in both neurons and astrocytes in human and mouse TBI, we quantified the number of p-Tau positive cells across time points as stratified by neuronal versus astrocytic p-Tau. Moreover, because we observed the earliest p-Tau accumulation in the corpus callosum with only later involvement of the cerebral cortex we additionally stratified these analyses according to the location in the superficial cortex (approximately cortical layers I-III), deep cortex (approximately cortical layers IV-VI), and the corpus callosum.
To determine the extent of neuronal loss, chromogen stained NeuN-positive cells were assessed in each coronal section. Images of 16 nonoverlapping fields of view (FOV; 8 per hemisphere; 659 × 439 µm, each) covering the dorsal cerebral cortex [11] were taken at 20 × magnification. NeuN positive cells were semiautomatically quantified using ImageJ [60] as previously described [15]. First, 16-bit color images were converted to 8-bit grayscale followed by automatic thresholding. The Analyze Particle tool was then used to count all particles with a circularity of 0.1–1.0 and a size of 25 to 250 µm2. Results were adjusted by manually counting all overlapping cells (ie, particle size greater than 250 µm2).
To assess microglia and astroglia in the dorsal cortex we used fluorescence staining. For GFAP, images of 16 nonoverlapping FOVs (8 per hemisphere; 667 × 667 µm, each) covering the dorsal cerebral cortex were taken at 20 × magnification. First, 16-bit color images were split into the individual color channels (red, green, blue) followed by automatic thresholding of the 8-bit green channel and black-white color inversion. The Analyze Particle tool was then used to quantify the total thresholded area (µm2). For Iba-1, TDP-43, and pTDP-43 images of 16 FOVs (8 per hemisphere; 211 × 211 µm, each) centered within the corresponding FOV used for the GFAP analyses were taken at 63 × magnification and analyzed as described for GFAP.
To assess the impact of rTBI on axonal integrity in the injured cortex, we used fluorescence staining for the myelin marker MBP and pan-axonal neurofilament marker SMI312. Images of one FOV (206 × 206 µm) per section were taken at 63 × magnification and analyzed as described for GFAP.
Assessment of chronic traumatic encephalopathy (CTE) related pathology
With respect to assessing CTE-like pathology in our model, we defined histopathological features according to recently published consensus group criteria developed by an NINDS/NIBIB panel [8, 46] with modifications for use in mice (Table 1). Specifically, a defining criterion for human CTE includes the presence of perivascular foci of p-Tau immunoreactive neurofibrillary tangles (NFTs) and abnormal neurites, with or without p-Tau immunoreactive astrocytes, in an irregular pattern in the cerebral cortex, with a tendency to involve the sulcal depths. Because the lissencephalic brains of mice lack sulci, we considered the presence of perivascular p-Tau in the cerebral cortex a pathognomonic criterion. With respect to supportive features, we assessed for several supportive tau- and nontau-related histopathological features (Table 1). We did not assess for macroscopic pathology such as dilation of ventricles, septal pathology, atrophy, contusions, or other signs of previous trauma [8, 46]. Finally, for additional context we also assessed several pathological features that have been repeatedly described in human TBI but are not specific to CTE and may be shared with TBI and other neurodegenerative conditions including the presence of reactive microglia, astrogliosis, neuronal and axonal loss, presence of beta amyloid and alpha-synuclein depositions as well as evidence of prior blood brain barrier (BBB) disruption and microvascular injury/cerebral microbleeds [45, 49].
Neurologic evaluation
Return of the righting reflex was measured as the time (s) from TBI/sham injury to righting from a supine to prone position after discontinuation of anesthesia. The neurological severity score (NSS) was assessed on a scale from 0 (no deficit) to 10 (maximal deficit) prior to TBI as well as serially until sacrifice as previously described in detail [31].
Statistical analysis
Unless otherwise stated, continuous variables are reported as mean ± standard error of the mean. Normality of data was examined using the Shapiro–Wilk test. One-way analysis of variance (ANOVA) on Ranks with post-hoc Dunn’s test was used to assess for between-group differences in histopathology (p-Tau, TDP-43, pTDP-43, NeuN, Iba-1, GFAP, MBP, SMI-312). Between-group comparisons of continuous variables over time (body weight, return of the righting reflex, NSS) were conducted using longitudinal mixed models. Time was treated as a categorical variable. The models included group (Sham versus rTBI) and time as fixed covariates, as well as the group × time interactions. Two-sided significance tests were used throughout and unless stated otherwise a two-sided P < 0.05 was considered statistically significant. All statistical analyses were performed using SigmaPlot 12.5 (Systat Software, Inc., Germany) or IBM® SPSS® Statistics Version 26 (IBM®-Armonk, NY).
Systematic review
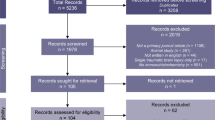
We conducted a systematic review of the literature by searching PubMed and Scopus using search criteria that were established to be specific for mouse models of closed head injury (including blast injuries) and that contained reference to tau protein assessment in wild type mouse strains. In addition to characterizing model characteristics of included studies, we collected histological outcomes that have been associated with CTE, including assessment of axonal and neuronal injury, astrogliosis, microglial activation, TDP-43 pathology, as well as presence of amyloid pathology, α-synuclein, cerebral microbleeds, and evidence of BBB disruption. Overall, we identified 1940 articles in PubMed and 2057 articles in Scopus. After removal of duplicates (n = 241), 3756 papers were included for screening. Details of the review methodology including the search strategy, inclusion and exclusion criteria, and retrieval of information from the full-text articles is summarized in the Additional file 1: Supplementary information.
Results
Mouse rTBI model
Similar to sham operated mice, brains of rTBI mice appeared macroscopically intact and without evidence for macroscopic cerebral hemorrhages at all time points post injury. Microscopically, the cerebral cortex appeared normal at 1 week after rTBI. However, at 4 and 24 weeks after rTBI we observed focal tissue disruption within the superficial cerebral cortex approximately corresponding to cortical layer I (Fig. 1c-e).
Consistent with human pathology [28], Prussian blue staining showed the presence of hemosiderin laden macrophages indicative of microvascular injury in the superficial layers of the ipsilateral cortex, at the depth of the superior longitudinal fissure, and grey-white matter junction between cortex and corpus callosum (Additional file 1: Figure S1). There was evidence of subtle BBB disruption as assessed by IgG staining at 1 week and 4 weeks post rTBI (Additional file 1: Figure S2).
p-Tau spreads from the corpus callosum to superficial cortical layers between 1 to 24 weeks after rTBI
The primary goal of this study was to determine the spatial and temporal evolution of p-Tau pathology over 24 weeks after murine closed head-injury. We used both AT8 and AT180 to detect p-Tau. AT180 staining yielded similar results as AT8 staining (Additional file 1: Figure S3) and we exclusively refer to AT8 staining to describe p-Tau pathology below. Importantly, we found no AT8 positive cells in sham operated mice that survived for 4 weeks and 12 months after surgery, respectively (Additional file 1: Figure S4).
