
Abstract
Iron is essential for neurons and glial cells, playing key roles in neurotransmitter synthesis, energy production and myelination. In contrast, high concentrations of free iron can be detrimental and contribute to neurodegeneration, through promotion of oxidative stress. Particularly in Parkinson’s disease (PD) changes in iron concentrations in the substantia nigra (SN) was suggested to play a key role in degeneration of dopaminergic neurons in nigrosome 1. However, the cellular iron pathways and the mechanisms of the pathogenic role of iron in PD are not well understood, mainly due to the lack of quantitative analytical techniques for iron quantification with subcellular resolution. Here, we quantified cellular iron concentrations and subcellular iron distributions in dopaminergic neurons and different types of glial cells in the SN both in brains of PD patients and in non-neurodegenerative control brains (Co). To this end, we combined spatially resolved quantitative element mapping using micro particle induced X-ray emission (µPIXE) with nickel-enhanced immunocytochemical detection of cell type-specific antigens allowing to allocate element-related signals to specific cell types. Distinct patterns of iron accumulation were observed across different cell populations. In the control (Co) SNc, oligodendroglial and astroglial cells hold the highest cellular iron concentration whereas in PD, the iron concentration was increased in most cell types in the substantia nigra except for astroglial cells and ferritin-positive oligodendroglial cells. While iron levels in astroglial cells remain unchanged, ferritin in oligodendroglial cells seems to be depleted by almost half in PD. The highest cellular iron levels in neurons were located in the cytoplasm, which might increase the source of non-chelated Fe3+, implicating a critical increase in the labile iron pool. Indeed, neuromelanin is characterised by a significantly higher loading of iron including most probable the occupancy of low-affinity iron binding sites. Quantitative trace element analysis is essential to characterise iron in oxidative processes in PD. The quantification of iron provides deeper insights into changes of cellular iron levels in PD and may contribute to the research in iron-chelating disease-modifying drugs.
Similar content being viewed by others


Introduction
Iron is essential for a proper CNS function. It plays an important role as cofactor of numerous enzymes and is involved in ATP production, myelination and synthesis of DNA, RNA, proteins, and neurotransmitters.
In the brain, variations in iron levels correlate with its structural integrity [42], and there is no other organ but the CNS that is in such a constant need for readily available iron [85]. Any mismatch in the demand and regional-temporal distribution of iron may result in neurological and/or mental dysfunction. Iron deficiency, for example, is a well-established cause for impaired motor and cognitive development [2, 70, 83, 95, 101]. On the other hand, increased levels of iron are harmful and iron accumulations are typical hallmarks of brain ageing and several neurodegenerative disorders particularly PD [34, 37, 38, 44, 72, 85, 91, 98]. Various factors have been suggested to account for increased iron accumulation in the SN of patients with PD such as, for example, dysfunction of the blood–brain barrier, altered cellular iron transport, an increased pro-inflammatory state and mutations in genes of iron transport, storage and binding [10, 13, 27, 53]. However, all these processes will differently affect different cell populations.
Still, the local accumulation of iron in the SN in patients with PD remains a controversial issue. Numerous studies on post mortem tissue report on an increased amount of total iron in the SN [22, 26, 51, 84] also supported by large body of in vivo findings from Magnetic Resonance Imaging (MRI) [9, 16, 46,47,48, 69, 82]. The increasing importance and approaches for in vivo brain iron assessment using multiparametric MRI is featured in a review by Moeller et al. [58]. Quantitative MRI may provide useful biomarkers for brain integrity assessment in iron-related neurodegeneration. Particularly, a prominent change in iron-sensitive T2* MRI contrast within the sub areas of the SN overlapping with nigrosome 1 were shown to be a hallmark of PD with high diagnostic power. Other studies failed to detect any disease-related differences [33, 49, 87]. Of note, more in-depth knowledge on whether changes in intracellular iron pools preferentially affect neurons or glial cells is limited by the lack of quantitative analytical techniques with sufficient spatial resolution for in-situ cellular analysis.
To glean much needed information about cellular and subcellular compartmentalisation of iron in health and disease, we have developed a powerful analytical approach, which we call immuno-micro particle induced X-ray emission (µPIXE). It allows for quantitative determining the iron concentrations of specific target structures in tissue sections with a spatial resolution in the µm range. To this end, quantitative element mapping using µPIXE was combined with nickel-enhanced immunocytochemical detection of cell type-specific antigens to specifically allocate element-related signals to specific cell types. In the present study, this method was applied to quantify the cell specific iron concentrations in SN nigrosome 1 in controls and PD.
Materials and methods
Tissue preparation
Midbrains comprising the nigrosome 1 of the SNc were obtained from individuals of both sexes with no signs of neuropathological alterations (8 cases; healthy controls, HC; mean age 66 ± 16 years) and from patients who died with a clinical diagnosis of idiopathic Parkinson’s disease (8 cases; PD; mean age 75 ± 7 years) (for detailed case profile see Table 1). The definite diagnosis of PD has been verified by neurohistopathological examination based on a severe loss of neuromelanin containing neurons in the nigrosome 1 of the SNc, the presence of extraneuronal melanin and Lewy bodies (Table 2). Although the appearance of Lewy bodies might be also a hallmark of dementia with Lewy bodies (DLB), a detailed synopsis of neuropathological examination and clinical presentation of the PD individuals included in our experimental analysis allowed a strict inclusion of patients that suffered exclusively from PD and did not present any comorbidity of dementia. Brains were fixed by immersion in 4% formaldehyde in 0.1% phosphate buffered saline (PBS, pH 7.4). Small blocks (25 mm × 25 mm) comprising the substantia nigra were dissected and embedded in Histowax following conventional protocols for paraffin embedding and block mounting. Subsequently, the tissue was sectioned (10 µm) and attached to microscope glass slides. Transverse sections throughout the midbrain were cut using a microtome (Jung Histoslide 2000, Leica).
Brains were provided by the Brain Banking Centre Leipzig of the German Brain-Net (GZ 01GI9999-01GI0299), operated by the Paul Flechsig Institute of Brain Research. The entire procedure of case recruitment, acquisition of the patients’ personal data, the protocols and the informed consent forms, performing the autopsy and handling the autopsy material have been approved by the responsible authorities (Approval # 282-02 and Approval # 205/17-ek).
Identification of anatomical regions and applied nomenclature
Anatomical regions were identified on Nissl- and anti-HuCD-stained sections using the atlas of the human brain by Mai et al. [50]. Delineations and nomenclature of substructures of SN followed the detailed anatomical descriptions of the SN given by Braak and Braak [11], Fearnley and Lees [49], van Domburg and ten Donkelaar [88], McRitchie et al. [54] and Damier et al. [17, 18].
Histochemistry
Histowax sections were deparaffinised, rehydrated in a descending alcohol series and incubated for 2 h at 37 °C in a mixture of 5% potassium ferrocyanide and 5% hydrochloric acid (Perls' stain). Subsequently, sections were rinsed in PBS (pH 7.4) and pre-incubated for 10 min in 0.5 mg/ml 3,3′-diaminobenzidine (DAB, Sigma) Tris–HCl. Subsequently, an incubation in DAB for 15 min at room temperature (0.5 mg DAB/ml Tris–HCl and 0.05% H2O2) was performed (DAB amplification of Perls’ stain). After further washing the sections successively in Tris–HCl (pH 8.0), PBS and purified water, sections were dehydrated, incubated in toluol and coverslipped with Entellan (Merck Millipore).
Immunohistochemistry
For immunohistochemistry, sections were deparaffinised in xylene, rehydrated in a descending alcohol series and transferred to PBS (pH 7.4). To block endogenous peroxidase-activity, samples were incubated in 60% methanol containing 2% H2O2 (for 1 h at room temperature). Antigen retrieval pre-treatment prior to the staining process was performed as follows. Sections prepared for detection of CNP, IBA-1 and Olig2 were pre-treated with citric acid sodium citrate buffer (pH 6.0) and sections prepared for the detection of Hu C/D were pre-treated with Tris–HCl (pH 8.0), respectively for 20 min at 90 °C. After washing, sections were incubated overnight at room temperature in a solution (phosphate-buffered saline with 2% BSA, 0.3% milk powder and 0.5% donkey serum) containing the following primary antibodies: (1) mouse anti-human neuronal protein HuCD (1:400, Thermo Fisher Scientific) for neurons, (2) rabbit anti IBA-1 (1:800, Wako) for microglial cells, (3) rabbit anti-GFAP (1:500, Dako) for astroglial cells, (4) rabbit anti-Olig2 (1:100, Immuno Biological Laboratories) for oligodendroglial cells, (5) mouse anti-CNP (1:300, BioLegend) for oligodendroglial cells, (6) goat anti-ferritin heavy chain Y-16 (1:200, Santa Cruz Biotechnology) for ferritin and (7) rat anti-myelin basic protein (1:400, abcam) for myelin.
