
Abstract
Microsatellites, or simple tandem repeat sequences, occur naturally in the human genome and have important roles in genome evolution and function. However, the expansion of microsatellites is associated with over two dozen neurological diseases. A common denominator among the majority of these disorders is the expression of expanded tandem repeat-containing RNA, referred to as xtrRNA in this review, which can mediate molecular disease pathology in multiple ways. This review focuses on the potential impact that simple tandem repeat expansions can have on the biology and metabolism of RNA that contain them and underscores important gaps in understanding. Merging the molecular biology of repeat expansion disorders with the current understanding of RNA biology, including splicing, transcription, transport, turnover and translation, will help clarify mechanisms of disease and improve therapeutic development.
Similar content being viewed by others


Introduction
In 1991 it was discovered that a microsatellite sequence expansion is the cause of two distinct neurological disorders, Fragile X syndrome (FXS) [303] and spinal bulbar muscular atrophy (SBMA), or Kennedy's disease [167]. Since then, simple repeat sequence expansions have been associated with over twenty more neurological disorders [166, 300, 333] (Table 1). What has been learned is that microsatellite expansions may cause disease in multiple ways. For nearly all of these neurological disorders, however, disease includes production of RNA that contains the aberrant repeat expansion sequence. Accordingly, the leading disease mechanisms involve repeat expansion RNA-mediated sequestration of critical RNA-binding proteins and translation of repeat expansion RNA into toxic repetitive polypeptides.
Tremendous progress has been made in understanding the metabolism of expanded tandem repeat-containing RNA (xtrRNA). Nonetheless, various gaps in our understanding of the underlying molecular biology and pathology remain, which in turn limits identification of promising therapeutic approaches. The goal of this review is to help address these gaps by discussing the potential impact of xtrRNA on cellular RNA metabolism. We begin with an overview that covers microsatellite origin, evolution, and expansion. We then follow xtrRNA through its life cycle, beginning with transcription and continuing through splicing, folding, protein interactions, localization, turnover, and translation. We rationalize the logic of current molecular disease models, note where important mechanistic information is lacking, and emphasize new pathways to consider for mechanistic insight. We use this discussion to also highlight areas where therapeutic intervention may be useful.
Origin and expansion of microsatellites in human disease
Simple tandem repeat sequences in the human genome
Microsatellite sequences comprise approximately 3% of the human genome, about twice as much as protein coding sequence [1, 171]. Microsatellites, interchangeably known as simple or short tandem repeats (STRs), are usually defined as simple sequence motifs of one to six nucleotides that are contiguously repeated at least a few times [24, 69]. Microsatellites occur throughout the genome, but are predominantly found in noncoding promoters, introns, 5' and 3' untranslated regions (UTRs), and intergenic regions [236, 257, 291]. Intergenic microsatellites seem to fit neutral evolution models, although not perfectly [69], and are among the most variable genomic sequences [32]. Therefore, they serve as the basis for forensic DNA analyses and as markers for population genetics studies [24, 61, 65, 236].
The origin and evolution of microsatellites is incompletely understood. They may have derived from simple and repetitive sequence motifs found in mobile genetic elements, such as non-LTR (long terminal repeat) retrotransposons like Alu and L1 [6, 51, 95]. Transposable elements have colonized the human genome extensively and their remains have undergone mutation and replication, providing the starting material for simple tandem repeats [69]. The dense repetitive sequence of centromeres and telomeres is proposed to originate from incorporation of mobile genetic elements during early eukaryotic evolution [87, 304]. Small sequence duplications of these simple sequences can further produce microsatellites with multiple repeats. The STRs that are expanded in Friedreich ataxia (FRDA) and myotonic dystrophy type 2 (DM2), for example, have been traced to an Alu element origin [41, 163]. In contrast, de novo genesis by events like random mutation, replication slippage, and duplication of unique sequence may also account for the birth of microsatellite sequences [28, 69]. STRs have been shown to have positive roles in evolution, such as bacterial resistance to antibiotics and circadian clock adaptation to the environment in Neurospora crassa and Drosophila melanogaster [83, 133, 211, 258, 279, 307]. The placement of microsatellite sequences in and near regulatory and coding regions of the genome also implicate them in control of gene expression and genetic interactions [133, 331].
Expansion of simple tandem repeats
There are several distinct mechanisms that can contribute to the expansion of naturally occurring microsatellites. In this section we provide a brief overview of these mechanisms. Many excellent reviews on this topic are cited in this section and recommended for further reading (see [84, 130, 149, 206, 212, 260, 297, 331, 333]).
A major source of microsatellite expansion in dividing cells is DNA replication, although mitotic recombination is also recognized as a contributing factor [84, 149, 212, 242]. During replication, repetitive sequences can cause problems at the replication fork and result in fork reversals or template switching, which can insert extra repeats [76, 144, 149, 212]. At the strand level, polymerase slipping can cause expansions in the leading or lagging strand [84, 137, 144, 149]. Repeats may also induce imperfect Okazaki fragment ligation and add repeats in the lagging strand [81, 93, 278]. The pathway followed for expansion of a repeat has been proposed to be a balancing act between several factors [84, 149, 212]. These include relative repeat length, the stability and types of non-canonical structures the repeat sequence can form, and nearby flanking sequences. After repeat sequences are added to one or both strands, the daughter strands reanneal. Misalignment and slippage will occur and extra sequences will bulge out to form non-canonical (non-B-form) structures like hairpins or quadruplexes [237, 331]. If these structures persist to the next round of replication, or if they undergo flawed repair, they can result in permanent expansions [130, 149, 212, 260, 297]. During DNA recombination, which repairs single-end or double-strand breaks, unequal crossing over or template switching can cause misalignments and introduction of additional repeats [208, 242, 306].
Repeat expansion events are intimately tied to the repair of non-canonical DNA structures and DNA damage. Multiple DNA damage control pathways have been implicated, including mechanisms that replace DNA bases, like base excision repair (BER) or nucleotide excision repair (NER), especially as sources for repeat expansion in non-dividing cells [206]. However, mismatch repair (MMR) has been argued to be a primary driver of repeat expansion [75, 106, 130, 260, 271]. MMR expands repeats through recognition and processing of unusual DNA structures, such as small bulges and hairpins [260], via the enzyme MutSβ (MSH2-MSH3 complex) [130, 260, 334]. The processing and damage rectification steps are carried out by MutSβ and associated proteins, including the MutLα (MLH1-PMS2 complex) or MutLγ (MLH1-MLH3 complex) endonucleases that help remove DNA lesions [106, 130, 241]. Polymerases like Polβ are then recruited, which can insert extra repeats due to flawed priming or templating [33, 190].
An important question is how repeats are able to expand out of control, sometimes into the hundreds or thousands of perfect tandem copies, without accumulating significant interruptions? Microsatellites that are evolutionarily neutral, typically in intergenic regions, become highly mutable when they exceed thresholds above just a few tandem repeats [68, 95, 320]. Therefore, the likelihood of remaining as a perfect tandem repeat without interruption is expected to decrease with tandem repeat length. This suggests that accumulation of large expansions must either occur quickly, before mutations can accumulate, or their disruption must be guarded against [320]. Genic regions of the genome, where all currently known disease-associated repeat expansions occur [31, 236] (Table 1), seem to enjoy special favor through positive evolutionary selection processes that protect sequence fidelity [191, 236, 284]. However, it seems unlikely that this would contribute significantly to large repeat expansions. For example, non-repetitive codons would presumably be preferred and selected over unstable repeat codons.
Mechanisms have been proposed that could provide large expansions in a single step, including template switching replication models where repeats are already sufficiently large enough [225, 266] and out-of-register synthesis during homologous recombination-based repair of double-strand breaks (DSBs) [212, 242, 249, 250, 283]. One intriguing mechanism for rapid and large repeat accumulation is break-induced replication (BIR) [148, 176]. BIR is a homologous recombination pathway that can rescue collapsed or broken replication forks [195]. It is induced when a replisome collides with a broken single-end DSB [189]. BIR is also believed to be selective for structure-prone or GC-rich repeats that are long enough to form stable structures [148]. In this mechanism of expansion, stable structures would cause fork reversals. Resolution of these four-way junction structures would result in a one-ended DSB. To restart the fork, the one-ended DSB invades the sister chromatid to form a D-loop, but likely does so out-of-register because of the repetitive sequence, thus leading to expansion. While this BIR study was performed in yeast, the results are expected to translate to human cells [176].
Incremental expansions, such as those caused by MMR, are typically on the order of 1-3 repeats at a time [260]. Could these events generate large uninterrupted expansions? Rapid accumulation of expansions via MMR or other DNA damage repair pathways might be facilitated by transcription across the repeat. It has been shown that transcription is required for expansion of the CGG repeat in a mouse model of FXS [2, 333]. Several studies have shown that transcription at repeat expansions is associated with repeat instability, possibly via formation of DNA-RNA hybrids, or R-loops [180, 181, 183, 195, 223, 246, 333]. It is possible that these events could allow DNA damage. One report has shown a correlation between R-loop formation and replication fork stalling, offering a familiar mechanism for repeat expansion through DNA replication [86, 109]. Alternative mechanisms might involve oxidation of free DNA strands, or simple misalignment upon strand reannealing, to signal DNA damage repair [180]. The latter model suggests that GC-rich or structure-prone repeats would be more susceptible to expansion during transcription, which might explain why transcription levels alone are not predictive of expansion [333]. Thus, cycles of transcription and R-loop formation might accelerate repeat expansion for structure-prone repeats via ongoing DNA damage repair [180]. A cell-based model where transcription levels or R-loop formation could be controlled, repeat sequence and size altered, expansions monitored, and DNA damage repair mechanisms systematically tested (perhaps building on HeLa cell models recently described [187]) might allow more direct testing of these ideas.
A common theme among sources of repeat instability and expansion is DNA metabolism associated with strand separation and reannealing at microsatellite sequences. These events can lead to formation of non-canonical structures and recruitment of DNA damage responses that ultimately and inadvertently add more repeats. Thus, mechanisms meant to maintain and protect the genome can also lead to large tandem repeat expansions and cause human disease [11, 130, 333].
Microsatellite repeat expansion disorders
Since it was first discovered that microsatellite expansions can cause disease, at least two dozen microsatellite repeat expansion disorders have been subsequently reported (Table 1). The latest discoveries are autism spectrum disorders caused by expansions in fragile 7A (FRA7A) and fragile 2A (FRA2A) fragile site loci [209, 210]. Comparing and contrasting these disorders can highlight several trends. Almost half of the microsatellite expansion disorders result from CAG trinucleotide expansions, mostly occurring in coding exons. All STRs for known repeat expansion disorders are GC-rich except for the trinucleotide GAA repeat of FRDA and the ATTCT and TGGAA pentanucleotide repeats of spinocerebellar ataxia 10 (SCA10) and 31 (SCA31), respectively. In this review we focus on large microsatellite repeat expansions that are transcribed into RNA, a feature that is shared by nearly all repeat expansion disorders (Table 1).
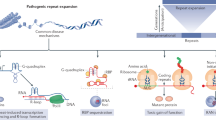
Microsatellite expansions cause disease through two broad molecular mechanisms (Fig. 1): loss-of-function for the associated gene or gain-of-function for the repeat expansion sequence. In loss of function mechanisms, gene expression can be silenced at the transcriptional level, such as by epigenetic modification, resulting in the complete loss of that gene's normal functions [70, 112]. Alternatively, the affected gene may lose function at the protein level by the introduction of unusually long polypeptide tracts in the translated protein product (Fig. 1) [168, 268]. In gain-of-function mechanisms the repetitive polypeptide can take on new roles, such as protein aggregation. Many of these mutant misfolded proteins cannot be degraded efficiently and will accumulate in cellular aggregates or inclusions [48, 168, 332]. Aggregation also tends to sequester proteins and critical cellular components and is taxing on cellular proteostasis [48]. The xtrRNA can also acquire gain of function mechanisms, primarily through interaction with nucleic acid-binding proteins (Fig. 1). The repetitive xtrRNA forms length-dependent focal aggregates in cell nuclei in several diseases [35, 59, 196, 262, 311]. Loss-of-function and gain-of-function mechanisms can result in complicated molecular disease pathologies and some disorders can simultaneously exhibit multiple mechanisms (Table 1).

Distinct loss-of-function and gain-of-function mechanisms of disease for various repeat expansion disorders. Repeat expansions can occur in 5’ or 3’ UTRs, exons, or introns. Expanded tandem repeat-containing RNA (xtrRNA) may not be transcribed due to epigenetic silencing, thereby causing loss of gene function. If transcribed, xtrRNA may become trapped in the cell nucleus where it can form focal aggregates and functionally deplete important RNA binding proteins. The xtrRNA may also be exported to the cytoplasm where it can undergo translation to produce repeat-containing polypeptides that disrupt cellular processes. In some cases, xtrRNA can form focal nuclear aggregates and also be translated into repeat-containing polypeptides. Repeat-containing polypeptides can be toxic in multiple ways, including insoluble aggregation, blocking normal host protein function, inhibiting nucleocytoplasmic transport, and disrupting other critical cellular functions
Transcription and splicing at simple tandem repeat expansions
Transcribing repeat expansion sequences
Repeat expansion sequences are known to inhibit or impede RNA Polymerase II (Pol II) initiation or elongation either directly or via induction of a repressed chromatin state [100]. Expansions like the GAA repeat in FRDA [19, 94, 97, 162, 231], the CTG repeat in myotonic dystrophy type 1 (DM1) [25], the GGGGCC repeat in C9ORF72-associated frontotemporal dementia and amyotrophic lateral sclerosis (C9FTD/ALS) [108], and the CGG repeat in FXS (also known as FRAXA) [44, 285] have all been implicated in reduced or silenced transcription. For FXS [230, 298], Fragile XE (FRAXE) [18], FRDA [97], FRA2A [210] and FRA7A [209], transcription appears to be blocked or significantly reduced by DNA methylation of the repeat expansion or nearby CpG islands. However, although transcription may be well below basal levels, it is possible that xtrRNA can still contribute to disease in some cases [44].
Slowed or stalled transcription across repeat expansions may lead to R-loops, which further slow transcription [123] and inadvertently contribute to deposition of repressive chromatin marks and silence transcription (Fig. 2) [44, 99, 316, 317]. R-loops play important roles in biology, such as immunoglobulin class switching [323], keeping CpG islands unmethylated [91, 254], and defining transcription termination signals [254, 270]. R-loop formation is common in transcription of C-rich template sequences [324], which most disease-associated repeat expansion genomic loci possess. The impact of R-loop formation on disease at repeat expansions is still unclear. Whether R-loop formation will trigger DNA methylation, transcriptional silencing, or other events may be dependent upon a number of factors specific to the affected gene or locus.

Effects of repeat expansion sequence on transcription. Repeat expansion sequences can perturb transcription by a epigenetic silencing, b inducing or facilitating bidirectional transcription, c reduced transcription kinetics, or d generating transcripts that can potentially be processed into small RNAs that could guide degradation or silencing of various complementary RNAs, including the xtrRNA itself
Bidirectional transcription of repeat expansions
Bidirectional transcription has been reported to occur in DM1, C9FTD/ALS, Huntington's disease (HD), spinocerebellar ataxia 8 (SCA8), and Huntington's disease-like 2 (HDL2), among other diseases [26, 40, 126, 312]. Slowed transcription across a repeat may also be able to induce antisense transcription of the non-template DNA strand via R-loop formation [270]. For example, FRDA-associated GAA repeat expansion sequences were shown to initiate transcription and act as promoters in yeast [330]. However, many genes exhibit bidirectional transcription [293] and in microsatellite diseases bidirectional transcription typically initiates outside of the repeat (Fig. 2) [26, 113]. Bidirectional transcription across repeats can also result in double R-loops that amplify repeat instability and accelerate methylation and transcriptional silencing [181, 183, 223]. Antisense transcription can often interfere with transcription of the coding gene [145]. Most relevant to this review is the production of two xtrRNAs from bidirectional transcription and the potential to synthesize repetitive polypeptides from both xtrRNA. For example, in C9FTD/ALS both xtrRNAs form nuclear foci [59, 90, 248, 340] that sequester RNA-binding proteins [47, 175, 217] and are translated into repetitive polypeptides [9, 216], highlighting the importance of bidirectional transcription to molecular disease pathology.
The role of Supt4h in xtrRNA transcription
Transcribing microsatellite expansions into xtrRNA requires processivity across repetitive sequence tracts that can have very high GC content. The 5,6-dichloro-1-β-D-ribofuranosylbenzimidazole (DRB) sensitivity-inducing factor (DSIF), composed of Supt4h and Supt5h proteins (Spt4 and Spt5 in yeast), aids RNA Polymerase II (Pol II) in transcription elongation and transcription rate [305, 308]. The DSIF complex is important for traversing sequences that elicit pausing of RNA Pol II [305] and has been identified as a factor involved in the transcription of RNA containing large simple repeat sequences. For example, transcription of repeat-containing RNA from the huntingtin and C9ORF72 genes significantly decreases when Supt4h is deleted or knocked-down [159, 186]. Supt5h is a conserved transcription factor with a homolog known as NusG in bacteria that is important for elongation and processivity [185, 202]. Supt5h binds directly to the clamp coiled-coil domain of RNA Pol II while Supt4h interacts through contact with Supt5h [17, 119, 201]. Together, the DSIF complex interacts with the DNA template outside of the transcription bubble [17, 50, 151, 185]. Supt4h has a zinc-finger domain that may be important for modulating DNA interactions of DSIF [308], and thereby improve processivity by maintaining RNA Pol II template interaction during periods of extended pausing [50, 151, 309]. Long repetitive sequences prone to formation of secondary structure in the transcription bubble, such as repeat-induced hairpin or R-loop structures, may represent prime sites for pausing or backtracking [251, 260, 333].