We found that 1 week after rTBI, 4 mice (n = 50%) showed faint AT8 staining restricted to the corpus callosum without any p-Tau accumulation in the overlying cerebral cortex (Figs. 1c, f, 2f). P-Tau pathology did not substantially progress in the corpus callosum between 1 and 24 weeks and AT8 positive cells were present in only a subset of mice (n = 3 [38%] at 4 weeks, n = 2 [50%] at 24 weeks), typically within the section adjacent to the impact center (Figs. 1b, f–h, 2f). Conversely, by 4 and 24 weeks we observed significant accumulation of AT8-stained cells in all investigated mice, particularly in subpial locations and the superficial cortex (approximately cortical layers I-III) in the traumatized hemisphere (Figs. 1d–e, 2e–f). Figure 1b depicts the approximate distribution of AT8-positive cells within the traumatized hemisphere at 4 weeks after rTBI. Interestingly, staining was most abundant in the cerebral cortex adjacent to the impact site (Bregma − 1.67 mm) rather than directly below the impact center (Bregma −3.07).

Temporal and spatial distribution of p-Tau pathology in neurons and astroglia. (a,b) Double staining indicates colocalization (white arrows) of hyperphosphorylated tau (p-Tau; AT8) with (a) neurons (NeuN) and (b) astrocytes (GFAP) in the cerebral cortex at 4 weeks after repetitive traumatic brain injury (rTBI). White scale bars correspond to 50 µm in low power and 17 µm in high power magnified panels. DAPI (blue) channel omitted from high power magnifications. (c) Dotted lines delineate cortical layers I-III (superficial cortex) from cortical layers IV-VI (deep cortex) and corpus callosum used to assess the presence of p-Tau stained cells in the ipsilateral hemisphere. Asterisk denotes the approximate location of the images taken for a-b. (d) Proportion of AT8 positive neurons and astrocytes at the investigated time points after rTBI. (e) Distribution of AT8 stained cells in the cerebral cortex (superficial + deep combined) relative to the impact center (black bars). There was no difference in the number and distribution of p-Tau positive cells at 4 weeks and 24 weeks after rTBI (P > 0.05). Because sham and 1-week rTBI animals had no cortical AT8-positive cells they were omitted from this analysis. Each bar corresponds to one cortical field of view (FOV) arranged from left, contralateral (FOV 1) to right, ipsilateral (FOV 16), whereby corresponding FOVs in the three investigated sections s1–s3 were summed. Data are mean ± SD. (f) Number of p-Tau positive neurons (green shades) and astrocytes (blue shades) in the traumatized hemisphere stratified by location in the superficial cortex, deep cortex, and corpus callosum over time (total number of cells counted in the three investigated sections s1–s3). *P < 0.05. Data are mean ± SEM. n = 8 mice for sham, 1 week, and 4 weeks, n = 4 for 24 weeks (there were no p-Tau positive cells in sham operated mice). All analyses were done using one-way ANOVA on Ranks with post-hoc Dunn's
p-Tau accumulation after murine rTBI occurs in cerebral locations reported for human TBI-associated tauopathy
At 4 weeks post rTBI we observed perivascular p-Tau accumulation (Fig. 1i, m), a pathognomonic histological feature of CTE. Additional, though less common, sites of p-Tau accumulation included the mamillary body (Fig. 1l) and periventricular tissues (Fig. 1k, l), which are considered histological features supporting a CTE diagnosis. Notably, in all examined rTBI mice p-Tau accumulation was present in the subpial cortex adjoining the superficial longitudinal fissure (Fig. 1b, m). We did not find any AT8 stained cells in the hippocampus, thalamus, and amygdala (Table 1).
p-Tau accumulates in cortical neurons and astrocytes
We found distinct co-localization of AT8 with both NeuN and GFAP in the traumatized hemisphere consistent with neuronal and astrocytic tau accumulation (Fig. 2a, b). Interestingly, colocalization of AT8 and GFAP was only observed at 4 and 24 weeks after rTBI and restricted to the superficial cerebral cortex without overt co-staining in deeper cortical layers or the corpus callosum (Fig. 2d, f). We found that perivascular p-Tau was present in both neurons and astrocytes (Additional file 1: Figure S5). The majority of AT8 positive cells co-stained with NeuN, and less than 10% of cells were positive for both AT8 and GFAP (Fig. 2d, f).
rTBI causes persistent nuclear loss and cytoplasmatic localization of TDP-43 and pTDP-43
Compared to sham and 1 week post rTBI mice, we noted an overall increase in TDP-43 and pTDP-43 in the ipsilateral cerebral cortex, particularly adjacent to the impact center, at 4 weeks and 24 weeks after rTBI (Fig. 3). On a cellular level, there was nuclear loss and cytoplasmatic localization of TDP-43 and pTDP-43 at the same time points (Fig. 3). However, while conspicuous aggregation of cytoplasmatic TDP-43 was present at 4 weeks (Fig. 3g), this was only observed at 24 weeks for pTDP-43 (Fig. 3q).

Patterns and evolution of TDP43 (a–h) and pTDP43 (j–q) pathology in the cerebral cortex after repetitive closed head traumatic brain injury (rTBI) in the mouse. (f) Increased linear pattern of TDP-43 reactivity suggesting neuritic distribution at 1 week after rTBI. Nuclear loss (red arrowheads) and cytoplasmatic localization (white arrowheads) of TDP-43 (g-h) and pTDP-43 (p-q) at 4 and 24 weeks. Aggregation of cytoplasmatic localized TDP-43 (g-h) and pTDP-43 (q). (r) Apparent reduction in signal intensity of pTDP-43 in the cortex next to the impact center (§). Scale bars correspond to 240 µm for low power and 20 µm for high power magnified images. In each group, bars correspond to one cortical field of view (FOV) arranged from left, contralateral (contra), to right, ipsilateral (ipsi). Data are shown as mean (+ SD). n = 8 for sham, 1 week, and 4 week post rTBI and n = 4 for 24 week post rTBI. Analyses were done at Bregma − 1.67 mm (corresponding to s2 in Fig. 1) *P < 0.05. All analyses were done using one-way ANOVA on Ranks with post-hoc Dunn's
In the contralateral (non-traumatized) hemisphere staining intensity for TDP-43 and pTDP-43 in the corpus callosum, subcortical nuclei, and hippocampus was overall similar to sham operated animals at all time points after rTBI (not shown). Nonetheless, by 24 weeks after rTBI, nuclear loss and cytoplasmic localization of TDP-43 (Additional file 1: Figure S6) and pTDP-43 (not shown) was present in the contralateral cerebral cortex and bilateral CA1 of the hippocampus.
rTBI causes astroglial and microglial activation and progressive neurodegeneration
In addition to assessing pertinent p-Tau and TDP-43 related histological features of human CTE (Table 1), we sought to determine pathology that occurs in human TBI-associated neurodegenerative disease, including the presence of reactive migroglia and astroglia, neuronal and axonal loss, and accumulation of βAPP and α-synuclein.
Sham and 1-week rTBI animals had few GFAP positive cells in the cerebral cortex and corpus callosum (< 1% staining signal coverage) without difference between hemispheres (Fig. 4c). We observed substantial astroglial activation at 4 weeks after rTBI as indicated by the presence of hypertrophied GFAP stained astrocytes and overall significantly increased GFAP staining in the cerebral cortex (Fig. 4a, c). Although GFAP staining remained significantly elevated by 24 weeks as compared to sham operated animals, overall staining intensity appeared attenuated, and cells appeared less hypertrophied when compared to 4 weeks (Fig. 4a, c).