Subsequently, sections were rinsed in PBS-Tween (pH 7.4) and incubated in biotinylated secondary antibody solution (containing a mixture of PBS-T and phosphate-buffered saline with 2% BSA, 0.3% milk powder and 0.5% donkey serum, 2:1, for 1 h at room temperature) using donkey anti-mouse IgG, donkey anti-rat IgG, donkey anti-rabbit IgG and donkey anti-goat IgG (1:1000, Dianova, Germany). Binding sides were revealed by incubation for 1 h with peroxidase-labeled streptavidin (Extravidin®, 1:2000, Sigma Aldrich). Further, sections were rinsed in PBS-T and Tris–HCl (pH 8.0). Following treatment with peroxidase complex, the colour reaction was developed using DAB and nickel-ammonium sulphate (Sigma Aldrich, 99.999% purity grade). Finally, sections were rinsed in Tris–HCL and PBS-T, dehydrated in an ascending alcohol series, incubated in toluol and coverslipped with Entellan (Merck Millipore).
Fluorescent immunohistochemistry
Histowax sections were deparaffinised, rehydrated and incubated in 60% methanol containing 2% H2O2 (for 1 h at room temperature), washed in PBS-T (pH 7.4) and incubated for 48 h at 4 °C in a solution (phosphate-buffered saline with 2% BSA, 0.3% milk powder and 0.5% donkey serum) with one of the following combinations of primary antibodies: (1) rabbit anti-IBA-1 (1:800, Wako) and goat anti-ferritin heavy chain Y-16 (1:200, Santa Cruz Biotechnology), (2) rabbit anti-GFAP (1:500, Dako) and goat anti-ferritin heavy chain Y-16 (1:200, Santa Cruz Biotechnology), (3) rabbit anti-Olig2 (1:100, Immuno Biological Laboratories) and goat anti-ferritin heavy chain Y-16 (1:200, Santa Cruz Biotechnology). Any pre-treatment of sections was performed corresponding to immunohistochemically procedures. Subsequently, brain sections were washed and incubated in a combination of fluorochrome-conjugated secondary antibodies (for 90 min at room temperature) using donkey anti-rabbit IgG (Dianova; Cy3, Cy5) and donkey anti-goat IgG (Dianova; Cy3, Cy5) for detection. Sections were washed and treated for 1 min with Sudan black B (Merck Millipore) dissolved in 70% EtOH to suppress auto-fluorescence and then differentiated in 70% EtOH for another 3 min. After rinsing in purified water, sections were coverslipped using aqua poly/mount mounting medium (Polysciences).
Light microscopy: image acquisition and processing
Stacked brightfield and fluorescence overview images were collected with 10 × , 40 × or 63 × Plan-Apochromat objectives using an automated slide scanner microscope (Zeiss AxioScan Z1) or a fluorescence-phase contrast microscope (Keyence BZ 9000). Stacks were collected at 1–5 µm intervals, merged and post-processed using the corresponding software (ZEN 2.3 or BZ-Analyzer). Shading correction and white balance were carried out. High magnification fluorescence images were obtained with a 63 × Plan-Apochromat (1.2 numerical aperture) objective using a confocal laser scanning microscope (Zeiss LSM 510 Meta or LSM 880 fast Airyscan) with lasers excitation at 543 and 633 nm and emission was detected using a BP 565–615 and a BP 650–710 filter.
Quantification of neuronal loss in the substantia nigra [stereological analysis]
The staging of PD according to Braak [11] and the compartmental organisation of the SN into matrix and nigrosomes according to Damier [17, 18] were the neuroanatomical basis of the experimental analysis. All PD cases could be assigned to Braak stages 3 to 6 with two PD brains characterized by an almost complete loss of dopaminergic neurons. In all Parkinsonian brains, highest neuronal cell loss was consistently observed within nigrosome 1.
Stereological analysis was performed using the optical fractionator method according to West et al. [92] as described by Morawski et al. [62]. Numerical densities of TH-positive neurons were determined using a Zeiss (Jena, Germany) Axioskop 2 plus microscope equipped with a motorised stage (Märzhäuser, Wetzlar, Germany), a Ludl MAC 5000 (LEP, Hawthorne, NY, USA) and a digital camera CX 9000 (MicroBrightField, Williston, VT, USA). Stereo Investigator software 7 (MicroBrightField, Williston, VT, USA) was used to analyse serial Sects. (10–12 µm thick) cut frontally through the midbrain encompassing the SN on the level of red nucleus, oculomotor nucleus and superior colliculus. A final section thickness of 10 ± 2 µm was received on average. This permitted for a dissector height of 10 µm using a guard zone of 2 µm on either side of the section. The contour of the SN/nigrosome 1 was outlined in a Stereo Investigator programme using a 5 × lens and the number of cells marked with the primary antibody was counted using an oil-immersion 63 × lens (1.4 numerical aperture). All neurons explicitly stained with at least fifty percent of their soma and one dendritic branch visible within the counting frame were considered. Neurons, which did not fulfil these criteria, were excluded. The coefficient of error (CE) was estimated with a one-stage systematic sampling procedure that has been described by Schaeffer et al. [80]. In parallel, the total number of neurons was determined on Nissl-stained sections by counting all neurons with visible nucleus. All cell counting and density measurements were performed blind to diagnosis by one rater. Statistical analysis by F-test and Student’s t-test was performed on absolute cell numbers (p < 0.05 significance threshold).
Quantitative iron mapping by immuno-µPIXE elemental analysis
Tissue preparation
Sections were treated following the immunohistochemical protocol for nickel-enhanced colour reaction of the target structures with the exception that all buffers and reagents were prepared with ultrapure and fresh substances (to avoid undesired signals in µPIXE), filtered using a pressure filtration system (Sarstedt) with a cellulose acetate membrane without surface-active agents and a pore size of 0.2 µm to prevent iron contamination. The colour reaction was performed using DAB and nickel (99.999% purity grade nickel ammonium sulphate) in Tris–HCl (pH 8.0) (Fig. 1). Sections were dehydrated and incubated overnight in xylol, followed by embedding in a thin layer of mounting medium DePeX (Serva Electrophoresis). After 4 h of drying at room temperature, an area of 15 × 20 mm2 was selected, cut out and pulled off the object slide for mounting on an aluminium frame using double sided adhesive carbon tape (G3939, Plano GmbH, Germany). Brightfield microscopy (Keyence BZ 9000) was performed before the µPIXE analysis for orientation, selection and documentation of the regions to be examined.

Sample preparation for proton beam analysis. The antibody (Ab)-complex, bound to a cell-type-specific antigen is enhanced with high-purity nickel (violet spheres). Since nickel is specifically identifiable in the PIXE map, it can be used as a marker for any antigen of interest. This combination of antigen labelling with subsequent nickel amplification allows for the application of quantitative PIXE analysis to any immunocytochemical preparation
Immuno-µPIXE quantitative element mapping
Quantitative element mapping using µPIXE was performed at the ion beam laboratory LIPSION at the Leipzig University (Germany) [75]. The proton induced X-rays were recorded with an energy dispersive high purity germanium detector (Canberra GUL0110) that allows simultaneous detection of characteristic X-rays from all elements above atomic number 11. Since the beam position during the scan is known, quantitative element maps can be projected from the data. The quantification of absolute element concentrations is based on the dynamic analysis method which was implemented in the GeoPIXE analysis software package [79]. It allows spectral deconvolution by fitting element peaks and background, yield calculation from fundamental parameters for X-ray production, matrix effects, geometric parameters and efficiency of the detector. While production cross sections and absorption coefficients for X-rays are precisely known from atomic physics, the detector geometry and efficiency are set-up-specific and need to be determined. The geometric parameters are known from the detector data sheet and from the installation geometry. The detector efficiency which depends on the X-ray energy, is fully described by a model function, based on the underlaying physical processes and their parameters. For the employed X-ray detector, the parameters were calibrated by measuring a set of elements with known concentrations within certified reference materials (SPEC25-53+FC, Astimex Scientific Ltd., Toronto, Canada).
