
Abstract
Multiple myeloma (MM) is an incurable hematological cancer with high spatial- and temporal-heterogeneity. Invasive single-point bone marrow sampling cannot capture the tumor heterogeneity and is difficult to repeat for serial assessments. Liquid biopsy is a technique for identifying and analyzing circulating MM cells and cell products produced by tumors and released into the circulation, allowing for the minimally invasive and comprehensive detection of disease burden and molecular alterations in MM and monitoring treatment response and disease progression. Furthermore, liquid biopsy can provide complementary information to conventional detection approaches and improve their prognostic values. This article reviewed the technologies and applications of liquid biopsy in MM.
Similar content being viewed by others


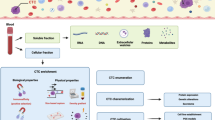
Introduction
Multiple myeloma (MM) is an incurable hematological cancer that is characterized by the abnormal proliferation of malignant plasma cells (PCs) in the bone marrow (BM). Currently, the diagnosis and evaluation of MM highly rely on BM sampling, which is invasive, painful, and difficult to repeat for serial assessments, highlighting the need for less invasive methods. The development of “liquid biopsies” opens up new avenues for noninvasive MM assessment and monitoring. Liquid biopsy is a diagnostic technique for identifying and analyzing circulating MM cells (CMMCs) and cell products produced by tumors and released into the peripheral blood (PB), particularly circulating cell-free nucleic acids (cf-NAs) (Fig. 1). Cell-free DNA (cfDNA) is primarily released into the circulation through cell death [1]. The first study on liquid biopsy in MM was published in 1993 and proposed that CMMCs were a measure of disease activity [2].

Schematic illustration of liquid biopsy in multiple myeloma (MM). We illustrated the role of circulating myeloma cells (CMMCs) in disease dissemination to distant bone marrow (BM) or extramedullary (EM) sites, and the utility of liquid biopsy (including CMMC and cell-free nucleic acids) in MM
CMMCs or cf-NAs allow for the minimally invasive detection of disease burden and molecular alterations in MM and repeated sampling for disease monitoring. MM is characterized by intra-clonal heterogeneity and multifocal tumor deposition; occasionally, extramedullary (EM) lesions were inaccessible, limiting their molecule analysis. A single-site BM aspirate would be unrepresentative of disease infiltration and mutational profile. Liquid biopsy allows for the capture of comprehensive tumor heterogeneity. To date, the clinical use of liquid biopsy has been suggested for several malignancies, including hematological malignancies and solid tumors (e.g., breast and lung tumors). Patients with MM had greater cell-free tumor DNA (ctDNA) concentrations and CMMC counts per blood tube than those with advanced solid tumors [3], which supports the use of liquid biopsy in MM. This article provides a review of the technologies and applications of liquid biopsy in MM.
Approaches for selection, enrichment, and isolation
The level of myeloma cells in the blood is much lower than that found in the BM [4, 5]. Next-generation flow cytometry (NGF)-based quantification detected only a median of approximately 1–1.9 CMMCs/uL in the PB from newly diagnosed multiple myeloma (NDMM) [6, 7]. The cfDNA level was low and variable in patients with MM (approximately 20.1–25.2 ng/mL of plasma) [3, 8,9,10]. cfDNA in the PB mainly originates from hematopoietic cells [11] and can be present in very low concentrations [12]. Different methods have been used to detect CMMCs and ctDNA with varying sensitivities and specificities (Table 1).
Wright–Giemsa-stained PB smear is the conventional method for identifying and counting PCs in the blood [13, 14, 50]. While this approach was simple and inexpensive, it could not identify cell clonality and was less sensitive than other methods. Slide-based immunofluorescence identified CMMCs using morphology and fluorescence staining [15, 16]. Single CMMC could further be isolated using fluorescence microscopy of CD138 + CD45 − cells, which was highly sensitive and specific although time- and labor-consuming [51]. The Epic Platform is an automated digital system that uses immunofluorescence to detect and characterize CMMCs based on morphological factors and levels of CD138 and CD45 expression. This test accurately identified all populations with MM CMMC with variable marker expression (positive or negative CD138) and could be further multiplexed with secondary biomarkers, including the phosphor-ribosomal protein S6 or CD56 [40, 52].
Multicolor flow cytometry (MFC)/fluorescence-activated cell sorting, alone or combined with magnetic cell sorting (MACS) enrichment (mainly with anti-CD138 antibody), is the most commonly used method for CMMC detection and isolation. The number of CMMCs detected by MFC correlated well with that detected by slide-based immunofluorescence microscopy [19]. However, there are some limitations. The sensitivity of MFC remained relatively low for the detection of extremely rare CMMCs, and a pre-enrichment step required sufficient starting CMMC concentrations. The heterogeneity of MFC instruments and detection markers resulted in variations in detection efficacy and the cut-off for CMMCs. The advent of NGF provides a possibility to adopt a standardized method for detecting CMMCs [7]. The purity of the NGF-sorted CMMCs has been confirmed by analysis of shared clonal mutations in BMPCs and CMMCs [37]. The CellSearch platform, which has received Food and Drug Administration approval for clinical use in several types of cancers, provided a more sensitive (when compared with MFC), highly reproducible, easily standardized, and high-throughput approach in CMMC detection. By combining with the DEPArray, the CellSearch–DEPArray system enabled the capture of a single CMMC [39].
Microfluidic devices were designed with microtraps whose sizes were tuned to physically capture a single CMMC with small volumes of samples and antibodies in a short time [53]. The device has high sensitivity (approximately < 10 CMMCs/mL of blood) [41] and meets the requirement for precise single-cell diagnostics using CMMCs. By combining the immunophenotypic and physical selections, anti-CD138 antibody-coated microfluidic channels were designed. The microfluidic-based CMMC counts and MFC analyses showed excellent correlation [41]. Another anti-CD138 antibody-coated microfluidic system permitted reversible cell capture. The antibody was attached via a linker, which could be degraded by enzymatic cleavage [42].
The nucleic acid-based method primarily detects CMMCs and ctDNA by identifying tumor-specific immunoglobulin (Ig) rearrangements or genetic abnormalities. The variable regions in Ig genes were transcribed in a patient-specific manner [35]. CMMCs and ctDNA from patients with MM had the same clonotypic Ig gene rearrangement as matched BM clonal PCs [12, 36, 54, 55]. Clonal Ig rearrangements were tracked using polymerase chain reaction (PCR)-based approaches, including quantitative PCR (qPCR) along with allele-specific oligonucleotides (ASOs) [4, 5, 8, 43], droplet digital PCR (ddPCR) [43], and next-generation sequencing (NGS) [12, 36]. A moderate agreement (approximately 80%) was observed between NGS of Ig rearrangement in cfDNA and MFC of CMMCs, indicating that cfDNA and CMMC analysis provided complementary information [36]. The main limitation of Ig rearrangement-based approaches was that they relied on the previously identified tumor clone.
Non-targeted approaches, including whole-genome sequencing (WGS), whole-exome sequencing (WES), and ultra-low pass WGS (ULP-WGS), allowed for genome-wide analysis. ULP-WGS (approximately 0.1 × coverage) provided a cost-effective approach for estimating genome-wide tumor fraction (TF) based on copy number aberration (CNA) profiles independent of prior knowledge of a patient’s tumor mutations [46]. However, other genetic aberrations (e.g., translocations) could not be assessed owing to the nature of the ULP-WGS analysis. These genome-wide analyses had lower assay sensitivity, which limits their use in patients with small TF (e.g., patients in the asymptomatic or pre-relapse stage) [56]. Deep-targeted sequencing approaches (e.g., NGS with a specific panel) have high sensitivity and can detect mutations in cfDNA that ULP-WGS or WES would miss [56]. One limitation of the targeted method is the requirement for prior identification of mutations in the primary tumor. A 14-gene cancer personalized profiling sequencing could detect all tumor PC-confirmed mutations in cfDNA when the variant allelic frequency (VAF) was ≥ 5% of mutations in BM tumor cells [56, 57]. Another 5-gene NGS panel that targeted all protein-coding exons of genes allowed for the detection of tumor-specific mutations in cfDNA at VAF as low as 0.25% (median 3.2%) [3]. The low DNA input hampered the utility of cfDNA with NGS in minimal residual disease (MRD) evaluation. A cross-platform evaluation of NGS-based ctDNA assays showed that, when the VAF was more than 0.5%, ctDNAs were detected with high sensitivity, precision, and reproducibility by all methods [58]. Generally, ddPCR and qPCR, as well as ddPCR and an NGS-based approach, demonstrated excellent correlation in mutation identification [10, 43]. However, in some studies, ddPCR was more sensitive (can detect mutation frequencies as low as 0.005%) than NGS and identified some mutations in cfDNA missed by NGS [10, 59,60,61].
Mechanisms explaining multiple myeloma trafficking through peripheral blood disease dissemination
It was hypothesized that as the disease progressed, myeloma cells developed the ability to grow independently of BM niches, translocate into the blood, and re-home at distant sites in the BM and other tissues. The mechanisms underlying the migration of PCs from the BM to the circulation and EM spread through PB dissemination remained unclear. Although, in general, CMMCs displayed overlapping immunophenotypic [7, 35, 62, 63], genomic [37], and transcriptomic [64] profiles with BM tumor PCs, there could be minor but consistent differences between myeloma cells in the PB and BM that could indicate hallmarks associated with cell translocation and disease dissemination.