DSIF is also used by RNA Pol I to presumably ensure robust transcription of abundant and repetitive ribosomal RNA [122, 309]. It is worth noting that repeat expansions might occur in ribosomal RNA genes but they have either not been characterized or have not been associated with disease [122]. In contrast, RNA Pol III, which only transcribes relatively small noncoding RNA genes, does not interact with the DSIF complex [309]. Thus, transcription is unlikely to be successful if large microsatellite expansions occur in the small RNA genes transcribed by RNA Pol III. These observations may lend some rationale as to why all disease-associated repeat expansions to date are associated with Pol II-transcribed genic regions [7, 31, 236].
Splicing of xtrRNA
Splicing involves several regulated steps, many accessory factors and the spliceosome, a complex multi-component enzyme. There is currently a lack of mechanistic insight regarding how the splicing apparatus reacts when encountering pre-mRNA containing large repetitive sequence tracts [14]. Since introns can be excessively large while still allowing productive and accurate splicing [263], the size of the repeat expansion itself is not expected to significantly impede splicing. However, transcription rates across microsatellite expansions can be reduced, which can influence alternative splicing [58, 270], and stem loop structures in large pre-mRNA introns have been predicted to affect splicing [263].
Examples of microsatellite repeat expansions modulating splicing include the GAA repeat expansion associated with FRDA. When placed near reporter gene exons or in the first intron of a frataxin minigene system, the GAA repeat caused complex splicing defects and accumulation of aberrant splice products [15]. The mechanism proposed involved binding of various splicing factors to the GAA repeat-containing transcripts [15]. In C9FTD/ALS, the intronic GGGGCC repeat has been implicated in splicing by favoring retention of the intron-containing repeat, suggesting a mechanism by which C9ORF72 xtrRNA can escape to the cytoplasm for translation [227]. Expanded CAG repeats of HD are also linked to production of short alternatively spliced forms of the huntingtin mRNA that contain the CAG repeat expansion and add to the production of toxic polyglutamine protein [255].
Potential impact on splicing factors
If repeat expansion sequences can mimic the binding motif of splicing regulators, they could recruit splicing factors and affect splice site selection. In DM1 the MBNL family of splicing factors and CUG binding proteins (CUGBPs) have an affinity for repetitive CUG and CAG sequence. Although the splicing of DM1 protein kinase (DMPK) mRNA does not appear to be affected by the CUG repeat expansion that it contains, the splicing pattern of an antisense transcript across the DMPK repeat, which contains a CAG repeat, appears to be altered by the expansion [105]. In HD the expanded CAG repeats have been proposed to interact with the splicing factor SRSF6, which is believed to contribute to altered splicing to generate truncated repeat-containing huntingtin mRNA [255].
Repeat expansion sequences in xtrRNA could also alter splicing by recruiting factors that are not typically involved in splicing. These factors might modulate splice site selection or spliceosome activity by changing local ribonucleoprotein (RNP) structure or access to splice signals [67]. The repetitive structural nature of repeat expansions could also sterically hinder access to splice signals, depending on their proximity to splice enhancer or silencer elements. Alternative splicing is a complicated interplay of modular protein and RNA interactions that are difficult to predict at present and local sequence and context will likely be important for understanding the impact of expanded repeats on splicing [14].
Therapeutic approaches to control xtrRNA transcription and splicing
Characterizing the effect of microsatellite expansions on transcription and splicing will directly benefit therapeutic approaches for repeat expansion disorders. Proof-of-principle methods to locally disrupt the interactions of xtrRNA at repeat expansion loci, such as R-loops, have been demonstrated for FXS and FRDA using small molecules and nucleic acids [44, 177]. Disrupting the interaction of Spt4 and Spt5, or modulating Spt4 function, could provide a therapeutic avenue for a number of repeat expansion disorders by reducing xtrRNA expression. This has been demonstrated for CAG and GGGGCC repeat expansions [159, 186] and might be particularly valuable in disorders exhibiting bidirectional transcription across the repeat expansion. For splicing-based therapeutics, blocking inclusion of repeat expansion-containing introns, such as with splice-modulating antisense oligonucleotides or small RNAs, could prove to be useful for disorders like FRDA and C9FTD/ALS.
With the emergence of gene editing technologies, the direct removal of repeat expansions from the genome may also be possible. Removal of genomic repeat expansions could eliminate the possibility of xtrRNA expression or reverse repressive epigenetic states. Careful SNP selection followed by targeting with CRISPR-Cas9 has been shown to block promoter function and silence the mutant expanded allele in HD [215] or completely delete large portions of the mutant HD allele [265]. Targeting CRISPR-Cas9 to sequences flanking the CTG repeat in DM1 also caused large repeat deletions [299]. In model cells of DM1, CRISPR-Cas9 was used to introduce a poly-A signal upstream of the CTG expansion in the DMPK gene to prevent CUG repeat transcription, which led to a reversal of molecular disease [318]. While potential CRISPR-based therapeutics are exciting, precautions must be taken to address potential pitfalls and challenges like off-target effects, delivery, and cell-type specific mechanisms of DNA damage repair [16, 54, 71, 85, 229, 238, 240, 322].
Structure, protein interactions, and localization of xtrRNA
Structure of xtrRNAs and targeting with small molecules
During and after the synthesis and processing of xtrRNA, the repetitive RNA will fold into repetitive and unique structures and interact with proteins that have an affinity for its sequence or structure. Watson-Crick pairing apparently dominates folding since all atomic resolution investigations of disease-associated xtrRNA to date, including CAG, CUG, CCG, CGG, CCUG, AUUCU, and CCCCGG, are imperfect A-form-like duplexes [39, 62, 147, 234, 341]. These structures possess repeating units of Watson-Crick and mismatch paired nucleotides [147, 341]. While some studies have identified G-quadruplexes or tetraplexes [45, 79, 194, 247], other reports suggest that xtrRNA either do not form quadruplexes or are transient and interconvert readily with Watson-Crick paired conformations, especially as the number of repeating units increase [62, 108, 281, 329, 341]. Some reports of tetraplex structure may be the result of unusual interactions like dimerization between imperfect repeat duplex RNA, as was observed for CGG repeat RNAs [103]. Convincing evidence for the presence or biological significance of RNA G-quadruplexes inside human cells is still lacking [20, 107, 164, 194], therefore direct roles for quadruplex RNA in repeat expansion disease remain unclear.
Available structures of short repeat RNAs reveal A-form-like conformations with unique mismatches that may be targeted with artificial molecules to selectively bind repeat expansion RNA structure. Small molecule screening and structure-guided synthesis have experimentally identified a variety of small molecules that can bind xtrRNA, such as the CUG, CCUG, CGG, and GGGGCC repeat RNAs associated with DM1, DM2, FXS or FXTAS (Fragile X-associated tremor/ataxia syndrome), and C9FTD/ALS, respectively [38, 39, 226, 281, 292, 314, 325]. These molecules have been shown to stabilize repetitive structure or disrupt protein binding, which can correct molecular disease markers like nuclear RNA foci and repetitive polypeptide translation, or improve pathology in cells and animal models. Although promising, their eventual therapeutic application will need to demonstrate exquisite specificity for the RNA target, minimal non-specific interactions, and pharmacologic safety and efficacy [102, 252].
Protein interactions and localization of xtrRNA
Both sequence specific and structure specific interactions likely underlie protein binding to xtrRNA. The repetitive nature of xtrRNA can result in multiple tandem binding sites for proteins. In DM1, the disease-associated xtrRNA contains hundreds or thousands of CUG repeats that bind and recruit possibly as many copies of MBNL-1 protein and potentially other CUG-binding proteins [173, 197, 235]. MBNL-1 recognizes CG dinucleotides separated by 1-17 nucleotides [92], which include motifs in pre-mRNA where MBNL-1 helps to regulate splicing [245]. Examples include the pre-mRNA of sarcoplasmic/endoplasmic reticulum Ca2+-ATPase 1 (SERCA1), which contains several YGCU(U/G)Y motifs downstream from exon 22. MBNL-1 usually interacts with these motifs to cause inclusion of exon 22 but in DM1 exon 22 is excluded during splicing [118, 34]. Blocking the interaction of proteins with DM1 xtrRNA by using morpholino oligonucleotides rescued splicing defects and molecular pathology [310]. Thus, a major contributor to disease mechanism in DM1 is the sequestration of splicing factors, particularly MBNL proteins.
A number of diseases are characterized by binding of specific proteins to xtrRNA or colocalization of proteins with xtrRNA focal aggregates (Table 1) [327]. These include proteins like MBNL-1 in DM1, DM2, HD, spinocerebellar ataxia 3 (SCA3), SCA8 and HDL2 [197, 282, 327], hnRNP K in SCA10 and C9FTD/ALS [46, 311], Pur-α, hnRNP F and SRSF2 in C9FTD/ALS [47, 108, 319], and Sam68 and hnRNP A2/B1 in FXTAS [262, 276]. As such, protein interactions with xtrRNA play key roles in disease mechanism and are expected to be important mediators of aberrant xtrRNA localization and aggregation [214]. Foci containing xtrRNA are believed to be the result of RNA-binding protein sequestration that can functionally deplete those proteins and partially protect the xtrRNA from degradation [214, 327].
Sequence specific interactions may not entirely explain xtrRNA localization or foci formation. While certain proteins that prefer to bind G-rich sequence, like hnRNP H/F, have been found to associate strongly with the GGGGCC repeats of C9FTD/ALS, other interacting proteins do not appear to have strong GGGGCC sequence-binding specificity, such as ALY/REF, SC-35, SF2, and nucleolin [47, 108, 175]. Imperfect A-form-like duplexes, or duplexes inter-converting with tetraplex conformations, may attract proteins that recognize the unique structures of xtrRNA rather than the specific sequence. Glycine-arginine-rich (GAR) proteins containing RGG/RG motifs, for example, are believed to recognize the structure of their nucleic acid partners rather than sequence [289]. The GAR domain-containing proteins FUS (fused in sarcoma), FMRP, and hnRNP U all recognize structured guanine-rich RNA sequences with an apparent preference for transitions between canonical duplexes and non-canonical structures like quadruplexes [233]. One explanation for foci is that proteins bind specifically to repeat sequence or structural elements of xtrRNA and then seed aggregation that recruits additional secondary interacting factors. Thus, xtrRNA may form foci by either merging with existing nuclear bodies or else establishing their own novel versions of RNA granules. While focal aggregation of xtrRNA can be detrimental to sequestered protein function, it may also protect the cell by preventing nuclear escape and translation of repeat RNAs [150].
xtrRNA localization and membrane-free cellular organelles
Whether there is a specific localization pattern of xtrRNA inside cell nuclei is not entirely clear. Foci might be expected to nucleate at the site of transcription. DMPK mRNA usually localizes to SC-35 splicing speckles after transcription. However, when containing CUG repeat expansions, the DMPK mRNA has been shown to localize peri-transcriptionally outside of SC-35 splicing speckles [274]. RNA containing CAG, CUG and GGGGCC repeats were also shown to localize to SC-35 splicing speckles and nuclear speckles [132, 295]. However, in other studies the xtrRNAs, specifically CUG and CGG RNAs, appeared to form foci stochastically [243, 280]. Live cell imaging of Spinach2 aptamer-tagged CGG repeat xtrRNA revealed rapid aggregation and formation of very stable foci [280]. CGG xtrRNA foci were additionally found to be mobile and dynamic and colocalized with Sam68 protein. They migrated around the nucleus over time and could be seen to merge into larger foci or disaggregate into smaller foci. Live cell imaging of CUG repeat xtrRNA tagged with the MS2-GFP system found similar effects for aggregation, foci formation and dynamics [243]. CUG repeat RNA foci formation depended on the presence of MBNL-1 protein. In live-cell experimental approaches the xtrRNA is likely to be over-expressed from an artificial genetic context and may not represent the true dynamics or localization of endogenous repeat expansions. Nonetheless, live and fixed cell imaging have revealed that xtrRNA foci are dynamic, stable aggregates that likely depend on protein interactions and may co-localize with known nuclear bodies.
Nuclear bodies can be built around RNA and the molecular forces that govern nuclear body formation may help explain xtrRNA foci formation and localization. For example, nuclear paraspeckles depend on the long noncoding RNA NEAT1 (nuclear paraspeckle assembly transcript 1) [321]. Nuclear bodies are essentially membrane-free organelles that are held together by transient or dynamic protein-protein and protein-RNA interactions. These interactions collectively provide a type of phase separation to organize and compartmentalize cellular processes [336]. It was recently demonstrated that CAG, CUG and GGGGCC repeat containing RNAs form soluble aggregates with sol-gel phase separation properties and behave similar to liquid-like droplets [132]. These properties were dependent on the repeat expansion length and base-pairing interactions. In contrast, CCCCGG repeats did not form phase transitions, suggesting that not all xtrRNA will possess these properties. Interestingly, guanine-rich nucleic acids are less soluble than other nucleic acids and appear to be intrinsically aggregate-prone apart from protein, especially when packing into quartets or higher-order quadruplex structures [21, 89, 179]. The disruption of membrane-free organelles, which are abundant in the nucleus, is linked to disease [198, 228, 272]. In fact, the disruption of membrane-free organelle assembly and dynamics by repetitive poly-glycine-arginine (poly-GR) and poly-proline-arginine (poly-PR) translation products has emerged as a leading molecular disease mechanism for C9FTD/ALS [165, 174, 182]. Association of certain proteins with xtrRNA, dependent upon RNA sequence and structure, may strongly influence the subsequent localization of xtrRNA with membrane-free cellular compartments.
Abundance and turnover of xtrRNA
Abundance of foci-forming xtrRNA
Understanding the biology of an RNA includes knowing the effective concentration or abundance of that RNA and its turnover and decay pathways. Three current studies highlight the importance of characterizing cellular xtrRNA abundance. The cellular abundance of CUG repeat-containing transcripts was recently measured using transgenes and endogenous DMPK RNA in mouse models of DM1 and human tissues from DM1 patients [104]. Surprisingly, a large 1000-fold discrepancy for transcript number was discovered across mouse models. In human samples only a few dozen DMPK mRNA molecules were detected per cell, with only half of those expected to contain the repeat expansion. In a similar study looking at the abundance and processing of an antisense transcript across the DMPK repeat expansion, only a handful of repeat containing antisense transcripts were quantified per cell [105]. Quantification of the repeat-containing intron of C9ORF72 in C9FTD/ALS patient cells found only a few copies per cell, concluding that each foci might be composed of as few as one xtrRNA transcript [188]. Therefore, one or a few copies of xtrRNA may be enough to generate focal aggregates. Importantly, the stochastic nature of foci formation, where many cells contain no foci but some contain several, suggests that there may be a disproportionate contribution to disease for xtrRNA at the individual cell level [188]. These reports indicate that knowing the number and type of xtrRNA species inside of cells will be important for correct interpretation of data and for understanding the role of xtrRNA in disease.
Nuclear xtrRNA retention and surveillance mechanisms
The nuclease enzymes primarily responsible for degrading nuclear RNA are the exosome complex (3'-5' exoribonuclease activity) and 5'-3' exoribonuclease 2 (XRN2) [67]. These enzymes act as part of a nuclear RNA quality control and surveillance pathway that monitors transcription, splicing, and 3'-end formation of pre-mRNAs, as well as their packaging into mRNP particles (Fig. 3) [67, 146, 338]. Instead of degradation, these pathways can also signal for retention of aberrant transcripts in the nucleus, typically at the site of transcription [52, 220] but sometimes near nuclear pores [67]. Retention at the site of transcription is coupled to nuclear exosome activity, particularly the Rrp6p subunit [66, 220]. The TPR protein, a mammalian ortholog of yeast Mlp1/2p, is implicated in retention at nuclear pores for mRNAs with retained introns that normally exit the nucleus through the nuclear export factor 1 (NXF1) pathway [49]. Both of these mechanisms may be relevant to xtrRNA, especially when repeat expansions are found in retained introns [43, 110, 227].

Possible mechanisms of nuclear and cytoplasmic RNA surveillance, nuclear export, and translation of xtrRNA. RNA containing large repeat expansion sequences may be subject to nuclear RNA surveillance mechanisms, including degradation by the nuclear exosome (1) or the XRN2 5'-3' exoribonuclease (1). Export of xtrRNA likely involves bulk mRNA transport via NXF1 (2b), but may also include alternative mechanisms like CRM1-mediated export (2a) or possibly nuclear envelope budding (2c). Cytoplasmic RNA surveillance mechanisms that may control xtrRNA levels and translation include nonsense-mediated decay (NMD) (3a), no-go decay (NGD) (3b), or nonstop decay (NSD) (3c). Translation of xtrRNA is likely to follow canonical cap-dependent translation (4), especially when repeat expansions are embedded in normal coding regions of an mRNA, but may potentially involve internal ribosome entry site (IRES)-like mechanisms (4). RAN translation has been shown to be cap-dependent for some repeat expansions, but complete mechanistic details remain to be determined
Surveillance mechanisms are also related to transcription or splicing of xtrRNA since these might be expected to trigger degradation [67, 146]. However, the existence of foci and nuclear export of xtrRNA argue that surveillance mechanisms are incomplete or inefficient for xtrRNA removal. At present it is unknown how many molecules of any repeat expansion-containing RNA are synthesized versus how many survive to form foci or exit the nucleus for translation. It is likely that repeat expansion-containing RNAs survive due to protection by protein binding, such as hnRNP proteins [125, 327]. Alternatively, factors responsible for recruiting RNA to the nuclear exosome, such as the TRAMP (Trf4/Air2/Mtr4p Polyadenylation) complex or NEXT (nuclear exosome targeting) complex, are unable to efficiently recognize and bind the xtrRNA [146, 259]. Thus, xtrRNA transcripts may escape degradation if they appear as "normal" mRNPs, having undergone proper RNA processing like capping, splicing and polyadenylation and are associated with appropriate post-processing factors.