Progression of cortical neuronal loss and glial activation after repetitive traumatic brain injury (rTBI). (a) Cortical neuronal loss after rTBI as shown by NeuN staining. (b) Astroglial activation at 4 weeks as indicated by (a) the presence of numerous hypertrophied GFAP stained astrocytes (inset) and (c) a significant increase in GFAP staining signal. Decrease in astroglial activation at 24 weeks after rTBI as shown by reduced cell hypertrophy (inset in (a)) and attenuated GFAP staining (c). Microglial activation was observed at 4 weeks as indicated by the presence of numerous bipolar/rod shaped Iba-1 stained microglia (inset in (a)) and significantly increased in Iba-1 stained area (d). Normalization of microglial activation at 24 weeks after rTBI as noted by increased ramification of Iba-1 stained cells (inset in (a)) and similar Iba-1 staining intensity as compared to sham operated mice (d). Each bar corresponds to one cortical field of view (FOV) arranged from left, contralateral (contra), to right, ipsilateral (ipsi). Data are shown as mean (+ SD). n = 8 (sham, 1 week and 4 week post rTBI, each) and n = 4 (24 week post rTBI) mice. Analyses are based on three coronal sections (corresponding to s1–s3 in Fig. 1). *P < 0.05. All analyses were done using one-way ANOVA on Ranks with post-hoc Dunn's
There was significant microglial activation in the cerebral cortex at 4 weeks after rTBI as indicated by the presence of bipolar/rod shaped Iba-1-stained microglia and overall increased Iba-1 staining (Fig. 4a, d). Activation subsided by 24 weeks as indicated by a more ramified appearance of Iba-1-stained cells and return of Iba-1 staining intensity to sham levels (Fig. 4a, d).
At 1 week after rTBI, neuronal density in the ipsilateral cortex was similar to sham operated mice (Fig. 4a, b). However, by 4 weeks after rTBI, we observed a significant loss of NeuN profiles, which worsened by 24 weeks indicative of progressive neurodegeneration (Fig. 4a, b). Loss of NeuN positive cells was most pronounced within the ipsilateral cerebral cortex adjacent to the impact site (rather than beneath the impact center) corresponding to the area of maximal p-Tau accumulation (compare with Fig. 1a).
At 1 week after rTBI, MBP and SMI-312 staining in the traumatized cerebral cortex appeared morphologically similar without significant difference in the quantified signal intensity as compared to sham operated mice (Fig. 5b). Concurrent with neuronal injury, we observed a significant loss of MBP and SMI-312 stained profiles indicating axonal degeneration by 4 weeks and 24 weeks after rTBI (Fig. 5c, d).

Axonal loss in cortex following repetitive traumatic brain injury (rTBI). (a) Cartoon depicting the 3 sections s1–s3 used for analysis. Black boxes indicate the location of the three fields of view (FOVs) used to quantify the immunofluorescent imaging signal for the myelin marker myelin basic protein (MBP) and the pan-axonal neurofilament marker SMI-312. (b) Representative photomicrographs showing progressive loss of the MBP and SMI-312 staining signal from 1 to 24 weeks after rTBI. Quantified staining signal for (c) MBP and (d) SMI-312. Data are mean ± SEM. n = 8 mice for sham, 1 week, and 4 weeks, n = 4 for 24 weeks. *P < 0.05. n.s. indicates not significant. All analyses were done using one-way ANOVA on Ranks with post-hoc Dunn's. Scale bar is 50 µm
Lastly, we found no βAPP and α-synuclein depositions in any of the operated mice (not shown).
rTBI causes long-term functional deficits
Sham operated mice regained their righting reflex within a median of 24 s (interquartile range 14–32 s) after discontinuation of anesthesia. In contrast, righting reflex was significantly suppressed in rTBI mice and only returned after a median of 135 s (interquartile range 79–229 s) (Fig. 6a).

Persistent neurological deficits in mice subjected to repetitive traumatic brain injury (rTBI). (a) Significantly prolonged time to the return of the righting reflex (RR) after discontinuation of anesthesia in mice subjected to repetitive TBI (rTBI) versus sham operated animals. There was as a significant group (P < 0.001) effect but no significant time (P = 0.958) effect or a group x time (P = 0.574) interaction (*P < 0.05, **P < 0.01, ***P < 0.001 versus sham). (b). There were significant group (P = 0.003) and time (P < 0.001) effects as well as presence of a significant group x time interaction (P < 0.001) for the composite neurological severity score (**P < 0.01, ***P < 0.001 versus baseline). (c) There were significant group and time effects as well as presence of a significant group x time interaction (P < 0.001, each) for the change in body weight during the first 4 weeks after surgery (*P < 0.05, **P < 0.01, ***P < 0.001 versus sham). Data are mean ± SEM. All statistical comparisons were made using mixed effects models (n = 8 for sham; n = 20 for baseline to 1 week after rTBI, n = 12 for 2 to 4 weeks after rTBI, and n = 4 for 24 weeks after rTBI). For clarity in the figure, post-hoc pairwise comparisons are only shown for significant differences between groups (a, c) and versus baseline (b)
We used the NSS, which is a composite of ratings measuring a combination of overall inquisitiveness, postural stability, and motor function, to examine the temporal evolution of functional deficits in rTBI animals versus controls. Whereas sham operated animals had no change in the NSS over the observation period, rTBI mice had significant neurological deficits at 2 h after the last impact (day 5) when compared to baseline and controls. Although deficits partially improved over time, persistent residual neurological deficits were present by 24 weeks (Fig. 6b).
Although both sham and rTBI mice lost weight after surgery, sham injured animals regained their baseline weight by 1 week whereas rTBI mice had a persistent weight loss up to 4 weeks after rTBI (Fig. 6c).
Systematic literature review
p-Tau pathology is infrequently reported in murine closed head models
From searches on both PubMed and Scopus, a total of 3997 articles were initially included. After removal of 241 duplicates 3756 articles were screened for inclusion and exclusion criteria (Additional file 1: Figure S7a). From this screening, we excluded 2394 articles because they did not include the predetermined keywords and 411 studies that did not use mice, leaving 951 articles included in the full text search. After full text review, we excluded studies that were not published in a peer-reviewed journal (n = 5); review articles (n = 235); conference proceeding without primary data (n = 30); studies using transgenic animals without wild-type mice (n = 106); lacking assessment of cerebral tau (n = 507); and penetrating head injury (n = 12). Lastly, two studies identified from references were added, resulting in a total of 58 studies (1.5% of screened publications) that investigated the presence of pathological tau in the brains of mice subjected to closed-head injury.
Characteristics of included studies reporting tau pathology assessment
Among the 58 included studies, 114 different injury paradigms were used (42 single TBI and 72 repetitive TBI models). Most models utilized male mice (112/114 [98%]), the C57BL/6 strain (111/114 [97%]), and anesthesia (111/114 [97%]). The median mouse age at the time of injury was 12 weeks (range 5 to 48 weeks). The most commonly used models were weight drop (n = 51), piston driven (n = 42), blast injury (n = 12), and rotational impact acceleration (n = 9) models.
