
Abstract
Background
Amyotrophic lateral sclerosis (ALS) is a primary progressive neurodegenerative disease characterised by neuronal loss of lower motor neurons (in the spinal cord and brainstem) and/or upper motor neurons (in the motor cortex) and subsequent denervation atrophy of skeletal muscle.
Aim
A comprehensive examination of muscle pathology from a cohort of clinically confirmed ALS patients, including an investigation of inflammation, complement activation, and deposition of abnormal proteins in order to compare them with findings from an age-matched, control group.
Material and methods
31 muscle biopsies from clinically confirmed ALS patients and 20 normal controls underwent a comprehensive protocol of histochemical and immunohistochemical stains, including HLA-ABC, C5b-9, p62, and TDP-43.
Results
Neurogenic changes were confirmed in 30/31 ALS cases. In one case, no neurogenic changes could be detected. Muscle fibre necrosis was seen in 5/31 cases and chronic mononuclear inflammatory cell infiltration in 5/31 (2 of them overlapped with those showing muscle necrosis). In four biopsies there was an increase in the proportion of cytochrome oxidase (COX) negative fibres (2-3%). p62 faintly stained cytoplasmic bodies in eight cases and none were immunoreactive to TDP-43.
Conclusion
This large series of muscle biopsies from patients with ALS demonstrates neurogenic atrophy is a nearly uniform finding and that mild mitochondrial abnormalities and low-grade inflammation can be seen and do not rule out the diagnosis of ALS. These findings could lend support to the notion that ALS is a complex and heterogeneous disorder.
Similar content being viewed by others

Introduction
Amyotrophic lateral sclerosis (ALS) is a progressive neurodegenerative disease characterised primarily by degeneration of upper and lower motor neurons (LMN) in the cerebral cortex, spinal cord and brainstem. This leads to relentlessly progressive weakness and widespread flaccid paralysis and/or spasticity, depending on the predominance of upper versus LMN disease [1],[2].
The presentation is heterogeneous and may begin with appendicular weakness, bulbar weakness or respiratory muscle weakness. Neuro-imaging is useful in excluding other diseases. Electromyography (EMG) is used to support the clinical findings, possibly allowing a more accurate and early diagnosis, as it can detect denervation sometimes before clinical signs are evident. (El Escorial and Awaji criteria) [3].
Pathologically, ALS is characterised by neuronal loss in the anterior horn of the spinal cord, brainstem nuclei, and Betz cells of the motor cortex, and also by the deposition of abnormal ubiquitinated inclusions immunoreactive to TDP-43 [4]. But these central nervous system changes are only accessible post mortem. In contrast, the muscle pathology of denervation atrophy, such as angular, atrophic fibres, grouped atrophy and fibre type grouping can be assessed pre-mortem and may lead to earlier diagnostic certainty or provide a means to assess possible treatment impact on disease progression. It is therefore very important to understand muscle pathology in ALS for clinical trials in this field.
The aetiology and pathogenesis of the neuronal death in ALS remains poorly understood. The majority of ALS cases are sporadic while 10-15% are familial. Mutations have been identified in genes encoding for Cu/Zn superoxide dismutase (SOD1), VAMP (vesicle associated membrane protein), angiogenin (ANG), TDP-43 and Fused in sarcoma (FUS), Optineurin (OPTN), and more recently C9orf72 (linked to chromosome 9) [5]-[10].
There are several theories to explain the neurodegenerative processes including glutamate-induced excitotoxicity, axonal transport impairment, proteasome dysfunction, aberrant functioning of glial cells, alterations in muscle and neuromuscular junction [11],[12], and mitochondrial dysfunction [13]-[18]. There are reports of abnormal aggregation of mitochondria and impaired respiratory chain function (particularly in complex 1) in experimental mice with SOD1 mutation [5],[19]-[21]. However, the role of mitochondrial abnormalities in the pathogenesis of ALS is unknown [22]. Although some reports suggest mitochondrial dysfunction in the skeletal muscle and a significant increase in mitochondrial DNA deletions indicate a primary defect in muscles of ALS patients; others suggest that mitochondrial dysfunction is a consequence of the motor neuron cell death [8],[11],[23]-[28]. The majority of reports describing the muscle pathology of ALS patients are case reports; systematic investigations of muscle pathology in a large cohort of patient with ALS are few [29]-[33].
We report the pathological changes in the skeletal muscle in a cohort of 31 patients with ALS according to El Escorial criteria and compare them with muscle biopsies from 20 age-matched controls.
Material and methods
Study populations
A total of 32 patients with ALS (age range 27–75 years, mean age 54.8 years) were recruited as part of a methodology study (NOG111329) to assess biomarkers of ALS; 31 subjects provided muscle biopsy samples. ALS patients with a diagnosis of clinically definite or probable ALS (according to El Escorial diagnostic criteria, revised according to the Airlie House Conference 1998) were recruited at 3 ALS centres. Inclusion criteria included onset of muscle weakness within 24 months of study entry and MRC score in the deltoid muscle of MRC grade 3 or 4. Subjects with evidence of other neuromuscular disorders were excluded. Twenty healthy volunteers (age range 41–69 years, mean age 55.7 years) were recruited at the Guy’s Drug Research Unit (GDRU) in London to assess levels of potential ALS biomarkers in muscle samples from healthy subjects (NOG113240).