Turnover and decay of xtrRNA
An unanswered question remains as to whether foci might contain partially degraded fragments of repeat RNA in addition to larger intact transcripts. A case in point is C9FTD/ALS where the microsatellite expansion occurs in an intron but nuclear RNA foci and cytoplasmic translation are both observed. When introns are spliced out of pre-mRNA transcripts they are typically destined for rapid degradation unless they contain a stably folding RNA element or recruit RNA binding proteins [115]. Examples include small nucleolar RNAs and microRNAs [56, 203]. It is possible that the structures that repeat expansion RNAs form, as well as the proteins that they bind, allow them to persist, accumulate, and aggregate as foci [3]. At present the exact type and number of xtrRNA species that are trapped in foci versus free and soluble for any disease is unknown. It is also not known whether partially degraded xtrRNA fragments are stable enough to accumulate. Furthermore, there is no distinction between what are the major species or which are nuclear versus cytoplasmic.
When mRNAs that contain microsatellite repeat expansions reach the cytoplasm they may encounter additional quality control mechanisms designed to eliminate aberrant mRNA and prevent its translation. These include nonsense-mediated decay (NMD), no-go decay (NGD), and non-stop decay (NSD) (Fig. 3) [267]. NMD recognizes premature stop codons through possibly multiple mechanisms. NMD can be triggered if exon-junction complexes (EJCs), which are deposited near splicing junctions, are encountered downstream of a stop codon [172, 199]. Another mechanism of NMD may involve the relative length of sequence 3' to the stop codon [4, 172]. Repeat expansions could conceivably alter the positioning of a stop codon or extend the 3' UTR region and trigger NMD. When ribosome translation is significantly slowed or stalled then NGD can be triggered. Stalling is thought to be initiated by unusually stable RNA structures or protein-binding motifs and result in endonucleolytic cleavage [63]. Since repeat expansions are believed to fold into stable hairpins or tetraplexes they could possibly trigger NGD. In the case of NSD, the ribosome can become stuck on mRNA that does not possess a stop codon. These complexes must be resolved by cleavage and degradation of the mRNA [82, 302]. NSD could become activated if repeat expansions alter the reading frame or cause loss of the stop codon, such as via mis-splicing.
For xtrRNA embedded in mRNAs, either as exons or introns, mRNA surveillance mechanisms should act to reduce translation. Activation of these pathways will lead to degradation by the cytoplasmic version of the exosome and XRN1, a 5'-3' exoribonuclease. Accessory factors that guide cleavage include the Ski complex (composed of Ski2, Ski3, and Ski8 proteins) and the Ski7 protein [5, 267]. However, similar to nuclear RNA surveillance mechanisms, the cytoplasmic pathways seem unable to detect or completely remove xtrRNA and prevent translation. Methods to enhance RNA surveillance mechanisms might represent reasonable targets for therapeutic intervention. For example ataluren (PTC124) is a small molecule drug that increases NMD and might sensitize mRNA surveillance to repeat expansions [73].
Some studies have uncovered potential RNA turnover mechanisms associated with repeat expansions. In a recent RNAi screen several RNA processing factors were identified as suppressors of toxicity in C. elegans expressing a (CUG)123 repeat expansion in a 3'-UTR of GFP [88]. These factors included RNases, helicases and RNA binding proteins that, when knocked-down, caused increased toxicity and enhanced nuclear foci formation. A nuclear pore complex (NPC) protein, npp-4, was also a suppressor in this screen. Interestingly, smg-2, a conserved helicase and central component of the NMD pathway, was a strong suppressor. Knock-down of NMD resulted in a several-fold increase in GFP-(CUG)123 RNA expression levels and increased GFP translation. The substantial increase in 3'-UTR GC-content was identified as the likely trigger for NMD of the GFP-(CUG)123 RNA [88]. Smg-2 was also identified as a suppressor of poly-glutamine aggregation previously, likely through NMD of aberrant repeat-containing HTT transcripts [328]. These results provide one example of the role of RNA turnover in controlling toxicity of xtrRNA in repeat expansion disorders.
Nuclear export and translation of xtrRNA
Canonical mRNA export pathways
RNA export from the nucleus involves several distinct pathways depending on the RNA and the various protein factors that constitute the RNP particle [273]. For nuclear mRNA there are two main export pathways: NXF1-mediated and chromosome region maintenance 1 (CRM1)-mediated, although NXF1 is the primary transport system for bulk mRNAs (Fig. 3) [60]. Both mechanisms rely on adapter proteins to specify the RNA cargo for export. Many of the factors required for successful export are deposited co-transcriptionally and post-transcriptionally during mRNP assembly [22]. The C-terminal domain of RNA Pol II serves as a docking platform for a wide variety of mRNA processing and mRNP assembly proteins and plays critical roles in establishing mRNP composition [139]. Correct processing of mRNA, such as capping, splicing and 3'-end formation, determines the ability of these factors to bind mRNA and assemble export-competent mRNP particles [36, 60, 335].
The mRNA-associated adapter proteins and complexes represent a complicated matrix of possible interactions that dictate export efficiency [22, 60]. NXF1 can specifically bind the constitutive transport element found in some mRNAs and viral RNAs, like that of the Mason Pfizer monkey virus, to directly facilitate export [101, 178]. However, for bulk cellular mRNA transport NXF1 uses adapters like TREX (transcription export complex) [60, 111]. Although the NXF1 protein interacts loosely with RNA, TREX helps mediate specific binding through its subunit ALY/REF [124], a protein previously reported to interact with C9ORF72 GGGGCC RNA repeats [47]. TREX associates with mRNA during synthesis and processing via mRNA capping and splicing events [138, 204] and appears to be primarily recruited to the 5' ends of mRNAs in human cells via interaction between ALY/REF and the cap binding complex component CBP80 [36]. In addition to ALY/REF, TREX is composed of the THO complex, CIP29, and UAP56, a component of the EJC [37, 53, 157]. For repeat expansion disorders, NXF1 seems to be the most likely pathway since disease-associated xtrRNA are transcribed from coding gene loci and TREX is deposited onto mRNAs early during transcription [204].
Nuclear export of xtrRNA
A recent study connected NXF1 transport of C9FTD/ALS intronic xtrRNA via interaction with the export adapter SR-rich splicing factor 1 (SRSF1) [110]. SRSF1 appeared to interact and colocalize with C9ORF72 xtrRNA. Depletion of SRSF1 prevented neurodegeneration in a fly model and suppressed cell death in patient-derived motor neurons and astrocytes. Depleting SRSF1 or preventing interaction with NXF1 inhibited nuclear export of repeat-containing C9ORF72 transcripts and blocked RAN translation. Thus, SRSF1 might serve as a therapeutic target in C9FTD/ALS. This report highlights the value of understanding RNA biology in the context of repeat expansion disorders.
Most disease-associated xtrRNA is embedded in exonic or untranslated regions (Table 1) and therefore likely exits the nucleus via mRNA export pathways. CRM1 exports proteins and their associated RNAs via interaction with nuclear export signal sequences and Ran-GTP (Fig. 3) [60, 74, 77]. CRM1 interacts directly with the NPC at the nuclear periphery and commonly exports noncoding RNAs like spliceosomal RNA (snRNA) [10, 74, 131]. There is no reported RNA binding affinity of CRM1 so selective export of mRNAs depends on the RNA-binding properties of its cargo proteins [60]. Export of xtrRNA by CRM1 might only require that the repeat expansion sequence or structure somehow recruit a CRM1 cargo protein.
Export of intronic xtrRNA would be expected to require aberrant splicing that resulted in its retention in mRNA, as has been implicated for the intronic C9FTD/ALS repeat expansion [227]. Alternative export pathways exist but seem unlikely given their very specific nature. For example, transfer RNA (tRNA) undergoes multiple maturation phases that cumulatively result in two separate import and export steps [273]. These export pathways involve specific RNA-protein interactions, such as EXP-t and EXP5 [8, 29], that are unlikely to mediate xtrRNA export. For any export pathway through the NPC, xtrRNA must somehow establish RNP complexes that pass the requisite tests for licensing of export.
Nuclear exit of RNP granules, such as nuclear xtrRNA foci, might also be possible through nuclear envelope budding (Fig. 3). This mechanism involves TorsinA, nuclear lamina, and other uncharacterized factors. Nuclear budding was discovered as part of the nuclear egress mechanism of large nucleocapsid particles of Herpes viruses [55, 78, 200, 221, 277]. Nuclear envelope budding has been found to be a natural process for nuclear release of large RNP complexes during development of neuromuscular junctions in Drosophila melanogaster [135, 277]. However, knock-out of TorsinA in HeLa cells had little impact on Herpes virus production [294], suggesting alternative factors or mechanisms in human cells. If xtrRNA is exported by nuclear envelope budding it would have to mimic specific RNP granule formation that elicits nuclear envelope budding, which at present involves mechanisms that are largely uncharacterized [78].
Translation of xtrRNA
If xtrRNA can successfully exit the nucleus it is a potential candidate for translation. However, mRNAs that contain expanded tandem repeats are possibly the only practical source for translation of repeat expansion polypeptides since they contain the prerequisite sequence elements and protein factors to mediate canonical cap-dependent translation. These typically include 5' cap structures, bound eukaryotic initiation factors (eIFs), a poly-A tail, and appropriate mRNP complexes like the EJC [116, 301, 315]. Translation of xtrRNA sequence embedded within the coding exon of a gene, such as is found in SBMA, HD, DRPLA (dentatorubral-pallidoluysian atrophy), OPMD (oculopharyngeal muscular dystrophy) and several of the SCA disorders (Table 1), are translated by canonical mechanisms. Most repeat expansions form stable secondary structures that have been shown to reduce the amount of overall translation by presumably inducing stalling, frame-shifting or abortive translation [72, 222, 244, 313]. In contrast, the specific binding of MID1 protein to huntingtin mRNA, which contains CAG repeat expansions, has been reported to enhance translation and lead to greater levels of aberrant protein [160]. This mechanism has also been proposed to enhance translation of other CAG repeat expansion-containing genes that cause disease [98]. Canonical translation of repeat expansions that are found in-frame in coding sequences is expected to generate otherwise normal protein that simply contain long tracts of repetitive polypeptide [296].
Repeat-associated non-AUG translation
The translation of noncoding xtrRNA irrespective of a canonical start codon was recently discovered and termed repeat-associated non-AUG (RAN) translation [42, 96, 290, 339]. Repeat expansion diseases where this mechanism has been observed now include SCA2, SCA8, SCA31, HD, FXTAS/FXPOI, and C9FTD/ALS [9, 13, 27, 96, 129, 218, 261, 290, 339, 340]. RAN translation of xtrRNA sequence can occur in many contexts, including repeat expansions found in untranslated regions, retained introns, and even those embedded in coding exons [96]. The mechanisms of RAN translation remain poorly understood and could involve several scenarios, possibly even internal ribosome entry site (IRES)-like mechanisms (Fig. 3) [96, 339]. For the CGG repeats of FMR1 that cause FXTAS a more straightforward mechanism is emerging. In this case, RAN translation is m7G cap-dependent where a pre-initiation complex scans the RNA looking for a start codon [96, 141]. When the CGG repeats are present and stable structures are presumably encountered then stalling occurs and significantly enhances the ability of the ribosome to select a near-cognate start codon, or possibly any codon, to initiate translation [141, 154, 158, 339]. A similar mechanism is favored for the CAG and CUG repeats of sense and antisense transcripts in SCA8 [339]. This mechanism is proposed to allow translation initiation upstream of a repeat expansion in multiple reading frames [42, 96]. The sequence context, such as the leader sequence during scanning, the types of potential near-cognate start codons, and the repeat expansion sequence and size all appear to modulate the degree of RAN translation [12, 141, 261, 339].
The mechanism of RAN translation may be related to translation of upstream open reading frames (uORFs), a widespread phenomena revealed through high-throughput ribosomal footprint profiling [128]. RAN translation could even represent a specialized form of uORF translation that is triggered by stable xtrRNA structures. Both mechanisms can initiate at near-cognate start codons (although RAN translation may use other codons or other mechanisms, like frameshifting) and are influenced by surrounding sequence context that might impact RNA folding or protein interactions [96, 117].
Recent investigations have demonstrated that certain RAN translation products of C9FTD/ALS disrupt the function of membrane-free cellular organelles, such as stress granules, Cajal bodies and the nucleolus [174, 182]. These polypeptides seem to block the formation or critical interaction dynamics of membrane-free organelles and RNA granules, which are important for neuronal cell signaling and health [269, 288]. Transport of macromolecules through the nuclear pore complex depends on interactions that resemble membrane-free organelle structure [207, 219]. They are organized by dynamic protein interactions of low complexity domain proteins, including phenylalanine-glycine (FG) repeats, which may explain why certain C9FTD/ALS RAN translation products are reported to disrupt nucleocytoplasmic transport [80, 136, 264, 326]. RAN translation products can also aggregate and are implicated in the disruption of a variety of other pathways [12, 42, 96, 142, 286, 339].
Several important questions remain concerning the mechanisms of RAN translation. For example, how similar are the mechanisms of RAN translation across diverse repeat expansion and sequence contexts [42, 96]? RAN translation maybe a spectrum of related mechanisms based upon modulation of ribosomal scanning, translation initiation, and translation elongation [301]. RAN translation can initiate just upstream from the repeat expansion, but how often can RAN translation initiate within the repeat sequence itself [339]? In vitro and cell-based model systems suggest that RAN translation can proceed uninterrupted through an entire repeat expansion [141, 213, 339, 340]. Yet some expansions are massive in size. Therefore, how often do repeat expansions induce frame-shifting or possibly even early translation termination [313]? Also, what factors are unique to RAN translation? Finding answers to these mechanistic questions may be critical for developing future therapeutic molecules that can target and selectively block xtrRNA translation.
Conclusion
RNA species that contain simple tandem repeat sequences occupy an underexplored world of RNA biology. Recent studies have begun to revisit the transcription and translation of repeat expansions. However, significant gaps remain for processes like cellular transport and turnover of xtrRNA. Placing repeat expansion disease mechanism studies in the context of current RNA biology will help reveal a better understanding of how the cell deals with xtrRNA and identify mechanisms unique to repeat expansions.
Investigations into the biology of xtrRNA promise to unlock new approaches to therapeutics. Transcription across repeat expansions has opportunities for therapeutic development, such as modulating the function of Supt4h. Likewise, translation of repeat expansions, especially RAN translation, may become more targetable as molecular mechanisms become better characterized and specific factors identified. Selectively blocking both the synthesis of xtrRNA or its translation are attractive therapeutic approaches since they could extrapolate to multiple repeat expansion disorders. Turnover of xtrRNA should become increasingly important since several potential therapeutic strategies employ targeted and selective degradation of repeat expansion-containing RNA, such as antisense oligonucleotides and small interfering RNAs [64, 134, 169, 239, 275].