Additional file 1: Figure S7b shows the primary methodologies used for p-Tau detection and the frequency of p-Tau detection. Of the included studies, 40 (68%) reported p-Tau pathology and 25 (44%) described p-Tau pathology by immunostaining (Additional file 1: Figure S7c-d). The most commonly used antibody to detect p-Tau was AT8 (8/25 [32%] of studies). Of the 25 studies that observed tau pathology, 22 found p-Tau in the cerebral cortex, hippocampus, or subcortical structures and 3 studies reported the presence of p-Tau in the optic tract (n = 2) and cerebellum (n = 1). Table 2 summarizes key characteristics of the 25 TBI models (15 repetitive TBI and 10 single TBI) that were associated with cortical or hippocampal p-Tau, as assessed by immunohistochemistry.
Presence of CTE-like pathology in mouse models reporting tau pathology
With respect to the presence of CTE-like pathology, only 3 studies (5.2% of included and 0.1% of all screened papers) reported perivascular p-Tau, a defining feature of human CTE. One of these studies specifically mentioned the presence of both neuronal and astroglial p-Tau (Additional file 1: Figure S7c). One additional study reported concurrent p-Tau pathology in both astroglia and neurons (located in the cortex and hippocampus) but did not report on potential perivascular location.
In terms of non-p-Tau related supportive histopathological CTE-like features, 3 studies reported the presence of cytoplasmatic localization of TDP-43 pathology (none specifically commented on pTDP-43) (Additional file 1: Figure S7c).
Forty-six (79%) of the included studies reported non-specific CTE-associated pathologies (Additional file 1: Figure S7d). Most commonly, these included neuronal loss (23%), axonal loss (43%), micro- and astrogliosis (50%, each), and beta amyloid deposition (29%) (Additional file 1: Figure S7c). Interestingly, no study reported cerebral microbleeds.
Discussion
Pathological hyperphosphorylation and aggregation of Tau protein is observed in a wide range of neurodegenerative disorders and is the key defining feature of a heterogeneous class of diseases called tauopathies. TBI has been identified as a strong risk factor for many of these tauopathies, highlighting the potential lifelong consequences of TBI exposure. Recently, CTE has been described as the prototypical TBI-associated tauopathy. Its diagnosis presently rests with the unique distribution of tau pathologies on a macroscopic and cellular level, yet little is known to what extent these pathologies are replicated by mouse closed-head TBI. To close this knowledge gap we described the pertinent histopathological features of human TBI-associated tauopathy in our rTBI model as well as by conducting a systematic review of the literature.
Our concussive mouse rTBI model showed p-Tau accumulation within the corpus callosum of the traumatized hemisphere as early as 1 week after injury with subsequent involvement of the cerebral cortex, mimicking the pathological tau progression described in human disease. Importantly, we found p-Tau in multiple cerebral locations that are frequently involved in human disease and are included in the CTE consensus criteria. These included, superficial cortical layers, the hippocampus, periventricular tissues, mamillary bodies, and perivascular locations [8, 46]. Nevertheless, we defined vessels based on morphology only and future studies should include staining for vascular markers to determine the specific association of p-Tau with the vascular compartment.
In this regard, it should be noted that human CTE criteria include the location of perivascular p-Tau at the depth of cerebral sulci [8, 46]. Because mice are lisencephalic, this criterion cannot be fulfilled in a murine model. However, we consistently observed p-Tau within the subpial cerebral cortex lining the superficial longitudinal fissure, as well as in the adjacent corpus callosum. This is consistent with computational models of the biomechanical forces predicting that cerebral areas with a change in morphology (such as at the site of sulci and the superior longitudinal fissure) represent locations of high stress and strain and thus greatest susceptibility to axonal and vascular injury [12, 16, 23].
Given the perivascular location of many neurodegenerative features it has been suggested that TBI triggers the neurodegenerative cascade by damaging the neurovascular unit. Indeed, consistent with human data [28, 39], we observed microvascular injury in our model, particularly in locations with p-Tau accumulation. Moreover, using IgG staining, we found evidence for BBB-disruption adding to the notion that BBB hyperpermeability is an important aspect of murine TBI-associated tauopathy. Nevertheless, IgG staining was faint and restricted to the optic tract. Injury to the visual pathway including BBB-disruption and p-Tau accumulation in the optic tract has been shown after murine closed head injury [3, 13, 14, 19]. Yet, we did not observe previously reported perivascular IgG staining in the cerebral cortex [69]. This is inconsistent with the observed microvascular injury as assessed by Prussian blue staining in our study. In this respect it is noteworthy that we used the cyclooxygenase-2 (COX-2) inhibitor carprofen as a post-operative analgesic. Carprofen has been shown to attenuate microglial activation, inflammation, and brain edema formation at therapeutic doses in mice, which may have attenuated BBB-disruption in our model [72]. Thus, while IgG-staining provided proof-of-principle that BBB-integrity was impaired in our model, further studies using different BBB-integrity markers as well as avoidance of anti-inflammatory drugs is required to better characterize the extent of BBB permeability and its relationship to p-Tau and other observed pathologies.
In addition to pathological p-Tau accumulation, several other proteinopathies have been described following human TBI. In particular, widespread TDP-43 inclusions in the neocortex of patients with CTE have been reported and the presence of TDP-43 pathology has been used to support the diagnosis of CTE [47, 49]. Nevertheless, TDP-43-related pathology is not specific to CTE. It has been described in a range of conditions, and colocalization of tau and TDP-43 is often limited. For this reason, it has yet to be determined exactly how TDP-43 aggregates coincide and interact with pathological p-Tau accumulation. Overall, few mouse studies have sought to evaluate TDP-43 pathology after closed-head TBI [1, 13, 73, 78]. Here, we found widespread TDP-43 expression following rTBI with a persistent presence of cytoplasmatic mislocalization by 6 months post rTBI. This is consistent with prior studies reporting persistent TDP-43 pathology in murine TBI associated with tauopathy [1, 13, 78]. We found long-term accumulation of pTDP-43 expression with associated nuclear loss and cytoplasmatic aggregation up to 24 weeks after rTBI. This is an important extension of previous studies that were limited to shorter (1 week) observation periods [55, 70, 73] and in light of reported transient alterations [55, 73]. Our observation that pathological TDP-43, but not tau, accumulation was present in the hippocampus adds to the notion that while these proteinopathies share a common pathophysiology, there may exist cell-specific susceptibilities [1, 68].
In contrast to pathological tau and TDP-43 accumulation after TBI, persistent α-synuclein and β-amyloid proteinopathy represent less consistent histopathological features after TBI [7, 8, 22, 38, 67]. Although several prior animal studies reported increased β-amyloid and α-synuclein after TBI, we did not find this present in our rTBI model [1, 13, 56, 62, 75, 78].
Mixed neuronal and astroglial tau pathology is considered a hallmark of CTE; as such, it is important to evaluate the precise cell types involved with TBI-associated tauopathy. For example, astrocyte activation is common after TBI, accompanies virtually all neurodegenerative tauopathies, and astrocytes may serve as a source for tau and thus could conceivably contribute to pathological tau accumulation [34, 41, 65, 69]. Yet, whether astroglial tau expression serves as a driver for injury-associated tauopathy remains uncertain [41]. Many groups hypothesize that astrocytes may promote neurodegeneration because astroglial tau pathology has been observed in the absence of neuronal tau inclusions, possibly through pro-inflammatory mechanisms [41]. We observed significant neuronal loss by 4 weeks after rTBI, which coincided with significant astroglial and microglial activation. However, we noted that neuronal p-Tau expression occurred prior to significant microglial and astroglial activation. Moreover, neuronal p-Tau expression was present earlier and progressed more widespread than astroglial p-Tau expression. Lastly, p-Tau-stained astrocytes were located in the superficial cortex, which had the greatest burden of neuronal p-Tau, but there were no p-Tau expressing astrocytes in deeper cortical layers and the corpus callosum. Together, these observations are consistent with the hypothesis that astroglial p-Tau expression after closed-head TBI is a secondary event rather than primary driver of tau pathology, possibly related to internalization of p-Tau into astrocytes from neighboring neurons and synapses [41].