A more immature and less proliferative immunophenotype was displayed on CMMCs. CMMCs expressed significantly lower levels of CD28, CD38, CD138, CD81, CD27, CD52, CD117, Vs38c, and Ki67 [7, 36, 62,63,64,65]. Virtually all CMMCs were in the sub-G0/G1 phase of the cell cycle [62], and the gene expression (e.g., CENPF or CDC6) and pathways (DNA repair, mitotic spindle formation, and G2M checkpoint) involved in the cell cycle were significantly downregulated in CMMCs [65]. Furthermore, CMMCs displayed lower expression of integrin and adhesion molecules, including CD11a, CD11c, CD29, CD33, CD49d, CD49e, [62] and CD56 [7, 35, 42, 52, 62, 63], which potentially enhanced its capacity to exit into the PB. Sphingosine 1-phosphate receptor 2 (S1RP2), whose inhibition significantly promoted cell migration and invasion via NF-kB pathway phosphorylation, was expressed at a significantly lower level in CMMCs [66]. The expression of adhesion-related genes (CD44 and galectin 1) and the pathway involved in epithelial–mesenchymal transition (EMT) were significantly upregulated in CMMCs. CD44 knockdown impaired cell migration and adhesion to fibronectin, whereas EMT is a significant process in tumor metastasis [65]. Furthermore, compared with BM clonal PCs, CMMCs demonstrated greater clonogenic potential in the colony and cluster formation in vitro and exhibited a circadian distribution by actively migrating to PB and metastasizing to other sites during the patients’ resting period [62].
It is unclear whether myeloma cells with distinct genetic features are more prone to spread the disease. Some data indicated that the CMMC population represented a more genetically abnormal subclone than the BM clonal PC or CMMC population from the early disease stage and that an appreciable number of mutations were identified in EM clones although absent in BM clones were identified in CMMC [37, 46, 51]. By comparing the degree of genomic similarity between BMPC, CMMC, and PC from EM, it can be determined that CMMCs are the most likely precursor of EM plasmacytomas and may act as a cellular bridge between BM and EM lesions [67]. Another hypothesis suggested that the spread of MM was driven by differential gene expression rather than unique genetic alterations. Some studies found that CMMCs had considerably increased levels of altered genes and pathways associated with hypoxia, inflammation, tumor migration, invasiveness, and metastasis, suggesting that the hypoxic and inflammatory microenvironment in BM niches would inhibit myeloma cell proliferation, forcing their migration into the PB and invasion of other niches [65]. Another possible mechanism is increased auto-secretion and self-feeding of myeloma cells. Chemokine CXCL12, which is normally expressed in BM stromal cells and is involved in CXCR4-dependent BM retention, was found to be significantly upregulated in MM CMMCs, suggesting that CMMCs generated a self-feed loop and released themselves from BM retention, thereby promoting egress to the PB [35].
Disease burden assessment
Sequential liquid biopsy examinations may provide a noninvasive real-time measure of tumor burden and a more comprehensive quantification of whole-body tumor burden than single-site BM biopsy examinations. The detection rate, the absolute number of CMMCs [7, 23, 39, 42, 68, 69], and the TF in CMMCs [46] were correlated with the disease status, which progressed from solitary plasmacytoma to monoclonal gammopathy of unknown significance (MGUS), smoldering multiple myeloma (SMM), and NDMM/MM at relapse. CMMCs were more frequently found in patients with active-relapsing MM than in those with stable disease (SD) [25]. The absolute number of CMMCs was significantly higher at baseline and relapse than that in MM undergoing treatment, and further decreased correlating to the depth of response, that is, partial response (PR), very good partial response (VGPR), and complete response (CR) [23, 26, 39, 41, 42]. The presence of CMMCs, the absolute number of CMMCs, and the TF in CMMCs were all significant predictors of clinical scores or indicators of high disease burden, including advanced Mayo Clinic Index and Spanish criteria of MGUS [7], high risk and ultra-high risk SMM [39], advanced Durie–Salmon (DS) stage [13, 23, 33], International Staging System (ISS) stage [6, 13, 23, 27, 33, 34, 39, 63, 70, 71] and Revised-ISS (R-ISS) stage [7, 33, 34, 46, 70], higher serum levels of beta2-microglobulin (β2-MG) [13, 20, 23, 25, 33, 63, 70] and lactate dehydrogenase (LDH) [20, 25, 26, 28, 33, 34, 70, 71], lower serum level of albumin [70], lower hemoglobin [13, 23, 24, 33, 63, 70] and platelet counts [14, 24], higher serum creatinine (Scr) [23, 25, 33], and advanced bone destruction [23, 33]. Chromosomal abnormalities (CAs) play a significant role for predicting the risk of patients with MM. CMMC levels were correlated with a higher incidence of high-risk cytogenetic abnormalities [6, 13, 20, 27, 28, 34, 63, 70,71,72], a lower incidence of hyperdiploidy [26], and standard-risk cytogenetic abnormalities [34]. CMMC abundance was associated with disease burden in the BM, including the tumor cell involvement [6, 7, 13, 14, 20, 23,24,25, 28, 33, 34, 39, 63, 70, 71] and the myeloma clone levels of Ig rearrangements in the BM [44]. The correlation between the percentage of tumor cells in the PB and BM adjusted better to a nonlinear rather than a linear trend [6]. The cancer cell fraction (CCF) of clonal mutations in CMMCs was only modestly correlated with the CCF of clonal mutations in myeloma cells in the BM owing to the presence of mutations that were clonal in one compartment but subclonal in another [37].
The cfDNA concentrations and the TF in cfDNA were correlated with the disease status and revealed significant differences between patients with MGUS, SMM, NDMM/MM at relapse, and post-treatment MM [3, 46, 49, 56, 57]. A previous study found that the TF in cfDNA from MM was 4.5 times higher than that in cfDNA from MGUS and SMM [56]. The cfDNA levels were observed to be significant predictors of clinical scores or markers of high disease burden, including advanced ISS stage [9, 57, 73] and R-ISS stage [46, 73], elevated levels of LDH [3, 9, 47, 73] and β2-MG in serum [9], more EM disease in positron emission tomography-computed tomography (PET-CT) [47, 74], or osteolytic lesions [48, 74]. Most patients showed a positive correlation between the frequencies and VAF of mutations [57, 73, 75, 76], the TF based on CNAs [3, 49, 74, 76], and the frequencies of MM clones (Ig rearrangements) [36] in paired myeloma cells in the BM and cfDNA. However, the ctDNA level only showed a conditional correlation with myeloma cell infiltration in the BM. Although some studies found that patients with a high ctDNA level had more BM infiltrations [47, 48, 57, 73], no quantificational correlation was found between the VAF of tumor-related mutations in cfDNA and BM MM cell infiltration [10], which could be explained by BM heterogeneity and the presence of EM lesions. According to a previous report, patients with short progression-free survival (PFS) and high tumor burden by cfDNA were observed to have inconsistently low BM infiltration. This suggests that cfDNA is less prone to spatial and technical bias than a BM biopsy and can assess a more thorough disease burden than a single-site BM biopsy [47].
There have been few studies that directly compare the disease burden mirrored by CMMCs and cfDNA. Patients with higher molecular tumor burden index levels in ctDNA had higher percentages of CPCs [73]. A comparison of the frequency of MM clones by IGK or IGL rearrangement in cfDNA by NGS and CMMC levels by MFC revealed 80% concordance, and the cell-based approach achieved greater patient coverage than the NGS assay [36]. Another study found a 30% discordance in the frequencies of MM clones by IGH and light chain (LC) rearrangement in cfDNA and CMMCs, indicating that cfDNA may not be entirely generated by CMMCs and may reflect overall tumor burden [45]. Studies focusing on the TF evaluated using ULP-WGS found that the TF in CMMCs was higher than that in paired cfDNA. Moreover, they showed a significant difference in the TF from matched cfDNA and CMMCs in a specific individual, suggesting that analyzing both cfDNA and CMMCs may broaden the applicability of liquid biopsies [46, 56].