References
Abecasis GR, Auton A, Brooks LD, DePristo MA, Durbin RM, Handsaker RE, Kang HM, Marth GT, McVean GA (2012) An integrated map of genetic variation from 1,092 human genomes. Nature 491:56–65. doi:10.1038/nature11632
Adihe Lokanga R, Zhao XN, Entezam A, Usdin K (2014) X inactivation plays a major role in the gender bias in somatic expansion in a mouse model of the fragile X-related disorders: implications for the mechanism of repeat expansion. Hum Mol Genet 23:4985–4994. doi:10.1093/hmg/ddu213
Akiyama BM, Eiler D, Kieft JS (2016) Structured RNAs that evade or confound exonucleases: function follows form. Curr Opin Struct Biol 36:40–47. doi:10.1016/j.sbi.2015.12.006
Amrani N, Ganesan R, Kervestin S, Mangus DA, Ghosh S, Jacobson A (2004) A faux 3'-UTR promotes aberrant termination and triggers nonsense-mediated mRNA decay. Nature 432:112–118. doi:10.1038/nature03060
Anderson JS, Parker RP (1998) The 3' to 5' degradation of yeast mRNAs is a general mechanism for mRNA turnover that requires the SKI2 DEVH box protein and 3' to 5' exonucleases of the exosome complex. EMBO J 17:1497–1506. doi:10.1093/emboj/17.5.1497
Arcot SS, Wang Z, Weber JL, Deininger PL, Batzer MA (1995) Alu repeats: a source for the genesis of primate microsatellites. Genomics 29:136–144. doi:10.1006/geno.1995.1224
Arimbasseri AG, Rijal K, Maraia RJ (2014) Comparative overview of RNA polymerase II and III transcription cycles, with focus on RNA polymerase III termination and reinitiation. Transcription 5:e27639. doi:10.4161/trns.27369
Arts GJ, Kuersten S, Romby P, Ehresmann B, Mattaj IW (1998) The role of exportin-t in selective nuclear export of mature tRNAs. EMBO J 17:7430–7441. doi:10.1093/emboj/17.24.7430
Ash PE, Bieniek KF, Gendron TF, Caulfield T, Lin WL, Dejesus-Hernandez M, van Blitterswijk MM, Jansen-West K, Paul JW, 3rd, Rademakers Ret al (2013) Unconventional translation of C9ORF72 GGGGCC expansion generates insoluble polypeptides specific to c9FTD/ALS. Neuron 77: 639-646. doi:10.1016/j.neuron.2013.02.004
Bai B, Moore HM, Laiho M (2013) CRM1 and its ribosome export adaptor NMD3 localize to the nucleolus and affect rRNA synthesis. Nucleus 4:315–325. doi:10.4161/nucl.25342
Bak ST, Sakellariou D, Pena-Diaz J (2014) The dual nature of mismatch repair as antimutator and mutator: for better or for worse. Front Genet 5:287. doi:10.3389/fgene.2014.00287
Banez-Coronel M, Ayhan F, Tarabochia AD, Zu T, Perez BA, Tusi SK, Pletnikova O, Borchelt DR, Ross CA, Margolis RL et al (2015) RAN Translation in Huntington Disease. Neuron 88:667–677. doi:10.1016/j.neuron.2015.10.038
Banez-Coronel M, Porta S, Kagerbauer B, Mateu-Huertas E, Pantano L, Ferrer I, Guzman M, Estivill X, Marti E (2012) A pathogenic mechanism in Huntington's disease involves small CAG-repeated RNAs with neurotoxic activity. PLoS Genet 8:e1002481. doi:10.1371/journal.pgen.1002481
Baralle D, Buratti E (2017) RNA splicing in human disease and in the clinic. Clin Sci (Lond) 131:355–368. doi:10.1042/CS20160211
Baralle M, Pastor T, Bussani E, Pagani F (2008) Influence of Friedreich ataxia GAA noncoding repeat expansions on pre-mRNA processing. Am J Hum Genet 83:77–88. doi:10.1016/j.ajhg.2008.06.018
Beaudet AL, Meng L (2016) Gene-targeting pharmaceuticals for single-gene disorders. Hum Mol Genet 25:R18–R26. doi:10.1093/hmg/ddv476
Bernecky C, Herzog F, Baumeister W, Plitzko JM, Cramer P (2016) Structure of transcribing mammalian RNA polymerase II. Nature 529:551–554. doi:10.1038/nature16482
Biancalana V, Taine L, Bouix JC, Finck S, Chauvin A, De Verneuil H, Knight SJ, Stoll C, Lacombe D, Mandel JL (1996) Expansion and methylation status at FRAXE can be detected on EcoRI blots used for FRAXA diagnosis: analysis of four FRAXE families with mild mental retardation in males. Am J Hum Genet 59:847–854
Bidichandani SI, Ashizawa T, Patel PI (1998) The GAA triplet-repeat expansion in Friedreich ataxia interferes with transcription and may be associated with an unusual DNA structure. Am J Hum Genet 62:111–121. doi:10.1086/301680
Biffi G, Di Antonio M, Tannahill D, Balasubramanian S (2014) Visualization and selective chemical targeting of RNA G-quadruplex structures in the cytoplasm of human cells. Nat Chem 6:75–80. doi:10.1038/nchem.1805
Biyani M, Nishigaki K (2005) Structural characterization of ultra-stable higher-ordered aggregates generated by novel guanine-rich DNA sequences. Gene 364:130–138. doi:10.1016/j.gene.2005.05.041
Bjork P, Wieslander L (2014) Mechanisms of mRNA export. Semin Cell Dev Biol 32:47–54. doi:10.1016/j.semcdb.2014.04.027
Brais B, Bouchard JP, Xie YG, Rochefort DL, Chretien N, Tome FM, Lafreniere RG, Rommens JM, Uyama E, Nohira Oet al (1998) Short GCG expansions in the PABP2 gene cause oculopharyngeal muscular dystrophy. Nat Genet 18:164–167. doi:10.1038/ng0298-164
Brinkmann B, Klintschar M, Neuhuber F, Huhne J, Rolf B (1998) Mutation rate in human microsatellites: influence of the structure and length of the tandem repeat. Am J Hum Genet 62:1408–1415. doi:10.1086/301869
Brouwer JR, Huguet A, Nicole A, Munnich A, Gourdon G (2013) Transcriptionally Repressive Chromatin Remodelling and CpG Methylation in the Presence of Expanded CTG-Repeats at the DM1 Locus. J Nucleic Acids 2013:567435. doi:10.1155/2013/567435
Budworth H, McMurray CT (2013) Bidirectional transcription of trinucleotide repeats: roles for excision repair. DNA repair 12:672–684. doi:10.1016/j.dnarep.2013.04.019
Buijsen RA, Visser JA, Kramer P, Severijnen EA, Gearing M, Charlet-Berguerand N, Sherman SL, Berman RF, Willemsen R, Hukema RK (2016) Presence of inclusions positive for polyglycine containing protein, FMRpolyG, indicates that repeat-associated non-AUG translation plays a role in fragile X-associated primary ovarian insufficiency. Hum Reprod 31:158–168. doi:10.1093/humrep/dev280
Buschiazzo E, Gemmell NJ (2006) The rise, fall and renaissance of microsatellites in eukaryotic genomes. BioEssays : news and reviews in molecular, cellular and developmental biology 28:1040–1050. doi:10.1002/bies.20470
Calado A, Treichel N, Muller EC, Otto A, Kutay U (2002) Exportin-5-mediated nuclear export of eukaryotic elongation factor 1A and tRNA. EMBO J 21:6216–6224
Campuzano V, Montermini L, Molto MD, Pianese L, Cossee M, Cavalcanti F, Monros E, Rodius F, Duclos F, Monticelli A et al (1996) Friedreich's ataxia: autosomal recessive disease caused by an intronic GAA triplet repeat expansion. Science 271:1423–1427
Chaisson MJ, Huddleston J, Dennis MY, Sudmant PH, Malig M, Hormozdiari F, Antonacci F, Surti U, Sandstrom R, Boitano M et al (2015) Resolving the complexity of the human genome using single-molecule sequencing. Nature 517:608–611. doi:10.1038/nature13907
Chakraborty R, Kimmel M, Stivers DN, Davison LJ, Deka R (1997) Relative mutation rates at di-, tri-, and tetranucleotide microsatellite loci. Proc Natl Acad Sci U S A 94:1041–1046
Chan NL, Guo J, Zhang T, Mao G, Hou C, Yuan F, Huang J, Zhang Y, Wu J, Gu L et al (2013) Coordinated processing of 3' slipped (CAG)n/(CTG)n hairpins by DNA polymerases beta and delta preferentially induces repeat expansions. J Biol Chem 288:15015–15022. doi:10.1074/jbc.M113.464370
Charlet BN, Savkur RS, Singh G, Philips AV, Grice EA, Cooper TA (2002) Loss of the muscle-specific chloride channel in type 1 myotonic dystrophy due to misregulated alternative splicing. Mol Cell 10:45–53
Chen IC, Lin HY, Lee GC, Kao SH, Chen CM, Wu YR, Hsieh-Li HM, Su MT, Lee-Chen GJ (2009) Spinocerebellar ataxia type 8 larger triplet expansion alters histone modification and induces RNA foci. BMC Mol Biol 10:9. doi:10.1186/1471-2199-10-9
Cheng H, Dufu K, Lee CS, Hsu JL, Dias A, Reed R (2006) Human mRNA export machinery recruited to the 5' end of mRNA. Cell 127:1389–1400. doi:10.1016/j.cell.2006.10.044
Chi B, Wang Q, Wu G, Tan M, Wang L, Shi M, Chang X, Cheng H (2013) Aly and THO are required for assembly of the human TREX complex and association of TREX components with the spliced mRNA. Nucleic Acids Res 41:1294–1306. doi:10.1093/nar/gks1188
Childs-Disney JL, Hoskins J, Rzuczek SG, Thornton CA, Disney MD (2012) Rationally designed small molecules targeting the RNA that causes myotonic dystrophy type 1 are potently bioactive. Acs Chem Biol 7:856–862. doi:10.1021/cb200408a
Childs-Disney JL, Yildirim I, Park H, Lohman JR, Guan L, Tran T, Sarkar P, Schatz GC, Disney MD (2014) Structure of the myotonic dystrophy type 2 RNA and designed small molecules that reduce toxicity. Acs Chem Biol 9:538–550. doi:10.1021/cb4007387
Chung DW, Rudnicki DD, Yu L, Margolis RL (2011) A natural antisense transcript at the Huntington's disease repeat locus regulates HTT expression. Hum Mol Genet 20:3467–3477. doi:10.1093/hmg/ddr263
Clark RM, Dalgliesh GL, Endres D, Gomez M, Taylor J, Bidichandani SI (2004) Expansion of GAA triplet repeats in the human genome: unique origin of the FRDA mutation at the center of an Alu. Genomics 83:373–383. doi:10.1016/j.ygeno.2003.09.001
Cleary JD, Ranum LP (2014) Repeat associated non-ATG (RAN) translation: new starts in microsatellite expansion disorders. Curr Opin Genet Dev 26C:6–15. doi:10.1016/j.gde.2014.03.002
Cohen-Hadad Y, Altarescu G, Eldar-Geva T, Levi-Lahad E, Zhang M, Rogaeva E, Gotkine M, Bartok O, Ashwal-Fluss R, Kadener S et al (2016) Marked Differences in C9orf72 Methylation Status and Isoform Expression between C9/ALS Human Embryonic and Induced Pluripotent Stem Cells. Stem Cell Rep 7:927–940. doi:10.1016/j.stemcr.2016.09.011
Colak D, Zaninovic N, Cohen MS, Rosenwaks Z, Yang WY, Gerhardt J, Disney MD, Jaffrey SR (2014) Promoter-bound trinucleotide repeat mRNA drives epigenetic silencing in fragile X syndrome. Science 343:1002–1005. doi:10.1126/science.1245831
Conlon EG, Lu L, Sharma A, Yamazaki T, Tang T, Shneider NA, Manley JL (2016) The C9ORF72 GGGGCC expansion forms RNA G-quadruplex inclusions and sequesters hnRNP H to disrupt splicing in ALS brains. eLife 5. doi:10.7554/eLife.17820
Cooper-Knock J, Higginbottom A, Stopford MJ, Highley JR, Ince PG, Wharton SB, Pickering-Brown S, Kirby J, Hautbergue GM, Shaw PJ (2015) Antisense RNA foci in the motor neurons of C9ORF72-ALS patients are associated with TDP-43 proteinopathy. Acta Neuropathol 130:63–75. doi:10.1007/s00401-015-1429-9
Cooper-Knock J, Walsh MJ, Higginbottom A, Robin Highley J, Dickman MJ, Edbauer D, Ince PG, Wharton SB, Wilson SA, Kirby J et al (2014) Sequestration of multiple RNA recognition motif-containing proteins by C9orf72 repeat expansions. Brain 137:2040-2051. doi:10.1093/brain/awu120
Cortes CJ, La Spada AR (2015) Autophagy in polyglutamine disease: Imposing order on disorder or contributing to the chaos? Mol Cell Neurosci 66:53–61. doi:10.1016/j.mcn.2015.03.010
Coyle JH, Bor YC, Rekosh D, Hammarskjold ML (2011) The Tpr protein regulates export of mRNAs with retained introns that traffic through the Nxf1 pathway. RNA 17:1344–1356. doi:10.1261/rna.2616111
Crickard JB, Fu J, Reese JC (2016) Biochemical Analysis of Yeast Suppressor of Ty 4/5 (Spt4/5) Reveals the Importance of Nucleic Acid Interactions in the Prevention of RNA Polymerase II Arrest. J Biol Chem 291:9853–9870. doi:10.1074/jbc.M116.716001
Crouau-Roy B, Clisson I (2000) Evolution of an Alu DNA element of type Sx in the lineage of primates and the origin of an associated tetranucleotide microsatellite. Genome 43:642–648
Custodio N, Carmo-Fonseca M, Geraghty F, Pereira HS, Grosveld F, Antoniou M (1999) Inefficient processing impairs release of RNA from the site of transcription. EMBO J 18:2855–2866. doi:10.1093/emboj/18.10.2855
Custodio N, Carvalho C, Condado I, Antoniou M, Blencowe BJ, Carmo-Fonseca M (2004) In vivo recruitment of exon junction complex proteins to transcription sites in mammalian cell nuclei. RNA 10:622–633
Dai WJ, Zhu LY, Yan ZY, Xu Y, Wang QL, Lu XJ (2016) CRISPR-Cas9 for in vivo Gene Therapy: Promise and Hurdles. Mol Ther Nucleic Acids 5:e349. doi:10.1038/mtna.2016.58
Darlington RW, Moss LH 3rd (1968) Herpesvirus envelopment. J Virol 2:48–55
Daugaard I, Hansen TB (2017) Biogenesis and Function of Ago-Associated RNAs. Trends Genet. doi:10.1016/j.tig.2017.01.003
David G, Abbas N, Stevanin G, Durr A, Yvert G, Cancel G, Weber C, Imbert G, Saudou F, Antoniou Eet al (1997) Cloning of the SCA7 gene reveals a highly unstable CAG repeat expansion. Nat Genet 17:65–70. doi:10.1038/ng0997-65
de la Mata M, Alonso CR, Kadener S, Fededa JP, Blaustein M, Pelisch F, Cramer P, Bentley D, Kornblihtt AR (2003) A slow RNA polymerase II affects alternative splicing in vivo. Mol Cell 12:525–532
DeJesus-Hernandez M, Mackenzie IR, Boeve BF, Boxer AL, Baker M, Rutherford NJ, Nicholson AM, Finch NA, Flynn H, Adamson J et al (2011) Expanded GGGGCC Hexanucleotide Repeat in Noncoding Region of C9ORF72 Causes Chromosome 9p-Linked FTD and ALS. Neuron 72:245–256. doi:10.1016/j.neuron.2011.09.011
Delaleau M, Borden KL (2015) Multiple Export Mechanisms for mRNAs. Cells 4:452–473. doi:10.3390/cells4030452
Diegoli TM (2015) Forensic typing of short tandem repeat markers on the X and Y chromosomes. Forensic Sci Int Genet 18:140–151. doi:10.1016/j.fsigen.2015.03.013
Dodd DW, Tomchick DR, Corey DR, Gagnon KT (2016) Pathogenic C9ORF72 Antisense Repeat RNA Forms a Double Helix with Tandem C:C Mismatches. Biochemistry 55:1283–1286. doi:10.1021/acs.biochem.6b00136
Doma MK, Parker R (2006) Endonucleolytic cleavage of eukaryotic mRNAs with stalls in translation elongation. Nature 440:561–564. doi:10.1038/nature04530
Donnelly CJ, Zhang PW, Pham JT, Haeusler AR, Mistry NA, Vidensky S, Daley EL, Poth EM, Hoover B, Fines DM et al (2013) RNA toxicity from the ALS/FTD C9ORF72 expansion is mitigated by antisense intervention. Neuron 80:415–428. doi:10.1016/j.neuron.2013.10.015
Dumache R, Ciocan V, Muresan C, Enache A (2016) Molecular DNA Analysis in Forensic Identification. Clin Lab 62:245–248
Eberle AB, Hessle V, Helbig R, Dantoft W, Gimber N, Visa N (2010) Splice-site mutations cause Rrp6-mediated nuclear retention of the unspliced RNAs and transcriptional down-regulation of the splicing-defective genes. PLoS One 5:e11540. doi:10.1371/journal.pone.0011540
Eberle AB, Visa N (2014) Quality control of mRNP biogenesis: networking at the transcription site. Semin Cell Dev Biol 32:37–46. doi:10.1016/j.semcdb.2014.03.033
Ellegren H (2000) Heterogeneous mutation processes in human microsatellite DNA sequences. Nat Genet 24:400–402. doi:10.1038/74249
