
Abstract
Oculopharyngodistal myopathy (OPDM) is a rare hereditary muscle disease characterized by progressive distal limb weakness, ptosis, ophthalmoplegia, bulbar muscle weakness and rimmed vacuoles on muscle biopsy. Recently, CGG repeat expansions in the noncoding regions of two genes, LRP12 and GIPC1, have been reported to be causative for OPDM. Furthermore, neuronal intranuclear inclusion disease (NIID) has been recently reported to be caused by CGG repeat expansions in NOTCH2NLC. We aimed to identify and to clinicopathologically characterize patients with OPDM who have CGG repeat expansions in NOTCH2NLC (OPDM_NOTCH2NLC). Note that 211 patients from 201 families, who were clinically or clinicopathologically diagnosed with OPDM or oculopharyngeal muscular dystrophy, were screened for CGG expansions in NOTCH2NLC by repeat primed-PCR. Clinical information and muscle pathology slides of identified patients with OPDM_NOTCH2NLC were re-reviewed. Intra-myonuclear inclusions were evaluated using immunohistochemistry and electron microscopy (EM). Seven Japanese OPDM patients had CGG repeat expansions in NOTCH2NLC. All seven patients clinically demonstrated ptosis, ophthalmoplegia, dysarthria and muscle weakness; they myopathologically had intra-myonuclear inclusions stained with anti-poly-ubiquitinated proteins, anti-SUMO1 and anti-p62 antibodies, which were diagnostic of NIID (typically on skin biopsy), in addition to rimmed vacuoles. The sample for EM was available only from one patient, which demonstrated intranuclear inclusions of 12.6 ± 1.6 nm in diameter. We identified seven patients with OPDM_NOTCH2NLC. Our patients had various additional central and/or peripheral nervous system involvement, although all were clinicopathologically compatible; thus, they were diagnosed as having OPDM and expanding a phenotype of the neuromyodegenerative disease caused by CGG repeat expansions in NOTCH2NLC.
Similar content being viewed by others


Introduction
Oculopharyngodistal myopathy (OPDM) is a rare adult-onset hereditary muscle disease clinically characterized by progressive ocular, pharyngeal, and distal limb muscle involvement and pathologically by rimmed vacuoles in muscle fibers [11, 20]. To date, two causative genes, LRP12 and GIPC1, were identified for OPDM (thereby, we refer to them as OPDM_LRP12 and OPDM_GIPC1, respectively). In both diseases, CGG repeat expansions in the noncoding regions of the corresponding genes were believed to be the cause, although the pathogenesis remained largely unclear [2, 6]. Recently, CGG repeat expansions in the noncoding region of NOTCH2NLC were reported as causative for neuronal intranuclear inclusion disease (NIID), a slowly progressive neurodegenerative disorder with eosinophilic intranuclear inclusions in the central and peripheral nervous systems and various organs [6, 13, 19]. Interestingly, certain patients with NIID manifested muscle weakness, dysarthria and dysphagia, and not fully but partially mimicking OPDM. Nevertheless, a diagnosis of OPDM has never been made in these patients most possibly because ocular symptoms, an essential feature of OPDM, were lacking [9, 14]. However, patients with OPDM were rarely reported to have accompanying sensorineural hearing loss and demyelinating neuropathy [3]. The presence of such patients with NIID and OPDM raised a possibility that OPDM in certain patients, particularly those with additional neurological manifestations, could be caused by the same pathogenic mechanism as NIID. Therefore, we evaluated CGG expansions in NOTCH2NLC in patients who were suspected to have OPDM.
Materials
Inclusion criteria of patients
The National Center of Neurology and Psychiatry (NCNP) functions as a referral center for muscle diseases in Japan, thus providing pathological and genetic diagnoses. Among the samples that were sent to the NCNP for diagnostic purposes from 1978 to 2020, we included 211 Japanese patients from 201 families who were clinically or clinicopathologically diagnosed with OPDM or oculopharyngeal muscular dystrophy (OPMD) based on a combination of at least two of ptosis, bulbar symptoms, or rimmed vacuoles on muscle biopsy but did not have the GCN repeat expansion in the polyadenylate binding protein nuclear 1 (PABPN1) gene, the causative factor for OPMD [1].