Utility in risk stratification and disease prognosis
Several studies have confirmed the CMMC level at diagnosis, after treatment, and at remission before/after autologous stem cell transplantation (ASCT) as a prognostic factor for therapeutic response and progression (or early relapse) in MGUS, SMM, and MM, independent of several known risk factors, including ISS/R-ISS stage and high-risk cytogenetics (Table 2). In 2005, it was first proposed that CMMCs had a prognostic value, independent of age, albumin, and β2-MG [19]. When CMMCs were modeled as a continuous predictor, the risk of progression and relapse continuously increased in patients with MM with undetectable CMMCs and those with increasing CMMC percentages [6]. However, the cut-off that separated patients with different prognosis in several trials using various quantitative approaches was different, thereby limiting their clinical utilization. Ravi et al. and Granell et al. observed that survival was similar between NDMM with 5%–19% and ≥ 20% CMMCs measured on a blood smear stained with Wright–Giemsa, which was significantly poorer than those with < 5% CMMCs. Those with ≥ 5% CMMCs had significantly poorer survival than those with standard-risk cytogenetics MM and high-risk MM [14, 50]. Based on these two studies, the International Myeloma Working Group (IMWG) revised the definition of plasma cell leukemia (PCL) to include the presence of 5% or more CMMCs in blood smears [77]. Moreover, the dynamic of CMMCs at different time points showed a great prognostic value. Patients with undetectable CMMCs at the last follow-up in sequential monitoring showed better outcomes than those with CMMCs at the last follow-up [38]. By evaluating the CMMC status at diagnosis and before ASCT, undetectable CMMCs at both time points were a biomarker predicting a high rate of post-transplant stringent CR. The presence of CMMCs following induction therapy was a factor in inferior survival [26, 72], and this adverse impact was not overcome by maintenance therapy [26, 31]. Regarding cfDNA, the level of the tumor-associated IGH sequence (≥ 4.7% of total reads) in cfDNA before therapy was a prognostic factor for inferior PFS [12]. The high ctDNA level (≥ 10% TF in cfDNA) at screening and after two cycles of treatment (C3D1) was an independent factor for inferior PFS [47]. Furthermore, the high cfDNA concentration (> 25.2 ng/mL of plasma) was an independent factor for inferior PFS and overall survival (OS) [9]. The numbers and VAF of driver genes in cfDNA were independent factors for inferior OS, and its changes after treatment (C1D5) were associated with PFS [10, 78].
The CMMC assay defined high-risk disease independently of cytogenetics by fluorescence in situ hybridization (FISH) and ISS, and its quantification improved the stratification of these traditional parameters. Several prognostic models that combine conventional variables/scoring systems and perform well in prognostic stratification have been developed (Table 3). For example, a nomogram that included CMMC, Scr, and LDH showed better risk-stratifying ability than the DS stage, ISS, and R-ISS stage [33]. The CMMC level at diagnosis in NDMM was observed to increase the stratification of cases with standard-risk cytogenetic changes [21, 29]. The presence of CMMCs before ASCT increased the stratification of high-risk cytogenetic changes [20, 27]. Furthermore, to predict prognosis independent of the R-ISS stage, age, and high-risk cytogenetics and improve the risk-stratifying ability of the R-ISS stage or cytogenetics, Abe et al. developed a PET-CMMC staging system that combined CMMC and imaging characteristics from PET-CT (presence of more than three focal lesions with or without EM disease) [32]. Based on the genetic profile of ctDNA in MM, a three-factor nomogram (age ≥ 65 years, DNA repair pathway mutation, and/or transcriptional regulation pathway mutation in ctDNA) was constructed to predict the PFS of patients with NDMM [73].
The ISS is the most reliable staging system in MM, and CA was integrated into the R-ISS. However, great heterogeneity in clinical characteristics and outcomes was observed in cases within identical R-ISS risk groups, particularly in the R-ISS II group [82,83,84], indicating the need for new parameters. Several studies presented in Table 3 have confirmed that the presence and quantification of CMMCs further improved the risk stratification of patients with different prognosis in the identical ISS and R-ISS stages [6, 19, 22, 28, 34, 68, 71]. A previous study defined an ultra-high-risk group by combining R-ISS stage III and CMMC ≥ 0.105% at diagnosis. They observed a trend for better survival in patients in the R-ISS III stage with CMMC < 0.105% than those in the R-ISS II stage and even those in the R-ISS I stage with a high level of CMMCs [72]. Deshpande et al. observed that gene expression profiling 70-gene (GEP70) high-risk patients had significantly higher cfDNA concentrations and TF in ctDNA with ULP-WGS than low-risk patients [9].
Furthermore, CMMC and cfDNA quantification could predict prognostic risk regardless of BM MRD and serum immunofixation electrophoresis (sIF) status [7, 20, 27] and discriminate between patients with different prognosis despite identical IMWG response depth or BM MRD status. The presence of CMMC at diagnosis further discriminated patients with inferior PFS in patients with identical post-treatment status (both in the ≥ VGPR and < VGPR groups) [7]. The presence of CMMC before ASCT discriminated patients with inferior PFS in patients who achieved VGPR or better [31]. Another scoring system that combined the CMMC status after treatment and the sIF status divided patients into three groups. Patients with persistent negative CMMC had the best prognosis regardless of their sIF status, whereas persistent positive CMMC was a predictor for inferior prognosis even in patients with persistent negative sIF [38]. In patients who achieved CR, those with > 10−4 tumor-associated Ig rearrangement in cfDNA showed the worst PFS [12]. In patients with PR or SD, those with ≥ 10% TF in cfDNA by ULP-WGS after two cycles of treatment showed inferior PFS [47]. A prognostic model combining the CMMC level at diagnosis and the BM MRD status at premaintenance showed the best prognosis in the CMMC − /MRD − group [34, 71]. Furthermore, the CMMC + /MRD − group had a better prognosis than the CMMC − /MRD + group, implying that BM MRD negativity could partially revoke the adverse effect of a high CMMC level [34]. Furthermore, other studies found that the presence of CMMC at diagnosis and after treatment further distinguished patients with poor PFS regardless of the BM MRD status [7, 38]. Only attaining MRD negativity (rather than CR) resulted in a statistically significant increase in PFS [6].
Risk stratification of precursor conditions
MGUS and SMM are heterogeneous precursor states of MM. The rates of transformation from MGUS and SMM to active MM are approximately 1% and 10% annually, respectively [85]. A previous study identified the following two distinct entities of patients with MGUS: a group of patients destined to progress and another group remaining in a stable condition for a long time [86]. Identification of patients with a high risk of progression and detection of the progression at early stages would allow earlier intervention and improve the outcome. The noninvasive nature of liquid biopsy made it feasible in the routine screening of MM transformation. Compared with those without CMMCs, it was observed that patients with MGUS with CMMCs were twice as likely to experience progression to a more aggressive PC disease. A model predicting the progression risk of MGUS was constructed by combining the CMMC status, the type of heavy chain, and the level of monoclonal protein (M protein) [15]. Compared with those with no risk factors, the risk of progression in 2–3 years was 2.2 times higher in patients with SMM with ≥ 5,000 × 106/L CMMCs or M protein level of ≥ 3 g/dL and 14 times higher in those with SMM with high M protein and CMMC levels [16]. Gonsalves et al. found ≥ 150 CMMCs as the biomarker of SMM for predicting 2-year progression with 97% specificity and 78% positive predictive value (PPV), which was better than the Mayo Clinic risk model [30]. Vasco-Mogorrón et al. observed that CMMC > 0.0035% was an independent adverse factor for PFS and OS in MGUS and SMM. By combining the level of CMMC, β2-MG, and LDH in serum, they constructed a prognostic model for MGUS and SMM and found that the annual progress rate was three times lower in low-risk MGUS patients with CMMCs < 0.0035% and 10 times higher in high-risk patients with CMMCs > 0.0035% than the average annual progress rate (approximately 1%) in MGUS [68]. Sanoja-Flores et al. observed that significantly higher rates of MGUS with ≥ 0.058 CMMC/µL progress to SMM and MM at 30 months, and SMM with ≥ 0.1 CMMC/μL progress to MM at approximately 2 years [7]. Foulk et al. reported that the CMMC level at baseline was a good predictor of disease progression of MM, corresponding to M protein, BMPCs, and the serum free light chain (sFLC) ratio [39]. By analyzing the cell-free messenger RNA (cf-mRNA) using RNA sequencing (RNA-seq), a selected cf-mRNA panel recapitulated the transition from MGUS to MM and distinguished normal controls and patients with MGUS from those with MM [81].
Minimal residual disease evaluation
MRD evaluation has been accepted as a sufficient endpoint in disease assessment in MM, whose presence was considered as the source of recurrence for MM, and BM examination was the best indicator for detecting MRD. MRD evaluation in MM has been evaluated using MFC, NGS, and the NG of BM samples or image evaluation using PET-CT [87]. Although the utility of liquid biopsy (CMMC and ctDNA) in MRD evaluation has been confirmed in multiple types of solid tumors (e.g., tumors in the breast, prostate, bladder, colorectum, or lungs) [88], whether the persistence of CMMC/ctDNA in patients with MM could be a surrogate of BM MRD positivity remained unknown. In a study with a small sample size by Biancon et al., MRD evaluated by MFC with BM samples showed complete concordance with ctDNA analysis by NGS of IGH rearrangements [12]. However, in most studies shown in Table 4, undetectable CMMC/ctDNA has been observed in a significant proportion of patients with positive BM MRD (low negative predictive value), whereas MRD in the PB is constantly positive in patients with positive BM MRD (high PPV). These observations suggested that negative MRD in the PB may still not serve as a sufficient surrogate for negative BM MRD in MM, whereas persistent positive PB MRD may reflect the positive BM MRD and avoid invasive BM evaluation. According to International harmonization in performing and reporting minimal residual disease assessment in multiple myeloma trials proposed in 2021, although MRD evaluation in the PB is convenient and may overcome limitations of patchy BM involvement or EM disease, further investigation and cross-validation using BM-based MRD assays are required to achieve similar sensitivity with BM MRD evaluation [89].