The tissue sections were scanned at 2 µm resolution using a focused 2.25 MeV proton beam at a current between 200 and 800 pA for about 3 h per scan. Cellular iron concentrations were extracted by one rater by encircling the cells manually as a region of interest (Fig. 2) using the wide range of graphical tools in GeoPIXE. The detection limit for iron in the cellular regions was about 20 µmol/l [63].

Selection of regions of interest for cellular iron quantification. In the nickel (Ni) map of the immunocytochemical preparation of Hu C/D, the region of interest (ROI) is defined by a spline (in green) marking the cell shape. This ROI applies also to the other element maps from which the concentrations can be extracted. HU C/D: neuronal marker, S: sulphur, Fe: iron. Scale bar 20 μm, applies to all images
In order to verify that samples were not contaminated with iron due to sample preparation and staining procedure, additional sections were deparaffinised and rehydrated, again dehydrated and embedded in DePeX and analysed. The iron concentrations did not differ significantly between stained and unstained sections.
MRI
MRI measurements of human midbrain on two samples (one Co and one PD patient from the same pool) were carried out on a whole body 7 T MRI scanner (Magnetom, Siemens, Erlangen, Germany) using a custom-made 2-channel radio-frequency (RF) transmit-receive coil. The brainstem was fixed for six weeks in phosphate buffered 4% formaldehyde solution (pH 7.4). One week prior to MR scanning, the brainstem was transferred into 0.1 M phosphate buffer solution (PBS, pH 7.4) to washout any formaldehyde from the tissue. For scanning procedures, the brain tissue was transferred into fomblin (Fomblin® Y, Sigma-Aldrich). High-resolution quantitative maps of effective transverse (R2*) relaxation rates were obtained using a 3D multi-echo fast low-angle shot (FLASH) acquisition with isotropic resolution of 230 μm, matrix size of 128 × 128 × 60, and five equidistant echo times with bipolar readout and TE1-TE5 = 7.81 ms, 18.11 ms, 27.76 ms, 37.41 ms, 47.06 ms, repetition time TR = 60 ms, flip angle of 27° and bandwidth of 344 Hz/Px Quantitative R2* maps were obtained by a mono-exponential fit of the MR signal decay across different echo times (TEs).
Statistical analysis
Statistical analysis was performed using the scientific graphic and statistic software SigmaPlot (version 11.0) of Systat Software Inc. (San Jose, CA, USA), and values with p < 0.05 were regarded as statistically significant (Group differences in the Mann–Whitney rank sum test). All values are given as the median [1st quartile, 3rd quartile]. Iron concentrations were compared between controls (Co) and Parkinsonian subjects (PD) and analysed for neurons (Co n = 175; PD n = 111), microglial cells (Co n = 62; PD n = 109), astroglial cells (Co n = 88; PD n = 142), oligodendroglial cells (Olig2: Co n = 44; PD n = 79), ferritin-rich domains within oligodendroglial cells (Co n = 70; PD n = 87) and neuromelanin (Co n = 37; PD n = 30). Group differences were assessed by a Mann–Whitney rank sum test. Significant differences between two groups are marked as follows: * p < 0.05; ** p < 0.005; *** p < 0.001.
Results
Stereological quantification of cell loss in the substantia nigra
Neuronal loss in the SN of patients with PD was stereologically quantified on Nissl-stained sections as well as on immunocytochemical preparations for tyrosine hydroxylase (TH), detecting dopaminergic neurons (Table 2).
In controls, the SN appeared as a region in the midbrain with high neuronal density. Out of a total of eight cases with PD, we identified three cases with severe [80–95%] neuronal loss, another three cases with moderate neuronal loss (70–80%) and two cases with a mild neuronal loss [50–60%]. Throughout the SN, loss of neurons was most pronounced in nigrosome 1 in all cases with PD [20, 29] (Fig. 3). Thus, we selected nigrosome 1 as defined by Damier [19, 20] for the subsequent more in-depth analysis.

Neuronal distribution in the substantia nigra in a control (Co, left) and a patient with PD (right) immunohistochemically detected by an anti-Hu C/D antibody. Note the most pronounced loss of neurons in nigrosome 1 (N1) CP: cerebral peduncle, M: matrix, ML: medial lemniscus, N: nigrosome, SNpd: dorsal part of SN, SNpl: lateral part of SN, VTA: ventral tegmental area, SCP: superior cerebellar peduncle. Scale bar 500 µm
Macro-imaging of iron distribution in the substantia nigra with MRI and histochemistry
To analyse the iron distribution in the SN and its alterations in PD, we first estimated a semi-quantitative map of non-chelated Fe3+ using a DAB-amplified Perls’ stain (Fig. 4a, b). We obtained a very dense staining which clearly defined the SN as an area with high iron precipitation against a pale background (Fig. 4a). While overall, colour precipitation within the SN was rather homogeneous, a ‘comma’-like shape hypodense area surrounded by an upper and lower band of hyperdense staining was clearly delineated near nigrosome 1. This typical staining pattern is also clearly visible on R2* maps of non-neurodegenerative control (Fig. 4c), where it has been described as a typical “swallow tail” shaped sign. It is considered as a specific marker to identify healthy substantia nigra unaffected by degenerative changes [50]. In PD, the typical “swallow tail” sign disappears in R2* images (Fig. 4c) and in the Perls’ staining pattern and the distribution of iron becomes more homogeneous throughout the SN (Fig. 4b). The remarkable similarity of the Perls’ iron staining pattern and the MRI R2* map, both forming the “swallow tail” sign, which disappeared in PD, adds additional support for the diagnostic value of this structural feature. This observation points towards increase of overall iron content in SN of PD patients in areas close to nigrosome 1.

Macroscopic distribution of iron in the substantia nigra obtained by Perls ‘ staining and R2* mapping (MRI). In controls (Co), the area near nigrosome 1 can be delineated by a comma-like hypodense signal (arrows) in both Perls’ staining (A) and MRI R2* maps (C), surrounded by two tails of hyperdense signal, giving rise to the typical “swallow tail” structure. This “swallow tail” pattern disappeared in PD (B, C) because the iron is distributed rather homogeneously, where iron distribution obtained by Perls’ staining (B) shows a more homogeneous distribution throughout the SN. Scale bar 1000 µm
Iron rich cellular structures on Perls’ reaction
A higher magnification of DAB-amplified Perls’ stain allows for identifying cell types that are characterised by a high accumulation of non-chelated Fe3+ (Fig. 5a, d). Above all, oligodendroglial cells showed an intense cytosolic Perls’ reaction for non-chelated Fe3+ (Fig. 5a, d). The surrounding parenchyma appears inhomogeneously coloured and shows fibre-like morphological characteristics. Oligodendroglial cells detected with an anti-ferritin antibody also showed a higher immunoreactivity in cytosolic regions than in the nucleus (Fig. 5b, e). In PD, oligodendroglial cells (Fig. 5c, f, red arrow) with a positive immunoreaction for ferritin appear tighter in nigrosome 1 supporting reactive changes potentially leading to oligodendrogliosis (Fig. 5b, e).

Perls' reaction (a, d), anti-ferritin- (b, e) and anti-Olig2- (c, f) immunoreactivity of oligodendroglia cells (arrows) in human SN nigrosome 1 in controls (Co, a–c) and patients with PD (d–f). Scale bar 10 µm
Cell-type specific quantitative mapping of iron by immuno-µPIXE
In order to bridge the gap between macro and micro-imaging in PD and to further specify the iron distribution and its disease-associated changes, we applied immuno-µPIXE. Typical examples of microscopic brightfield images of immunocytochemical preparations and corresponding µPIXE element maps are shown in Fig. 6.