Methods
Genetic analysis
Note that 211 patients were screened for CGG expansions in NOTCH2NLC by repeat primed-PCR (RP-PCR), and the CGG repeat length in patients who had expanded CGG repeats was determined by fragment analysis and/or Southern blotting, as previously described [6, 13]. The CGG repeats in LRP12 and GIPC1 were also evaluated by RP-PCR, fragment analysis, and/or Southern blotting [2, 6].
Muscle histology
We re-reviewed the muscle pathology slides of a battery of histochemical stains that were prepared at the time of diagnosis. Immunohistochemical analysis of 8-µm-thick serial frozen sections was performed using the following primary antibodies: anti-poly-ubiquitinated proteins (FK1, BML-PW8805, 1:100; Enzo Life Sciences), anti-SUMO-1 (D-11:sc-5308, 1:50; Santa Cruz Biotechnology), anti-phospho-p62/SQSTM1 (Ser351) (PM074, 1:500; Medical & Biological Laboratories), and anti-caveolin-3 (sc7665, 1:200; Santa Cruz Biotechnology). After incubation with primary antibodies, the sections were incubated with DAPI (Cellstain DAPI, 1:1000; Fujifilm) and Alexa Fluor 488-, 568-, and 647-conjugated secondary antibodies (1:600; Invitrogen). Images were obtained using Keyence CCD camera (Keyence, Osaka, Japan). For evaluating regenerating fibers, we performed immunohistochemical analysis of serial frozen sections using neonatal myosin heavy chain (nMHC, NCL-MHCn, 1:20; Leica Biosystems Newcastle). We analyzed the frequency of regenerating fibers, fibers with internal nuclei, fibers with rimmed vacuoles, small angular fibers, and type 2C fibers in 200 randomly selected muscle fibers in each patient. Moreover, we analyzed the frequency of myonuclei positive with anti-p62, anti-poly-ubiquitinated protein, and anti-SUMO antibodies in 200 randomly selected myonuclei. Glutaraldehyde-fixed muscle sample for EM was available only from patient 2. On EM, we measured the diameter of 75 randomly selected intra-myonuclear filaments, which were straight, non-overlapped and well-demarcated.
Skin histology
Skin biopsy samples were available from two patients (patients 1 and 7). Serial sections that had a thickness of 4 µm were fixed in formalin and stained by hematoxylin and eosin (H&E) and anti-p62/SQSTM1 antibody (sc-28359, 1;200, Santa Cruz Biotechnology).
Results
We screened 211 patients with a clinical or clinicopathological diagnosis of OPDM or OPMD and identified seven unrelated patients with OPDM with CGG expansions in NOTCH2NLC (OPDM_NOTCH2NLC) using RP-PCR. Among them, five were sporadic while two had affected siblings (Fig. 1a). Repeat units were determined by fragment analysis in five patients (patients 1, 4, 5, 6, and 7) and by Southern blotting in two patients (patients 2 and 3) (Fig. 1a–c). All patients had > 100 repeat expansions, and patient 2 had two long CGG expansions, the longer one being approximately 674 repeats (Fig. 1a–c and Table 1). None of the seven patients had expanded CGG repeats in LRP12 and GIPC1.

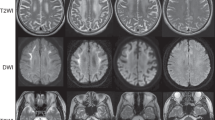
Clinical information of patients with CGG expansion in NOTCH2NLC. a Family trees of seven families. White circles and squares show unaffected indivisuals. Black circles and squares show patients. * shows probands. b Repeat primed PCR and fragment analysis for patient 1 and control. c Southern blotting analysis for CGG expansion in NOTCH2NLC. The arrows and arrowheads indicate the expanded alleles in patients 2 and 3, respectively. M, DNA marker. d Imaging of muscle CT from patient 2 shows asymmetric muscle atrophy. Right gluteus maximus, left vastus lateralis, adductor magnus, and rectus femoris are severely affected. e DWI of patient 1. f FLAIR image of patient 1. g DWI of patient 5. h FLAIR image of patient 5. i Marked fiber size variation, moderate endomysial fibrosis, and fibers with internal nuclei are seen on H&E. j Rimmed vacuole is present in a muscle fiber on mGT. k Some myofibers express neonatal myosin heavy chain. i–k Scale bars denote 50 µm
Table 1 summarizes the clinical information of the seven patients with CGG expansions in NOTCH2NLC. The age at onset extensively varied from infancy to 67 years. All seven patients had ptosis, ophthalmoplegia, dysarthria, limb muscle weakness, and decreased deep tendon reflex. Dysphagia and facial muscle weakness were detected in five (71%) patients. Five (71%) patients had predominantly distal limb muscle weakness and muscle atrophy, whereas one (14%) patient had proximal upper limb muscle weakness. In addition to these clinical manifestations typical of OPDM, all patients had central or peripheral nervous system abnormalities such as leukoencephalopathy, retinal pigmentary degeneration, ataxia, tremor, deafness, peripheral neuropathy, and increased CSF protein levels. The CK level was moderately increased (436–1886 U/L) in five (71%) patients. Patient 6 had left ventricular hypertrophy with first-degree atrioventricular block, whereas patient 2 had long QT syndrome.