Genetic and transcriptional profile identification
Spatial genomic heterogeneity in MM has been confirmed by multi-region sequencing in BM samples and even at different EM lesion sites [90, 91]. Liquid biopsies could offer a more thorough clonal heterogeneity profile in MM. It would be helpful to sequence matched CMMCs, cf-NAs, and BM and EM samples from patients with MM to validate the use of liquid biopsies in noninvasive molecular screening (Table 5).
Circulating myeloma cells
According to the conventional FISH method, the cytogenetic alterations between CMMCs and BM clonal PCs were substantially correlated [39, 42, 62]. In recent years, to identify the molecular alterations in CMMCs, high-throughput techniques (e.g., microarray, WES, and WGS) and single-cell sequencing were employed. A high concordance (approximately 92%–95%) of arm-level CNAs was observed between matched BM and PB tumor cells across paired samples by WES [37, 92]. Most paired BM clonal PCs and CMMCs had high-risk CNAs in MM, including 1q21 amplification and 13q deletion [37, 46, 52, 92, 94]. There is insufficient evidence for the feasibility of IGH translocation evaluation with CMMCs by high-throughput methods. A small sample study found that translocation, including t(11; 14) and t(6; 14), was shared by BM clonal PCs and paired CMMCs when comparing the IGH translocation in BM using FISH and related oncogene (CCND1 and CCND3) in CMMCs [51]. When PB and BM samples from the plasma of patients with leukemia were compared using single-cell RNA sequencing (scRNA-seq), the status of the IGH-WHSC1 gene fusion was frequently consistent in both samples, with more fusions being found in the BM than that in the PB [96]. However, the WES concordance for translocations between matched BM and PB tumor cells was only approximately 39%, which was likely because of the operating process’ random DNA fragmentation [37].
The matched tumor samples from separate compartments (BM, EM, and CMMC) had a high degree of concordance regarding the number, type, and protein effects of mutations [37]. CMMCs were observed to have the majority of mutations (92%–93%) and approximately all clonal mutations (CCF > 0.9) that were altered in BM or EM tumor cells [37, 46, 92]. The most recurrent and potentially driver mutations in genes (e.g., KRAS, NRAS, BRAF, and TP53) were shared by tumor cells from the BM, circulation, and EM [37, 46, 51, 67, 92].
However, the existence of mutation heterogeneity was observed in the tumor clones from different compartments. Generally, CMMCs had a higher frequency of somatic mutations than BM clonal PCs [51]. Some clones identified in CMMCs are not present at the BM or EM biopsy site or only present at the EM biopsy site although not at the BM biopsy site [37, 46, 51]. Private mutations had a significantly lower CCF than shared mutations [37, 92]. The discordance could be the consequence of a population of MM cells whose VAF was too low in the BM biopsy sample to be detected or a population that was not present at the BM biopsy site but rather only in a distant BM or EM site. These findings suggested that CMMC analysis may reveal other molecular alterations that single-point biopsies missed, although reflecting multiple tumor sites in the body.
Generally, the CMMC transcriptional signatures highly resembled the BM transcriptional states at single-cell and bulk levels in each patient [35, 64, 65]. A microarray-based study also found that the GEP signatures of BM clonal PCs could be appropriately reflected by CMMCs [69]. However, the discordance of gene expression was still observed between tumor cells from the different compartments, likely resulting from the different environments (e.g., expression of CRIP1 and KLF6) [64] or tumor cell aggressiveness and dissemination (detailed explanation in Mechanisms explaining MM trafficking through PB disease dissemination) [35, 65, 66].
Circulating cell-free DNAs
Since CtDNA contains the dominant clones that are generated from numerous separate foci, its presence in the circulation may represent a comprehensive tumor genome. Overall, an average of approximately 83%–93% of clonal mutations discovered in BM clonal PCs were confirmed in cfDNA, and approximately 88%–91% of clonal mutations discovered in cfDNA were confirmed in BM clonal PCs [46, 49]. Most recurrently mutated genes in MM and pan-cancer mutations were shared by matched cfDNA and BM samples [3, 10, 46, 49, 57, 73, 75, 76, 79, 80]. Furthermore, identical subclonal hierarchies were observed in paired BM and plasma samples from patients with MM with ≥ 3 mutations or several mutations in the same gene [3]. In patients with MM, cfDNA and BM samples showed high concordance of CNAs (86.4%–90.5%) [49, 74], and most MM-related sCNAs (e.g., 1q gain and 13q deletion) were shared by two samples [46, 95]. The can profile from cfDNA produced a corresponding risk classification in 78% of patients with MM as the one obtained from BM clonal PCs based on 1q21 gain and 17p13 deletion [48]. A previous study used cfDNA from the circulation to identify IGH translocation. Detection rates of IGH translocation, which was identified in the BM by FISH, were similar in ctDNA (approximately 73.7%) and BM samples (approximately 78.9%) by NGS. Some IGH translocations missed by BM-FISH could also be identified in ctDNA by FISH [73]. Another study reported that CCND1 mutation was detected in cfDNA from a patient with MM with t(11; 14) in BM tumor cells. Monosomy 13 was reliably identified in cfDNA despite only being equivocally detectable in the BM compartment in this instance [95].
In general, cfDNA had higher VAF and detection rate of the mutations in driver genes than BM samples [78,79,80]. Additionally, cfDNA carried some unique mutations overlooked by a single-site BM biopsy, which were presumably from a distant BM or EM site [3, 10, 46, 56, 73, 76, 78, 80]. Matched cfDNA samples (66.67%) were observed to have more EM lesion-related mutations than matched BM samples (31.25%), indicating that cfDNA may be a superior alternative to BM samples when EM lesion biopsies are unavailable [59]. However, the molecular profile of tumor PCs in the BM was not frequently accurately replicated by ctDNA sequencing, and several BM-specific molecular alterations were observed [10, 56, 78, 80]. Compared with other shared mutations, the VAF of these BM-exclusive mutations was relatively low [3, 56, 57, 78]. One possible explanation for these missed mutations by cfDNA is that tumor-related mutations in cfDNA had significantly lower VAF than DNA from BM clonal PCs [56, 76]. A higher TF of tumor-related CNAs and VAF of tumor-related mutations in BM clonal PCs increased the likelihood of discovering tumor-specific mutations and CNAs in cfDNA [56, 57, 78]. Genetic analysis using both BM and plasma samples revealed more mutations (approximately 80%) than using BM samples alone (approximately 60%) [80].
Comparison between circulating myeloma cells and cell-free DNAs
To date, only one study systematically compared the molecule profiles of cfDNA and patients with CMMCs [46]. According to the study, CMMCs and cfDNA had high concordance in exome-wide somatic single-nucleotide variants and sCNAs. Overall, approximately 96% of non-silent clonal mutations found in cfDNA were confirmed in CMMC, whereas approximately 84% of non-silent clonal mutations found in CMMC were confirmed in cfDNA. They further proposed that both approaches provided distinct but complementary information. The combination of CMMCs and cfDNA detected almost all clonal mutations identified in the BM sample and uncovered other subclones that were missed in a single-site BM biopsy. TF evaluation in both CMMCs and cfDNA resulted in a higher proportion of patients who had at least one sample with sufficient tumor abundance for further deep sequencing (e.g., WES).
Circulating cell-free RNAs
Circulating RNAs are generated via the following two main mechanisms: cell death and active secretion of vesicles containing RNAs [54]. According to a whole transcriptome study of extracellular RNA (exRNA) in the PB of MM patients and healthy controls, approximately 45% of the exRNA genes were protein-coding, and 85% of the identified genes were covered more than 70%, indicating that a sizable collection of gene transcripts was complete in the exRNA profile [97]. The researchers also discovered that the differentially expressed genes in the exRNA profile could be distinguished between MM patients and healthy controls. These findings suggested that exRNA profiles in the PB from MM patients could be potential biomarkers for MM detection and monitoring. The role of circulating non-coding RNA (primarily miRNA) in PB from MM patients have been comprehensively summarized by several reviews [98,99,100]. Here, we introduced the applications of cell-free messenger RNA (cf-mRNA) in PB in MM. With current MM therapeutics relying not only on direct anti-MM cell effects but also on immune cell response modulation, evaluating cfRNA could reflect a more comprehensive therapeutic response. Cf-mRNA analysis with a selected panel for MM noted that a high cf-mRNA level of CRBN and a low cf-mRNA level of IKZF1/3 at baseline were associated with a high risk of early disease progression [80]. According to another study, longitudinal cf-mRNA profiling of tumor-specific Ig rearrangement reflected the response to ASCT. Moreover, sequential monitoring of hematopoietic lineage-specific transcripts (e.g., erythrocytes and neutrophils) in cf-mRNA reflected hematopoietic reconstitution following ASCT and therapeutic response to stimulation with growth factors (e.g., EPO, G-CSF) [54]. A recent cf-mRNA global profiling in exosomes recapitulated the transition from MGUS to MM. This cf-mRNA panel, which contains a small number of genes (most of which have relatively high expression in the BM compared with other tissues and cell types), differentiated MM from premalignant conditions and healthy individuals [81]. These observations indicated that cf-mRNA may potentially provide a real-time approach to noninvasively evaluate BM function.