The study protocol, protocol amendments, informed consent and any other information that required pre-approval were reviewed and approved by a national, regional or investigational centre ethics committee or an institutional review board. This study was conducted in accordance with Good Clinical Practice and the guiding principles of the Declaration of Helsinki, and all subjects provided written informed consent.
Muscle biopsy procedure
Thirty-one ALS patients and 20 healthy volunteers underwent open muscle biopsy from the deltoid muscle according to standard procedures. For ALS patients, the weaker of the two deltoid muscles was selected. A muscle specimen of an approximate size of 1.0 cm × 0.5 cm × 0.5 cm was taken per subject.
Processing muscle biopsy samples
The muscle biopsies were processed freshly (i.e. unfixed) in the laboratory soon after the surgical removal to minimize preparative artefact. The muscle biopsy sample was divided perpendicular to the muscle fibres into two equally large pieces, one piece for pathological assessment, while the second piece was processed for other biomarkers (not reported here). The pathology sample was oriented and fixed transversely to cork discs with OCT and then snap frozen in iso-pentane cooled on dry ice. Tissue was transferred to pre-cooled cryotubes and stored frozen at -80°C.
Histology and immunohistochemistry
Serial cryostat sections were cut from the frozen biopsy material (7 mm thickness) and stained for H&E, ATPase 9.4, ATPase 4.6, ATPase 4.2, nicotinamide adenine dinucleotide tetrazolium reductase (NADH-TR), succinic dehydrogenase (SDH), cytochrome oxidase (COX) with and without SDH, Gomori trichrome (GT), acid phosphatase (AP), Periodic acid-Schiff (PAS), Periodic acid-Schiff with diastase (PAS-D), myophosphorylase and Sudan Black.
Further immunohistochemistry stains were performed for p62 (BD transduction Labs, clone: 3/p62 LCK Ligand, dilution 1:150), pTDP-43 (Cosmo Bio Co. LTD, code TIP-PTD-P02, dilution1:1500), HLA-ABC (Dako, clone: W6/32, dilution 1:1200) and C5b-9 “membrane attack antigen” (Novocastra, clone: aE11, dilution 1:200). CD3(Dako, code M7254M dilution 1:50), CD20(Dako code M075501, dilution 1:200), CD8(Novocastra code CD8-295-CE, dilution 1:20), CD 4 (Novocastracode CD4-1f6-CE-S, dilution 1:20), neonatal myosin (Novocastra code NCL-MHCn, dilution 1:5) and Utrophin (Novocastra code NCL-DRP2, dilution 1:100).
Cryostat sections were processed according to the protocol at the Department of Clinical Neuropathology, King’s College Hospital London. Slides were interpreted by a senior Neuropathologist (SALS) blind to clinical information and findings are summarised in Tables 1 and 2.
Laboratory assessments
Plasma (EDTA) samples were collected from ALS subjects prior to collection of open muscle biopsy samples and aliquots dispensed and frozen at -80°C. Frozen samples were shipped to The Doctors Laboratory (TDL) and an exploratory analysis performed for lactate dehydrogenase (LDHI2), creatinine (CREJ2) and creatine kinase (CK) using the Roche Cobas system. Note that sample stability data was not available to cover the storage time (>1 year) or conditions (−80°C) for these samples.
Non parametric Mann Whitney test was carried out to find the correlation between increased CK levels and muscle fibre necrosis and inflammation.
Results
Thirty of 31 ALS muscle biopsies showed evidence of neurogenic atrophy with various stages of denervation and re-innervation of muscle fibres. These included early changes such as scattered angular, atrophic fibres which are densely stained with NADH-TR as well as more chronic changes with small and large grouped atrophy and fibre type grouping. One case showed no clear features of neurogenic changes but definite ALS on clinical grounds. There were occasional small fibres and lymphocyte infiltration limited to the perimysium, however, there was no endomysial inflammation, the HLA-ABC was not over-expressed and creatinine kinase was low (CK: 47 IU) suggesting that the focus of inflammatory cells was a non-specific finding and that inflammatory myopathy was unlikely.
Neurogenic changes
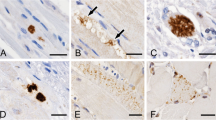
The extent of neurogenic atrophy differed between patients: small angulated fibres were observed in 29/31 (93.5%) cases, small grouped atrophy in 22/31 (71%) cases, (Figure 1A), pyknotic nuclear clumps in 13/31 (41.9%), increased internalized nuclei in 8/31 (25.8%) cases and fibre type grouping in 18/31 (58%) cases. There were NADH densely stained fibres in 27/31 (87%) cases; targetoid fibres in 20/31 (64.5%) cases (Figure 1B); and moth-eaten fibres in 13/31 (41.9%) cases.