Ellegren H (2004) Microsatellites: simple sequences with complex evolution. Nat Rev Genet 5:435–445. doi:10.1038/nrg1348
Evans-Galea MV, Hannan AJ, Carrodus N, Delatycki MB, Saffery R (2013) Epigenetic modifications in trinucleotide repeat diseases. Trends Mol Med 19:655–663. doi:10.1016/j.molmed.2013.07.007
Fellmann C, Gowen BG, Lin PC, Doudna JA, Corn JE (2017) Cornerstones of CRISPR-Cas in drug discovery and therapy. Nat Rev Drug Discov 16:89–100. doi:10.1038/nrd.2016.238
Feng Y, Zhang F, Lokey LK, Chastain JL, Lakkis L, Eberhart D, Warren ST (1995) Translational suppression by trinucleotide repeat expansion at FMR1. Science 268:731–734
Finkel RS (2010) Read-through strategies for suppression of nonsense mutations in Duchenne/ Becker muscular dystrophy: aminoglycosides and ataluren (PTC124). J Child Neurol 25:1158–1164. doi:10.1177/0883073810371129
Floer M, Blobel G (1999) Putative reaction intermediates in Crm1-mediated nuclear protein export. J Biol Chem 274:16279–16286
Foiry L, Dong L, Savouret C, Hubert L, te Riele H, Junien C, Gourdon G (2006) Msh3 is a limiting factor in the formation of intergenerational CTG expansions in DM1 transgenic mice. Hum Genet 119:520–526. doi:10.1007/s00439-006-0164-7
Follonier C, Oehler J, Herrador R, Lopes M (2013) Friedreich's ataxia-associated GAA repeats induce replication-fork reversal and unusual molecular junctions. Nat Struct Mol Biol 20:486–494. doi:10.1038/nsmb.2520
Fornerod M, Ohno M, Yoshida M, Mattaj IW (1997) CRM1 is an export receptor for leucine-rich nuclear export signals. Cell 90:1051–1060
Fradkin LG, Budnik V (2016) This bud's for you: mechanisms of cellular nucleocytoplasmic trafficking via nuclear envelope budding. Curr Opin Cell Biol 41:125–131. doi:10.1016/j.ceb.2016.05.001
Fratta P, Mizielinska S, Nicoll AJ, Zloh M, Fisher EMC, Parkinson G, Isaacs AM (2012) C9orf72 hexanucleotide repeat associated with amyotrophic lateral sclerosis and frontotemporal dementia forms RNA G-quadruplexes. Sci Rep-Uk 2:1016. doi:10.1038/Srep01016
Freibaum BD, Lu Y, Lopez-Gonzalez R, Kim NC, Almeida S, Lee KH, Badders N, Valentine M, Miller BL, Wong PC et al (2015) GGGGCC repeat expansion in C9orf72 compromises nucleocytoplasmic transport. Nature 525:129–133. doi:10.1038/nature14974
Freudenreich CH, Kantrow SM, Zakian VA (1998) Expansion and length-dependent fragility of CTG repeats in yeast. Science 279:853–856
Frischmeyer PA, van Hoof A, O'Donnell K, Guerrerio AL, Parker R, Dietz HC (2002) An mRNA surveillance mechanism that eliminates transcripts lacking termination codons. Science 295:2258–2261. doi:10.1126/science.1067338
Froehlich AC, Liu Y, Loros JJ, Dunlap JC (2002) White Collar-1, a circadian blue light photoreceptor, binding to the frequency promoter. Science 297:815–819. doi:10.1126/science.1073681
Gadgil R, Barthelemy J, Lewis T, Leffak M (2017) Replication stalling and DNA microsatellite instability. Biophys Chem 225:38–48. doi:10.1016/j.bpc.2016.11.007
Gagnon KT, Corey DR (2015) Stepping toward therapeutic CRISPR. Proc Natl Acad Sci U S A 112:15536–15537. doi:10.1073/pnas.1521670112
Gan W, Guan Z, Liu J, Gui T, Shen K, Manley JL, Li X (2011) R-loop-mediated genomic instability is caused by impairment of replication fork progression. Genes Dev 25:2041–2056. doi:10.1101/gad.17010011
Garavis M, Gonzalez C, Villasante A (2013) On the origin of the eukaryotic chromosome: the role of noncanonical DNA structures in telomere evolution. Genome Biol Evol 5:1142–1150. doi:10.1093/gbe/evt079
Garcia SM, Tabach Y, Lourenco GF, Armakola M, Ruvkun G (2014) Identification of genes in toxicity pathways of trinucleotide-repeat RNA in C. elegans. Nat Struct Mol Biol 21:712–720. doi:10.1038/nsmb.2858
Gellert M, Lipsett MN, Davies DR (1962) Helix formation by guanylic acid. Proc Natl Acad Sci U S A 48:2013–2018
Gendron TF, Bieniek KF, Zhang YJ, Jansen-West K, Ash PE, Caulfield T, Daughrity L, Dunmore JH, Castanedes-Casey M, Chew J et al (2013) Antisense transcripts of the expanded C9ORF72 hexanucleotide repeat form nuclear RNA foci and undergo repeat-associated non-ATG translation in c9FTD/ALS. Acta Neuropathol 126:829–844. doi:10.1007/s00401-013-1192-8
Ginno PA, Lott PL, Christensen HC, Korf I, Chedin F (2012) R-loop formation is a distinctive characteristic of unmethylated human CpG island promoters. Mol Cell 45:814–825. doi:10.1016/j.molcel.2012.01.017
Goers ES, Purcell J, Voelker RB, Gates DP, Berglund JA (2010) MBNL1 binds GC motifs embedded in pyrimidines to regulate alternative splicing. Nucleic Acids Res 38:2467–2484. doi:10.1093/nar/gkp1209
Gordenin DA, Kunkel TA, Resnick MA (1997) Repeat expansion--all in a flap? Nat Genet 16:116–118. doi:10.1038/ng0697-116
Grabczyk E, Usdin K (2000) The GAA*TTC triplet repeat expanded in Friedreich's ataxia impedes transcription elongation by T7 RNA polymerase in a length and supercoil dependent manner. Nucleic Acids Res 28:2815–2822
Grandi FC, An W (2013) Non-LTR retrotransposons and microsatellites: Partners in genomic variation. Mobile Genet Elem 3:e25674. doi:10.4161/mge.25674
Green KM, Linsalata AE, Todd PK (2016) RAN translation-What makes it run? Brain Res. doi:10.1016/j.brainres.2016.04.003
Greene E, Mahishi L, Entezam A, Kumari D, Usdin K (2007) Repeat-induced epigenetic changes in intron 1 of the frataxin gene and its consequences in Friedreich ataxia. Nucleic Acids Res 35:3383–3390. doi:10.1093/nar/gkm271
Griesche N, Schilling J, Weber S, Rohm M, Pesch V, Matthes F, Auburger G, Krauss S (2016) Regulation of mRNA Translation by MID1: A Common Mechanism of Expanded CAG Repeat RNAs. Front Cell Neurosci 10:226. doi:10.3389/fncel.2016.00226
Groh M, Lufino MM, Wade-Martins R, Gromak N (2014) R-loops associated with triplet repeat expansions promote gene silencing in Friedreich ataxia and fragile X syndrome. PLoS Genet 10:e1004318. doi:10.1371/journal.pgen.1004318
Groh M, Silva LM, Gromak N (2014) Mechanisms of transcriptional dysregulation in repeat expansion disorders. Biochem Soc Trans 42:1123–1128. doi:10.1042/BST20140049
Gruter P, Tabernero C, von Kobbe C, Schmitt C, Saavedra C, Bachi A, Wilm M, Felber BK, Izaurralde E (1998) TAP, the human homolog of Mex67p, mediates CTE-dependent RNA export from the nucleus. Mol Cell 1:649–659
Guan L, Disney MD (2012) Recent advances in developing small molecules targeting RNA. Acs Chem Biol 7:73–86. doi:10.1021/cb200447r
Gudanis D, Popenda L, Szpotkowski K, Kierzek R, Gdaniec Z (2016) Structural characterization of a dimer of RNA duplexes composed of 8-bromoguanosine modified CGG trinucleotide repeats: a novel architecture of RNA quadruplexes. Nucleic Acids Res 44:2409–2416. doi:10.1093/nar/gkv1534
Gudde AE, Gonzalez-Barriga A, van den Broek WJ, Wieringa B, Wansink DG (2016) A low absolute number of expanded transcripts is involved in myotonic dystrophy type 1 manifestation in muscle. Hum Mol Genet 25:1648–1662. doi:10.1093/hmg/ddw042
Gudde AE, van Heeringen SJ, de Oude AI, van Kessel ID, Estabrook J, Wang ET, Wieringa B, Wansink DG (2017) Antisense transcription of the myotonic dystrophy locus yields low-abundant RNAs with and without (CAG)n repeat. RNA Biol 0. doi:10.1080/15476286.2017.1279787
Guo J, Gu L, Leffak M, Li GM (2016) MutSbeta promotes trinucleotide repeat expansion by recruiting DNA polymerase beta to nascent (CAG)n or (CTG)n hairpins for error-prone DNA synthesis. Cell Res 26:775–786. doi:10.1038/cr.2016.66
Guo JU, Bartel DP (2016) RNA G-quadruplexes are globally unfolded in eukaryotic cells and depleted in bacteria. Science 353. doi:10.1126/science.aaf5371
Haeusler AR, Donnelly CJ, Periz G, Simko EA, Shaw PG, Kim MS, Maragakis NJ, Troncoso JC, Pandey A, Sattler Ret al (2014) C9orf72 nucleotide repeat structures initiate molecular cascades of disease. Nature 507:195–200. doi:10.1038/nature13124
Hamperl S, Cimprich KA (2016) Conflict Resolution in the Genome: How Transcription and Replication Make It Work. Cell 167:1455–1467. doi:10.1016/j.cell.2016.09.053
Hautbergue GM, Castelli LM, Ferraiuolo L, Sanchez-Martinez A, Cooper-Knock J, Higginbottom A, Lin YH, Bauer CS, Dodd JE, Myszczynska MA et al (2017) SRSF1-dependent nuclear export inhibition of C9ORF72 repeat transcripts prevents neurodegeneration and associated motor deficits. Nat Commun 8:16063. doi:10.1038/ncomms16063
Hautbergue GM, Hung ML, Golovanov AP, Lian LY, Wilson SA (2008) Mutually exclusive interactions drive handover of mRNA from export adaptors to TAP. Proc Natl Acad Sci U S A 105:5154–5159. doi:10.1073/pnas.0709167105
He F, Todd PK (2011) Epigenetics in nucleotide repeat expansion disorders. Semin Neurol 31:470–483. doi:10.1055/s-0031-1299786
He Y, Vogelstein B, Velculescu VE, Papadopoulos N, Kinzler KW (2008) The antisense transcriptomes of human cells. Science 322:1855–1857. doi:10.1126/science.1163853
Heitz D, Rousseau F, Devys D, Saccone S, Abderrahim H, Le Paslier D, Cohen D, Vincent A, Toniolo D, Della Valle G et al (1991) Isolation of sequences that span the fragile X and identification of a fragile X-related CpG island. Science 251:1236–1239
Hesselberth JR (2013) Lives that introns lead after splicing. Wiley Interdiscip Rev RNA 4:677–691. doi:10.1002/wrna.1187
Hinnebusch AG (2014) The scanning mechanism of eukaryotic translation initiation. Annu Rev Biochem 83:779–812. doi:10.1146/annurev-biochem-060713-035802
Hinnebusch AG, Ivanov IP, Sonenberg N (2016) Translational control by 5'-untranslated regions of eukaryotic mRNAs. Science 352:1413–1416. doi:10.1126/science.aad9868
Hino S, Kondo S, Sekiya H, Saito A, Kanemoto S, Murakami T, Chihara K, Aoki Y, Nakamori M, Takahashi MP et al (2007) Molecular mechanisms responsible for aberrant splicing of SERCA1 in myotonic dystrophy type 1. Hum Mol Genet 16:2834–2843 doi:10.1093/hmg/ddm239
Hirtreiter A, Damsma GE, Cheung AC, Klose D, Grohmann D, Vojnic E, Martin AC, Cramer P, Werner F (2010) Spt4/5 stimulates transcription elongation through the RNA polymerase clamp coiled-coil motif. Nucleic Acids Res 38:4040–4051. doi:10.1093/nar/gkq135
Holmes SE, O'Hearn E, Rosenblatt A, Callahan C, Hwang HS, Ingersoll-Ashworth RG, Fleisher A, Stevanin G, Brice A, Potter NT et al (2001) A repeat expansion in the gene encoding junctophilin-3 is associated with Huntington disease-like 2. Nat Genet 29:377–378. doi:10.1038/ng760
Holmes SE, O'Hearn EE, McInnis MG, Gorelick-Feldman DA, Kleiderlein JJ, Callahan C, Kwak NG, Ingersoll-Ashworth RG, Sherr M, Sumner AJ et al (1999) Expansion of a novel CAG trinucleotide repeat in the 5' region of PPP2R2B is associated with SCA12. Nat Genet 23:391–392. doi:10.1038/70493
Houseley J, Tollervey D (2011) Repeat expansion in the budding yeast ribosomal DNA can occur independently of the canonical homologous recombination machinery. Nucleic Acids Res 39:8778–8791. doi:10.1093/nar/gkr589
Huertas P, Aguilera A (2003) Cotranscriptionally formed DNA:RNA hybrids mediate transcription elongation impairment and transcription-associated recombination. Mol Cell 12:711–721
Hung ML, Hautbergue GM, Snijders AP, Dickman MJ, Wilson SA (2010) Arginine methylation of REF/ALY promotes efficient handover of mRNA to TAP/NXF1. Nucleic Acids Res 38:3351–3361. doi:10.1093/nar/gkq033
Iglesias N, Stutz F (2008) Regulation of mRNP dynamics along the export pathway. FEBS letters 582:1987–1996. doi:10.1016/j.febslet.2008.03.038
Ikeda Y, Daughters RS, Ranum LP (2008) Bidirectional expression of the SCA8 expansion mutation: one mutation, two genes. Cerebellum 7:150–158. doi:10.1007/s12311-008-0010-7
Imbert G, Saudou F, Yvert G, Devys D, Trottier Y, Garnier JM, Weber C, Mandel JL, Cancel G, Abbas N et al (1996) Cloning of the gene for spinocerebellar ataxia 2 reveals a locus with high sensitivity to expanded CAG/glutamine repeats. Nat Genet 14:285–291. doi:10.1038/ng1196-285
Ingolia NT, Ghaemmaghami S, Newman JR, Weissman JS (2009) Genome-wide analysis in vivo of translation with nucleotide resolution using ribosome profiling. Science 324:218–223. doi:10.1126/science.1168978
Ishiguro T, Sato N, Ueyama M, Fujikake N, Sellier C, Kanegami A, Tokuda E, Zamiri B, Gall-Duncan T, Mirceta M et al (2017) Regulatory Role of RNA Chaperone TDP-43 for RNA Misfolding and Repeat-Associated Translation in SCA31. Neuron. doi:10.1016/j.neuron.2017.02.046
Iyer RR, Pluciennik A, Napierala M, Wells RD (2015) DNA Triplet Repeat Expansion and Mismatch Repair. Annu Rev Biochem. doi:10.1146/annurev-biochem-060614-034010
Izumi H, McCloskey A, Shinmyozu K, Ohno M (2014) p54nrb/NonO and PSF promote U snRNA nuclear export by accelerating its export complex assembly. Nucleic Acids Res 42:3998–4007. doi:10.1093/nar/gkt1365
Jain A, Vale RD (2017) RNA phase transitions in repeat expansion disorders. Nature 546:243–247. doi:10.1038/nature22386
Jansen A, Gemayel R, Verstrepen KJ (2012) Unstable microsatellite repeats facilitate rapid evolution of coding and regulatory sequences. Genome Dyn 7:108–125. doi:10.1159/000337121
Jiang J, Zhu Q, Gendron TF, Saberi S, McAlonis-Downes M, Seelman A, Stauffer JE, Jafar-Nejad P, Drenner K, Schulte D et al (2016) Gain of Toxicity from ALS/FTD-Linked Repeat Expansions in C9ORF72 Is Alleviated by Antisense Oligonucleotides Targeting GGGGCC-Containing RNAs. Neuron 90:535–550. doi:10.1016/j.neuron.2016.04.006
Jokhi V, Ashley J, Nunnari J, Noma A, Ito N, Wakabayashi-Ito N, Moore MJ, Budnik V (2013) Torsin mediates primary envelopment of large ribonucleoprotein granules at the nuclear envelope. Cell Rep 3:988–995. doi:10.1016/j.celrep.2013.03.015
Jovicic A, Mertens J, Boeynaems S, Bogaert E, Chai N, Yamada SB, Paul JW, 3rd, Sun S, Herdy JR, Bieri Get al (2015) Modifiers of C9orf72 dipeptide repeat toxicity connect nucleocytoplasmic transport defects to FTD/ALS. Nat Neurosci 18: 1226-1229 Doi doi:10.1038/nn.4085
Kang S, Jaworski A, Ohshima K, Wells RD (1995) Expansion and deletion of CTG repeats from human disease genes are determined by the direction of replication in E. coli. Nat Genet 10:213–218. doi:10.1038/ng0695-213
Katahira J (2012) mRNA export and the TREX complex. Biochimica Et Biophysica Acta 1819:507–513. doi:10.1016/j.bbagrm.2011.12.001
Katahira J (2015) Nuclear export of messenger RNA. Genes 6:163–184. doi:10.3390/genes6020163
Kawaguchi Y, Okamoto T, Taniwaki M, Aizawa M, Inoue M, Katayama S, Kawakami H, Nakamura S, Nishimura M, Akiguchi I et al (1994) CAG expansions in a novel gene for Machado-Joseph disease at chromosome 14q32.1 Nat Genet 8:221–228. doi:10.1038/ng1194-221