Liquid biopsy of methylation biomarkers in cell-free DNAs
In addition to genetic information, cfDNA carries cancer-specific nongenetic information such as epigenetic information. One of the most frequent epigenetic alterations is aberrant DNA methylation. In recent years, many studies have revealed that detecting cfDNA methylation was a good approach for the screening and localization of cancer [101, 102]. The Circulating Cell-free Genome Atlas (CCGA), a population-based cancer screening program, is currently underway to develop a blood-based test for multi-cancer early detection, including MM [103]. The research found that methylation patterns evaluated by whole-genome bisulfite sequencing (WGBS) outperformed WGS and targeted sequencing in cancer detection and localization [104]. Across more than 50 cancer types, the false-positive rate of this methylation approach in cancer detection was less than 1% [103]. In a CCGA sub-study, the sensitivity of the WGBS was 73% in MM detection [104]. Furthermore, the methylation signature accurately predicted the origin of cfDNA in 92% ~ 100% of participants with plasma cell neoplasm [103, 105]. These observations suggested the potential value of cfDNA methylation profile in MM detection and monitoring, which still need to be confirmed by more studies. The cfDNA 5-hydroxymethylcytosine (5hmC) pattern could also be potential biomarkers for MM-related researches and clinical applications. Recently, a study that profiled genome‑wide 5hmC in circulating cfDNA from patients with NDMM and precursor states found that African Americans and European Americans had different 5hmC modifications, which correlated with their survival [106, 107].
Disease monitoring
Sequential monitoring would help to early identify disease progression and recurrence before patients experience symptoms from overt relapse disease. Conventional monitoring, including PET-CT and single-site BM biopsy, cannot frequently perform in a timely manner, whereas serological markers are occasionally inadequate and nontrackable in some patients with MM. Given liquid sampling over multiple time points allowed the disease burden to be frequently tracked, liquid biopsy could be utilized as a dynamic tool to track tumor kinetics and define response or progression (Table 6).
Disease monitoring using serologic assays versus liquid biopsy
Sequential serologic assays, including serum M protein, sFLC, and sIF, played significant roles in disease monitoring and response evaluation. In disease status evaluation at a single time point, concordance was observed between the CMMC level and serological measures in most studies [39, 41, 44,45,46]. CMMC assays represented as a more sensitive parameter than serological assays in some cases [44] and were detected in some cases who achieved CR [5, 23, 27, 36, 38, 39, 41]. For cfDNA assays, a good correlation was not frequently observed between the ctDNA level and serological measures [8,9,10, 36, 45,46,47,48]. A previous study showed that although the TF in cfDNA after treatment was concordant with IMWG responses in most patients, considerable variation in individual cfDNA TF was still observed, including several cases with very high TF despite apparent response according to the IMWG criteria [47]. However, it was also observed that a small group of cases with persistent M protein had complete clearance of CMMCs/cfDNA [5, 36, 38, 44, 45, 49, 75]. In sequential monitoring, the CMMC/ctDNA levels were generally concordant with tumor dynamics evaluated using the IMWG criteria (BMPCs, M protein levels, and sFLC ratios) [5, 10, 12, 46, 49, 59, 75, 80, 108]. However, in some studies, conventional serologic monitoring appeared insufficient and delayed for response assessment and progression and relapse prediction compared with early detection using liquid biopsy [5, 10, 47, 59, 75, 91, 108]. Therefore, liquid biopsy may complement the longitudinal evaluation of serologic parameters and help with the early detection of imminent progression/relapse, particularly in patients with serologically nontrackable diseases (e.g., LC escape, oligo-, or nonsecretory myeloma) [10, 36, 75, 91].
Possible causes of discordance between serologic assays and liquid biopsy may include the following: 1) M protein had a long half-life for days [109], whereas the half-life of cfDNA ranged from minutes to a few hours [110]. cfDNA may represent a prompt measure for the tracking of MM. 2) Serologic assays may be interfered with by therapeutic antibodies. 3) Serologic assays failed in evaluating disease status in patients with serologically nontrackable diseases, including nonsecretory MM [36]. 4) MM in the PB could be biased when the molecular properties were not involved in the detection panel. 5) The tumor cell burden in the PB was significantly lower than that in the BM (approximately 40–100 times lower) [5, 23, 41, 44]. Furthermore, cfDNA had significantly lower TF and VAF of tumor-related mutations than the BM [74]. MM in the PB could be missed when the disease burden did not reach the lower limit of the detection method.
Response evaluation using the specific target of targeted treatment
The sequential analysis of target mutations in liquid samples could track the response to targeted therapies more frequently and comprehensively than BM biopsy. In a study of trametinib (a MEK inhibitor) in patients with MM with BRAF, NRAS, or KRAS mutations, researchers observed that the clinical disease progression was associated with an increase in VAFs of NRAS and KRAS mutations, indicating the involvement of a MAPK pathway-dependent mechanism in the resistance to trametinib. The authors further observed some inconstancies between serological response and the dynamics of the specific mutation in cfDNA during the therapy, indicating the existence of clones with differential responses to treatment [3]. Another study based on a cohort treated with lenalidomide and CC-486 (oral azacitidine) proposed that the cf-mRNA dynamics of CRBN, IKZF1, and IKZF3 could act as a biomarker of response to lenalidomide-based therapy. Low CRBN expression and high IKZF1 and IKZF3 expression in cf-mRNA at baseline could be indicative of patients more sensitive to lenalidomide. By comparing the expression level at baseline and on C1D5, increased IKZF1 expression was found to be an early marker of response to therapy [80].
Tracking clonal evolution and identification of drug resistance
MM is a highly heterogeneous and dynamic disease. Liquid biopsy can be a noninvasive and dynamic method for capturing real-time genetic events, reevaluating disease risk over time, and identifying potentially targetable oncogenes for precision therapeutics.
Genomic and transcriptional heterogeneity exists among different individuals with MM [51, 64]. A high degree of heterogeneity in CMMC CNA profiles was observed among different patients with MM by single-cell DNA sequencing (scDNA-seq) [93]. The transcriptional profile of CMMCs between the two patients also substantially differed based on scRNA-seq [51]. Resistance to a specific drug could be the result of the presence of one or more driver mutations in oncogenes and/or tumor suppressors. Patients with relapsed/refractory MM had a significantly higher absolute number and frequency of plasma-exclusive mutations in cfDNA than those with NDMM [10, 78]. Moreover, patients with different IMWG responses showed different mutation patterns in ctDNA after treatment. Patients who achieved CR/VGPR had enriched pathways only in clonal mutations, whereas those who achieved PD/PR had enriched pathways only in subclonal mutations [73]. Liquid biopsy could identify potential genes involved in drug resistance and guide personalized therapy in MM by comparing the molecular profile of patients with differential therapeutic responses.
Approximately all patients with MM eventually acquired drug resistance. Clonal heterogeneity and constant clonal evolution of individuals with MM are likely explanations for the emergence of drug resistance. Longitudinal monitoring of BMMC and cfDNA showed that distinct populations carried different mutations and acquired new mutations through the progression, and the dominance of the population changed, with the more resistant clones possessing a growth advantage [10, 91, 111]. cfDNA analysis provided information on subclonal architecture in MM and constructed the evolution tree using relative fractions of mutations from the blood [3]. For example, the RAS–RAF pathway mutations appeared to be dominant in BM samples, whereas the DNA repair gene variants had a more predominant presence in cfDNA, indicating that the RAS mutations may be ancestral to the DNA repair gene mutations [78].
Serial cfDNA sequencing further monitored the dynamic changes in mutation fractions. The CNA profile and mutational hierarchy of cfDNA were persistently concordant with the profile of BM clonal PCs in serial samples, suggesting that cfDNA provides a good source for reflecting the sequential molecule characterization of BMPCs [3, 49]. Potential drivers and pathways (e.g., protein kinase A signaling and Wnt/β-catenin signaling) involved in disease progression and drug resistance in a specific individual were identified by comparing the genetic profile in ctDNA samples at different time points during disease progression and relapse [49, 91]. Furthermore, clonal outgrowth and subclone disappearance co-existent with disease progression identified different clones with different therapeutic responses and provided critical clues regarding therapy selection and the predominant driver mutation involved in resistance to therapy [10, 75, 80, 91].
Serial CMMC monitoring may reflect the clonal evolution of PCs developing anchorage independence and growth potential outside of the BM microenvironment under therapeutic and immunological pressure. For example, a sequential sequencing of CMMCs using scDNA-seq revealed that CNA patterns were overall conserved in individuals, along with subclonal divergent alterations and convergent lesions, indicating the co-existence of branched tumor evolution and convergent alternations [93, 94]. Determining genomic alterations during disease progression and detecting the emergence of a drug-resistant MM clone could be achieved by comparing the mutational profile of CMMCs at several time points [26]. An obvious similarity existed in the clonal architecture of CMMCs during remission and at relapse, suggesting that the treatment did not selectively kill particular resistant subclones and that these subclones could be the source of disease relapse [51].