Pathological changes in the muscles from ALS patients. A: Low power view of muscle with neurogenic changes showing small and large group atrophy (arrow). B: NADH-TR stain showing atrophic and hyperdense fibres and target fibres (arrow). C: Fibre necrosis (arrow). D: Inflammatory cell infiltration in the endomysium (arrow) and E: in the perimysium (arrow). F: Cytochrome oxidase negative fibres (arrow). G: Patchy over expression of HLA) - ABC in muscle fibres. H: Deposition of compliment (C5-9) in the endomysial capillaries.
Muscle fibre necrosis and COX negative fibres
Muscle fibre necrosis was seen in five biopsies (16%) (Figure 1C); only one patient showed increased in CK level to 1386, two patients with inflammatory infiltrate and three with uniform over expression of HLA-ABC. The chronic mononuclear inflammatory cell infiltration was observed in five (16%) patients biopsies (Figure 1D and E), two of which also showed fibre necrosis, two with HLA-ABC over expression and two patient with increased CK level to 936 and 1386. In four biopsies there was more than 1% increase in the proportion of COX negative fibres (Figure 1F) with rates of 2- 3% . These cases also contain 1–3 ragged red fibres in the entire biopsy of each case. Although ragged red fibres are useful in showing abnormal accumulation of mitochondria, the percentage of COX negative/SDH positive fibres was more sensitive in identifying fibres with mitochondrial abnormality in muscle biopsy.
Other changes
Cytoplasmic bodies presented in the form of globular, irregular, homogenous bodies, best demonstrated on the Gomori trichrome stain were present in 13/31 (41.9%) cases.
Immunohistochemistry
The inflammatory infiltrate is mainly composed of lymphocytes and few macrophages. The lymphocytes are predominantly CD3 positive (T lymphocytes). The amount of inflammatory infiltrate was small in the deeper sections for the immunostainings for proper quantitative assessment but there is slight increase of CD4 over CD8 positive T lymphocytes Immunohistochemistry showed uniform and sarcolemmal over-expression of HLA-ABC (Figure 1G) in 5/31 (16%) but only few scattered small fibres positive with neonatal neomysin; therefore excluding HLA-ABC over expression was due to fibre regeneration and degeneration. Those five muscles with HLA over expression include two cases necrosis and one case with a predominantly chronic mononuclear inflammatory cell infiltration.
In 21/31 (67.7%) muscle biopsies there was granular deposition of the compliment (C5b-9) in the sarcolemma of the atrophic fibres consistent with fibre degeneration. However there were 3/31 cases where the C5b-9 antibody deposition was intense in the endomysial capillaries (Figure 1H) in addition to the presence of an inflammatory cell infiltration suggesting activation of compliments.
In the immunohistochemical stains, p62 was positive in 8/31 (25.8%) cases, displayed as faint, irregular and granular deposits at the sites of cytoplasmic bodies. None of the cases show abnormal TDP-43 immunoreactivity or over expression of lysosomal enzyme such as acid phosphatase; therefore the appearances of p62 reflected a non-specific deposition in fibre degeneration due to neurogenic changes.
Plasma samples were collected at the time of biopsy, but assessed for CK levels only after prolonged storage of the samples (>1 year), so results have an indicative value rather than providing an absolute quantification.
In three patients there was a marked increase in the serum CK level (807 IU/L, 1386 IU/L and 963 IU/L). In two of these cases, the muscle biopsy contained inflammatory cells and myofibre necrosis. The CK was mildly increased (200–500 IU/L) in 12/31 patients; in one of these patients an inflammatory cell infiltrate was present. The remaining patients had normal serum CK levels.
The Mann Whitney test shows no significant association between CK levels and inflammation (P = 0.33) and muscle fibre necrosis (P = 0.35).
In the controls all 20 of the muscle biopsies appeared normal. There was no evidence of neurogenic atrophy, myofiber necrosis or inflammation. The immunohistochemical staining for HLA Class I and C5b-9 was also negative in all fibres. In 3/20 biopsies there were occasional COX negative fibres amounting to less than 1% of total fibres, within normal limits attributed to age-related changes. Immunohistochemical stains for p62 and TDP-43 were negative.
Discussion
In this large collection of muscle biopsies from patients with ALS, the predominantn pathological finding is neurogenic atrophy with a spectrum of findings due to denervation and re-innervation [34]. The most common feature of such neurogenic changes is small angulated and NADH-TR hyper-dense fibres. Additional features are fibre type grouping in nearly 50% of the biopsies and target fibres in 60% of the biopsies. In many muscle biopsies there is also evidence of disruption of intermyofibrillary architecture. Fibre type grouping resulting from re-innervations of fibres is only present in approximately 50% of our cases. On the other hand the NADH-TR stain appears to be useful in establishing the neurogenic changes in showing hyper-dense fibres and targetoid appearances which is present in 60% of the cases (Figure 1B). Other findings are an increased number of fibres with internalized nuclei and pyknotic nuclear clumps. The ATPase stains confirmed the atrophic fibres are of both types and not selectively type 2. Though it has been reported that type 2 fibres are affected by denervation more rapidly than type 1 fibres in early stages of the disease process [34], the lack of selective involvement of fibre type 2 in our cohort may reflect more chronic disease.