Element maps in human SN nigrosome 1 in controls (Co) and patients with PD, obtained by µPIXE. Brightfield microscope images (top row) show the immunocytochemical reaction for cell-type specific antigens providing the basis for definition of regions of interest for the subsequent PIXE imaging (below). The ultrapure nickel-enhancement of the immunoreaction allows to allocate the µPIXE element signals to specific cell types. The iron map was used to quantify the local concentrations of iron in different cell types (see Fig. 7). Scale bar top left 20 µm applies to all images
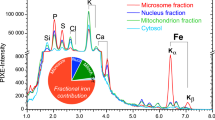
The distribution of sulphur in the PIXE map largely corresponds to the neuronal pigment neuromelanin, which, due to its iron-sulphur-clusters, is enriched in sulphur (approx. 10 × 103 µg/g). Extracellular sulphur (approx. 2.5 × 103 µg/g) mainly represents chondroitin sulphate proteoglycans (CSPG), the main components of the extracellular matrix. Iron was found to be concentrated in neurons and glial cells, which both showed higher cellular cytosolic iron loads than their extracellular environment (Fig. 6, bottom). Neuronal iron deposits typically co-located with neuromelanin which has two iron binding sides, with high and low affinity for iron, respectively.
PD, as compared to controls was associated with both changes in the total content of cellular iron and a shift in the cellular distribution of iron (Fig. 7a, b). The average total cellular iron of cells in nigrosome 1 in healthy controls amounts to 2.7 mM [1.5, 4.6] (n = 369) and to 4.3 mM [3.2, 6.4] (n = 441) in PD which corresponds to an increase by about 62% (Fig. 7a). Of note, total cellular iron content showed a very large individual variability which in healthy controls reached from 0.30 mM to 29 mM.

Quantification of iron in nigrosome 1 neurons in controls (Co) and patients with PD (a). The cellular compartmentation of iron in the SN nigrosome 1 in controls (Co) and patients with PD for different cells types, ferritin positive cells and in the pigment neuromelanin (b). Cellular concentrations of iron were obtained through quantitative element mapping by µPIXE in combination with nickel-enhanced immunocytochemical detection of cell type-specific antigens. Data are given as median values ± SD. n: analyzed cell number; Neurons (Hu C/D), Oligodendrocytes (Olig2), Microglia (IBA-1), Astroglia (GFAP), Ferritin (Fer) and Neuromelanin (NM)
The cellular compartmentalisation of iron in the SN was altered in PD patients and iron upregulation in PD showed differences in the variability for different cell types (Fig. 7b). In anti-Hu C/D reactive neurons, iron concentration was about doubled in PD compared to controls. Glial cells, where iron concentrations in controls tended to be higher than in neurons, showed more complex changes in PD with an increase in most cell types (Fig. 7b).
In oligodendroglial cells, that were subdivided according to the presence of specific antigens into functionally different, i.e. myelinating and non-myelinating (satellite) subtypes, iron concentrations in anti-Olig2-immunoreactive oligodendroglial cells were increased from 2.8 mM [1.9, 4.0] (n = 44) by about 150% up to 6.8 mM [4.7, 8.3] (n = 79). In anti-ferritin-immunoreactive oligodendroglial cells, that showed the highest iron concentration among all cell types in controls, iron concentrations in PD cases dropped to 51% (controls: 6.0 mM [2.8, 10.8] (n = 70); PD: 3.2 mM [1.8, 5.5] (n = 87)). This reduction can be caused by a depletion of ferritin, the main form of iron storage in glial cells, or by a reduction in ferritin loading. Iron concentrations in PD in anti-IBA-1 reactive microglial cells were increased by about one quarter (26%, controls: 3.2 mM [2.2, 4.3] (n = 62); PD: 4.0 mM [2.8, 6.0] (n = 109)) but remained unaffected in anti GFAP reactive astroglial cells (controls: 4.3 mM [2.0, 6.9] (n = 88); PD: 4.1 mM [3.3, 5.4] (n = 142)).
In addition to the specific changes in the cellular compartmentalisation of iron in PD, the iron loading of neuromelanin as a structure with significant iron binding capacity must be considered. Contrary to controls, where neuromelanin is restricted to dopaminergic neurons, it is found in PD predominantly extracellularly which could be due to remains of disintegrated neurons and therefore a consequence of neurodegeneration. The iron concentration of neuromelanin in PD was increased by about two thirds (62%, controls: 8.7 mM [6.6, 12.1] (n = 37); PD: 14.2 mM [10.4, 19.0] (n = 30)).
Iron storage proteins, oligodendroglial cells and myelin
To assign the strong ferritin-associated µPIXE iron signal to defined cellular structures, we performed double-label fluorescent immunohistochemistry with anti-ferritin and anti-glial cell markers. As illustrated in Fig. 8, ferritin immunoreactivity both in controls and PD was largely restricted to oligodendroglial cells (yellow colour overlap, Fig. 8a, d) while astroglial or microglial cells showed no co-immunoreaction (Fig. 8b, e and c, f).

Double-label fluorescent immunohistochemistry with anti-ferritin and anti-glial cell markers in nigrosome 1 in controls (Co, a–c) and patients with PD (d–f). Anti-ferritin antibody (red) in combination with anti-Olig2 antibody (green) (a, d), anti-ferritin antibody (red) combined with anti-GFAP antibody (green) (b, e) and anti-ferritin antibody (red) combined with anti-IBA-1 antibody (green) (c, f) did not show significant spatial co-localization whereas immunoreactivity in oligodendrocytes (a, d) could be confirmed. Immunohistochemical observations with anti-ferritin antibody show positive reactions in oligodendrocytes. In addition, (e) and (f) show strong activation signs of glial cells including a hypertrophic morphology and an increase in cell density. Territories of neurons surrounded by glia cell processes (a) and (d) are labelled with *. Sections were treated with Sudan black B to suppress tissue auto fluorescence. Scale bar 50 µm
Overall, oligodendroglial cells showed the most pronounced changes in iron compartmentalisation in PD. To further characterise pathology related changes in oligodendroglial cells, myelinating and non-myelinating (satellite) subtypes were investigated differentially. To this end, sections were immunolabelled with anti-Olig2 and antibodies against myelin basic protein (MBP) (Fig. 9). Morphological appearance and immunoreactivity for both antigens differed between controls and PD. While myelinated fibres in controls are more homogeneously distributed (Fig. 9a), they appeared rarefied, more delicate and partially agglomerated in PD (Fig. 9c). Sections labelled with an anti-Olig2 antibody show an increase in cell density of stained oligodendroglial cells in PD (Fig. 9d). Observed reactive morphological changes could be due to previous neurodegeneration leading to a secondary degenerative shift of myelin or may show direct evidence of oligodendrogliosis.

Oligodendroglia activation and degenerative change of myelin in nigrosome 1 of controls (Co, a, b) and PD (c, d) patients. Brightfield microscopy of myelin basic protein (MBP) shows a dense and rather homogeneous network of myelinated fibres in controls (a). In PD, myelinated fibres appear less dense (c). Additionally, in PD an increase of anti-Olig2 positive oligodendrocytes is visible (arrows, d). Scale bar 50 µm
Discussion
Iron is one of the key factors that most likely plays a major role in PD although its precise pathogenic role is not fully understood [98]. While several studies report on increased levels of total iron in the SN [21, 22, 26, 40, 43, 51, 77, 84], others did not [33, 49, 87]. This increase of total nigral iron may reflect an interference of multiple pathological processes including inflammation, myelination, perturbed iron uptake as well as vascular damage, each affecting distinct cell populations. In addition, there are various pre-analytical and analytical factors of iron determination including tissue acervation and sample preparation that may account for such discrepancies.
An incubation of brain tissue in formalin for four years was shown to decrease iron levels down to 57% compared with similar analysed fresh frozen tissue [81]. It is reasonable to assume that iron leaching depends primarily on the fixation time, chemical form and to a minor extend on the pathology. Thus, the relative increase or decrease in iron concentrations of PD brains compared to control brains is only marginally affected in our study. Different or larger extends of iron leaching are not expected since the sample preparation processes followed the same protocol with fixation times less than half a year. To test the extend of preparatory effects, we have analysed the iron concentrations of nine neuromelanin deposits in three sections of a cryofixed and unstained substantia nigra from control brain. The average iron concentration of neuromelanin from the cryofixed tissue was 8.0 mM [6.4, 11.2] (n = 9), which is in agreement with the results of 8.7 mM [6.6, 12.1] (n = 37) for neuromelanin of short-term paraformaldehyde-fixed substantia nigra from control brains.