Conclusion and prospect
Liquid biopsy allows for the minimally invasive detection of disease burden and molecular alterations in MM, as well as repeated sampling for monitoring treatment response, drug resistance, and the appearance of potential molecular targets. The use of liquid biopsy would improve disease evaluation, particularly in patients with precursor diseases, EM diseases, or serologically nontrackable diseases. For further clinical transformation, approaches of liquid biopsy in MM should be standardized to establish consistency to compare clinical trial data. To date, the sensitivity of liquid biopsy in MRD evaluation remains lower than that of BM-based MRD assay in MM. Methods with high sensitivity of liquid biopsy for MRD evaluation need to be further explored. For example, a method called phased variant enrichment and detection sequencing has been confirmed with extremely high sensitivity and specificity in MRD detection in the PB in lymphoma, which allowed for ctDNA detection in the ppm range in samples [112]. In recent years, nongenetic features of cfDNAs, such as DNA epigenetics (primarily methylation), fragmentation, and topology, have broadened the utility of cfDNA [101]. So far, only a few studies have shown that cfDNA methylation can be used to detect MM. Further researches are required to exploring the cfDNA methylation pattern and other epigenetic patterns, and their utility in predicting therapeutic responses and disease prognosis in MM. In the future, the introduction of liquid biopsy into the disease evaluation of MM would provide a tool for the comprehensive and real-time assessment complementary to conventional methods, promoting the development of new risk stratification systems and individual therapy options.
Availability of data and materials
Not applicable.
Abbreviations
- ASCT:
-
Autologous stem cell transplantation
- ASO:
-
Allele-specific oligonucleotide
- β2-MG:
-
Beta2-microglobulin
- BM:
-
Bone marrow
- BMPC:
-
Bone marrow plasma cell
- CA:
-
Chromosomal abnormality
- CCF:
-
Cancer cell fraction
- CCGA:
-
Circulating Cell-free Genome Atlas
- CDC6:
-
Cell division cycle 6
- CENPF:
-
Centromere protein F
- cfDNA:
-
Cell-free DNA
- cf-mRNA:
-
Cell-free messenger RNA
- cf-NA:
-
Cell-free nucleic acid
- CMMC:
-
Circulating multiple myeloma cell
- CNA:
-
Copy number aberration
- CR:
-
Complete response
- CRIP1:
-
Cysteine-rich intestinal protein 1
- ctDNA:
-
Cell-free tumor DNA
- CXCL12:
-
C-X-C motif chemokine ligand 12
- CXCR4:
-
C-X-C motif chemokine receptor 4
- ddPCR:
-
Droplet digital polymerase chain reaction
- DS:
-
Durie–Salmon
- EM:
-
Extramedullary
- EMT:
-
Epithelial–mesenchymal transition
- EPO:
-
Erythropoietin
- exRNA:
-
Extracellular RNA
- FACS:
-
Fluorescence-activated cell sorting
- FISH:
-
Fluorescence in situ hybridization
- G-CSF:
-
Granulocyte colony-stimulating factor
- GEP:
-
Gene expression profiling
- Ig:
-
Immunoglobulin
- IGH:
-
Immunoglobulin heavy chain
- IMWG:
-
International Myeloma Working Group
- ISS:
-
International Staging System
- KLF6:
-
Kruppel-like factor 6
- LC:
-
Light chain
- LDH:
-
Lactate dehydrogenase
- MACS:
-
Magnetic cell sorting
- MAPK:
-
Mitogen-activated protein kinase
- MFC:
-
Multicolor flow cytometry
- MGUS:
-
Monoclonal gammopathy of unknown significance
- MRD:
-
Minimal residual disease
- MM:
-
Multiple myeloma
- M protein:
-
Monoclonal protein
- NDMM:
-
Newly diagnosed multiple myeloma
- NGF:
-
Next-generation flow cytometry
- NGS:
-
Next-generation sequencing
- PB:
-
Peripheral blood
- PC:
-
Plasma cell
- PCR:
-
Polymerase chain reaction
- OS:
-
Overall survival
- PET-CT:
-
Positron emission tomography-computed tomography
- PFS:
-
Progression-free survival
- PPV:
-
Positive predictive value
- PR:
-
Partial response
- qPCR:
-
Quantitative polymerase chain reaction
- R-ISS:
-
Revised-International Staging System
- Scr:
-
Serum creatinine
- scDNA-seq:
-
Single-cell DNA sequencing
- scRNA-seq:
-
Single-cell RNA sequencing
- SD:
-
Stable disease
- sFLC:
-
Serum free light chain
- sIF:
-
Serum immunofixation electrophoresis
- SMM:
-
Smoldering multiple myeloma
- S1RP2:
-
Sphingosine 1-phosphate receptor 2
- sSNV:
-
Somatic single-nucleotide variant
- TF:
-
Tumor fraction
- ULP-WGS:
-
Ultra-low pass whole-genome sequencing
- VAF:
-
Variant allelic frequency
- VGPR:
-
Very good partial response
- WES:
-
Whole-exome sequencing
- WGBS:
-
Whole-genome bisulfite sequencing
- WGS:
-
Whole-genome sequencing
- WHSC1:
-
Wolf-Hirschhorn syndrome candidate gene-1
References
Wan JCM, Massie C, Garcia-Corbacho J, Mouliere F, Brenton JD, Caldas C, et al. Liquid biopsies come of age: towards implementation of circulating tumour DNA. Nat Rev Cancer. 2017;17(4):223–38.
Witzig TE, Dhodapkar MV, Kyle RA, Greipp PR. Quantitation of circulating peripheral blood plasma cells and their relationship to disease activity in patients with multiple myeloma. Cancer. 1993;72(1):108–13.
Kis O, Kaedbey R, Chow S, Danesh A, Dowar M, Li T, et al. Circulating tumour DNA sequence analysis as an alternative to multiple myeloma bone marrow aspirates. Nat Commun. 2017;8:15086.
Billadeau D, Van Ness B, Kimlinger T, Kyle RA, Therneau TM, Greipp PR, et al. Clonal circulating cells are common in plasma cell proliferative disorders: a comparison of monoclonal Gammopathy of undetermined significance, smoldering multiple Myeloma, and Active Myeloma. Blood. 1996;88(1):289–96.
Korthals M, Sehnke N, Kronenwett R, Schroeder T, Strapatsas T, Kobbe G, et al. Molecular monitoring of minimal residual disease in the peripheral blood of patients with multiple myeloma. Biol Blood Marrow Transplant. 2013;19(7):1109–15.
Garces JJ, Cedena MT, Puig N, Burgos L, Perez JJ, Cordon L, et al. Circulating Tumor Cells for the Staging of Patients With Newly Diagnosed Transplant-Eligible Multiple Myeloma. J Clin Oncol. 2022;40(27):3151-61.
Sanoja-Flores L, Flores-Montero J, Garces JJ, Paiva B, Puig N, Garcia-Mateo A, et al. Next generation flow for minimally-invasive blood characterization of MGUS and multiple myeloma at diagnosis based on circulating tumor plasma cells (CTPC). Blood Cancer J. 2018;8(12):117.
Vrabel D, Sedlarikova L, Besse L, Rihova L, Bezdekova R, Almasi M, et al. Dynamics of tumor-specific cfDNA in response to therapy in multiple myeloma patients. Eur J Haematol. 2019;104(3):190–7.
Deshpande S, Tytarenko RG, Wang Y, Boyle EM, Ashby C, Schinke CD, et al. Monitoring treatment response and disease progression in myeloma with circulating cell-free DNA. Eur J Haematol. 2021;106(2):230–40.
Mithraprabhu S, Khong T, Ramachandran M, Chow A, Klarica D, Mai L, et al. Circulating tumour DNA analysis demonstrates spatial mutational heterogeneity that coincides with disease relapse in myeloma. Leukemia. 2017;31(8):1695–705.
Snyder MW, Kircher M, Hill AJ, Daza RM, Shendure J. Cell-free DNA Comprises an In Vivo Nucleosome Footprint that Informs Its Tissues-Of-Origin. Cell. 2016;164(1–2):57–68.
Biancon G, Gimondi S, Vendramin A, Carniti C, Corradini P. Noninvasive Molecular Monitoring in Multiple Myeloma Patients Using Cell-Free Tumor DNA: A Pilot Study. J Mol Diagn. 2018;20(6):859–70.
An G, Qin X, Acharya C, Xu Y, Deng S, Shi L, et al. Multiple myeloma patients with low proportion of circulating plasma cells had similar survival with primary plasma cell leukemia patients. Ann Hematol. 2015;94(2):257–64.
Granell M, Calvo X, Garcia-Guinon A, Escoda L, Abella E, Martinez CM, et al. Prognostic impact of circulating plasma cells in patients with multiple myeloma: implications for plasma cell leukemia definition. Haematologica. 2017;102(6):1099–104.
Kumar S, Rajkumar SV, Kyle RA, Lacy MQ, Dispenzieri A, Fonseca R, et al. Prognostic value of circulating plasma cells in monoclonal gammopathy of undetermined significance. J Clin Oncol. 2005;23(24):5668–74.
Bianchi G, Kyle RA, Larson DR, Witzig TE, Kumar S, Dispenzieri A, et al. High levels of peripheral blood circulating plasma cells as a specific risk factor for progression of smoldering multiple myeloma. Leukemia. 2013;27(3):680–5.