In addition to acute and chronic neurogenic atrophy, a few cases in our cohort demonstrated additional pathology less typical of denervation, such as fibre necrosis, low grade inflammation, and mitochondrial abnormalities. Albeit infrequent, these changes were not seen in our cohort of age-matched control biopsies. They are, nonetheless interesting as alternative theories are raised regarding the pathogenesis of ALS and involve the peripheral rather than the central nervous system. The neuronal degeneration in the upper and lower motor neurons in the CNS in ALS may not be the only abnormality and other cell abnormalities, such as in the muscles or changes at the neuromuscular junction may play a pathogenic role [1],[22],[35]-[39].
Four of our cases show evidence of mitochondrial abnormalities with an increased proportion of cytochrome oxidase negative fibres (Figure 1F) and few ragged red fibres beyond that seen in the age-match controls. Mitochondrial alteration has been reported in association with aging [27],[40]; however in these four patients, the number of CXO negative fibres is seen in higher proportion (2-3%) expected in aging process and three of patients were in their 60s. This finding is in keeping with other reports of mitochondrial abnormality in the muscle of sporadic ALS patients and do not correlate with aging [15],[17],[23],[41],[42]. Mitochondria are known to be involved in the cellular production of oxygen radicals which play an important role in cell damage via oxygen/free radicals in many neurodegenerative diseases [1],[16],[43]-[45]. There is some evidence of involvement of mitochondria in the pathogenesis of the ALS. For instance, in experimental SOD1 mutant mice, there are reports of abnormal aggregates of mitochondria with vacuolation in skeletal muscle as well as in the anterior horn cells [18] and dysfunction of mitochondria in muscle, spinal cord and liver. Mitochondrial function has an important role to play in the pathogenesis of the some of the familial ALS like SOD1 mutation which appears to have toxic gene of function mutation in copper zinc super-oxide dysmutase, cytcolic and mitochondrial protein [36],[43],[46]. However, the nature of any contribution of mitochondrial abnormalities to human ALS remains uncertain. Abnormal mitochondrial functions have been reported in lymphocytes in ALS [11] and mitochondrial DNA deletions in ALS patients have been described [29],[42]. There are also case reports of ALS and mitochondrial myopathy [12],[16],[32],[41]. However Echaniz-laguna et al., [23] and Grehl et al. [47] reported that normal mitochondrial function is progressively altered with the disease’s progression secondarily. Recently, Bradley et al. [37] found no difference in respiratory chain enzyme and deleted mitochondrial DNA between ALS and controls. The finding of mitochondrial abnormalities in the biopsies of few of the ALS patients in this cohort and other published studies suggest some link whether primary or secondary to the pathogenesis of ALS or the ultimate motor neuron injury [44],[48]. It is possible that mitochondrial abnormality in the skeletal muscle of sub-group of ALS patients which does not correlate with the age and may support the hypothesis that at least in such cases, mitochondrial dysfunction plays a role in the pathogenesis of the disease. Subclinical mitochondrial dysfunction in subgroups of ALS could also be present which is otherwise undetectable in the resting condition and become manifested with further external and environmental stress such as inflammation and may contribute to defective axonal transport at neuro muscular level and to retrograde neuronal degeneration in few patients.. This suggestion is supported by evidence from Siniliano et al. who reported increased lactate levels in patient with ALS after exercise [45].
Another interesting finding in muscle biopsies from our cohort of ALS cases is the presence of fibre necrosis and low grade inflammatory changes (Figure 1C, D and E). In five cases, there was evidence of muscle fibre necrosis; a rare but reported finding in neurogenic atrophy. Interestingly two of these cases showed an inflammatory cell infiltration and relatively high serum CK level. However we did not find association between presence of mild inflammation, increased CK levels and clinical manifestation and progress of the patient. The inflammatory process, including the up regulation of HLA- ABC and compliment deposition (C5b-9) could also be secondary to neurogenic changes and secondary myofiber necrosis. These cases show no evidence for endomysial inflammation with invasion of non-necrotic fibres as seen in inflammatory myopathies or inclusion body myositis IBM. However, a finding of low grade inflammation in the muscle is intriguing given evidence that inflammation in the central nervous system may be a contributory factor in ALS pathology. There is mounting evidence that activated microglia and astrocytes, endothelium and lymphocytes trafficking in the CNS of ALS patients may play an etiologic role through the production of molecules, like IL-6 and TNFalpha and increased oxidative stress [49]-[52]. Our results may not indicate a primary muscle disease or inflammation, but it support the view that the low grade inflammation and sub clinical mitochondrial alteration in the muscle of some ALS patients may add further burden in damaging the neuromuscular junctions and contribute to “dying back” of motor neutrons. The interaction between the peripheral and central inflammatory process is critical to understand the its pathogenesis It has been theorized that ALS pathology may not necessarily be directly due to motor neuron degeneration but may start more distally at the neuromuscular junction. Fischer et al. [53] showed that the alteration of the neuromuscular junction led to axonal degeneration and ultimately motor neuron degeneration in the CNS. Evidence showed that a reduction in NOGO-A, a neurite outgrowth inhibitor led to increase in the lifespan of SOD1 mice [54],[55]. In addition Bradley et al. [37] showed that myoblasts from ALS patients exhibit an enhanced sensitivity to oxidative stress. This evidence suggests that ALS muscle may be particularly vulnerable to additional stress and activate an apoptotic process [37],[56]. Such oxidative stress may come through different routes such as environmental or even inflammatory myopathy [39],[49],[57]. It is possible that in some ALS patients’ alterations in muscle and the neuromuscular junction may be pathogenic contributors resulting in retrograde degeneration of the motor neurons. ALS is a complex and multifactorial disorder with many potential etiologic and pathogenic mechanisms resulting in dysfunction at the neuromuscular, axonal and neuronal levels [1].