Semi-quantitative histochemical methods were able to demonstrate that iron deposits were present in both neurons and glial cells of the SN, with an increase of ferritin-loaded microglial cells in the SN [43]. Still, precise quantitative data on cellular iron compartmentalisation in the brain and its changes in PD are still lacking.
Here, we close this gap in knowledge by applying immuno-µPIXE to the human brain, a newly developed technique which allows a quantitative determination of major and trace elements and their assignment to specific cell types. In particular it is suitable to analyse iron in brain sections with a spatial resolution in the µm-range and a detection limit of approximately 10 µM [31, 57, 59,60,61, 73, 74, 76].
We determined a tissue concentration of total iron in nigrosome 1 in controls of 2.7 mM [1.5, 4.6] (n = 369) and of 4.3 mM [3.2, 6.4] (n = 441) in PD. To convert this for comparison into ng/mg wet tissue weight, the values amount to 180 ng/mg in control and of 300 ng/mg in PD. This in in good agreement with values reported by Friedman et al. [32], Dexter et al. [22] and Riederer et al. [77].
In PD, our results show an increase of total iron content within the nigrosome 1 as compared to controls by about two thirds (62%) which is in line with previous investigations that reported an increase in total iron in PD [23, 36, 71, 84]. However, we for the first time demonstrate that the increase of total iron differs across cell types in PD.
Most strikingly was the twofold increase (207%) of cellular iron within the dopaminergic neurons of nigrosome 1 in PD, which is in line with previous reports [1, 60, 65, 66]. This finding may provide an insight in the pathological role of iron accumulation in PD. Due to the rather unaffected sulphur content, our results confirm that neuromelanin is unsaturated with an Fe/S ratio of 10 at% under physiological conditions. A similar physiological Fe/S ratio of 13 at% was obtained with high resolution analysis directly on single neuromelanin granules [7]. Sulphur in the granules originate from pheomelanin, which derives from 5-S-cysteinyldopamin. Structurally, pheomelanin seem to favour the formation of the low-affinity mononuclear iron centre [30]. Eumelanin, the other melanin moiety, forms the high-affinity site where iron remains redox-inactive. Thus, neuromelanin has physiologically a protective function, since iron, bound preferentially to its high-affinity binding sides, is segregated from supporting any redox processes [96, 102]. Under conditions of increasing iron overload, which may result from alterations in iron metabolism [5, 6, 45, 78], high-affinity eumelanin becomes saturated and the low-affinity binding sites of pheomelanin are increasingly occupied. Then loosely bound iron can be released easily and trigger further toxic reactions that may lead to progressive neurodegeneration [28]. A pathogenic role of elevated iron concentrations is also supported by reports of direct correlations between the severity of neuronal loss in PD and the amount of iron accumulation [52, 93].
However, despite the high increase in neuronal iron concentration, it only contributes little to the overall increase of iron in nigrosome 1. Of note, both in the normal brain and in PD, the major iron pool (about 80%) is localised in glial cells. In the normal brain, the population of ferritin-stained oligodendroglial cells contain most iron, followed by astroglial cells, microglial cells and another population of Olig2-stained oligodendroglial cells (ferritin-stained oligodendroglial cells > astroglial cells > microglial cells > Olig2-stained oligodendroglial cells). This pattern is inverted in PD (Olig2-stained oligodendroglial cells > microglial cells > astroglial cells > ferritin-stained oligodendroglial cells). While the iron content of astroglial cells in PD remained unchanged, microglial cells showed an iron accumulation of about one quarter (26%) and Olig2-stained oligodendroglial cells by about 150%. Descriptions of the physiological and pathophysiological role of iron need to take into account the iron redistribution between neurons and glial cells.
The increase of iron in microglial cells has been attributed to its activation [6, 86, 99] and might be due to phagocytosis of neuromelanin fragments [94, 97, 100, 102] and increased iron uptake via transferrin-receptor [91] or non-transferrin bound iron [8]. Accordingly, to our morphological observations, microglial cells have the potential to degrade and solubilize neuromelanin that is released by degenerating neurons [100]. As a result of this, soluble iron is generated that could be internalized in other surrounding cells, promoting neuroinflammation and ultimately neurodegeneration. Extracellular neuromelanin goes hand in hand with a release of proinflammatory and toxic molecules that can maintain neuroinflammation as well as degeneration processes [100]. The iron pool of activated microglial cells may again be subject to cellular redistribution. Apoptosis of exhausted and damaged microglial cells might result in release of iron into the extracellular space [12] and circulating toxic iron may be taken by neurons or glial cells through transferrin-dependent and independent mechanisms [5, 8, 78, 86]. Activation of microglial cells, furthermore, is associated by a reduction of ferritin-de-novo-synthesis and H-ferritin levels, which may occur as a consequence of increased oxidative stress [55, 56]. Although the concentration of L-ferritin in the SN is lower than that of H-ferritin, levels of L-ferritin increased as well as H-ferritin levels during the normal ageing process [98]. Instead, in PD, decreasing levels of H-ferritin are described by Zecca et al. [98]. In accordance to these investigations, we equally observed a reduction of ferritin deposits in glial cells in PD down to nearly 50%.
Strikingly, changes of the iron content in PD were most pronounced in oligodendroglial cells. Oligodendroglial cells may hold one of the highest oxidative metabolism of all brain cells [64] and may thus be particularly vulnerable to sequelae of oxidative stress. Due to their involvement in myelin synthesis, oligodendroglial cells may show high concentrations of cellular iron and ferritin [14, 15, 66]. Since oxidative stress in microglial cells contributes to a decrease in H-ferritin levels [55, 56], there is reason to suppose that cellular stress may be reflected as well in a reduction of H-ferritin in oligodendroglial cells. The reduction of both neuromelanin-bound iron and ferritin-bound iron might result in an increase of the labile iron pool, which will further promote oxidative damage.
Astroglial cells even in the normal brain contain a comparatively large cellular iron pool that may play a role in neurotransmitter homeostasis [90]. Astrocytic processes form parts of the blood brain barrier [4, 25] and regulate the uptake and distribution of metal ions within the brain [24, 41, 67, 89, 90], which makes them the first parenchymal cell type that will get in contact with iron after having crossed the blood brain barrier [41]. Due to their large intracellular iron pool together with their close contacts to various other brain cell types [3, 35, 39, 67, 89], they play a key role in processes of iron distribution within the neuropil [68]. The cellular iron concentration in astrocytes remains unchanged in PD, which may indicate that they are largely able to maintain their homeostatic pathways, even under inflammatory conditions.
Taken together, we demonstrated that the total increase of iron concentration in PD is a result of multiple physiological processes differentially affecting different cell populations.
Since iron is involved in both physiological and pathophysiological processes, in order to identify potentially modifiable therapeutic targets, it is essential to accurately determine its cellular compartmentalisation, intercellular re-distribution or differential accumulation both under normal and diseased conditions. While, the pathogenic role of elevated iron in PD remains unquestionable in general, more in-depth knowledge on iron pools and iron fluxes is still lacking. Our immuno-µPIXE approach allows for a highly sensitive quantification of iron at the subcellular level in well preserved cellular architectures. We believe that this knowledge will eventually contribute to the development of disease modifying strategies.
Availability of supporting data
The data that support the findings of this study are available from the corresponding author M.M. upon reasonable request.