To put our results in context with ongoing work in the field, we conducted a systematic review of the literature. Our analysis showed a striking paucity of studies that sought to determine tau pathology in experimental closed-head mouse TBI. Fewer than 2% of screened studies (58/3,756) sought to assess pathological tau accumulation. Nevertheless, of these studies, approximately 40% found evidence of p-Tau pathology by immunohistochemistry. Despite the overall scarcity of investigations, these and our observations provide cumulative evidence that murine closed head TBI replicates the critical histopathological aspects of human TBI-associated tau pathology with specific features of CTE. Reflecting human disease, pathology was seen across a wide variety of TBI-paradigms, highlighting that tau pathology after murine closed head TBI is reproducible and robust. This indicates that model differences could be leveraged to study the association of pathophysiological mechanisms with varying mechanics of injury [9] as well as the ability to control for possible confounders. For example, similar to other studies reporting p-Tau accumulation after murine closed-head rTBI [56, 62, 75, 78] we exposed the skull for precise impact delivery to the same coordinates across animals. Yet, this is inconsistent with the clinical situation, and our systematic review showed that this approach is not critical for inducing pertinent CTE-like neuropathological features.
Finally, though accumulation of p-Tau is a critical early event in the cascade leading to tauopathy, formation of small, soluble oligomeric tau species as well as the aggregation into larger insoluble filaments known as neurofibrillary tangles (NFTs), represent the hallmark of tauopathies including CTE [5, 30, 33]. There is strong evidence that pathogenic tau, sometimes referred to as a tau prion or prion-like tau, self-templates to progressively spread disease in tauopathy patients. In a sub-set of rTBI patients, the repeated injury gives rise to the formation of tau prions, which include a toxic species responsible for neuronal death. However, further studies are needed to determine if murine closed-head models are able to replicate the formation of tau prions in mice [24, 25, 33].
In addition to ongoing efforts to better understand the effects of TBI in wild-type mice, several groups have used transgenic mouse models expressing human tau to investigate the link between traumatic injury, tau misfolding, and p-Tau pathology. For example, the rTg4510 mouse model, which expresses a doxycycline-repressible isoform of tau containing the P301L mutation [59] developed elevated p-Tau levels in the cortex 7 days after either a single or double closed-head injury [6]. Investigating the longer-term effects of TBI on tau aggregation in Tg mice, others have used the PS19 mouse model [77], which expresses human tau with the P301S mutation, and performed a semi-quantitative analysis of p-Tau pathology up to 7 months post-injury [71]. While these studies have helped address several questions about the link between TBI and tau spreading, there are also important caveats to consider with regard to these mouse models. A careful analysis of the transgene insertion sites in the rTg4510 model revealed that disruption of essential genes is at least partially responsible for the p-Tau pathology that results from overexpressing the human protein [21]. And extensive variability in the PS19 model, with regard to both the rate of tau spreading and p-Tau formation [74], limits the interpretations that can be drawn from studies using the mouse line.
In conclusion, our study highlights the translational value of murine closed skull TBI to replicate the pertinent aspects of human TBI-associated tauopathy with a wide range of related histopathological features.
Data availability
Data supporting the findings of this study are available from the corresponding author upon reasonable request.
References
Albayram O, Kondo A, Mannix R, Smith C, Tsai CY, Li C, Herbert MK, Qiu J, Monuteaux M, Driver J et al (2017) Cis P-tau is induced in clinical and preclinical brain injury and contributes to post-injury sequelae. Nat Commun 8:1000. https://doi.org/10.1038/s41467-017-01068-4
Albayram O, MacIver B, Mathai J, Verstegen A, Baxley S, Qiu C, Bell C, Caldarone BJ, Zhou XZ, Lu KP et al (2019) Traumatic Brain Injury-related voiding dysfunction in mice is caused by damage to rostral pathways, altering inputs to the reflex pathways. Sci Rep 9:8646. https://doi.org/10.1038/s41598-019-45234-8
Angoa-Perez M, Zagorac B, Anneken JH, Briggs DI, Winters AD, Greenberg JM, Ahmad M, Theis KR, Kuhn DM (2020) Repetitive, mild traumatic brain injury results in a progressive white matter pathology, cognitive deterioration, and a transient gut microbiota dysbiosis. Sci Rep 10:8949. https://doi.org/10.1038/s41598-020-65972-4
Anonymous (1999) Consensus conference. Rehabilitation of persons with traumatic brain injury. NIH consensus development panel on rehabilitation of persons with traumatic brain injury. JAMA 282: 974-983
Arena JD, Smith DH, Lee EB, Gibbons GS, Irwin DJ, Robinson JL, Lee VM, Trojanowski JQ, Stewart W, Johnson VE (2020) Tau immunophenotypes in chronic traumatic encephalopathy recapitulate those of ageing and Alzheimer’s disease. Brain 143:1572–1587. https://doi.org/10.1093/brain/awaa071
Bachstetter AD, Morganti JM, Bodnar CN, Webster SJ, Higgins EK, Roberts KN, Snider H, Meier SE, Nation GK, Goulding DS et al (2020) The effects of mild closed head injuries on tauopathy and cognitive deficits in rodents: Primary results in wild type and rTg4510 mice, and a systematic review. Exp Neurol 326:113180. https://doi.org/10.1016/j.expneurol.2020.113180
Baugh CM, Stamm JM, Riley DO, Gavett BE, Shenton ME, Lin A, Nowinski CJ, Cantu RC, McKee AC, Stern RA (2012) Chronic traumatic encephalopathy: neurodegeneration following repetitive concussive and subconcussive brain trauma. Brain Imaging Behav 6:244–254. https://doi.org/10.1007/s11682-012-9164-5
Bieniek KF, Cairns NJ, Crary JF, Dickson DW, Folkerth RD, Keene CD, Litvan I, Perl DP, Stein TD, Vonsattel JP et al (2021) The second NINDS/NIBIB consensus meeting to define neuropathological criteria for the diagnosis of chronic traumatic encephalopathy. J Neuropathol Exp Neurol 80:210–219. https://doi.org/10.1093/jnen/nlab001