Finally, many muscles in this series showed a cytoplasmic body which is a known alteration in the intermyofibrillary article true in long standing neurogenic changes expected in ALS patients. Also few cases showed p62 granular deposits but with no over expression lysosomal enzyme such as acid phosphatase. The appearances are not specific and reflect process of degeneration in the muscle fibres. There is no specific well define inclusions and all the sections were negative with TDP43 stain.
Conclusion
In conclusion, acute or chronic neurogenic atrophy is the nearly uniform finding in this large cohort of muscle biopsies of ALS patients. In addition, low grade endomysial and perimysial chronic inflammation and mitochondrial alterations are seen in a small proportion of muscle biopsies from patients with sporadic ALS beyond that seen in age-matched control biopsies. These findings do not exclude the diagnosis of ALS. Neither do these findings meet criteria for primary inflammatory muscle disease or mitochondrial cytopathy. However, these findings could reflect contributing pathogenic mechanisms whereby additional oxidative stress by inflammation or other factors could unmask subclinical mitochondrial dysfunction in the muscle of some ALS patients. These muscle biopsy findings from a relatively large ALS patient cohort, could provide additional clues to understanding the seemingly multifactorial, complex pathogenesis of ALS.
Acknowledgements
The authors thank Mrs Mary Davitt for her efforts to prepare the manuscript and Dr Istvan Bodi for his technical help to produce the photographs. We are grateful for all the ALS centres at, The Neurosciences clinical centre, Pitie-Salpetriere, Paris, The John Hopkins medical school, Baltimore, USA and the Neuroscience centre at Institute of Psychiatry, Psychology and Neurosciences ( IOPNN) at Kings College London, UK for recruiting the patients for this study.
References
Shaw PJ: Molecular and cellular pathology of neurodegeneration in motor neuron disease. J Neurol Neurosurg Psychiatry 2005, 76: 1046–1057. 10.1136/jnnp.2004.048652
Mitchell JD, Borasio GD: Amyotrophic lateral sclerosis. Lancet 2007, 369: 2031–2041. 10.1016/S0140-6736(07)60944-1
Pasinelli P, Brown RH: Molecular biology of amyotrophic lateral sclerosis; insight from genetics. Nat Rev Neuroscience 2006, 7: 710–723. 10.1038/nrn1971
Love S, Louis DN, Ellison DW. Disease of movement disorder and system degeneration; motor neurone disease. Greenfield’s Neuropathology, eight Edition 2008: Chapter 22, 947–961.