Kearse MG, Green KM, Krans A, Rodriguez CM, Linsalata AE, Goldstrohm AC, Todd PK (2016) CGG Repeat-Associated Non-AUG Translation Utilizes a Cap-Dependent Scanning Mechanism of Initiation to Produce Toxic Proteins. Mol Cell 62:314–322. doi:10.1016/j.molcel.2016.02.034
Kearse MG, Todd PK (2014) Repeat-associated non-AUG translation and its impact in neurodegenerative disease. Neurotherapeutics 11:721–731. doi:10.1007/s13311-014-0292-z
Kenneson A, Zhang F, Hagedorn CH, Warren ST (2001) Reduced FMRP and increased FMR1 transcription is proportionally associated with CGG repeat number in intermediate-length and premutation carriers. Hum Mol Genet 10:1449–1454
Kerrest A, Anand RP, Sundararajan R, Bermejo R, Liberi G, Dujon B, Freudenreich CH, Richard GF (2009) SRS2 and SGS1 prevent chromosomal breaks and stabilize triplet repeats by restraining recombination. Nat Struct Mol Biol 16:159–167. doi:10.1038/nsmb.1544
Khorkova O, Myers AJ, Hsiao J, Wahlestedt C (2014) Natural antisense transcripts. Hum Mol Genet 23:R54–R63. doi:10.1093/hmg/ddu207
Kilchert C, Wittmann S, Vasiljeva L (2016) The regulation and functions of the nuclear RNA exosome complex. Nat Rev Mol Cell Biol 17:227–239. doi:10.1038/nrm.2015.15
Kiliszek A, Rypniewski W (2014) Structural studies of CNG repeats. Nucleic Acids Res 42:8189–8199
Kim JC, Harris ST, Dinter T, Shah KA, Mirkin SM (2017) The role of break-induced replication in large-scale expansions of (CAG)n/(CTG)n repeats. Nat Struct Mol Biol 24:55–60. doi:10.1038/nsmb.3334
Kim JC, Mirkin SM (2013) The balancing act of DNA repeat expansions. Curr Opin Genet Dev 23:280–288. doi:10.1016/j.gde.2013.04.009
Kino Y, Washizu C, Kurosawa M, Oma Y, Hattori N, Ishiura S, Nukina N (2015) Nuclear localization of MBNL1: splicing-mediated autoregulation and repression of repeat-derived aberrant proteins. Hum Mol Genet 24:740–756. doi:10.1093/hmg/ddu492
Klein BJ, Bose D, Baker KJ, Yusoff ZM, Zhang X, Murakami KS (2011) RNA polymerase and transcription elongation factor Spt4/5 complex structure. Proc Natl Acad Sci U S A 108:546–550. doi:10.1073/pnas.1013828108
Knight SJ, Flannery AV, Hirst MC, Campbell L, Christodoulou Z, Phelps SR, Pointon J, Middleton-Price HR, Barnicoat A, Pembrey ME et al (1993) Trinucleotide repeat amplification and hypermethylation of a CpG island in FRAXE mental retardation. Cell 74:127–134
Kobayashi H, Abe K, Matsuura T, Ikeda Y, Hitomi T, Akechi Y, Habu T, Liu W, Okuda H, Koizumi A (2011) Expansion of intronic GGCCTG hexanucleotide repeat in NOP56 causes SCA36, a type of spinocerebellar ataxia accompanied by motor neuron involvement. Am J Hum Genet 89:121–130. doi:10.1016/j.ajhg.2011.05.015
Kochetov AV, Palyanov A, Titov II, Grigorovich D, Sarai A, Kolchanov NA (2007) AUG_hairpin: prediction of a downstream secondary structure influencing the recognition of a translation start site. BMC Bioinformatics 8:318. doi:10.1186/1471-2105-8-318
Koide R, Ikeuchi T, Onodera O, Tanaka H, Igarashi S, Endo K, Takahashi H, Kondo R, Ishikawa A, Hayashi T et al (1994) Unstable expansion of CAG repeat in hereditary dentatorubral-pallidoluysian atrophy (DRPLA). Nat Genet 6:9–13. doi:10.1038/ng0194-9
Koob MD, Moseley ML, Schut LJ, Benzow KA, Bird TD, Day JW, Ranum LP (1999) An untranslated CTG expansion causes a novel form of spinocerebellar ataxia (SCA8). Nat Genet 21:379–384. doi:10.1038/7710
Kota KP, Wagner SR, Huerta E, Underwood JM, Nickerson JA (2008) Binding of ATP to UAP56 is necessary for mRNA export. J Cell Sci 121:1526–1537. doi:10.1242/jcs.021055
Kozak M (1990) Downstream secondary structure facilitates recognition of initiator codons by eukaryotic ribosomes. Proc Natl Acad Sci U S A 87:8301–8305
Kramer NJ, Carlomagno Y, Zhang YJ, Almeida S, Cook CN, Gendron TF, Prudencio M, Van Blitterswijk M, Belzil V, Couthouis J et al (2016) Spt4 selectively regulates the expression of C9orf72 sense and antisense mutant transcripts. Science 353:708–712. doi:10.1126/science.aaf7791
Krauss S, Griesche N, Jastrzebska E, Chen C, Rutschow D, Achmuller C, Dorn S, Boesch SM, Lalowski M, Wanker E et al (2013) Translation of HTT mRNA with expanded CAG repeats is regulated by the MID1-PP2A protein complex. Nat Commun 4:1511. doi:10.1038/ncomms2514
Kremer EJ, Yu S, Pritchard M, Nagaraja R, Heitz D, Lynch M, Baker E, Hyland VJ, Little RD, Wada Met al (1991) Isolation of a human DNA sequence which spans the fragile X. Am J Hum Genet 49:656–661
Kumari D, Biacsi RE, Usdin K (2011) Repeat expansion affects both transcription initiation and elongation in friedreich ataxia cells. J Biol Chem 286:4209–4215. doi:10.1074/jbc.M110.194035
Kurosaki T, Ueda S, Ishida T, Abe K, Ohno K, Matsuura T (2012) The unstable CCTG repeat responsible for myotonic dystrophy type 2 originates from an AluSx element insertion into an early primate genome. PLoS One 7:e38379. doi:10.1371/journal.pone.0038379
Kwok CK, Marsico G, Sahakyan AB, Chambers VS, Balasubramanian S (2016) rG4-seq reveals widespread formation of G-quadruplex structures in the human transcriptome. Nat Methods 13:841–844. doi:10.1038/nmeth.3965
Kwon I, Xiang S, Kato M, Wu L, Theodoropoulous P, Wang T, Kim J, Yun J, Xie Y, McKnight SL (2014) Poly-dipeptides encoded by the C9orf72 repeats bind nucleoli, impede RNA biogenesis, and kill cells. Science 345:1139–1145
La Spada AR, Taylor JP (2010) Repeat expansion disease: progress and puzzles in disease pathogenesis. Nat Rev Genet 11:247–258. doi:10.1038/nrg2748
La Spada AR, Wilson EM, Lubahn DB, Harding AE, Fischbeck KH (1991) Androgen receptor gene mutations in X-linked spinal and bulbar muscular atrophy. Nature 352:77–79. doi:10.1038/352077a0
Labbadia J, Morimoto RI (2013) Huntington's disease: underlying molecular mechanisms and emerging concepts. Trends Biochem Sci 38:378–385. doi:10.1016/j.tibs.2013.05.003
Lagier-Tourenne C, Baughn M, Rigo F, Sun S, Liu P, Li HR, Jiang J, Watt AT, Chun S, Katz M et al (2013) Targeted degradation of sense and antisense C9orf72 RNA foci as therapy for ALS and frontotemporal degeneration. Proc Natl Acad Sci U S A 110:E4530–E4539. doi:10.1073/pnas.1318835110
Lalioti MD, Scott HS, Buresi C, Rossier C, Bottani A, Morris MA, Malafosse A, Antonarakis SE (1997) Dodecamer repeat expansion in cystatin B gene in progressive myoclonus epilepsy. Nature 386:847–851. doi:10.1038/386847a0
Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, Baldwin J, Devon K, Dewar K, Doyle M, FitzHugh W et al (2001) Initial sequencing and analysis of the human genome. Nature 409:860–921. doi:10.1038/35057062
Le Hir H, Izaurralde E, Maquat LE, Moore MJ (2000) The spliceosome deposits multiple proteins 20-24 nucleotides upstream of mRNA exon-exon junctions. EMBO J 19:6860–6869. doi:10.1093/emboj/19.24.6860
Lee JE, Cooper TA (2009) Pathogenic mechanisms of myotonic dystrophy. Biochem Soc Trans 37:1281–1286. doi:10.1042/BST0371281
Lee KH, Zhang P, Kim HJ, Mitrea DM, Sarkar M, Freibaum BD, Cika J, Coughlin M, Messing J, Molliex A et al (2016) C9orf72 Dipeptide Repeats Impair the Assembly, Dynamics, and Function of Membrane-Less Organelles. Cell 167(774-788):e717. doi:10.1016/j.cell.2016.10.002
Lee YB, Chen HJ, Peres JN, Gomez-Deza J, Attig J, Stalekar M, Troakes C, Nishimura AL, Scotter EL, Vance C et al (2013) Hexanucleotide Repeats in ALS/FTD Form Length-Dependent RNA Foci, Sequester RNA Binding Proteins, and Are Neurotoxic. Cell Rep 5:1178–1186. doi:10.1016/j.celrep.2013.10.049
Leffak M (2017) Break-induced replication links microsatellite expansion to complex genome rearrangements. Bioessays 39. doi:10.1002/bies.201700025
Li L, Matsui M, Corey DR (2016) Activating frataxin expression by repeat-targeted nucleic acids. Nat Commun 7:10606. doi:10.1038/ncomms10606
Li Y, Bor YC, Misawa Y, Xue Y, Rekosh D, Hammarskjold ML (2006) An intron with a constitutive transport element is retained in a Tap messenger RNA. Nature 443:234–237. doi:10.1038/nature05107
Li YY, Abu-Ghazalah R, Zamiri B, Macgregor RB Jr (2016) Concentration-dependent conformational changes in GQ-forming ODNs. Biophys Chem 211:70–75. doi:10.1016/j.bpc.2016.02.002
Lin Y, Hubert L Jr, Wilson JH (2009) Transcription destabilizes triplet repeats. Mol Carcinog 48:350–361. doi:10.1002/mc.20488
Lin Y, Leng M, Wan M, Wilson JH (2010) Convergent transcription through a long CAG tract destabilizes repeats and induces apoptosis. Mol Cell Biol 30:4435–4451. doi:10.1128/MCB.00332-10
Lin Y, Mori E, Kato M, Xiang S, Wu L, Kwon I, McKnight SL (2016) Toxic PR Poly-Dipeptides Encoded by the C9orf72 Repeat Expansion Target LC Domain Polymers. Cell 167:789–802 e712. doi:10.1016/j.cell.2016.10.003
Lin Y, Wilson JH (2012) Nucleotide excision repair, mismatch repair, and R-loops modulate convergent transcription-induced cell death and repeat instability. PLoS One 7:e46807. doi:10.1371/journal.pone.0046807
Liquori CL, Ricker K, Moseley ML, Jacobsen JF, Kress W, Naylor SL, Day JW, Ranum LP (2001) Myotonic dystrophy type 2 caused by a CCTG expansion in intron 1 of ZNF9. Science 293:864–867. doi:10.1126/science.1062125
Liu B, Steitz TA (2017) Structural insights into NusG regulating transcription elongation. Nucleic Acids Res 45:968–974. doi:10.1093/nar/gkw1159
Liu CR, Chang CR, Chern Y, Wang TH, Hsieh WC, Shen WC, Chang CY, Chu IC, Deng N, Cohen SN et al (2012) Spt4 is selectively required for transcription of extended trinucleotide repeats. Cell 148:690–701. doi:10.1016/j.cell.2011.12.032
Liu G, Chen X, Bissler JJ, Sinden RR, Leffak M (2010) Replication-dependent instability at (CTG) x (CAG) repeat hairpins in human cells. Nat Chem Biol 6:652–659. doi:10.1038/nchembio.416
Liu J, Hu J, Ludlow AT, Pham JT, Shay JW, Rothstein JD, Corey DR (2017) c9orf72 Disease-Related Foci Are Each Composed of One Mutant Expanded Repeat RNA. Cell Chem Biol 24:141–148. doi:10.1016/j.chembiol.2016.12.018
Llorente B, Smith CE, Symington LS (2008) Break-induced replication: what is it and what is it for? Cell Cycle 7:859–864. doi:10.4161/cc.7.7.5613
Lokanga RA, Senejani AG, Sweasy JB, Usdin K (2015) Heterozygosity for a hypomorphic Polbeta mutation reduces the expansion frequency in a mouse model of the Fragile X-related disorders. PLoS Genet 11:e1005181. doi:10.1371/journal.pgen.1005181
Lujan SA, Clausen AR, Clark AB, MacAlpine HK, MacAlpine DM, Malc EP, Mieczkowski PA, Burkholder AB, Fargo DC, Gordenin DA et al (2014) Heterogeneous polymerase fidelity and mismatch repair bias genome variation and composition. Genome Res 24:1751–1764. doi:10.1101/gr.178335.114
MacDonald ME, Barnes G, Srinidhi J, Duyao MP, Ambrose CM, Myers RH, Gray J, Conneally PM, Young A, Penney J et al (1993) Gametic but not somatic instability of CAG repeat length in Huntington's disease. J Med Genet 30:982–986
Mahadevan M, Tsilfidis C, Sabourin L, Shutler G, Amemiya C, Jansen G, Neville C, Narang M, Barcelo J, O'Hoy K et al (1992) Myotonic dystrophy mutation: an unstable CTG repeat in the 3' untranslated region of the gene. Science 255:1253–1255
Malgowska M, Gudanis D, Kierzek R, Wyszko E, Gabelica V, Gdaniec Z (2014) Distinctive structural motifs of RNA G-quadruplexes composed of AGG, CGG and UGG trinucleotide repeats. Nucleic Acids Res 42:10196–10207. doi:10.1093/nar/gku710
Malkova A, Haber JE (2012) Mutations arising during repair of chromosome breaks. Annu Rev Genet 46:455–473. doi:10.1146/annurev-genet-110711-155547
Mankodi A, Logigian E, Callahan L, McClain C, White R, Henderson D, Krym M, Thornton CA (2000) Myotonic dystrophy in transgenic mice expressing an expanded CUG repeat. Science 289:1769–1773
Mankodi A, Urbinati CR, Yuan QP, Moxley RT, Sansone V, Krym M, Henderson D, Schalling M, Swanson MS, Thornton CA (2001) Muscleblind localizes to nuclear foci of aberrant RNA in myotonic dystrophy types 1 and 2. Hum Mol Genet 10:2165–2170
Mao YS, Zhang B, Spector DL (2011) Biogenesis and function of nuclear bodies. Trends Genet 27:295–306. doi:10.1016/j.tig.2011.05.006
Maquat LE, Li X (2001) Mammalian heat shock p70 and histone H4 transcripts, which derive from naturally intronless genes, are immune to nonsense-mediated decay. RNA 7:445–456
Maric M, Shao J, Ryan RJ, Wong CS, Gonzalez-Alegre P, Roller RJ (2011) A functional role for TorsinA in herpes simplex virus 1 nuclear egress. J Virol 85:9667–9679. doi:10.1128/JVI.05314-11
Martinez-Rucobo FW, Sainsbury S, Cheung AC, Cramer P (2011) Architecture of the RNA polymerase-Spt4/5 complex and basis of universal transcription processivity. EMBO J 30:1302–1310. doi:10.1038/emboj.2011.64
Mason SW, Greenblatt J (1991) Assembly of transcription elongation complexes containing the N protein of phage lambda and the Escherichia coli elongation factors NusA, NusB, NusG, and S10. Genes Dev 5:1504–1512
Massenet S, Bertrand E, Verheggen C (2016) Assembly and trafficking of box C/D and H/ACA snoRNPs. RNA Biol 1–13. doi:10.1080/15476286.2016.1243646
Masuda S, Das R, Cheng H, Hurt E, Dorman N, Reed R (2005) Recruitment of the human TREX complex to mRNA during splicing. Genes Dev 19:1512–1517. doi:10.1101/gad.1302205
Matsuura T, Yamagata T, Burgess DL, Rasmussen A, Grewal RP, Watase K, Khajavi M, McCall AE, Davis CF, Zu L et al (2000) Large expansion of the ATTCT pentanucleotide repeat in spinocerebellar ataxia type 10. Nat Genet 26:191–194. doi:10.1038/79911
McMurray CT (2010) Mechanisms of trinucleotide repeat instability during human development. Nat Rev Genet 11:786–799. doi:10.1038/nrg2828
Meng F, Na I, Kurgan L, Uversky VN (2015) Compartmentalization and Functionality of Nuclear Disorder: Intrinsic Disorder and Protein-Protein Interactions in Intra-Nuclear Compartments. Int J Mol Sci 17. doi:10.3390/ijms17010024
Meservy JL, Sargent RG, Iyer RR, Chan F, McKenzie GJ, Wells RD, Wilson JH (2003) Long CTG tracts from the myotonic dystrophy gene induce deletions and rearrangements during recombination at the APRT locus in CHO cells. Mol Cell Biol 23:3152–3162
Metsu S, Rainger JK, Debacker K, Bernhard B, Rooms L, Grafodatskaya D, Weksberg R, Fombonne E, Taylor MS, Scherer SW et al (2014) A CGG-repeat expansion mutation in ZNF713 causes FRA7A: association with autistic spectrum disorder in two families. Hum Mutat 35:1295–1300. doi:10.1002/humu.22683