Muscle CT data were available for three patients. One of three patients had asymmetric muscle involvement with fat replacement markedly in the left adductor magnus and moderately in the right gluteus maximus, left vastus lateralis, and left rectus femoris (Fig. 1d). The other two patients showed muscle atrophy in calf muscles. Brain MR examinations were available in five patients (Fig. 1e–h). High-intensity signals were observed in the middle cerebellar peduncles on fluid-attenuated inversion recovery (FLAIR) images in three patients (patients 1, 5, and 7). Furthermore, high signals on FLAIR were observed in the medial part of the cerebellar hemisphere right beside the vermis and cerebral white matter in two patients (patients 1 and 5). High signals were noted along the corticomedullary junction on diffusion-weighted imaging (DWI) in two patients (patients 1 and 5). Moreover, moderate to marked ventricular enlargement was observed in three of five patients.
On muscle histology, all patients had fibers with rimmed vacuoles and small angular fibers (Fig. 1i, j, and Table 2). The variation in fiber size was moderate to marked in six patients and mild in one patient. Six patients had regenerating fibers, and one patient had no necrotic or regenerating fibers (Fig. 1k and Table 2). Only one patient (patient 3) had moderate endomysial fibrosis, whereas the other patients had no or only minimal endomysial fibrosis (Fig. 1i and Table 2). Intra-myonuclear inclusions, primarily located in the center of the myonuclei, were detected using anti-poly-ubiquitinated protein, anti-SUMO1, and anti-phospho-p62/SQSTM1 antibodies in all patients (Fig. 2a–l). EM analysis of the muscle from patient 2 revealed tubulofilamentous inclusions without limiting membrane but with low-electron density halo around the nucleus, located in the center of the myonuclei (Fig. 2m,n). The diameter of the 75 randomly chosen filaments was 12.6 ± 1.6 nm (M ± SD).

Pathology, revealed by electron microscopy and disease spectrum. Anti-poly-ubiquitinated protein antibody (a), anti-caveolin-3 antibody (b, f, j), DAPI (c, g, k), anti-SUMO-1 antibody (e), and anti-p62 (i) are stained. d, h, l show merged immunohistochemistry. a–l Scale bars denote 10 µm. On EM, a longitudinal section shows tubulofilamentous inclusions within the myonuclei together with markedly disorganized myofibrils (arrow) in the surrounding area (m, n). m Scale bar denotes 5 µm. n Scale bar shows 500 nm. o, q HE, p, r p62 are stained on serial sections of the skin sample in patient 1. Intranuclear inclusions with p62-positive in sweat gland cells (p) and adipocytes (r) are seen. o–r Scale bars denote 20 µm. s Disease spectrum caused by CGG expansion in NOTCH2NLC
Skin biopsies from patients 1 and 7 showed p62-positive intranuclear inclusions (Fig. 2o–r).
Discussion
NIID is a slowly progressive neurodegenerative disorder that is pathologically characterized by eosinophilic hyaline intranuclear inclusions in the central and peripheral nervous systems, as well as in the visceral organs and skin. This disorder has been considered to be a heterogeneous disease because of the highly variable clinical manifestations [14]. Recent studies reported noncoding CGG repeat expansions in NOTCH2NLC as the causative factor for NIID [6, 13, 19]. Subsequently, various diseases have been associated with CGG repeat expansions in NOTCH2NLC, including multiple system atrophy (MSA), leukoencephalopathy, Alzheimer’s disease and frontotemporal dementia (AD/FTD), tremor, and retinal dystrophy, suggesting that the spectrum of NOTCH2NLC diseases is, in fact, wide (Fig. 2s) [4, 5, 7, 10, 17].