References
Ayton S, Lei P, Hare DJ et al (2015) Parkinson’s disease iron deposition caused by nitric oxide-induced loss of β-amyloid precursor protein. J Neurosci 35:3591–3597. https://doi.org/10.1523/JNEUROSCI.3439-14.2015
Bastian TW, von Hohenberg WC, Mickelson DJ et al (2016) Iron Deficiency Impairs Developing Hippocampal Neuron Gene Expression, Energy Metabolism, and Dendrite Complexity. Dev Neurosci 38:264–276. https://doi.org/10.1159/000448514
Baxter PS, Hardingham GE (2016) Adaptive regulation of the brain’s antioxidant defences by neurons and astrocytes. Free Radic Biol Med 100:147–152. https://doi.org/10.1016/j.freeradbiomed.2016.06.027
Begley DJ, Brightman MW (2003) Structural and functional aspects of the blood-brain barrier. Prog Drug Res 61:39–78. https://doi.org/10.1007/978-3-0348-8049-7_2
Belaidi AA, Bush AI (2016) Iron neurochemistry in Alzheimer’s disease and Parkinson’s disease: targets for therapeutics. J Neurochem 139(Suppl 1):179–197. https://doi.org/10.1111/jnc.13425
Berg D, Gerlach M, Youdim M et al (2001) Brain iron pathways and their relevance to Parkinson’s disease. J Neurochem 79:225–236
Biesemeier A, Eibl O, Eswara S et al (2016) Elemental mapping of Neuromelanin organelles of human Substantia Nigra: correlative ultrastructural and chemical analysis by analytical transmission electron microscopy and nano-secondary ion mass spectrometry. J Neurochem 138:339–353. https://doi.org/10.1111/jnc.13648
Bishop GM, Dang TN, Dringen R et al (2011) Accumulation of non-transferrin-bound iron by neurons, astrocytes, and microglia. Neurotox Res 19:443–451. https://doi.org/10.1007/s12640-010-9195-x
Blazejewska AI, Schwarz ST, Pitiot A et al (2013) Visualization of nigrosome 1 and its loss in PD: pathoanatomical correlation and in vivo 7 T MRI. Neurology 81:534–540. https://doi.org/10.1212/WNL.0b013e31829e6fd2
Borie C, Gasparini F, Verpillat P et al (2002) Association study between iron-related genes polymorphisms and Parkinson’s disease. J Neurol 249:801–804. https://doi.org/10.1007/s00415-002-0704-6
Braak H, Braak E (1986) Nuclear configuration and neuronal types of the nucleus niger in the brain of the human adult. Hum Neurobiol 5:71–82
Colton CA, Gilbert DL (1987) Production of superoxide anions by a CNS macrophage, the microglia. FEBS Lett 223:284–288
Conde JR, Streit WJ (2006) Microglia in the aging brain. J Neuropathol Exp Neurol 65:199–203. https://doi.org/10.1097/01.jnen.0000202887.22082.63
Connor JR (1995) A quantitative analysis of isoferritins in select regions of aged, Parkinsonian, and Alzheimer’s diseased brains. J Neurochem 1995:717–724
Connor JR, Menzies SL (1996) Relationship of iron to oligondendrocytes and myelination. Glia 17:83–93
Cosottini M, Frosini D, Pesaresi I et al (2014) MR imaging of the substantia nigra at 7 T enables diagnosis of Parkinson disease. Radiology 271:831–838. https://doi.org/10.1148/radiol.14131448
Damier P (1999) The substantia nigra of the human brain. I. Nigrosomes and the nigral matrix, a compartmental organization based on calbindin D28K immunohistochemistry. Brain 122:1421–1436. https://doi.org/10.1093/brain/122.8.1421
Damier P (1999) The substantia nigra of the human brain. II. Patterns of loss of dopamine-containing neurons in Parkinson’s disease. Brain 122:1437–1448. https://doi.org/10.1093/brain/122.8.1437
Damier P, Hirsch EC, Agid Y et al (1999) The substantia nigra of the human brain. I. Nigrosomes and the nigral matrix, a compartmental organization based on calbindin D(28K) immunohistochemistry. Brain 122(Pt 8):1421–1436. https://doi.org/10.1093/brain/122.8.1421
Damier P, Hirsch EC, Agid Y et al (1999) The substantia nigra of the human brain. II. Patterns of loss of dopamine-containing neurons in Parkinson’s disease. Brain 122(Pt 8):1437–1448. https://doi.org/10.1093/brain/122.8.1437
Dexter DT, Wells FR, Agid F et al (1987) Increased nigral iron content in postmortem parkinsonian brain. The Lancet 330:1219–1220. https://doi.org/10.1016/s0140-6736(87)91361-4
Dexter DT, Wells FR, Lees AJ et al (1989) Increased nigral iron content and alterations in other metal ions occurring in brain in Parkinson’s disease. J Neurochem 52:1830–1836. https://doi.org/10.1111/j.1471-4159.1989.tb07264.x
Dexter DT, Carayon A, Javoy-Agid F et al (1991) Alterations in the levels of iron, ferritin and other trace metals in Parkinson’s disease and other neurodegenerative diseases affecting the basal ganglia. Brain 114(Pt 4):1953–1975. https://doi.org/10.1093/brain/114.4.1953
Dringen R, Bishop GM, Koeppe M et al (2007) The pivotal role of astrocytes in the metabolism of iron in the brain. Neurochem Res 32:1884–1890. https://doi.org/10.1007/s11064-007-9375-0
Dyrna F, Hanske S, Krueger M et al (2013) The blood-brain barrier. J Neuroimmune Pharmacol 8:763–773. https://doi.org/10.1007/s11481-013-9473-5
Earle KM (1968) Studies on Parkinson’s disease including x-ray fluorescent spectroscopy of formalin fixed brain tissue. J Neuropathol Exp Neurol 27:1–14. https://doi.org/10.1097/00005072-196801000-00001
Faucheux BA, Agid Y, Hirsch EC et al (1999) Blood vessels change in the mesencephalon of patients with Parkinson’s disease. The Lancet 353:981–982. https://doi.org/10.1016/S0140-6736(99)00641-8
Faucheux BA, Martin M-E, Beaumont C et al (2003) Neuromelanin associated redox-active iron is increased in the substantia nigra of patients with Parkinson’s disease. J Neurochem 86:1142–1148. https://doi.org/10.1046/j.1471-4159.2003.01923.x
Fearnley JM, Lees AJ (1991) Ageing and Parkinson’s disease: substantia nigra regional selectivity. Brain 114(Pt 5):2283–2301. https://doi.org/10.1093/brain/114.5.2283
Ferrari E, Engelen M, Monzani E et al (2013) Synthesis and structural characterization of soluble neuromelanin analogs provides important clues to its biosynthesis. J Biol Inorg Chem 18:81–93. https://doi.org/10.1007/s00775-012-0951-7
Fiedler A, Reinert T, Morawski M et al (2007) Intracellular iron concentration of neurons with and without perineuronal nets. Nucl Instrum Methods Phys Res, Sect B 260:153–158. https://doi.org/10.1016/j.nimb.2007.02.069
Friedman A, Galazka-Friedman J, Koziorowski D (2009) Iron as a cause of Parkinson disease – a myth or a well established hypothesis? Parkinsonism Relat Disord 15:S212–S214. https://doi.org/10.1016/S1353-8020(09)70817-X
Gałazka-Friedman J, Bauminger ER, Friedman A et al (1996) Iron in parkinsonian and control substantia nigra—a Mössbauer spectroscopy study. Mov Disord 11:8–16. https://doi.org/10.1002/mds.870110104
Gerlach M, Ben-Shachar D, Riederer P et al (1994) Altered brain metabolism of iron as a cause of neurodegenerative diseases? J Neurochem 63:793–807. https://doi.org/10.1046/j.1471-4159.1994.63030793.x
Giaume C, Venance L (1998) Intercellular calcium signaling and gap junctional communication in astrocytes. Glia 24:50–64
Griffiths PD, Dobson BR, Jones GR et al (1999) Iron in the basal ganglia in Parkinson’s disease. An in vitro study using extended X-ray absorption fine structure and cryo-electron microscopy. Brain 122(Pt 4):667–673. https://doi.org/10.1093/brain/122.4.667
Hallgren B, SOURANDER P, (1958) The effect of age on the non-haemin iron in the human brain. J Neurochem 3:41–51. https://doi.org/10.1111/j.1471-4159.1958.tb12607.x