Metsu S, Rooms L, Rainger J, Taylor MS, Bengani H, Wilson DI, Chilamakuri CS, Morrison H, Vandeweyer G, Reyniers E et al (2014) FRA2A is a CGG repeat expansion associated with silencing of AFF3. PLoS Genet 10:e1004242. doi:10.1371/journal.pgen.1004242
Michael TP, Park S, Kim TS, Booth J, Byer A, Sun Q, Chory J, Lee K (2007) Simple sequence repeats provide a substrate for phenotypic variation in the Neurospora crassa circadian clock. PLoS One 2:e795. doi:10.1371/journal.pone.0000795
Mirkin SM (2007) Expandable DNA repeats and human disease. Nature 447:932–940. doi:10.1038/nature05977
Mizielinska S, Gronke S, Niccoli T, Ridler CE, Clayton EL, Devoy A, Moens T, Norona FE, Woollacott IO, Pietrzyk J et al (2014) C9orf72 repeat expansions cause neurodegeneration in Drosophila through arginine-rich proteins. Science 345:1192–1194. doi:10.1126/science.1256800
Mohan A, Goodwin M, Swanson MS (2014) RNA-protein interactions in unstable microsatellite diseases. Brain Res 1584:3–14. doi:10.1016/j.brainres.2014.03.039
Monteys AM, Ebanks SA, Keiser MS, Davidson BL (2017) CRISPR/Cas9 Editing of the Mutant Huntingtin Allele In Vitro and In Vivo. Mol Ther 25:12–23. doi:10.1016/j.ymthe.2016.11.010
Mori K, Arzberger T, Grasser FA, Gijselinck I, May S, Rentzsch K, Weng SM, Schludi MH, van der Zee J, Cruts M et al (2013) Bidirectional transcripts of the expanded C9orf72 hexanucleotide repeat are translated into aggregating dipeptide repeat proteins. Acta Neuropathol 126:881–893. doi:10.1007/s00401-013-1189-3
Mori K, Lammich S, Mackenzie IR, Forne I, Zilow S, Kretzschmar H, Edbauer D, Janssens J, Kleinberger G, Cruts M et al (2013) hnRNP A3 binds to GGGGCC repeats and is a constituent of p62-positive/TDP43-negative inclusions in the hippocampus of patients with C9orf72 mutations. Acta Neuropathol 125:413–423. doi:10.1007/s00401-013-1088-7
Mori K, Weng SM, Arzberger T, May S, Rentzsch K, Kremmer E, Schmid B, Kretzschmar HA, Cruts M, Van Broeckhoven C et al (2013) The C9orf72 GGGGCC repeat is translated into aggregating dipeptide-repeat proteins in FTLD/ALS. Science 339:1335–1338. doi:10.1126/science.1232927
Moussavi-Baygi R, Mofrad MR (2016) Rapid Brownian Motion Primes Ultrafast Reconstruction of Intrinsically Disordered Phe-Gly Repeats Inside the Nuclear Pore Complex. Sci Rep 6:29991. doi:10.1038/srep29991
Muhlemann O, Mock-Casagrande CS, Wang J, Li S, Custodio N, Carmo-Fonseca M, Wilkinson MF, Moore MJ (2001) Precursor RNAs harboring nonsense codons accumulate near the site of transcription. Mol Cell 8:33–43
Muranyi W, Haas J, Wagner M, Krohne G, Koszinowski UH (2002) Cytomegalovirus recruitment of cellular kinases to dissolve the nuclear lamina. Science 297:854–857. doi:10.1126/science.1071506
Myers SJ, Huang Y, Genetta T, Dingledine R (2004) Inhibition of glutamate receptor 2 translation by a polymorphic repeat sequence in the 5'-untranslated leaders. J Neurosci 24:3489–3499. doi:10.1523/JNEUROSCI.4127-03.2004
Nakamori M, Pearson CE, Thornton CA (2011) Bidirectional transcription stimulates expansion and contraction of expanded (CTG)*(CAG) repeats. Hum Mol Genet 20:580–588. doi:10.1093/hmg/ddq501
Nakamura K, Jeong SY, Uchihara T, Anno M, Nagashima K, Nagashima T, Ikeda S, Tsuji S, Kanazawa I (2001) SCA17, a novel autosomal dominant cerebellar ataxia caused by an expanded polyglutamine in TATA-binding protein. Hum Mol Genet 10:1441–1448
Neil AJ, Kim JC, Mirkin SM (2017) Precarious maintenance of simple DNA repeats in eukaryotes. BioEssays. doi:10.1002/bies.201700077
Nguyen L, Luu LM, Peng S, Serrano JF, Chan HY, Zimmerman SC (2015) Rationally Designed Small Molecules That Target Both the DNA and RNA Causing Myotonic Dystrophy Type 1. J Am Chem Soc 137:14180–14189. doi:10.1021/jacs.5b09266
Niblock M, Smith BN, Lee YB, Sardone V, Topp S, Troakes C, Al-Sarraj S, Leblond CS, Dion PA, Rouleau GA et al (2016) Retention of hexanucleotide repeat-containing intron in C9orf72 mRNA: implications for the pathogenesis of ALS/FTD. Acta neuropathol Commun 4:18. doi:10.1186/s40478-016-0289-4
Nunes VS, Moretti NS (2017) Nuclear subcompartments: an overview. Cell Biol Int 41:2–7. doi:10.1002/cbin.10703
O'Geen H, Yu AS, Segal DJ (2015) How specific is CRISPR/Cas9 really? Curr Opin Chem Biol 29:72–78. doi:10.1016/j.cbpa.2015.10.001
Oberle I, Rousseau F, Heitz D, Kretz C, Devys D, Hanauer A, Boue J, Bertheas MF, Mandel JL (1991) Instability of a 550-base pair DNA segment and abnormal methylation in fragile X syndrome. Science 252:1097–1102
Ohshima K, Montermini L, Wells RD, Pandolfo M (1998) Inhibitory effects of expanded GAA.TTC triplet repeats from intron I of the Friedreich ataxia gene on transcription and replication in vivo. J Biol Chem 273:14588–14595
Orr HT, Chung MY, Banfi S, Kwiatkowski TJ Jr, Servadio A, Beaudet AL, McCall AE, Duvick LA, Ranum LP, Zoghbi HY (1993) Expansion of an unstable trinucleotide CAG repeat in spinocerebellar ataxia type 1. Nat Genet 4:221–226. doi:10.1038/ng0793-221
Ozdilek BA, Thompson VF, Ahmed NS, White CI, Batey RT, Schwartz JC (2017) Intrinsically disordered RGG/RG domains mediate degenerate specificity in RNA binding. Nucleic Acids Res. doi:10.1093/nar/gkx460
Park H, Gonzalez AL, Yildirim I, Tran T, Lohman JR, Fang P, Guo M, Disney MD (2015) Crystallographic and Computational Analyses of AUUCU Repeating RNA That Causes Spinocerebellar Ataxia Type 10 (SCA10). Biochemistry 54:3851–3859. doi:10.1021/acs.biochem.5b00551
Paul S, Dansithong W, Jog SP, Holt I, Mittal S, Brook JD, Morris GE, Comai L, Reddy S (2011) Expanded CUG repeats Dysregulate RNA splicing by altering the stoichiometry of the muscleblind 1 complex. J Biol Chem 286:38427–38438. doi:10.1074/jbc.M111.255224
Payseur BA, Jing P, Haasl RJ (2011) A genomic portrait of human microsatellite variation. Mol Biol Evol 28:303–312. doi:10.1093/molbev/msq198
Pearson CE, Sinden RR (1996) Alternative structures in duplex DNA formed within the trinucleotide repeats of the myotonic dystrophy and fragile X loci. Biochemistry 35:5041–5053. doi:10.1021/bi9601013
Peng R, Lin G, Li J (2015) Potential Pitfalls of CRISPR/Cas9-mediated Genome Editing. The FEBS J. doi:10.1111/febs.13586
Pfister EL, Kennington L, Straubhaar J, Wagh S, Liu W, DiFiglia M, Landwehrmeyer B, Vonsattel JP, Zamore PD, Aronin N (2009) Five siRNAs targeting three SNPs may provide therapy for three-quarters of Huntington's disease patients. Curr Biol 19:774–778. doi:10.1016/j.cub.2009.03.030
Pineda M, Moghadam F, Ebrahimkhani MR, Kiani S (2017) Engineered CRISPR Systems for Next Generation Gene Therapies. ACS Synthetic Biol. doi:10.1021/acssynbio.7b00011
Pluciennik A, Burdett V, Baitinger C, Iyer RR, Shi K, Modrich P (2013) Extrahelical (CAG)/(CTG) triplet repeat elements support proliferating cell nuclear antigen loading and MutLalpha endonuclease activation. Proc Natl Acad Sci U S A 110:12277–12282. doi:10.1073/pnas.1311325110
Polleys EJ, NCM H, Freudenreich CH (2017) Role of recombination and replication fork restart in repeat instability. DNA Repair 56:156–165. doi:10.1016/j.dnarep.2017.06.018
Querido E, Gallardo F, Beaudoin M, Menard C, Chartrand P (2011) Stochastic and reversible aggregation of mRNA with expanded CUG-triplet repeats. J Cell Sci 124:1703–1714 Doi 2461242/jcs.073270
Raca G, Siyanova EY, McMurray CT, Mirkin SM (2000) Expansion of the (CTG)(n) repeat in the 5'-UTR of a reporter gene impedes translation. Nucleic Acids Res 28:3943–3949
Ranum LP, Cooper TA (2006) RNA-mediated neuromuscular disorders. Annu Rev Neurosci 29:259–277. doi:10.1146/annurev.neuro.29.051605.113014
Reddy K, Schmidt MH, Geist JM, Thakkar NP, Panigrahi GB, Wang YH, Pearson CE (2014) Processing of double-R-loops in (CAG).(CTG) and C9orf72 (GGGGCC).(GGCCCC) repeats causes instability. Nucleic Acids Res 42:10473–10487. doi:10.1093/nar/gku658
Reddy K, Zamiri B, Stanley SYR, Macgregor RB, Pearson CE (2013) The Disease-associated r(GGGGCC)(n) Repeat from the C9orf72 Gene Forms Tract Length-dependent Uni- and Multimolecular RNA G-quadruplex Structures. J Biol Chem 288:9860–9866. doi:10.1074/jbc.C113.452532
Renton AE, Majounie E, Waite A, Simon-Sanchez J, Rollinson S, Gibbs JR, Schymick JC, Laaksovirta H, van Swieten JC, Myllykangas L et al (2011) A Hexanucleotide Repeat Expansion in C9ORF72 Is the Cause of Chromosome 9p21-Linked ALS-FTD. Neuron 72:257–268. doi:10.1016/j.neuron.2011.09.010
Richard GF, Cyncynatus C, Dujon B (2003) Contractions and expansions of CAG/CTG trinucleotide repeats occur during ectopic gene conversion in yeast, by a MUS81-independent mechanism. J Mol Biol 326:769–782
Richard GF, Goellner GM, McMurray CT, Haber JE (2000) Recombination-induced CAG trinucleotide repeat expansions in yeast involve the MRE11-RAD50-XRS2 complex. EMBO J 19:2381–2390. doi:10.1093/emboj/19.10.2381
Richard P, Manley JL (2016) R Loops and Links to Human Disease. J Mol Biol. doi:10.1016/j.jmb.2016.08.031
Rzuczek SG, Colgan LA, Nakai Y, Cameron MD, Furling D, Yasuda R, Disney MD (2017) Precise small-molecule recognition of a toxic CUG RNA repeat expansion. Nat Chem Biol 13:188–193. doi:10.1038/nchembio.2251
Sanpei K, Takano H, Igarashi S, Sato T, Oyake M, Sasaki H, Wakisaka A, Tashiro K, Ishida Y, Ikeuchi T et al (1996) Identification of the spinocerebellar ataxia type 2 gene using a direct identification of repeat expansion and cloning technique, DIRECT. Nat Genet 14:277–284. doi:10.1038/ng1196-277
Sanz LA, Hartono SR, Lim YW, Steyaert S, Rajpurkar A, Ginno PA, Xu X, Chedin F (2016) Prevalent, Dynamic, and Conserved R-Loop Structures Associate with Specific Epigenomic Signatures in Mammals. Mol Cell 63:167–178. doi:10.1016/j.molcel.2016.05.032
Sathasivam K, Neueder A, Gipson TA, Landles C, Benjamin AC, Bondulich MK, Smith DL, Faull RL, Roos RA, Howland D et al (2013) Aberrant splicing of HTT generates the pathogenic exon 1 protein in Huntington disease. Proc Natl Acad Sci U S A 110:2366–2370. doi:10.1073/pnas.1221891110
Sato N, Amino T, Kobayashi K, Asakawa S, Ishiguro T, Tsunemi T, Takahashi M, Matsuura T, Flanigan KM, Iwasaki S et al (2009) Spinocerebellar ataxia type 31 is associated with "inserted" penta-nucleotide repeats containing (TGGAA)n. Am J Hum Genet 85:544–557. doi:10.1016/j.ajhg.2009.09.019
Sawaya S, Bagshaw A, Buschiazzo E, Kumar P, Chowdhury S, Black MA, Gemmell N (2013) Microsatellite tandem repeats are abundant in human promoters and are associated with regulatory elements. PLoS One 8:e54710. doi:10.1371/journal.pone.0054710
Sawyer LA, Hennessy JM, Peixoto AA, Rosato E, Parkinson H, Costa R, Kyriacou CP (1997) Natural variation in a Drosophila clock gene and temperature compensation. Science 278:2117–2120
Schmidt K, Butler JS (2013) Nuclear RNA surveillance: role of TRAMP in controlling exosome specificity. Wiley Interdiscip Rev RNA 4:217–231. doi:10.1002/wrna.1155
Schmidt MH, Pearson CE (2016) Disease-associated repeat instability and mismatch repair. DNA Repair 38:117–126. doi:10.1016/j.dnarep.2015.11.008
Scoles DR, Ho MH, Dansithong W, Pflieger LT, Petersen LW, Thai KK, Pulst SM (2015) Repeat Associated Non-AUG Translation (RAN Translation) Dependent on Sequence Downstream of the ATXN2 CAG Repeat. PLoS One 10:e0128769. doi:10.1371/journal.pone.0128769
Sellier C, Rau F, Liu Y, Tassone F, Hukema RK, Gattoni R, Schneider A, Richard S, Willemsen R, Elliott DJ et al (2010) Sam68 sequestration and partial loss of function are associated with splicing alterations in FXTAS patients. EMBO J 29:1248–1261. doi:10.1038/emboj.2010.21
Shepard S, McCreary M, Fedorov A (2009) The peculiarities of large intron splicing in animals. PLoS One 4:e7853. doi:10.1371/journal.pone.0007853
Shi KY, Mori E, Nizami ZF, Lin Y, Kato M, Xiang S, Wu LC, Ding M, Yu Y, Gall JG et al (2017) Toxic PRn poly-dipeptides encoded by the C9orf72 repeat expansion block nuclear import and export. Proc Natl Acad Sci U S A 114:E1111–E1117. doi:10.1073/pnas.1620293114
Shin JW, Kim KH, Chao MJ, Atwal RS, Gillis T, MacDonald ME, Gusella JF, Lee JM (2016) Permanent inactivation of Huntington's disease mutation by personalized allele-specific CRISPR/Cas9. Hum Mol Genet 25:4566–4576. doi:10.1093/hmg/ddw286
Shishkin AA, Voineagu I, Matera R, Cherng N, Chernet BT, Krasilnikova MM, Narayanan V, Lobachev KS, Mirkin SM (2009) Large-scale expansions of Friedreich's ataxia GAA repeats in yeast. Mol Cell 35:82–92. doi:10.1016/j.molcel.2009.06.017
Shoemaker CJ, Green R (2012) Translation drives mRNA quality control. Nat Struct Mol Biol 19:594–601. doi:10.1038/nsmb.2301
Shoubridge C, Gecz J (2012) Polyalanine tract disorders and neurocognitive phenotypes. Adv Exp Med Biol 769:185–203
Shukla S, Parker R (2016) Hypo- and Hyper-Assembly Diseases of RNA-Protein Complexes. Trends Mol Med 22:615–628. doi:10.1016/j.molmed.2016.05.005
Skourti-Stathaki K, Proudfoot NJ, Gromak N (2011) Human senataxin resolves RNA/DNA hybrids formed at transcriptional pause sites to promote Xrn2-dependent termination. Mol Cell 42:794–805. doi:10.1016/j.molcel.2011.04.026
Slean MM, Panigrahi GB, Castel AL, Pearson AB, Tomkinson AE, Pearson CE (2016) Absence of MutSbeta leads to the formation of slipped-DNA for CTG/CAG contractions at primate replication forks. DNA repair 42:107–118. doi:10.1016/j.dnarep.2016.04.002
Sleeman JE, Trinkle-Mulcahy L (2014) Nuclear bodies: new insights into assembly/dynamics and disease relevance. Curr Opin Cell Biol 28:76–83. doi:10.1016/j.ceb.2014.03.004
Sloan KE, Gleizes PE, Bohnsack MT (2016) Nucleocytoplasmic Transport of RNAs and RNA-Protein Complexes. J Mol Biol 428:2040–2059. doi:10.1016/j.jmb.2015.09.023
Smith KP, Byron M, Johnson C, Xing Y, Lawrence JB (2007) Defining early steps in mRNA transport: mutant mRNA in myotonic dystrophy type I is blocked at entry into SC-35 domains. J Cell Biol 178:951–964. doi:10.1083/jcb.200706048
Sobczak K, Wheeler TM, Wang W, Thornton CA (2013) RNA interference targeting CUG repeats in a mouse model of myotonic dystrophy. Mol Ther 21:380–387. doi:10.1038/mt.2012.222
Sofola OA, Jin P, Qin Y, Duan R, Liu H, de Haro M, Nelson DL, Botas J (2007) RNA-binding proteins hnRNP A2/B1 and CUGBP1 suppress fragile X CGG premutation repeat-induced neurodegeneration in a Drosophila model of FXTAS. Neuron 55:565–571. doi:10.1016/j.neuron.2007.07.021
Speese SD, Ashley J, Jokhi V, Nunnari J, Barria R, Li Y, Ataman B, Koon A, Chang YT, Li Q et al (2012) Nuclear envelope budding enables large ribonucleoprotein particle export during synaptic Wnt signaling. Cell 149:832–846. doi:10.1016/j.cell.2012.03.032