Bodnar CN, Roberts KN, Higgins EK, Bachstetter AD (2019) A systematic review of closed head injury models of mild traumatic brain injury in mice and rats. J Neurotrauma 36:1683–1706. https://doi.org/10.1089/neu.2018.6127
Bolton-Hall AN, Hubbard WB, Saatman KE (2019) Experimental designs for repeated mild traumatic brain injury: challenges and considerations. J Neurotrauma 36:1203–1221. https://doi.org/10.1089/neu.2018.6096
Bouley J, Chung DY, Ayata C, Brown RH Jr, Henninger N (2019) Cortical spreading depression denotes concussion injury. J Neurotrauma 36:1008–1017. https://doi.org/10.1089/neu.2018.5844
Bradshaw DR, Ivarsson J, Morfey CL, Viano DC (2001) Simulation of acute subdural hematoma and diffuse axonal injury in coronal head impact. J Biomech 34:85–94. https://doi.org/10.1016/s0021-9290(00)00135-4
Briggs DI, Angoa-Perez M, Kuhn DM (2016) Prolonged repetitive head trauma induces a singular chronic traumatic encephalopathy-like pathology in white matter despite transient behavioral abnormalities. Am J Pathol 186:2869–2886. https://doi.org/10.1016/j.ajpath.2016.07.013
Cansler SM, Guilhaume-Correa F, Day D, Bedolla A, Evanson NK Indirect traumatic optic neuropathy after head trauma in adolescent male mice is associated with behavioral visual impairment, neurodegeneration, and elevated endoplasmic reticulum stress markers at acute and subacute times. BioRxiv [Preprint] June 12, 2020 (Cited 06/01/2021). https://doi.org/10.1101/2020.06.11.144766
Cheng WH, Martens KM, Bashir A, Cheung H, Stukas S, Gibbs E, Namjoshi DR, Button EB, Wilkinson A, Barron CJ et al (2019) CHIMERA repetitive mild traumatic brain injury induces chronic behavioural and neuropathological phenotypes in wild-type and APP/PS1 mice. Alzheimers Res Ther 11:6. https://doi.org/10.1186/s13195-018-0461-0
Cloots RJ, Gervaise HM, van Dommelen JA, Geers MG (2008) Biomechanics of traumatic brain injury: influences of the morphologic heterogeneities of the cerebral cortex. Ann Biomed Eng 36:1203–1215. https://doi.org/10.1007/s10439-008-9510-3
Crane PK, Gibbons LE, Dams-O’Connor K, Trittschuh E, Leverenz JB, Keene CD, Sonnen J, Montine TJ, Bennett DA, Leurgans S et al (2016) Association of traumatic brain injury with late-life neurodegenerative conditions and neuropathologic findings. JAMA Neurol 73:1062–1069. https://doi.org/10.1001/jamaneurol.2016.1948
DeWitt DS, Hawkins BE, Dixon CE, Kochanek PM, Armstead W, Bass CR, Bramlett HM, Buki A, Dietrich WD, Ferguson AR et al (2018) Pre-clinical testing of therapies for traumatic brain injury. J Neurotrauma 35:2737–2754. https://doi.org/10.1089/neu.2018.5778
Evanson NK, Guilhaume-Correa F, Herman JP, Goodman MD (2018) Optic tract injury after closed head traumatic brain injury in mice: A model of indirect traumatic optic neuropathy. PLoS ONE 13:e0197346. https://doi.org/10.1371/journal.pone.0197346
Fleminger S, Ponsford J (2005) Long term outcome after traumatic brain injury. BMJ 331:1419–1420. https://doi.org/10.1136/bmj.331.7530.1419
Gamache J, Benzow K, Forster C, Kemper L, Hlynialuk C, Furrow E, Ashe KH, Koob MD (2019) Factors other than hTau overexpression that contribute to tauopathy-like phenotype in rTg4510 mice. Nat Commun 10:2479. https://doi.org/10.1038/s41467-019-10428-1
Gavett BE, Stern RA, McKee AC (2011) Chronic traumatic encephalopathy: a potential late effect of sport-related concussive and subconcussive head trauma. Clin Sports Med 30:179–188. https://doi.org/10.1016/j.csm.2010.09.007
Ghajari M, Hellyer PJ, Sharp DJ (2017) Computational modelling of traumatic brain injury predicts the location of chronic traumatic encephalopathy pathology. Brain 140:333–343. https://doi.org/10.1093/brain/aww317
Goedert M, Eisenberg DS, Crowther RA (2017) Propagation of tau aggregates and neurodegeneration. Annu Rev Neurosci 40:189–210. https://doi.org/10.1146/annurev-neuro-072116-031153
Goedert M, Spillantini MG (2017) Propagation of tau aggregates. Mol. Brain 10:18. https://doi.org/10.1186/s13041-017-0298-7
Goldstein LE, Fisher AM, Tagge CA, Zhang XL, Velisek L, Sullivan JA, Upreti C, Kracht JM, Ericsson M, Wojnarowicz MW et al (2012) Chronic traumatic encephalopathy in blast-exposed military veterans and a blast neurotrauma mouse model. Sci Transl Med 4:134–160. https://doi.org/10.1126/scitranslmed.3003716
Graham NS, Sharp DJ (2019) Understanding neurodegeneration after traumatic brain injury: from mechanisms to clinical trials in dementia. J Neurol Neurosurg Psychiatry 90:1221–1233. https://doi.org/10.1136/jnnp-2017-317557
Griffin AD, Turtzo LC, Parikh GY, Tolpygo A, Lodato Z, Moses AD, Nair G, Perl DP, Edwards NA, Dardzinski BJ et al (2019) Traumatic microbleeds suggest vascular injury and predict disability in traumatic brain injury. Brain 142:3550–3564. https://doi.org/10.1093/brain/awz290
Guo Z, Cupples LA, Kurz A, Auerbach SH, Volicer L, Chui H, Green RC, Sadovnick AD, Duara R, DeCarli C et al (2000) Head injury and the risk of AD in the MIRAGE study. Neurology 54:1316–1323. https://doi.org/10.1212/wnl.54.6.1316
Gyparaki MT, Arab A, Sorokina EM, Santiago-Ruiz AN, Bohrer CH, Xiao J, Lakadamyali M (2021) Tau forms oligomeric complexes on microtubules that are distinct from tau aggregates. Proc Natl Acad Sci U S A. https://doi.org/10.1073/pnas.2021461118
Henninger N, Bouley J, Sikoglu EM, An J, Moore CM, King JA, Bowser R, Freeman MR, Brown RH Jr (2016) Attenuated traumatic axonal injury and improved functional outcome after traumatic brain injury in mice lacking Sarm1. Brain 139:1094–1105. https://doi.org/10.1093/brain/aww001
Hernandez F, Merchan-Rubira J, Valles-Saiz L, Rodriguez-Matellan A, Avila J (2020) Differences between human and murine tau at the N-terminal end. Front Aging Neurosci 12:11. https://doi.org/10.3389/fnagi.2020.00011
Holtzman DM, Carrillo MC, Hendrix JA, Bain LJ, Catafau AM, Gault LM, Goedert M, Mandelkow E, Mandelkow EM, Miller DS et al (2016) Tau: From research to clinical development. Alzheimers Dement 12:1033–1039. https://doi.org/10.1016/j.jalz.2016.03.018
Hsu ET, Gangolli M, Su S, Holleran L, Stein TD, Alvarez VE, McKee AC, Schmidt RE, Brody DL (2018) Astrocytic degeneration in chronic traumatic encephalopathy. Acta Neuropathol 136:955–972. https://doi.org/10.1007/s00401-018-1902-3