Interestingly, certain patients with NIID demonstrate additional myopathic features such as limb muscle weakness, dysphagia, and dysarthria [9, 14]. However, neither ptosis/ophthalmoplegia, a clinical hallmark of OPDM, nor rimmed vacuole, a pathological hallmark of OPDM, has never been described in any histologically or genetically confirmed NIID patient [14, 18]. Not surprisingly, to our knowledge, no patients with NIID have ever been clinicopathologically diagnosed with OPDM and vice versa.
Although our patients with OPDM_NOTCH2NLC were clinicopathologically diagnosed with OPDM, they had additional clinical manifestations that were partially reminiscent of NIID, including leukoencephalopathy and retinal degeneration. Moreover, one and three patients had tremor and ataxia. MRI results in two of five patients revealed high-intensity signals in the middle cerebellar peduncles and in the paravermal area on FLAIR images and in the corticomedullary junction on DWI, similar to those in patients with NIID (Fig. 1e–h) [6, 12, 16].
The identification of patients with OPDM_NOTCH2NLC suggests that CGG expansions in NOTCH2NLC result in at least two different diseases, namely NIID and OPDM. Nevertheless, because our patients had peripheral and/or central nervous system involvement together with the clinicopathological features of OPDM, which fills the phenotypic gap between the two diseases, they are most likely in a broad phenotypic spectrum of a single neuromyodegenerative disease rather than in two distinct diseases (Fig. 2s). The identification of the intranuclear inclusions in skin biopsy from two patients with OPDM_NOTCH2NLC, which is a diagnostic finding of NIID [15], as well as supports this notion further.
A recent study reported that patients with the muscle subtype of NIID had longer CGG repeat expansions from 118 to 517 repeats than those with other NIID subtypes [19]. Indeed, the CGG repeats in our patients ranged from 116 to 674. Interestingly, the patient (patient 2) carrying 674 repeats, although with mosaicism, had milder phenotype than the patient (patient 1) carrying 139 repeats who had an early-onset and severe phenotype, suggesting that there was no apparent correlation between the size of the CGG repeats and the clinical symptoms in OPDM_NOTCH2NLC.
Diagnostically, the presence of intranuclear inclusions stained with anti-poly-ubiquitinated protein, anti-SUMO1, and anti-p62 antibodies in the skin and other organs is pathognomonic of NIID [15]. In this study, we confirm the presence of essentially the same type of intranuclear inclusions in muscles, further suggesting that OPDM and NIID may be in the same spectrum with the identical degenerative process; although different organs, such as skeletal muscle, skin, and CNS, are affected in variable degrees. Interestingly, the average diameter of the intranuclear filaments was 12.6 nm on EM, which was similar to that observed in neuronal cells in patients with NIID (8–16 nm) [8, 21], suggesting that the underlying mechanism of the myodegeneration in OPDM_NOTCH2NLC and the neurodegeneration in NIID could be identical.
In terms of the pattern of muscle involvement on muscle imaging, one study described predominantly involved soleus and long head of the biceps femoris, relatively preserved rectus femoris, and asymmetric involvement pattern in OPDM (without genetic diagnosis) [22]. Another study reported markedly involved soleus in patients with OPDM_GIPC1 [2]. The results of muscle CT in the present three patients with OPDM_NOTCH2NLC are similar to previous reports [2, 22], suggesting that the myodegeneration mechanism for those three OPDM types may be similar.
In conclusion, our seven Japanese patients with OPDM_NOTCH2NLC exhibit distinct clinicopathological features, including the involvement of central and peripheral nervous systems. Our results widen the phenotypic spectrum of a neuromyodegenerative disease caused by CGG repeat expansions in NOTCH2NLC.
Availability of data and material
The data supporting the results of this study are available from the corresponding author upon request.