Witzig TE, Gertz MA, Lust JA, Kyle RA, O’Fallon WM, Greipp PR. Peripheral blood monoclonal plasma cells as a predictor of survival in patients with multiple myeloma. Blood. 1996;88(5):1780–7.
Witzig TE, Kyle RA, O’Fallon WM, Greipp PR. Detection of peripheral blood plasma cells as a predictor of disease course in patients with smouldering multiple myeloma. Br J Haematol. 1994;87(2):266–72.
Nowakowski GS, Witzig TE, Dingli D, Tracz MJ, Gertz MA, Lacy MQ, et al. Circulating plasma cells detected by flow cytometry as a predictor of survival in 302 patients with newly diagnosed multiple myeloma. Blood. 2005;106(7):2276–9.
Dingli D, Nowakowski GS, Dispenzieri A, Lacy MQ, Hayman SR, Rajkumar SV, et al. Flow cytometric detection of circulating myeloma cells before transplantation in patients with multiple myeloma: a simple risk stratification system. Blood. 2006;107(8):3384–8.
Vagnoni D, Travaglini F, Pezzoni V, Ruggieri M, Bigazzi C, Dalsass A, et al. Circulating plasma cells in newly diagnosed symptomatic multiple myeloma as a possible prognostic marker for patients with standard-risk cytogenetics. Br J Haematol. 2015;170(4):523–31.
Galieni P, Travaglini F, Vagnoni D, Ruggieri M, Caraffa P, Bigazzi C, et al. The detection of circulating plasma cells may improve the Revised International Staging System (R-ISS) risk stratification of patients with newly diagnosed multiple myeloma. Br J Haematol. 2021;193(3):542–50.
Wang N, Tesfaluul N, Li J, Gao X, Liu S, Yue B. Enrichment of circulating myeloma cells by immunomagnetic beads combined with flow cytometry for monitoring minimal residual disease and relapse in patients with multiple myeloma. Ann Hematol. 2019;98(12):2769–80.
Bae MH, Park CJ, Kim BH, Cho YU, Jang S, Lee DH, et al. Increased circulating plasma cells detected by flow cytometry predicts poor prognosis in patients with plasma cell myeloma. Cytometry B Clin Cytom. 2018;94(3):493–9.
Gonsalves WI, Morice WG, Rajkumar V, Gupta V, Timm MM, Dispenzieri A, et al. Quantification of clonal circulating plasma cells in relapsed multiple myeloma. Br J Haematol. 2014;167(4):500–5.
Chakraborty R, Muchtar E, Kumar SK, Jevremovic D, Buadi FK, Dingli D, et al. Serial measurements of circulating plasma cells before and after induction therapy have an independent prognostic impact in patients with multiple myeloma undergoing upfront autologous transplantation. Haematologica. 2017;102(8):1439–45.
Chakraborty R, Muchtar E, Kumar SK, Jevremovic D, Buadi FK, Dingli D, et al. Risk stratification in myeloma by detection of circulating plasma cells prior to autologous stem cell transplantation in the novel agent era. Blood Cancer J. 2016;6(12):e512.
Gonsalves WI, Jevremovic D, Nandakumar B, Dispenzieri A, Buadi FK, Dingli D, et al. Enhancing the R-ISS classification of newly diagnosed multiple myeloma by quantifying circulating clonal plasma cells. Am J Hematol. 2020;95(3):310–5.
Gonsalves WI, Rajkumar SV, Gupta V, Morice WG, Timm MM, Singh PP, et al. Quantification of clonal circulating plasma cells in newly diagnosed multiple myeloma: implications for redefining high-risk myeloma. Leukemia. 2014;28(10):2060–5.
Gonsalves WI, Rajkumar SV, Dispenzieri A, Dingli D, Timm MM, Morice WG, et al. Quantification of circulating clonal plasma cells via multiparametric flow cytometry identifies patients with smoldering multiple myeloma at high risk of progression. Leukemia. 2017;31(1):130–5.
Cowan AJ, Stevenson PA, Libby EN, Becker PS, Coffey DG, Green DJ, et al. Circulating Plasma Cells at the Time of Collection of Autologous PBSC for Transplant in Multiple Myeloma Patients is a Negative Prognostic Factor Even in the Age of Post-Transplant Maintenance Therapy. Biol Blood Marrow Transplant. 2018;24(7):1386–91.
Abe Y, Narita K, Kobayashi H, Kitadate A, Miura D, Takeuchi M, et al. Pretreatment (18)F-FDG PET/CT combined with quantification of clonal circulating plasma cells as a potential risk model in patients with newly diagnosed multiple myeloma. Eur J Nucl Med Mol Imaging. 2019;46(6):1325–33.
Cheng Q, Cai L, Zhang Y, Chen L, Hu Y, Sun C. Circulating Plasma Cells as a Biomarker to Predict Newly Diagnosed Multiple Myeloma Prognosis: Developing Nomogram Prognostic Models. Front Oncol. 2021;11:639528.
Bertamini L, Oliva S, Rota-Scalabrini D, Paris L, More S, Corradini P, et al. High Levels of Circulating Tumor Plasma Cells as a Key Hallmark of Aggressive Disease in Transplant-Eligible Patients With Newly Diagnosed Multiple Myeloma. J Clin Oncol. 2022;40(27):3120-31.
Geng S, Wang J, Zhang X, Zhang JJ, Wu F, Pang Y, et al. Single-cell RNA sequencing reveals chemokine self-feeding of myeloma cells promotes extramedullary metastasis. FEBS Lett. 2020;594(3):452–65.
Mack EKM, Hartmann S, Ross P, Wollmer E, Mann C, Neubauer A, et al. Monitoring multiple myeloma in the peripheral blood based on cell-free DNA and circulating plasma cells. Ann Hematol. 2022;101(4):811-24.
Garces JJ, Bretones G, Burgos L, Valdes-Mas R, Puig N, Cedena MT, et al. Circulating tumor cells for comprehensive and multiregional non-invasive genetic characterization of multiple myeloma. Leukemia. 2020;34(11):3007–18.
Sanoja-Flores L, Flores-Montero J, Puig N, Contreras-Sanfeliciano T, Pontes R, Corral-Mateos A, et al. Blood monitoring of circulating tumor plasma cells by next generation flow in multiple myeloma after therapy. Blood. 2019;134(24):2218–22.
Foulk B, Schaffer M, Gross S, Rao C, Smirnov D, Connelly MC, et al. Enumeration and characterization of circulating multiple myeloma cells in patients with plasma cell disorders. Br J Haematol. 2018;180(1):71–81.
Zhang L, Beasley S, Prigozhina NL, Higgins R, Ikeda S, Lee FY, et al. Detection and Characterization of Circulating Tumour Cells in Multiple Myeloma. J Circ Biomark. 2016;5:10.
Qasaimeh MA, Wu YC, Bose S, Menachery A, Talluri S, Gonzalez G, et al. Isolation of Circulating Plasma Cells in Multiple Myeloma Using CD138 Antibody-Based Capture in a Microfluidic Device. Sci Rep. 2017;7:45681.
Kamande JW, Lindell MAM, Witek MA, Voorhees PM, Soper SA. Isolation of circulating plasma cells from blood of patients diagnosed with clonal plasma cell disorders using cell selection microfluidics. Integr Biol (Camb). 2018;10(2):82–91.
Drandi D, Kubiczkova-Besse L, Ferrero S, Dani N, Passera R, Mantoan B, et al. Minimal Residual Disease Detection by Droplet Digital PCR in Multiple Myeloma, Mantle Cell Lymphoma, and Follicular Lymphoma: A Comparison with Real-Time PCR. J Mol Diagn. 2015;17(6):652–60.
Vij R, Mazumder A, Klinger M, O’Dea D, Paasch J, Martin T, et al. Deep sequencing reveals myeloma cells in peripheral blood in majority of multiple myeloma patients. Clin Lymphoma Myeloma Leuk. 2014;14(2):131-9 e1.
Oberle A, Brandt A, Voigtlaender M, Thiele B, Radloff J, Schulenkorf A, et al. Monitoring multiple myeloma by next-generation sequencing of V(D)J rearrangements from circulating myeloma cells and cell-free myeloma DNA. Haematologica. 2017;102(6):1105–11.
Manier S, Park J, Capelletti M, Bustoros M, Freeman SS, Ha G, et al. Whole-exome sequencing of cell-free DNA and circulating tumor cells in multiple myeloma. Nat Commun. 2018;9(1):1691.
Waldschmidt JM, Yee AJ, Vijaykumar T, Pinto RA, Frede J, Anand P, et al. Cell-free DNA for the detection of emerging treatment failure in relapsed/ refractory multiple myeloma. Leukemia. 2022;36(4):1078-87.
Rengifo LY, Smits S, Buedts L, Delforge M, Dehaspe L, Tousseyn T, et al. Ultra-low coverage whole genome sequencing of ccfDNA in multiple myeloma: a tool for laboratory routine? Cancer Treat Res Commun. 2021;28:100380.
Guo G, Raje NS, Seifer C, Kloeber J, Isenhart R, Ha G, et al. Genomic discovery and clonal tracking in multiple myeloma by cell-free DNA sequencing. Leukemia. 2018;32(8):1838–41.