Pasinelli P, Belford ME, Lennon N, Bacskai BJ, Hyman BT, Trotti D, Brown BH: Amyotrophic lateral sclerosis associated SOD1 binds and aggregate with Bcl2 in spinal cord mitochondria. Neuron 2004, 43: 19–30. 10.1016/j.neuron.2004.06.021
Al-Sarraj S, King A, Troackes C, Smith B, Maekawa S, Bodi I, Rogeli B, Al-Chalabi A, Hortobagyi T, Shaw CE: p62 positive, TDP43 negative neuronal cytoplasmic inclusion in the cerebellum and hippocampus define the pathology of C9orf72 FTLD-MND. Acta Neuropathol 2011, 122(6):691–702. 10.1007/s00401-011-0911-2
Sreedharan J, Blair IP, Tripathi VB, Hu X, Vance C, Rogelj B, Ackerley S, Durnall JC, Williams KL, Buratti E, Baralle F, De Belleroche J, Mitchell JD, Leigh PN, Al-Chalabi A, Miller CC, Nicholson G, Shaw CE: TDP43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science 2008, 319(5870):1668–1672. 10.1126/science.1154584
Krasnianski A, Deschauer M, Neudecker S, Gellerich FN, Müller T, Schoser BG, Krasnianski M, Zierz S: Mitochondrial changes in skeletal muscle in amyotrophic lateral sclerosis and other neurogenic atrophies. Brain 2005, 128: 1870–1876. 10.1093/brain/awh540
Vance C, Rogelj B, Hortobagyi T, De Vos KJ, Nishimura AL, Sreedharan J, Hu X, Smith B, Ruddy D, Wright P, Ganesalingam J, Williams KL, Tripathi V, Al-Saraj S, Al-Chalabi A, Leigh PN, Blair IP, Nicholson G, de Belleroche J, Gallo JM, Miller CC, Shaw CE: Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science 2009, 323(5918):1208–1211. 10.1126/science.1165942
Rosen DR, Teepu S, David P, Figlewicz DA, Peter S, Afif H, Deirdre D, Jun G, O'Regan JP, Han-Xiang D, Zohra R, Aldis K, Diane MK-Y, Annarueber C, Gaston SM, Ralph B, Tanzi RE, Halperin JJ, Brian H, Van den Bergh R, Wu-Yen H, Thomas B, Gang D, Mulder DW, Celestine S, Laing NG, Edwin S, Pericak–Vance MA, Jonathan H, Rouleau GA, Gusella JS, Robert Horvitz H, Brown Jr RH: Mutation in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 1993, 362(6415):59–62. 10.1038/362059a0
Vielhaber S, Kunz D, Winkler K, Wiedemann FR, Kirches E, Feistner H, Heinze HJ, Elger CE, Schubert W, Kunz WS: Mitochondrial DNA abnormalities in skeletal muscle of patients with sporadic amyotrophic lateral sclerosis. Brain 2000, 123: 1339–1348. 10.1093/brain/123.7.1339
Hirano M, Angelini C, Montagna P, Hays AP, Tanji K, Mitsumoto H, Gordon PH, Naini AB, DiMauro S, Rowland LP: Amyotrophic lateral sclerosis with ragged red fibres. Arch Neurol 2008, 65(3):403–406. 10.1001/archneurol.2007.65
Magrane J, Hervias I, Henning MS, Damiaso M, Kawamata H, Manfredi G: Mutant SOD1 in neuronal mitochondria causes toxicity and mitochondrial dynamic abnormalities. Hummol Genet 2009, 18: 4552–4558.
Dal Canto MC, Gurney ME: Neuropathological changes in two lines of mice carrying a transgene for mutant human CWZN SOD and are over-expressing wild human SOD; a model of familial amyotrophic lateral sclerosis. Brain Res 1995, 676: 25–40. 10.1016/0006-8993(95)00063-V
Wiedermann FR, Winkler K, Kuznetsov AV, Von Bossanyi P, Dietzmann K, Kunz WS: Impairment of mitochondrial function in skeletal muscle of patients with amyotrophic lateral sclerosis. J Neurol Sci 1998, 146: 65–72. 10.1016/S0022-510X(98)00008-2
Comi GP, Bordoni A, Salani S, Franceschina L, Sciacco M, Prelle A, Fortunato F, Zeviani M, Napoli L, Bresolin N, Moggio M, Ausenda CD, Taanman JW, Scarlato G: Cytochrome c oxidase subunit 1 microdeletion in a patient with motor neuron disease. Ann Neurol 1998, 43(1):110–116. 10.1002/ana.410430119
Crugnola V, Lamperti C, Lucchini V, Ronchi D, Peverelli L, Prelle A, Sciacco M, Bordoni A, Fassone E, Fortunato F, Corti S, Silani V, Bresolin N, Di Mauro S, Comi GP, Moggio M: Mitochondrial respiratory chain dysfunction in muscle from patient with amyotrophic lateral sclerosis. Arch Neurol 2010, 67(7):849–854. 10.1001/archneurol.2010.128
Xu Z, Jung C, Higgins C, Levine J, Kong J: Mitochondrial degeneration in amyotrophic lateral sclerosis. J Bioenerg Biomembr 2004, 36(4):395–399. 10.1023/B:JOBB.0000041774.12654.e1
Altoniemi T, Jaronen Y, Keksa-Goldstein V, Koistinaho J: Mutant SOD1 from spinal cord of G93A rats is destabilised and binds to inner mitochondria membrane. Neurobiol Dis 2008, 32: 479–485. 10.1016/j.nbd.2008.08.010
Ferri A, Cozzolino M, Crosio C, Nencini M, Casciati A: Familial ALS – superoxide dismutases associated with mitochondrias and shift redox potential. Proc Natl Acad Sci U S A 2006, 103: 13860–13865. 10.1073/pnas.0605814103
Rizzardini M, Mangolini A, Lupi M, Ubezio P, Bendotti C, Cantoni L: Low levels of ALS-linked Cu/Zn superoxide dismutase increase the production of reactive oxygen species and cause mitochondrial damage and death in motor neuron-like cells. J Neurol Sci 2005, 232(1–2):95–103. 10.1016/j.jns.2005.02.004
Duffy LM, Chapman AL, Shaw PJ, Grierson AJ: Review: the role of mitochondria in the pathogenesis of amyotrophic lateral sclerosis. Neuropathol Appl Neurobiol 2011, 37: 336–352. 10.1111/j.1365-2990.2011.01166.x