Abbreviations
- OPMD:
-
oculopharyngeal muscular dystrophy
- OPDM:
-
oculopharyngodistal myopathy
- OPDM_NOTCH2NLC:
-
OPDM patients who have CGG repeat expansions in NOTCH2NLC
- NCNP:
-
the National Center of Neurology and Psychiatry
- NIID:
-
neuronal intranuclear inclusion disease
- PABPN1:
-
polyadenylate binding protein nuclear 1
- MSA:
-
multiple system atrophy
- AD/FTD:
-
leukoencephalopathy, Alzheimer’s disease and frontotemporal dementia
References
Brais B, Bouchard JP, Xie YG, Rochefort DL, Chretien N, Tome FM, Lafreniere RG, Rommens JM, Uyama E, Nohira O et al (1998) Short GCG expansions in the PABP2 gene cause oculopharyngeal muscular dystrophy. Nat Genet 18:164–167. https://doi.org/10.1038/ng0298-164
Deng J, Yu J, Li P, Luan X, Cao L, Zhao J, Yu M, Zhang W, Lv H, Xie Z et al (2020) Expansion of GGC repeat in GIPC1 is associated with oculopharyngodistal myopathy. Am J Hum Genet 106:793–804. https://doi.org/10.1016/j.ajhg.2020.04.011
Durmus H, Laval SH, Deymeer F, Parman Y, Kiyan E, Gokyigiti M, Ertekin C, Ercan I, Solakoglu S, Karcagi V et al (2011) Oculopharyngodistal myopathy is a distinct entity: clinical and genetic features of 47 patients. Neurology 76:227–235. https://doi.org/10.1212/WNL.0b013e318207b043
Fang P, Yu Y, Yao S, Chen S, Zhu M, Chen Y, Zou K, Wang L, Wang H, Xin L et al (2020) Repeat expansion scanning of the NOTCH2NLC gene in patients with multiple system atrophy. Ann Clin Transl Neurol 7:517–526. https://doi.org/10.1002/acn3.51021
Hayashi T, Katagiri S, Mizobuchi K, Yoshitake K, Kameya S, Matsuura T, Iwata T, Nakano T (2020) Heterozygous GGC repeat expansion of NOTCH2NLC in a patient with neuronal intranuclear inclusion disease and progressive retinal dystrophy. Ophthal Genet 41:93–95. https://doi.org/10.1080/13816810.2020.1723119
Ishiura H, Shibata S, Yoshimura J, Suzuki Y, Qu W, Doi K et al (2019) Noncoding CGG repeat expansions in neuronal intranuclear inclusion disease, oculopharyngodistal myopathy and an overlapping disease. Nat Genet 51:1222–1232. https://doi.org/10.1038/s41588-019-0458-z
Jiao B, Zhou L, Zhou Y, Weng L, Liao X, Tian Y et al (2020) Identification of expanded repeats in NOTCH2NLC in neurodegenerative dementias. Neurobiol Aging 89(e141–2):e147. https://doi.org/10.1016/j.neurobiolaging.2020.01.010
Malandrini A, Villanova M, Tripodi S, Palmeri S, Sicurelli F, Parrotta E et al (1998) Neuronal intranuclear inclusion disease: neuropathologic study of a case. Brain Dev 20:290–294. https://doi.org/10.1016/s0387-7604(98)00032-1
O’Sullivan JD, Hanagasi HA, Daniel SE, Tidswell P, Davies SW, Lees AJ (2000) Neuronal intranuclear inclusion disease and juvenile parkinsonism. Mov Disord 15:990–995. https://doi.org/10.1002/1531-8257(200009)15:5%3c990:aid-mds1035%3e3.0.co;2-i
Okubo M, Doi H, Fukai R, Fujita A, Mitsuhashi S, Hashiguchi S et al (2019) GGC repeat expansion of NOTCH2NLC in adult patients with leukoencephalopathy. Ann Neurol 86:962–968. https://doi.org/10.1002/ana.25586
Satoyoshi E, Kinoshita M (1977) Oculopharyngodistal myopathy. Arch Neurol 34:89–92. https://doi.org/10.1001/archneur.1977.00500140043007
Sone J, Kitagawa N, Sugawara E, Iguchi M, Nakamura R, Koike H et al (2014) Neuronal intranuclear inclusion disease cases with leukoencephalopathy diagnosed via skin biopsy. J Neurol Neurosurg Psychiatry 85:354–356. https://doi.org/10.1136/jnnp-2013-306084
Sone J, Mitsuhashi S, Fujita A, Mizuguchi T, Hamanaka K, Mori K et al (2019) Long-read sequencing identifies GGC repeat expansions in NOTCH2NLC associated with neuronal intranuclear inclusion disease. Nat Genet 51:1215–1221. https://doi.org/10.1038/s41588-019-0459-y
Sone J, Mori K, Inagaki T, Katsumata R, Takagi S, Yokoi S et al (2016) Clinicopathological features of adult-onset neuronal intranuclear inclusion disease. Brain 139:3170–3186. https://doi.org/10.1093/brain/aww249