Spiro C, Pelletier R, Rolfsmeier ML, Dixon MJ, Lahue RS, Gupta G, Park MS, Chen X, Mariappan SV, McMurray CT (1999) Inhibition of FEN-1 processing by DNA secondary structure at trinucleotide repeats. Mol Cell 4:1079–1085
Stern A, Brown M, Nickel P, Meyer TF (1986) Opacity genes in Neisseria gonorrhoeae: control of phase and antigenic variation. Cell 47:61–71
Strack RL, Disney MD, Jaffrey SR (2013) A superfolding Spinach2 reveals the dynamic nature of trinucleotide repeat-containing RNA. Nat Methods 10:1219–1224. doi:10.1038/Nmeth.2701
Su Z, Zhang Y, Gendron TF, Bauer PO, Chew J, Yang WY, Fostvedt E, Jansen-West K, Belzil VV, Desaro P et al (2014) Discovery of a biomarker and lead small molecules to target r(GGGGCC)-associated defects in c9FTD/ALS. Neuron 83:1043–1050. doi:10.1016/j.neuron.2014.07.041
Sun X, Li PP, Zhu S, Cohen R, Marque LO, Ross CA, Pulst SM, Chan HY, Margolis RL, Rudnicki DD (2015) Nuclear retention of full-length HTT RNA is mediated by splicing factors MBNL1 and U2AF65. Sci Rep 5:12521. doi:10.1038/srep12521
Sundararajan R, Gellon L, Zunder RM, Freudenreich CH (2010) Double-strand break repair pathways protect against CAG/CTG repeat expansions, contractions and repeat-mediated chromosomal fragility in Saccharomyces cerevisiae. Genetics 184:65–77. doi:10.1534/genetics.109.111039
Supek F, Lehner B (2015) Differential DNA mismatch repair underlies mutation rate variation across the human genome. Nature 521:81–84. doi:10.1038/nature14173
Sutcliffe JS, Nelson DL, Zhang F, Pieretti M, Caskey CT, Saxe D, Warren ST (1992) DNA methylation represses FMR-1 transcription in fragile X syndrome. Hum Mol Genet 1:397–400
Tao Z, Wang H, Xia Q, Li K, Jiang X, Xu G, Wang G, Ying Z (2015) Nucleolar stress and impaired stress granule formation contribute to C9orf72 RAN translation-induced cytotoxicity. Hum Mol Genet. doi:10.1093/hmg/ddv005
Tassone F, Hagerman RJ, Taylor AK, Hagerman PJ (2001) A majority of fragile X males with methylated, full mutation alleles have significant levels of FMR1 messenger RNA. J Med Genet 38:453–456
Taylor JP, Brown RH Jr, Cleveland DW (2016) Decoding ALS: from genes to mechanism. Nature 539:197–206. doi:10.1038/nature20413
Thandapani P, O'Connor TR, Bailey TL, Richard S (2013) Defining the RGG/RG motif. Mol Cell 50:613–623. doi:10.1016/j.molcel.2013.05.021
Todd PK, Oh SY, Krans A, He F, Sellier C, Frazer M, Renoux AJ, Chen KC, Scaglione KM, Basrur V et al (2013) CGG repeat-associated translation mediates neurodegeneration in fragile X tremor ataxia syndrome. Neuron 78:440–455. doi:10.1016/j.neuron.2013.03.026
Toth G, Gaspari Z, Jurka J (2000) Microsatellites in different eukaryotic genomes: survey and analysis. Genome Res 10:967–981
Tran T, Childs-Disney JL, Liu B, Guan L, Rzuczek S, Disney MD (2014) Targeting the r(CGG) repeats that cause FXTAS with modularly assembled small molecules and oligonucleotides. Acs Chem Biol 9:904–912. doi:10.1021/cb400875u
Trinklein ND, Aldred SF, Hartman SJ, Schroeder DI, Otillar RP, Myers RM (2004) An abundance of bidirectional promoters in the human genome. Genome Res 14:62–66. doi:10.1101/gr.1982804
Turner EM, Brown RS, Laudermilch E, Tsai PL, Schlieker C (2015) The Torsin Activator LULL1 Is Required for Efficient Growth of Herpes Simplex Virus 1. J Virol 89:8444–8452. doi:10.1128/JVI.01143-15
Urbanek MO, Jazurek M, Switonski PM, Figura G, Krzyzosiak WJ (2016) Nuclear speckles are detention centers for transcripts containing expanded CAG repeats. Biochim Biophys Acta 1862:1513–1520. doi:10.1016/j.bbadis.2016.05.015
Usdin K (2008) The biological effects of simple tandem repeats: lessons from the repeat expansion diseases. Genome Res 18:1011–1019. doi:10.1101/gr.070409.107 18/7/1011
Usdin K, House NC, Freudenreich CH (2015) Repeat instability during DNA repair: Insights from model systems. Crit Rev Biochem Mol Biol 50:142–167. doi:10.3109/10409238.2014.999192
Usdin K, Kumari D (2015) Repeat-mediated epigenetic dysregulation of the FMR1 gene in the fragile X-related disorders. Front Genet 6:192. doi:10.3389/fgene.2015.00192
van Agtmaal EL, Andre LM, Willemse M, Cumming SA, van Kessel IDG, van den Broek W, Gourdon G, Furling D, Mouly V, Monckton DG et al (2017) CRISPR/Cas9-Induced (CTGCAG)n Repeat Instability in the Myotonic Dystrophy Type 1 Locus: Implications for Therapeutic Genome Editing. Mol Ther 25:24–43. doi:10.1016/j.ymthe.2016.10.014
van Blitterswijk M, DeJesus-Hernandez M, Rademakers R (2012) How do C9ORF72 repeat expansions cause amyotrophic lateral sclerosis and frontotemporal dementia: can we learn from other noncoding repeat expansion disorders? Curr Opin Neurol 25:689–700
Van Der Kelen K, Beyaert R, Inze D, De Veylder L (2009) Translational control of eukaryotic gene expression. Crit Rev Biochem Mol Biol 44:143–168. doi:10.1080/10409230902882090
van Hoof A, Frischmeyer PA, Dietz HC, Parker R (2002) Exosome-mediated recognition and degradation of mRNAs lacking a termination codon. Science 295:2262–2264. doi:10.1126/science.1067272
Verkerk AJ, Pieretti M, Sutcliffe JS, Fu YH, Kuhl DP, Pizzuti A, Reiner O, Richards S, Victoria MF, Zhang FP et al (1991) Identification of a gene (FMR-1) containing a CGG repeat coincident with a breakpoint cluster region exhibiting length variation in fragile X syndrome. Cell 65:905–914
Villasante A, Abad JP, Mendez-Lago M (2007) Centromeres were derived from telomeres during the evolution of the eukaryotic chromosome. Proc Natl Acad Sci U S A 104:10542–10547. doi:10.1073/pnas.0703808104
Wada T, Takagi T, Yamaguchi Y, Ferdous A, Imai T, Hirose S, Sugimoto S, Yano K, Hartzog GA, Winston F et al (1998) DSIF, a novel transcription elongation factor that regulates RNA polymerase II processivity, is composed of human Spt4 and Spt5 homologs. Genes Dev 12:343–356
Warren ST (1997) Polyalanine expansion in synpolydactyly might result from unequal crossing-over of HOXD13. Science 275:408–409
Weiser JN, Love JM, Moxon ER (1989) The molecular mechanism of phase variation of H. influenzae lipopolysaccharide. Cell 59:657–665
Wenzel S, Martins BM, Rosch P, Wohrl BM (2009) Crystal structure of the human transcription elongation factor DSIF hSpt4 subunit in complex with the hSpt5 dimerization interface. Biochem J 425:373–380. doi:10.1042/BJ20091422
Werner F (2012) A nexus for gene expression-molecular mechanisms of Spt5 and NusG in the three domains of life. J Mol Biol 417:13–27. doi:10.1016/j.jmb.2012.01.031
Wheeler TM, Sobczak K, Lueck JD, Osborne RJ, Lin X, Dirksen RT, Thornton CA (2009) Reversal of RNA dominance by displacement of protein sequestered on triplet repeat RNA. Science 325:336–339
White MC, Gao R, Xu W, Mandal SM, Lim JG, Hazra TK, Wakamiya M, Edwards SF, Raskin S, Teive HA et al (2010) Inactivation of hnRNP K by expanded intronic AUUCU repeat induces apoptosis via translocation of PKCdelta to mitochondria in spinocerebellar ataxia 10. PLoS Genet 6:e1000984. doi:10.1371/journal.pgen.1000984
Wilburn B, Rudnicki DD, Zhao J, Weitz TM, Cheng Y, Gu X, Greiner E, Park CS, Wang N, Sopher BL et al (2011) An antisense CAG repeat transcript at JPH3 locus mediates expanded polyglutamine protein toxicity in Huntington's disease-like 2 mice. Neuron 70:427–440. doi:10.1016/j.neuron.2011.03.021
Wojciechowska M, Olejniczak M, Galka-Marciniak P, Jazurek M, Krzyzosiak WJ (2014) RAN translation and frameshifting as translational challenges at simple repeats of human neurodegenerative disorders. Nucleic Acids Res 42:11849–11864. doi:10.1093/nar/gku794
Wong CH, Fu Y, Ramisetty SR, Baranger AM, Zimmerman SC (2011) Selective inhibition of MBNL1-CCUG interaction by small molecules toward potential therapeutic agents for myotonic dystrophy type 2 (DM2). Nucleic Acids Res 39:8881–8890. doi:10.1093/nar/gkr415
Woodward LA, Mabin JW, Gangras P, Singh G (2016) The exon junction complex: a lifelong guardian of mRNA fate. Wiley Int Rev RNA. doi:10.1002/wrna.1411
Xi Z, Zhang M, Bruni AC, Maletta RG, Colao R, Fratta P, Polke JM, Sweeney MG, Mudanohwo E, Nacmias B et al (2015) The C9orf72 repeat expansion itself is methylated in ALS and FTLD patients. Acta Neuropathol 129:715–727. doi:10.1007/s00401-015-1401-8
Xi Z, Zinman L, Moreno D, Schymick J, Liang Y, Sato C, Zheng Y, Ghani M, Dib S, Keith J et al (2013) Hypermethylation of the CpG island near the G4C2 repeat in ALS with a C9orf72 expansion. Am J Hum Genet 92:981–989. doi:10.1016/j.ajhg.2013.04.017
Xia G, Gao Y, Jin S, Subramony SH, Terada N, Ranum LP, Swanson MS, Ashizawa T (2015) Genome modification leads to phenotype reversal in human myotonic dystrophy type 1 induced pluripotent stem cell-derived neural stem cells. Stem Cells 33:1829–1838. doi:10.1002/stem.1970
Xu Z, Poidevin M, Li X, Li Y, Shu L, Nelson DL, Li H, Hales CM, Gearing M, Wingo TS et al (2013) Expanded GGGGCC repeat RNA associated with amyotrophic lateral sclerosis and frontotemporal dementia causes neurodegeneration. Proc Natl Acad Sci U S A 110:7778–7783. doi:10.1073/pnas.1219643110
Yamada NA, Smith GA, Castro A, Roques CN, Boyer JC, Farber RA (2002) Relative rates of insertion and deletion mutations in dinucleotide repeats of various lengths in mismatch repair proficient mouse and mismatch repair deficient human cells. Mutat Res 499:213–225
Yamazaki T, Hirose T (2015) The building process of the functional paraspeckle with long non-coding RNAs. Front Biosci (Elite Ed) 7:1–41
Yang W, Tu Z, Sun Q, Li XJ (2016) CRISPR/Cas9: Implications for Modeling and Therapy of Neurodegenerative Diseases. Front Mol Neurosci 9:30. doi:10.3389/fnmol.2016.00030
Yu K, Chedin F, Hsieh CL, Wilson TE, Lieber MR (2003) R-loops at immunoglobulin class switch regions in the chromosomes of stimulated B cells. Nat Immunol 4:442–451. doi:10.1038/ni919
Yu K, Roy D, Huang FT, Lieber MR (2006) Detection and structural analysis of R-loops. Methods Enzymol 409:316–329. doi:10.1016/S0076-6879(05)09018-X
Zamiri B, Reddy K, Macgregor RB Jr, Pearson CE (2014) TMPyP4 porphyrin distorts RNA G-quadruplex structures of the disease-associated r(GGGGCC)n repeat of the C9orf72 gene and blocks interaction of RNA-binding proteins. J Biol Chem 289:4653–4659. doi:10.1074/jbc.C113.502336
Zhang K, Donnelly CJ, Haeusler AR, Grima JC, Machamer JB, Steinwald P, Daley EL, Miller SJ, Cunningham KM, Vidensky S et al (2015) The C9orf72 repeat expansion disrupts nucleocytoplasmic transport. Nature 525:56–61. doi:10.1038/nature14973
Zhang N, Ashizawa T (2017) RNA toxicity and foci formation in microsatellite expansion diseases. Curr Opin Genet Dev 44:17–29. doi:10.1016/j.gde.2017.01.005
Zhang S, Binari R, Zhou R, Perrimon N (2010) A genomewide RNA interference screen for modifiers of aggregates formation by mutant Huntingtin in Drosophila. Genetics 184:1165–1179. doi:10.1534/genetics.109.112516
Zhang Y, Roland C, Sagui C (2016) Structure and Dynamics of DNA and RNA Double Helices Obtained from the GGGGCC and CCCCGG Hexanucleotide Repeats That Are the Hallmark of C9FTD/ALS Diseases. ACS Chem Neurosci. doi:10.1021/acschemneuro.6b00348
Zhang Y, Shishkin AA, Nishida Y, Marcinkowski-Desmond D, Saini N, Volkov KV, Mirkin SM, Lobachev KS (2012) Genome-wide screen identifies pathways that govern GAA/TTC repeat fragility and expansions in dividing and nondividing yeast cells. Mol Cell 48:254–265. doi:10.1016/j.molcel.2012.08.002
Zhao J, Bacolla A, Wang G, Vasquez KM (2010) Non-B DNA structure-induced genetic instability and evolution. Cell Mol Life Sci 67:43–62. doi:10.1007/s00018-009-0131-2
Zhao T, Hong Y, Li XJ, Li SH (2016) Subcellular Clearance and Accumulation of Huntington Disease Protein: A Mini-Review. Front Mol Neurosci 9:27. doi:10.3389/fnmol.2016.00027
Zhao X, Usdin K (2015) The Repeat Expansion Diseases: The dark side of DNA repair. DNA Repair 32:96–105
Zhao XN, Lokanga R, Allette K, Gazy I, Wu D, Usdin K (2016) A MutSbeta-Dependent Contribution of MutSalpha to Repeat Expansions in Fragile X Premutation Mice? PLoS Genet 12:e1006190. doi:10.1371/journal.pgen.1006190
Zhou Z, Luo MJ, Straesser K, Katahira J, Hurt E, Reed R (2000) The protein Aly links pre-messenger-RNA splicing to nuclear export in metazoans. Nature 407:401–405. doi:10.1038/35030160
Zhu L, Brangwynne CP (2015) Nuclear bodies: the emerging biophysics of nucleoplasmic phases. Curr Opin Cell Biol 34:23–30. doi:10.1016/j.ceb.2015.04.003
Zhuchenko O, Bailey J, Bonnen P, Ashizawa T, Stockton DW, Amos C, Dobyns WB, Subramony SH, Zoghbi HY, Lee CC (1997) Autosomal dominant cerebellar ataxia (SCA6) associated with small polyglutamine expansions in the alpha 1A-voltage-dependent calcium channel. Nat Genet 15:62–69. doi:10.1038/ng0197-62
Zinder JC, Lima CD (2017) Targeting RNA for processing or destruction by the eukaryotic RNA exosome and its cofactors. Genes Dev 31:88–100. doi:10.1101/gad.294769.116
Zu T, Gibbens B, Doty NS, Gomes-Pereira M, Huguet A, Stone MD, Margolis J, Peterson M, Markowski TW, Ingram MA et al (2011) Non-ATG-initiated translation directed by microsatellite expansions. Proc Natl Acad Sci U S A 108:260–265. doi:10.1073/pnas.1013343108
Zu T, Liu Y, Banez-Coronel M, Reid T, Pletnikova O, Lewis J, Miller TM, Harms MB, Falchook AE, Subramony SH et al (2013) RAN proteins and RNA foci from antisense transcripts in C9ORF72 ALS and frontotemporal dementia. Proc Natl Acad Sci U S A 110:E4968–E4977. doi:10.1073/pnas.1315438110
Zumwalt M, Ludwig A, Hagerman PJ, Dieckmann T (2007) Secondary structure and dynamics of the r(CGG) repeat in the mRNA of the fragile X mental retardation 1 (FMR1) gene. RNA Biol 4:93–100
Funding
This work was supported by a United States Department of Defense Amyotrophic Lateral Sclerosis Research Program (ALSRP) Grant and a Judith and Jean Pape Adams Charitable Foundation Grant to K.T.G.
Authors’ contributions
KJR performed an initial literature survey and draft of the manuscript and prepared tables and figures. KTG performed an in-depth literature survey, finalized tables and figures, and wrote the final draft of the manuscript. Both authors read and approved the final manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Rohilla, K.J., Gagnon, K.T. RNA biology of disease-associated microsatellite repeat expansions. acta neuropathol commun 5, 63 (2017). https://doi.org/10.1186/s40478-017-0468-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40478-017-0468-y