Hare D, Ayton S, Bush A et al (2013) A delicate balance: Iron metabolism and diseases of the brain. Front Aging Neurosci 5:34. https://doi.org/10.3389/fnagi.2013.00034
Hatton GI (2002) Glial-neuronal interactions in the mammalian brain. Adv Physiol Educ 26:225–237. https://doi.org/10.1152/advan.00038.2002
Hirsch EC, Brandel JP, Galle P et al (1991) Iron and aluminum increase in the substantia nigra of patients with Parkinson’s disease: an X-ray microanalysis. J Neurochem 56:446–451. https://doi.org/10.1111/j.1471-4159.1991.tb08170.x
Hohnholt MC, Dringen R (2013) Uptake and metabolism of iron and iron oxide nanoparticles in brain astrocytes. Biochem Soc Trans 41:1588–1592. https://doi.org/10.1042/BST20130114
Jahanshad N, Rajagopalan P, Thompson PM (2013) Neuroimaging, nutrition, and iron-related genes. Cell Mol Life Sci 70:4449–4461. https://doi.org/10.1007/s00018-013-1369-2
Jellinger K, Paulus W, Grundke-Iqbal I et al (1990) Brain iron and ferritin in Parkinson’s and Alzheimer’s diseases. J Neural Transm Park Dis Dement Sect 2:327–340. https://doi.org/10.1007/BF02252926
Jomova K, Vondrakova D, Lawson M et al (2010) Metals, oxidative stress and neurodegenerative disorders. Mol Cell Biochem 345:91–104. https://doi.org/10.1007/s11010-010-0563-x
Kaur D, Andersen J (2004) Does cellular iron dysregulation play a causative role in Parkinson’s disease? Ageing Res Rev 3:327–343. https://doi.org/10.1016/j.arr.2004.01.003
Kwon D-H, Kim J-M, Oh S-H et al (2012) Seven-Tesla magnetic resonance images of the substantia nigra in Parkinson disease. Ann Neurol 71:267–277. https://doi.org/10.1002/ana.22592
Langkammer C, Pirpamer L, Seiler S et al (2016) Quantitative Susceptibility Mapping in Parkinson’s Disease. PLoS ONE 11:e0162460. https://doi.org/10.1371/journal.pone.0162460
Lehéricy S, Bardinet E, Poupon C et al (2014) 7 Tesla magnetic resonance imaging: a closer look at substantia nigra anatomy in Parkinson’s disease. Mov Disord 29:1574–1581. https://doi.org/10.1002/mds.26043
Loeffler DA, Connor JR, Juneau PL et al (1995) Transferrin and iron in normal, Alzheimer’s disease, and Parkinson’s disease brain regions. J Neurochem 65:710–724. https://doi.org/10.1046/j.1471-4159.1995.65020710.x
Mai JK, Majtanik M, Paxinos G (2016) Atlas of the human brain, 4th edn. Elsevier Ltd, Amsterdam
Mann VM, Cooper JM, Daniel SE et al (1994) Complex I, iron, and ferritin in Parkinson’s disease substantia nigra. Ann Neurol 36:876–881. https://doi.org/10.1002/ana.410360612
Martin WRW, Wieler M, Gee M (2008) Midbrain iron content in early Parkinson disease: a potential biomarker of disease status. Neurology 70:1411–1417. https://doi.org/10.1212/01.wnl.0000286384.31050.b5
Mastroberardino PG, Hoffman EK, Horowitz MP et al (2009) A novel transferrin/TfR2-mediated mitochondrial iron transport system is disrupted in Parkinson’s disease. Neurobiol Dis 34:417–431. https://doi.org/10.1016/j.nbd.2009.02.009
McRitchie DA, Halliday GM, Cartwright H (1995) Quantitative analysis of the variability of substantia nigra pigmented cell clusters in the human. Neuroscience 68:539–551. https://doi.org/10.1016/0306-4522(95)00163-d
Mehlhase J, Sandig G, Pantopoulos K et al (2005) Oxidation-induced ferritin turnover in microglial cells: role of proteasome. Free Radic Biol Med 38:276–285. https://doi.org/10.1016/j.freeradbiomed.2004.10.025
Mehlhase J, Gieche J, Widmer R et al (2006) Ferritin levels in microglia depend upon activation: modulation by reactive oxygen species. Biochim Biophys Acta 1763:854–859. https://doi.org/10.1016/j.bbamcr.2006.04.012
Meinecke C, Morawski M, Reinert T et al (2006) Cellular distribution and localisation of iron in adult rat brain (substantia nigra). Nucl Instrum Methods Phys Res, Sect B 249:688–691. https://doi.org/10.1016/j.nimb.2006.03.118
Möller HE, Bossoni L, Connor JR et al (2019) Iron, Myelin, and the Brain: Neuroimaging Meets Neurobiology. Trends Neurosci 42:384–401. https://doi.org/10.1016/j.tins.2019.03.009
Morawski M, Reinert T, Meinecke C et al (2005) Antibody meets the microbeam – or how to find neurofibrillary tangles. Nucl Instrum Methods Phys Res, Sect B 231:229–233. https://doi.org/10.1016/j.nimb.2005.01.062
Morawski M, Meinecke C, Reinert T et al (2005) Determination of trace elements in the human substantia nigra. Nucl Instrum Methods Phys Res, Sect B 231:224–228. https://doi.org/10.1016/j.nimb.2005.01.061
Morawski M, Reinert T, Brückner G et al (2005) The Binding of Iron to Perineuronal Nets. A Combined Nuclear Microscopy and Mössbauer Study. Hyperfine Interact 159:285–291. https://doi.org/10.1007/s10751-005-9116-1
Morawski M, Brückner G, Arendt T et al (2012) Aggrecan: Beyond cartilage and into the brain. Int J Biochem Cell Biol 44:690–693. https://doi.org/10.1016/j.biocel.2012.01.010
Morawski M, Reinert T, Meyer-Klaucke W et al (2015) Ion exchanger in the brain: Quantitative analysis of perineuronally fixed anionic binding sites suggests diffusion barriers with ion sorting properties. Sci Rep 5:16471. https://doi.org/10.1038/srep16471
Norton WT (ed) (1984) Oligodendroglia. Advances in neurochemistry, vol 5. Springer, Boston, pp 199–232
Oakley AE, Collingwood JF, Dobson J et al (2007) Individual dopaminergic neurons show raised iron levels in Parkinson disease. Neurology 68:1820–1825. https://doi.org/10.1212/01.wnl.0000262033.01945.9a
Ortiz E, Pasquini JM, Thompson K et al (2004) Effect of manipulation of iron storage, transport, or availability on myelin composition and brain iron content in three different animal models. J Neurosci Res 77:681–689. https://doi.org/10.1002/jnr.20207
Parpura V, Heneka MT, Montana V et al (2012) Glial cells in (patho)physiology. J Neurochem 121:4–27. https://doi.org/10.1111/j.1471-4159.2012.07664.x
Pelizzoni I, Zacchetti D, Campanella A et al (2013) Iron uptake in quiescent and inflammation-activated astrocytes: a potentially neuroprotective control of iron burden. Biochim Biophys Acta 1832:1326–1333. https://doi.org/10.1016/j.bbadis.2013.04.007
Péran P, Cherubini A, Assogna F et al (2010) Magnetic resonance imaging markers of Parkinson’s disease nigrostriatal signature. Brain 133:3423–3433. https://doi.org/10.1093/brain/awq212
Pivina L, Semenova Y, Doşa MD et al (2019) Iron Deficiency, Cognitive Functions, and Neurobehavioral Disorders in Children. J Mol Neurosci 68:1–10. https://doi.org/10.1007/s12031-019-01276-1
Pyatigorskaya N, Sharman M, Corvol J-C et al (2015) High nigral iron deposition in LRRK2 and Parkin mutation carriers using R2* relaxometry. Mov Disord 30:1077–1084. https://doi.org/10.1002/mds.26218
Qian Z, Wang Q, Pu Y (1997) Brain iron and neurological disorders. Chin Med J (Engl) 110:455–458
Reinert T (2003) Quantitative microanalysis of perineuronal nets in brain tissue. Nucl Instrum Methods Phys Res, Sect B 210:395–400. https://doi.org/10.1016/S0168-583X(03)01041-3
Reinert T, Fiedler A, Morawski M et al (2007) High resolution quantitative element mapping of neuromelanin-containing neurons. Nucl Instrum Methods Phys Res, Sect B 260:227–230. https://doi.org/10.1016/j.nimb.2007.02.070
Reinert T, Andrea T, Barapatre N et al (2011) Biomedical research at LIPSION – Present state and future developments. Nucl Instrum Methods Phys Res, Sect B 269:2254–2259. https://doi.org/10.1016/j.nimb.2011.02.071