Huber BR, Meabon JS, Martin TJ, Mourad PD, Bennett R, Kraemer BC, Cernak I, Petrie EC, Emery MJ, Swenson ER et al (2013) Blast exposure causes early and persistent aberrant phospho- and cleaved-tau expression in a murine model of mild blast-induced traumatic brain injury. J Alzheimers Dis 37:309–323. https://doi.org/10.3233/JAD-130182
Hyder AA, Wunderlich CA, Puvanachandra P, Gururaj G, Kobusingye OC (2007) The impact of traumatic brain injuries: a global perspective. NeuroRehabilitation 22:341–353
Iliff JJ, Chen MJ, Plog BA, Zeppenfeld DM, Soltero M, Yang L, Singh I, Deane R, Nedergaard M (2014) Impairment of glymphatic pathway function promotes tau pathology after traumatic brain injury. J Neurosci 34:16180–16193. https://doi.org/10.1523/JNEUROSCI.3020-14.2014
Iverson GL, Gardner AJ, McCrory P, Zafonte R, Castellani RJ (2015) A critical review of chronic traumatic encephalopathy. Neurosci Biobehav Rev 56:276–293. https://doi.org/10.1016/j.neubiorev.2015.05.008
Kenney K, Amyot F, Haber M, Pronger A, Bogoslovsky T, Moore C, Diaz-Arrastia R (2016) Cerebral vascular injury in traumatic brain injury. Exp Neurol 275(Pt 3):353–366. https://doi.org/10.1016/j.expneurol.2015.05.019
Kondo A, Shahpasand K, Mannix R, Qiu J, Moncaster J, Chen CH, Yao Y, Lin YM, Driver JA, Sun Y et al (2015) Antibody against early driver of neurodegeneration cis P-tau blocks brain injury and tauopathy. Nature 523:431–436. https://doi.org/10.1038/nature14658
Kovacs GG (2020) Astroglia and Tau: new perspectives. Front Aging Neurosci 12:96. https://doi.org/10.3389/fnagi.2020.00096
Liu B, Cao Y, Shi F, Wang L, Li N, Cheng X, Du J, Tian Q, Zhou X (2020) The overexpression of RBM3 alleviates TBI-induced behaviour impairment and AD-like tauopathy in mice. J Cell Mol Med 24:9176–9188. https://doi.org/10.1111/jcmm.15555
Logsdon AF, Lucke-Wold BP, Turner RC, Li X, Adkins CE, Mohammad AS, Huber JD, Rosen CL, Lockman PR (2017) A mouse model of focal vascular injury induces astrocyte reactivity, tau oligomers, and aberrant behavior. Arch Neurosci. https://doi.org/10.5812/archneurosci.44254
Luo J, Nguyen A, Villeda S, Zhang H, Ding Z, Lindsey D, Bieri G, Castellano JM, Beaupre GS, Wyss-Coray T (2014) Long-term cognitive impairments and pathological alterations in a mouse model of repetitive mild traumatic brain injury. Front Neurol 5:12. https://doi.org/10.3389/fneur.2014.00012
McKee AC, Abdolmohammadi B, Stein TD (2018) The neuropathology of chronic traumatic encephalopathy. Handb Clin Neurol 158:297–307. https://doi.org/10.1016/B978-0-444-63954-7.00028-8
McKee AC, Cairns NJ, Dickson DW, Folkerth RD, Keene CD, Litvan I, Perl DP, Stein TD, Vonsattel JP, Stewart W et al (2016) The first NINDS/NIBIB consensus meeting to define neuropathological criteria for the diagnosis of chronic traumatic encephalopathy. Acta Neuropathol 131:75–86. https://doi.org/10.1007/s00401-015-1515-z
McKee AC, Gavett BE, Stern RA, Nowinski CJ, Cantu RC, Kowall NW, Perl DP, Hedley-Whyte ET, Price B, Sullivan C et al (2010) TDP-43 proteinopathy and motor neuron disease in chronic traumatic encephalopathy. J Neuropathol Exp Neurol 69:918–929. https://doi.org/10.1097/NEN.0b013e3181ee7d85
McKee AC, Robinson ME (2014) Military-related traumatic brain injury and neurodegeneration. Alzheimers Dement 10:S242-253. https://doi.org/10.1016/j.jalz.2014.04.003
Mez J, Daneshvar DH, Kiernan PT, Abdolmohammadi B, Alvarez VE, Huber BR, Alosco ML, Solomon TM, Nowinski CJ, McHale L et al (2017) Clinicopathological evaluation of chronic traumatic encephalopathy in players of american football. JAMA 318:360–370. https://doi.org/10.1001/jama.2017.8334
Niziolek GM, Boudreau RM, Baker J, Friend LA, Makley AT, Edwards MJ, Gulbins E, Goodman MD (2020) Acid sphingomyelinase inhibition mitigates histopathological and behavioral changes in a murine model of traumatic brain injury. J Neurotrauma 37:1902–1909. https://doi.org/10.1089/neu.2019.6436
Niziolek GM, Hoehn RS, Seitz AP, Jernigan PL, Makley AT, Gulbins E, Edwards MJ, Goodman MD (2021) The role of acid sphingomyelinase inhibition in repetitive mild traumatic brain injury. J Surg Res 259:296–304. https://doi.org/10.1016/j.jss.2020.09.034
Paxinos G, Franklin KBJ (2013) The mouse brain in stereotaxic coordinates. Elsevier Academic Press, Cambridge
Peeters W, van den Brande R, Polinder S, Brazinova A, Steyerberg EW, Lingsma HF, Maas AI (2015) Epidemiology of traumatic brain injury in Europe. Acta Neurochir (Wien) 157:1683–1696. https://doi.org/10.1007/s00701-015-2512-7
Petraglia AL, Plog BA, Dayawansa S, Dashnaw ML, Czerniecka K, Walker CT, Chen M, Hyrien O, Iliff JJ, Deane R et al (2014) The pathophysiology underlying repetitive mild traumatic brain injury in a novel mouse model of chronic traumatic encephalopathy. Surg Neurol Int 5:184. https://doi.org/10.4103/2152-7806.147566
Rajic Bumber J, Pilipovic K, Jankovic T, Dolenec P, Grzeta N, Kriz J, Zupan G (2021) Repetitive traumatic brain injury is associated with TDP-43 alterations, neurodegeneration, and glial activation in mice. J Neuropathol Exp Neurol 80:2–14. https://doi.org/10.1093/jnen/nlaa130
Rehman SU, Ahmad A, Yoon GH, Khan M, Abid MN, Kim MO (2018) Inhibition of c-Jun N-terminal kinase protects against brain damage and improves learning and memory after traumatic brain injury in adult mice. Cereb Cortex 28:2854–2872. https://doi.org/10.1093/cercor/bhx164
Sabbagh JJ, Fontaine SN, Shelton LB, Blair LJ, Hunt JB Jr, Zhang B, Gutmann JM, Lee DC, Lloyd JD, Dickey CA (2016) Noncontact rotational head injury produces transient cognitive deficits but lasting neuropathological changes. J Neurotrauma 33:1751–1760. https://doi.org/10.1089/neu.2015.4288
Sacramento CB, Sondhi D, Rosenberg JB, Chen A, Giordano S, Pey E, Lee V, Stiles KM, Havlicek DF, Leopold PL et al (2020) Anti-phospho-tau gene therapy for chronic traumatic encephalopathy. Hum Gene Ther 31:57–69. https://doi.org/10.1089/hum.2019.174
Santacruz K, Lewis J, Spires T, Paulson J, Kotilinek L, Ingelsson M, Guimaraes A, DeTure M, Ramsden M, McGowan E et al (2005) Tau suppression in a neurodegenerative mouse model improves memory function. Science 309:476–481. https://doi.org/10.1126/science.1113694
Schneider CA, Rasband WS, Eliceiri KW (2012) NIH Image to ImageJ: 25 years of image analysis. Nat Methods 9:671–675. https://doi.org/10.1038/nmeth.2089