Sone J, Tanaka F, Koike H, Inukai A, Katsuno M, Yoshida M et al (2011) Skin biopsy is useful for the antemortem diagnosis of neuronal intranuclear inclusion disease. Neurology 76:1372–1376. https://doi.org/10.1212/WNL.0b013e3182166e13
Sugiyama A, Sato N, Kimura Y, Maekawa T, Enokizono M, Saito Y et al (2017) MR imaging features of the cerebellum in adult-onset neuronal intranuclear inclusion disease: 8 cases. AJNR Am J Neuroradiol 38:2100–2104. https://doi.org/10.3174/ajnr.A5336
Sun QY, Xu Q, Tian Y, Hu ZM, Qin LX, Yang JX et al (2020) Expansion of GGC repeat in the human-specific NOTCH2NLC gene is associated with essential tremor. Brain 143:222–233. https://doi.org/10.1093/brain/awz372
Tateishi J, Nagara H, Ohta M, Matsumoto T, Fukunaga H, Shida K (1984) Intranuclear inclusions in muscle, nervous tissue, and adrenal gland. Acta Neuropathol 63:24–32. https://doi.org/10.1007/bf00688467
Tian Y, Wang JL, Huang W, Zeng S, Jiao B, Liu Z et al (2019) Expansion of human-specific GGC repeat in neuronal intranuclear inclusion disease-related disorders. Am J Hum Genet 105:166–176. https://doi.org/10.1016/j.ajhg.2019.05.013
van der Sluijs BM, ter Laak HJ, Scheffer H, van der Maarel SM, van Engelen BG (2004) Autosomal recessive oculopharyngodistal myopathy: a distinct phenotypical, histological, and genetic entity. J Neurol Neurosurg Psychiatry 75:1499–1501. https://doi.org/10.1136/jnnp.2003.025072
Woulfe JM (2007) Abnormalities of the nucleus and nuclear inclusions in neurodegenerative disease: a work in progress. Neuropathol Appl Neurobiol 33:2–42. https://doi.org/10.1111/j.1365-2990.2006.00819.x
Zhao J, Liu J, Xiao J, Du J, Que C, Shi X et al (2015) Clinical and muscle imaging findings in 14 mainland Chinese patients with oculopharyngodistal myopathy. PLoS ONE 10:e0128629. https://doi.org/10.1371/journal.pone.0128629
Acknowledgements
We thank the patients and their families whose help and participation made this work possible. We thank Dr Noriko Sato for the evaluation of MRI. We also thank Ms. Kaoru Tatezawa, Ms. Naho Fushimi, Ms. Kazu Iwasawa, Ms. Yoko Tsutsumi, and Ms. Kanae Kanna in NCNP for their technical assistance.
Funding
This study was supported partly by Intramural Research Grant (2-5 and 29-4 to IN; 2-5 and 30-9 to AI) for Neurological and Psychiatric Disorders of NCNP and by AMED under Grant Nos. 20ek0109490h0001 and JP19ek0109285h0003 (to IN) and Joint Usage and Joint Research Programs, the Institute of Advanced Medical Sciences, Tokushima University (2019, A9, 2020, 2A19 to AI).
Author information
Authors and Affiliations
Contributions
MO, TK, and IN designed studies; AI completed genetic analysis and analysis of data; ZW contributed to PCR methods; MO, HN, SH, SN, AO and IN evaluated muscle pathology; YO, HK, AN, RA, HT, RH, HD, and FT contributed to the clinical diagnosis and muscle biopsy of patients with OPDM; MO, AI, SH, SN and IN wrote and edited the manuscript; and IN supervised the project; All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
All patients provided informed consent for using their samples for research after the diagnosis. This study was approved by the ethical committees of the NCNP (A2019-123).
Competing interests
The authors report no competing interests.
Clinical information
For patients with CGG expansions in NOTCH2NLC, clinical information that included imaging data of muscle and brain provided by the physicians at the time of muscle biopsy and/or genetic analysis was reviewed.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Ogasawara, M., Iida, A., Kumutpongpanich, T. et al. CGG expansion in NOTCH2NLC is associated with oculopharyngodistal myopathy with neurological manifestations. acta neuropathol commun 8, 204 (2020). https://doi.org/10.1186/s40478-020-01084-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40478-020-01084-4