Echaniz-Laguna A, Zoll J, Ribera F, Tranchant C, Warter JM, Lonsdorfer J, Lampert E: Mitochondrial respiratory chain function in skeletal muscle of ALS patients. Ann Neurol 2002, 52(5):623–627. 10.1002/ana.10357
Echaniz-Laguna A, Zoll J, Ponsot E, N'guessan B, Tranchant C, Loeffler JP, Lampert E: Muscle mitochondrial function in amyotrophic lateral sclerosis is progressively altered as the disease develops: a temporal study in man. Exp Neurol 2006, 198: 25–30. 10.1016/j.expneurol.2005.07.020
Brierley EJ, Johnson MA, Lightowlers RN, James OF, Turnbull DM: Role of mitochondrial DNA in human ageing; implication of the central nervous system and muscle. Ann Neurol 1998, 43(2):217–223. 10.1002/ana.410430212
Brierley EJ, Johnson MA, James OF, Turnbull DM: Mitochondrial involvement in the ageing is fact and controversies. Mol Cell Biochem 1997, 174(102):325–328. 10.1023/A:1006847319162
Johnston W, Karpati G, Carpenter S, Arnold D, Shoubridge EA: Late-onset mitochondrial myopathy. Ann Neurol 1995, 37(1):16–23. 10.1002/ana.410370106
Magnus T, Beck M, Gless R, Puls I, Naumann M, Toyka KV: Disease progression in amyotrophic lateral sclerosis: predictors of survival. Muscle Nerve 2002, 25(5):709–714. 10.1002/mus.10090
Ro LS, Lai SL, Chen CM, Chen ST: Deleted 4977-bp mitochondrial DNA mutation is associated with sporadic amyotrophic lateral sclerosis: a hospital-based case–control study. Muscle Nerve 2003, 28: 737–743. 10.1002/mus.10504
Rubio-Gozalbo ME, Smeitink JA, Ruitenbeek W, Ter Laak H, Mullaart RA, Schuelke M, Mariman EC, Sengers RC, Gabreëls FJ: Spinal muscular atrophy-like picture, cardiomyopathy and cytochrome c oxidase deficiency. Neurology 1999, 52(2):383–386. 10.1212/WNL.52.2.383
Fetoni V, Brierm E, Carrara F, Mora M, Zeviani M: Monomelic amyotrophy associated with the 7472insC mutation in the mtDNA tRNA ser(UCN) gene. Neuromuscul Disord 2004, 14(11):723–726. 10.1016/j.nmd.2004.07.002
Finsterer J: Mitochondriopathy mimicking amyotrophic lateral sclerosis. Neurologist 2003, 9(1):45–48. 10.1097/01.nrl.0000038589.58012.a8
Finisterer J: Lactate stress testing in sporadic amyotrophic lateral sclerosis. Int J Neurosci 2005, 115: 583–591. 10.1080/00207450590522847
Karpati G: Structural and molecular basis of skeletal muscle diseases; effects of denervation on muscle. ISN Neuropathology Press Basal 2002, Chapter 13: 252–256.
Dupuis L, Oudart H, Rene F, de Aguilar JL G, Loeffler JP: Evidence for defective energy homeostasis in amyotrophic lateral sclerosis: enefit of a high-energy diet in a transgenic mouse model. Proc Natl Acad Sci U S A 2004, 101: 11159–11164. 10.1073/pnas.0402026101
Dupuis L, de Aguilar JL G, Oudart H, de Tapia M, Barbeito L, Loeffler JP: Mitochondria in amyotrophic lateral sclerosis: a trigger and a target. Neurodegener Dis 2004, 1: 245–254. 10.1159/000085063
Bradley J, Taanman JW, Kallis C, Orrel R: Increased sensitivity of myoblasts to oxidative stress in amyotrophic lateral sclerosis peripheral tissue. Exp Neurol 2009, 218: 92–97. 10.1016/j.expneurol.2009.04.007
Manfredi G, Xu Z: Mitochondrial function and its role in motor neuron degeneration in ALS. Mitochondrion 2005, 5: 77–87. 10.1016/j.mito.2005.01.002
Werydt P, Moler T: Neuro-inflammation in the pathogenesis of amyotrophic lateral sclerosis. Neuroreport 2005, 16(6):527–531. 10.1097/00001756-200504250-00001
Beal MF: Mitochondria take center stage in ageing and neurodegneneration. Ann Neurol 2005, 58: 49–505. 10.1002/ana.20624
Borthwick GM, Taylor RW, Walls TJ, Tonska K, Taylor GA, Shaw PJ, Ince PG, Turnbull DM: Motor neuron disease in a patient with a mitochondrial tRNA mutation. Ann Neurol 2006, 59: 570–574. 10.1002/ana.20758
Wiedermann FR, Manfredi G, Mawrin C, Beal MF, Schon EA: Mitochondrial DNA and respiratory chain function in spinal cords of ALS patients. J Neurochem 2002, 80(4):616–625. 10.1046/j.0022-3042.2001.00731.x
Sasaki S, Iwata M: Mitochondrial alterations in the spinal cord of patients with sporadic amyotrophic lateral sclerosis. J Neuropathol Exp Neurol 2007, 66: 10–16. 10.1097/nen.0b013e31802c396b
Simpson EP, Yen AA, Appel SH: Oxidative stress: a common denominator in the pathogenesis of amyotrophic lateral sclerosis. Curr Opin Rheumatol 2003, 15(6):730–736. 10.1097/00002281-200311000-00008
Siciliano G, Pastorini E, Pasquali L, Manca ML, Iudice A, Murri L: Impaired oxidative metabolism in exercising muscle from ALS patients. J Neurol Sci 2009, 191: 61–65. 10.1016/S0022-510X(01)00620-7
Mattiazzi M, D’Aurelio N, Gajewski CD, Martushova K, Kiaei M, Beal MF, Manfredi G: Mutated human SOD1 causes dysfunction of oxidative phosphorylation in mitochondria of transgenic mice. J Biol Chem 2002, 33: 29626–29633. 10.1074/jbc.M203065200
Grehl T, Fishcer S, Muller K, Malin JP, Zang J: A Prospective Study to Evaluate the Impact of 31P-MRS to Determinate. 2007.