Reinert A, Morawski M, Seeger J et al (2019) Iron concentrations in neurons and glial cells with estimates on ferritin concentrations. BMC Neurosci 20:25. https://doi.org/10.1186/s12868-019-0507-7
Riederer P, Sofic E, Rausch WD et al (1989) Transition metals, ferritin, glutathione, and ascorbic acid in parkinsonian brains. J Neurochem 52:515–520. https://doi.org/10.1111/j.1471-4159.1989.tb09150.x
Rouault TA (2013) Iron metabolism in the CNS. Implications for neurodegenerative diseases. Nat Rev Neurosci 14:551–564. https://doi.org/10.1038/nrn3453
Ryan CG (2001) Developments in Dynamic Analysis for quantitative PIXE true elemental imaging. Nucl Instrum Methods Phys Res, Sect B 181:170–179. https://doi.org/10.1016/S0168-583X(01)00374-3
Schaeffer RL, Mendenhall W, Ott L (1990) Elementary survey sampling
Schrag M, Dickson A, Jiffry A et al (2010) The effect of formalin fixation on the levels of brain transition metals in archived samples. Biometals 23:1123–1127. https://doi.org/10.1007/s10534-010-9359-4
Schwarz ST, Afzal M, Morgan PS et al (2014) The “swallow tail” appearance of the healthy nigrosome—a new accurate test of Parkinson’s disease: a case-control and retrospective cross-sectional MRI study at 3T. PLoS ONE 9:e93814. https://doi.org/10.1371/journal.pone.0093814
Shafir T, Angulo-Barroso R, Jing Y et al (2008) Iron deficiency and infant motor development. Early Hum Dev 84:479–485. https://doi.org/10.1016/j.earlhumdev.2007.12.009
Sofic E, Riederer P, Heinsen H et al (1988) Increased iron (III) and total iron content in post mortem substantia nigra of parkinsonian brain. J Neural Transm 74:199–205. https://doi.org/10.1007/BF01244786
Thompson KJ, Shoham S, Connor JR (2001) Iron and neurodegenerative disorders. Brain Res Bull 55:155–164. https://doi.org/10.1016/s0361-9230(01)00510-x
Thomsen MS, Andersen MV, Christoffersen PR et al (2015) Neurodegeneration with inflammation is accompanied by accumulation of iron and ferritin in microglia and neurons. Neurobiol Dis 81:108–118. https://doi.org/10.1016/j.nbd.2015.03.013
Uitti RJ, Rajput AH, Rozdilsky B et al (1989) Regional metal concentrations in Parkinson’s disease, other chronic neurological diseases, and control brains. Can J Neurol Sci 16:310–314. https://doi.org/10.1017/s0317167100029140
van Domburg PH, ten Donkelaar HJ (1991) The human substantia nigra and ventral tegmental area. A neuroanatomical study with notes on aging and aging diseases. Adv Anat Embryol Cell Biol 121:1–132
Verkhratsky A, Parpura V (2010) Recent advances in (patho)physiology of astroglia. Acta Pharmacol Sin 31:1044–1054. https://doi.org/10.1038/aps.2010.108
Verkhratsky A, Steinhäuser C (2000) Ion channels in glial cells. Brain Res Rev 32:380–412. https://doi.org/10.1016/S0165-0173(99)00093-4
Ward RJ, Zucca FA, Duyn JH et al (2014) The role of iron in brain ageing and neurodegenerative disorders. The Lancet Neurology 13:1045–1060. https://doi.org/10.1016/S1474-4422(14)70117-6
West MJ, Slomianka L, Gundersen HJ (1991) Unbiased stereological estimation of the total number of neurons in thesubdivisions of the rat hippocampus using the optical fractionator. Anat Rec 231:482–497. https://doi.org/10.1002/ar.1092310411
Wieler M, Gee M, Wayne Martin WR (2015) Longitudinal midbrain changes in early Parkinson’s disease: Iron content estimated from R2*/MRI. Parkinsonism Relat Disord 21:179–183
Wilms H, Rosenstiel P, Sievers J et al (2003) Activation of microglia by human neuromelanin is NF-kappaB dependent and involves p38 mitogen-activated protein kinase: implications for Parkinson’s disease. FASEB J 17:500–502. https://doi.org/10.1096/fj.02-0314fje
Wurzinger B, König P (2016) Eisenmangel, Müdigkeit und Restless-Legs-Syndrom (Iron deficiency, Fatigue and Restless-Legs-Syndrome). Wien Med Wochenschr 166:447–452. https://doi.org/10.1007/s10354-016-0497-3
Zecca L (2001) SN neuromelanin structure, synthesis and molecular behaviour. J Clin Pathol Mol Pathol 2001:414–418
Zecca L, Zucca FA, Costi P et al (2003) The neuromelanin of human substantia nigra: structure, synthesis and molecular behaviour. J Neural Transm Suppl 20:145–155. https://doi.org/10.1007/978-3-7091-0643-3_8
Zecca L, Stroppolo A, Gatti A et al (2004) The role of iron and copper molecules in the neuronal vulnerability of locus coeruleus and substantia nigra during aging. Proc Natl Acad Sci U S A 101:9843–9848. https://doi.org/10.1073/pnas.0403495101
Zhang X, Surguladze N, Slagle-Webb B et al (2006) Cellular iron status influences the functional relationship between microglia and oligodendrocytes. Glia 54:795–804. https://doi.org/10.1002/glia.20416
Zhang W, Phillips K, Wielgus AR et al (2011) Neuromelanin activates microglia and induces degeneration of dopaminergic neurons: implications for progression of Parkinson’s disease. Neurotox Res 19:63–72. https://doi.org/10.1007/s12640-009-9140-z
Zhang YL, Zheng SS, Zhu LY et al (2019) Impact of iron deficiency in early life stages on children’s motor development: a longitudinal follow-up. Zhonghua Er Ke Za Zhi 57:194–199. https://doi.org/10.3760/cma.j.issn.0578-1310.2019.03.007
Zucca FA, Segura-Aguilar J, Ferrari E et al (2017) Interactions of iron, dopamine and neuromelanin pathways in brain aging and Parkinson’s disease. Prog Neurobiol 155:96–119. https://doi.org/10.1016/j.pneurobio.2015.09.012
Acknowledgements
Not applicable.
Funding
Open Access funding enabled and organized by Projekt DEAL. I.F. received funding from a doctoral scholarship from the Leipzig Medical Faculty. The research leading to these results has received funding from the DFG, PP 2041 Computational Connectomics (MO 2249/3-1 and MO 2249/3-2) and the Alzheimer Forschung Initiative (AFI#18072) to M.M. N.W. was supported by the European Research Council under the European Union's Seventh Framework Programme (FP7/2007-2013)/ERC Grant Agreement No. 616905; the European Union's Horizon 2020 research and innovation programme under the Grant Agreement No. 681094; the BMBF (01EW1711A & B) in the framework of ERA-NET NEURON.
Author information
Authors and Affiliations
Contributions
M.M. and T.A. designed research; I.F., K.R., S.J., C.J., S.J., E.K., T.R. and M.M., performed research; I.F., T.R. E.K., S.J., E.K., M.S., J.S., J.M., N.W. and M.M. analysed data; and I.F., T.A., and M.M. wrote the paper. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Brains were provided by the Brain Banking Centre Leipzig of the German Brain-Net (GZ 01GI9999-01GI0299), operated by the Paul Flechsig Institute of Brain Research. The entire procedure of case recruitment, acquisition of the patients’ personal data, the protocols and the informed consent forms, performing the autopsy and handling the autopsy material have been approved by the responsible authorities (Approval # 282-02 and Approval # 205/17-ek).
Consent for publication
All authors have read the manuscript and indicated consent for publication.
Competing interests
The authors declare no competing financial interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Friedrich, I., Reimann, K., Jankuhn, S. et al. Cell specific quantitative iron mapping on brain slices by immuno-µPIXE in healthy elderly and Parkinson’s disease. acta neuropathol commun 9, 47 (2021). https://doi.org/10.1186/s40478-021-01145-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40478-021-01145-2