Schneider J, Jeon S, Gladman JT, Corriveau RA ADRD Summit 2019 Report to the National Advisory Neurological Disorders and Stroke Council. https://www.ninds.nih.gov/News-Events/Events-Proceedings/Events/Alzheimers-Disease-Related-Dementias-Summit-2019 accessed 05/13/2020
Selvaraj P, Wen J, Tanaka M, Zhang Y (2019) Therapeutic effect of a novel fatty acid amide hydrolase inhibitor PF04457845 in the repetitive closed head injury mouse model. J Neurotrauma 36:1655–1669. https://doi.org/10.1089/neu.2018.6226
Seo JS, Lee S, Shin JY, Hwang YJ, Cho H, Yoo SK, Kim Y, Lim S, Kim YK, Hwang EM et al (2017) Transcriptome analyses of chronic traumatic encephalopathy show alterations in protein phosphatase expression associated with tauopathy. Exp Mol Med 49:e333. https://doi.org/10.1038/emm.2017.56
Shin N, Kim HG, Shin HJ, Kim S, Kwon HH, Baek H, Yi MH, Zhang E, Kim JJ, Hong J et al (2019) Uncoupled endothelial nitric oxide synthase enhances p-Tau in chronic traumatic encephalopathy mouse model. Antioxid Redox Signal 30:1601–1620. https://doi.org/10.1089/ars.2017.7280
Sidoryk-Wegrzynowicz M, Struzynska L (2019) Astroglial contribution to tau-dependent neurodegeneration. Biochem J 476:3493–3504. https://doi.org/10.1042/BCJ20190506
Smith DH, Dolle JP, Ameen-Ali KE, Bretzin A, Cortes E, Crary JF, Dams-O’Connor K, Diaz-Arrastia R, Edlow BL, Folkerth R et al (2021) Collaborative neuropathology network characterizing outcomes of TBI (CONNECT-TBI). Acta Neuropathol Commun 9:32. https://doi.org/10.1186/s40478-021-01122-9
Smith DH, Johnson VE, Stewart W (2013) Chronic neuropathologies of single and repetitive TBI: substrates of dementia? Nat Rev Neurol 9:211–221. https://doi.org/10.1038/nrneurol.2013.29
Smith VD, Bachstetter AD, Ighodaro E, Roberts K, Abner EL, Fardo DW, Nelson PT (2018) Overlapping but distinct TDP-43 and tau pathologic patterns in aged hippocampi. Brain Pathol 28:264–273. https://doi.org/10.1111/bpa.12505
Tagge CA, Fisher AM, Minaeva OV, Gaudreau-Balderrama A, Moncaster JA, Zhang XL, Wojnarowicz MW, Casey N, Lu H, Kokiko-Cochran ON et al (2018) Concussion, microvascular injury, and early tauopathy in young athletes after impact head injury and an impact concussion mouse model. Brain 141:422–458. https://doi.org/10.1093/brain/awx350
Tan XL, Sun M, Brady RD, Liu S, Llanos R, Cheung S, Wright DK, Casillas-Espinosa PM, Sashindranath M, O'Brien TJ et al (2019) Transactive Response DNA-Binding Protein 43 Abnormalities after Traumatic Brain Injury. J Neurotrauma. 36:87–99. https://doi.org/10.1089/neu.2017.5491
Tang SJ, Fesharaki-Zadeh A, Takahashi H, Nies SH, Smith LM, Luo A, Chyung A, Chiasseu M, Strittmatter SM (2020) Fyn kinase inhibition reduces protein aggregation, increases synapse density and improves memory in transgenic and traumatic Tauopathy. Acta Neuropathol Commun 8:96. https://doi.org/10.1186/s40478-020-00976-9
Thau-Zuchman O, Shohami E, Alexandrovich AG, Trembovler V, Leker RR (2012) The anti-inflammatory drug carprofen improves long-term outcome and induces gliogenesis after traumatic brain injury. J Neurotrauma 29:375–384. https://doi.org/10.1089/neu.2010.1673
Wiesner D, Tar L, Linkus B, Chandrasekar A, Olde Heuvel F, Dupuis L, Tsao W, Wong PC, Ludolph A, Roselli F (2018) Reversible induction of TDP-43 granules in cortical neurons after traumatic injury. Exp Neurol 299:15–25. https://doi.org/10.1016/j.expneurol.2017.09.011
Woerman AL, Patel S, Kazmi SA, Oehler A, Freyman Y, Espiritu L, Cotter R, Castaneda JA, Olson SH, Prusiner SB (2017) Kinetics of human mutant tau prion formation in the brains of 2 transgenic mouse lines. JAMA Neurol 74:1464–1472. https://doi.org/10.1001/jamaneurol.2017.2822
Xu X, Cowan M, Beraldo F, Schranz A, McCunn P, Geremia N, Brown Z, Patel M, Nygard KL, Khazaee R et al (2021) Repetitive mild traumatic brain injury in mice triggers a slowly developing cascade of long-term and persistent behavioral deficits and pathological changes. Acta Neuropathol Commun 9:60. https://doi.org/10.1186/s40478-021-01161-2
Yang Z, Wang P, Morgan D, Lin D, Pan J, Lin F, Strang KH, Selig TM, Perez PD, Febo M et al (2015) Temporal MRI characterization, neurobiochemical and neurobehavioral changes in a mouse repetitive concussive head injury model. Sci Rep 5:11178. https://doi.org/10.1038/srep11178
Yoshiyama Y, Higuchi M, Zhang B, Huang SM, Iwata N, Saido TC, Maeda J, Suhara T, Trojanowski JQ, Lee VM (2007) Synapse loss and microglial activation precede tangles in a P301S tauopathy mouse model. Neuron 53:337–351. https://doi.org/10.1016/j.neuron.2007.01.010
Zhang J, Teng Z, Song Y, Hu M, Chen C (2015) Inhibition of monoacylglycerol lipase prevents chronic traumatic encephalopathy-like neuropathology in a mouse model of repetitive mild closed head injury. J Cereb Blood Flow Metab 35:706. https://doi.org/10.1038/jcbfm.2014.247
Funding
This study was supported by institutional Grants, National Institutes of Health Grant K08NS091499 to N.H.
Author information
Authors and Affiliations
Contributions
All authors contributed to the study conception and design. A.K. conducted histological preparations, histological analysis, data analyses. J.B. conducted animal surgery, behavioral testing, histological preparations, and genotyping. N.H. conceived and designed the study, conducted data analyses, animal surgery, behavioral testing, histological preparations, and wrote the paper. All authors discussed the results, commented on the manuscript for important intellectual content, read, and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval
All animal procedures were approved by the University of Massachusetts Medical School Institutional Animal Care and Use Committee. This manuscript was prepared in adherence to the ARRIVE and PRISMA guidelines (Additional file 1: Supplementary information).
Competing interests
The authors declare no competing financial interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1
. Supplementary information.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Kahriman, A., Bouley, J., Smith, T.W. et al. Mouse closed head traumatic brain injury replicates the histological tau pathology pattern of human disease: characterization of a novel model and systematic review of the literature. acta neuropathol commun 9, 118 (2021). https://doi.org/10.1186/s40478-021-01220-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40478-021-01220-8