Dupuis L, de Aguilar Jl G, Echaniz-Laguna A, Eschbach J, Rene F, Oudart H, Halter B, Huze C, Schaeffer L, Bouillaud F, Loeffler JP: Muscle mitochondrial uncoupling dismantles neuromuscular junction and triggers distal degeneration of motor neurons. PLoS One 2009, 4: e5390. 10.1371/journal.pone.0005390
Moissek K, Strong MJ: Innate immunity in amyotrophic lateral sclerosis. Biochem Biophys Acta 2006, 1176(11–12):1083–1093.
Sargsyan SA, Monk PN, Shau PJ: Microglia as potential contributors to motor neurone injury in amyotrophic lateral sclerosis. Glia 2005, 51(4):241–253. 10.1002/glia.20210
Dewil M, Van Den Bosch L, Robberecht W: Microglia in amyotrophic lateral sclerosis. Acta Neurol Belg 2007, 107(3):63–70.
Yamanaka K, Chun SJ, Boillee S, Fujimori-Tonou N, Yamashita H, Gutmann DH, Takahashi R, Misawa H, Cleveland DW: Astrocytes as determinants of disease progression in inherited amyotrophic lateral sclerosis. Nat Neurosci 2008, 11(3):251–253. 10.1038/nn2047
Fischer LR, Gulver DG, Tennat P, Davis AA, Wang M, Castellano-Sanchez A, Khan J: Amyotrophic lateral sclerosis is a distal axonopathy; evidence in mice and man. Exp Neurol 2004, 85: 232–240. 10.1016/j.expneurol.2003.10.004
Jokic N, de Aguilar JL G, Dimou L, Lin S, Fergania A: The neurite outgrowth inhibitor NOGO-A promotes degeneration in amyotrophic lateral sclerosis model. EMBO Rep 2006, 11: 1162–1167. 10.1038/sj.embor.7400826
Pradat PF, Bruneteau G, Gozalez de Aguilar JL, Dupuisis L, Jokic N, Salachas F, Le Forestier N, Echaniz-Laguna A, Dubourg O, Hauw JJ, Tranchant C, Loeffler JP, Meininger V: Muscle NOGO-A expression is prognostic marker in lower motor neuron syndrome. Ann Neurol 2007, 62: 15–20. 10.1002/ana.21122
Mancuso M, Confori FL, Rocchi A, Tessitore A, Muglia M, Tedeschi G, Panza D, Monsurrò M, Sola P, Mandrioli J, Choub A, DelCorona A, Manca ML, Mazzei R, Sprovieri T, Filosto M, Salviati A, Valentino P, Bono F, Caracciolo M, Simone IL, La Bella V, Majorana G, Siciliano G, Murri L, Quattrone A: Could mitochondrial haplogroups paly a role in sporadic amyotrophic lateral sclerosis? Neurosci Lett 2004, 371: 158–162. 10.1016/j.neulet.2004.08.060
Dupuis L, Loeffler JP: Neuromuscular junction destruction during amyotrophic lateral sclerosis: insights from transgenic models. Curr Opin Pharmacol 2009, 9: 31–346. 10.1016/j.coph.2009.03.007
Author information
Authors and Affiliations
Corresponding authors
Additional information
Competing interests
Jens Wuerthner is a former employee and owns some stock with GSK Safa Al-Sarraj, Andrew King, Nigel Leigh, Vincent Meininger, Jeffrey Rothstein have no competing interests.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

{kind=link}
Cite this article
Al-Sarraj, S., King, A., Cleveland, M. et al. Mitochondrial abnormalities and low grade inflammation are present in the skeletal muscle of a minority of patients with amyotrophic lateral sclerosis; an observational myopathology study. acta neuropathol commun 2, 165 (2014). https://doi.org/10.1186/s40478-014-0165-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40478-014-0165-z