Ravi P, Kumar SK, Roeker L, Gonsalves W, Buadi F, Lacy MQ, et al. Revised diagnostic criteria for plasma cell leukemia: results of a Mayo Clinic study with comparison of outcomes to multiple myeloma. Blood Cancer J. 2018;8(12):116.
Lohr JG, Kim S, Gould J, Knoechel B, Drier Y, Cotton MJ, et al. Genetic interrogation of circulating multiple myeloma cells at single-cell resolution. Sci Transl Med. 2016;8(363):363147.
Ndacayisaba LJ, Rappard KE, Shishido SN, Ruiz Velasco C, Matsumoto N, Navarez R, et al. Enrichment-Free Single-Cell Detection and Morphogenomic Profiling of Myeloma Patient Samples to Delineate Circulating Rare Plasma Cell Clones. Curr Oncol. 2022;29(5):2954–72.
MWeerakoon-Ratnayake K, Vaidyanathan S, Larky N, Dathathreya K, Hu M, Jose J, et al. Microfluidic Device for On-Chip Immunophenotyping and Cytogenetic Analysis of Rare Biological Cells. Cells. 2020;9(2):519.
Ibarra A, Zhuang J, Zhao Y, Salathia NS, Huang V, Acosta AD, et al. Non-invasive characterization of human bone marrow stimulation and reconstitution by cell-free messenger RNA sequencing. Nat Commun. 2020;11(1):400.
Mazzotti C, Buisson L, Maheo S, Perrot A, Chretien ML, Leleu X, et al. Myeloma MRD by deep sequencing from circulating tumor DNA does not correlate with results obtained in the bone marrow. Blood Adv. 2018;2(21):2811–3.
Manzoni M, Pompa A, Fabris S, Pelizzoni F, Ciceri G, Seia M, et al. Limits and Applications of Genomic Analysis of Circulating Tumor DNA as a Liquid Biopsy in Asymptomatic Forms of Multiple Myeloma. Hemasphere. 2020;4(4):e402.
Gerber B, Manzoni M, Spina V, Bruscaggin A, Lionetti M, Fabris S, et al. Circulating tumor DNA as a liquid biopsy in plasma cell dyscrasias. Haematologica. 2018;103(6):e245–8.
Deveson IW, Gong B, Lai K, LoCoco JS, Richmond TA, Schageman J, et al. Evaluating the analytical validity of circulating tumor DNA sequencing assays for precision oncology. Nat Biotechnol. 2021;39(9):1115–28.
Long X, Xu Q, Lou Y, Li C, Gu J, Cai H, et al. The utility of non-invasive liquid biopsy for mutational analysis and minimal residual disease assessment in extramedullary multiple myeloma. Br J Haematol. 2020;189(2):e45–8.
Bessi L, Viailly P-J, Bohers E, Ruminy P, Maingonnat C, Bertrand P, et al. Somatic mutations of cell-free circulating DNA detected by targeted next-generation sequencing and digital droplet PCR in classical Hodgkin lymphoma. Leuk Lymphoma. 2019;60(2):498–502.
Demuth C, Spindler KLG, Johansen JS, Pallisgaard N, Nielsen D, Hogdall E, et al. Measuring KRAS Mutations in Circulating Tumor DNA by Droplet Digital PCR and Next-Generation Sequencing. Transl Oncol. 2018;11(5):1220–4.
Paiva B, Paino T, Sayagues JM, Garayoa M, San-Segundo L, Martin M, et al. Detailed characterization of multiple myeloma circulating tumor cells shows unique phenotypic, cytogenetic, functional, and circadian distribution profile. Blood. 2013;122(22):3591–8.
Terpos E, Kostopoulos IV, Papanota AM, Papadimitriou K, Malandrakis P, Micheli P, et al. Next Generation Flow Cytometry Provides a Standardized, Highly Sensitive and Informative Method for the Analysis of Circulating Plasma Cells in Newly Diagnosed Multiple Myeloma: A Single Center Study in 182 Patients. Blood. 2019;134(Supplement_1):4338.
Ledergor G, Weiner A, Zada M, Wang SY, Cohen YC, Gatt ME, et al. Single cell dissection of plasma cell heterogeneity in symptomatic and asymptomatic myeloma. Nat Med. 2018;24(12):1867–76.
Garces JJ, Simicek M, Vicari M, Brozova L, Burgos L, Bezdekova R, et al. Transcriptional profiling of circulating tumor cells in multiple myeloma: a new model to understand disease dissemination. Leukemia. 2020;34(2):589–603.
Pang M, Li C, Zheng D, Wang Y, Wang J, Zhang W, et al. S1PR2 Knockdown Promotes Migration and Invasion in Multiple Myeloma Cells via NF-kappaB Activation. Cancer Manag Res. 2020;12:7857–65.
Bretones G, Paiva B, Valdes-Mas R, Alignani D, Garcia M, Burgos L, et al. Genomic Profiles of Bone Marrow (BM) Clonal Plasma Cells (PCs) Vs Circulating Tumor Cells (CTCs) and Extramedullary (EM) Plasmacytomas in Multiple Myeloma (MM). Blood. 2016;128(22):4442.
Vasco-Mogorrón MA, Campillo JA, Periago A, Cabañas V, Berenguer M, García-Garay MC, et al. Blood-based risk stratification for pre-malignant and symptomatic plasma cell neoplasms to improve patient management. Am J Cancer Res. 2021;11(6):2736–53.
Burgos L, Alignani D, Garces J-J, Ortiz L, Jelinek T, Segura V, et al. Non-Invasive Genetic Profiling Is Highly Applicable in Multiple Myeloma (MM) through Characterization of Circulating Tumor Cells (CTCs). Blood. 2016;128(22):801.
Muccio VE, Gilestro M, Saraci E, Capra A, Costa A, Ruggeri M, et al. Tumor Circulating Plasma Cells Detected By Flow Cytometric Single Platform Method Correlate with Clinical Response to Therapy and Unfavorable Patients’ Characteristics. Blood. 2019;134(Supplement_1):4357.
Bertamini L, Grasso M, D’Agostino M, Pascarella A, Tosi P, Monaco F, et al. Poor Prognosis of Multiple Myeloma Predicted By High Levels of Circulating Plasma Cells Is Independent from Other High-Risk Features but Is Modulated By the Achievement of Minimal Residual Disease Negativity. Blood. 2020;136(Supplement 1):12–3.
Han W, Jin Y, Xu M, Zhao SS, Shi Q, Qu X, et al. Prognostic value of circulating clonal plasma cells in newly diagnosed multiple myeloma. Hematology. 2021;26(1):510–7.
Liu Y, Guo J, Yi Y, Gao X, Wen L, Duan W, et al. Circulating Tumor DNA: Less Invasive, More Representative Method to Unveil the Genomic Landscape of Newly Diagnosed Multiple Myeloma Than Bone Marrow Aspirates. Cancers (Basel). 2022;14(19):4914.
Martello M, Poletti A, Borsi E, Taurisano B, Solli V, Armuzzi S, et al. Towards a comprehensive multimodal minimal residual disease assessment in multiple myeloma: the role of circulating cell-free DNA to define the extent of disease spreading. Clin LYMPHOMA MYELOMA LEUKEMIA. 2021;21:S37-S.
Rustad EH, Coward E, Skytoen ER, Misund K, Holien T, Standal T, et al. Monitoring multiple myeloma by quantification of recurrent mutations in serum. Haematologica. 2017;102(7):1266–72.
Quivoron C, Lecourt H, Michot J-M, Lazarovici J, Rossignol J, Ghez D, et al. Molecular Profiling Feasibility on Cell-Free Tumoral DNA in Relapse/Refractory (R/R) Multiple Myeloma (MM) Patients Screened for Phase I Trials. Blood. 2021;138(Supplement 1):3763.
Fernandez de Larrea C, Kyle R, Rosinol L, Paiva B, Engelhardt M, Usmani S, et al. Primary plasma cell leukemia: consensus definition by the International Myeloma Working Group according to peripheral blood plasma cell percentage. Blood Cancer J. 2021;11(12):192.
Mithraprabhu S, Hocking J, Ramachandran M, Choi K, Klarica D, Khong T, et al. DNA-Repair Gene Mutations Are Highly Prevalent in Circulating Tumour DNA from Multiple Myeloma Patients. Cancers (Basel). 2019;11(7):917.
Li Q, Huang HJ, Ma J, Wang Y, Cao Z, Karlin-Neumann G, et al. RAS/RAF mutations in tumor samples and cell-free DNA from plasma and bone marrow aspirates in multiple myeloma patients. J Cancer. 2020;11(12):3543–50.
Mithraprabhu S, Morley R, Khong T, Kalff A, Bergin K, Hocking J, et al. Monitoring tumour burden and therapeutic response through analysis of circulating tumour DNA and extracellular RNA in multiple myeloma patients. Leukemia. 2019;33(8):2022–33.
Roskams-Hieter B, Kim HJ, Anur P, Wagner JT, Callahan R, Spiliotopoulos E, et al. Plasma cell-free RNA profiling distinguishes cancers from pre-malignant conditions in solid and hematologic malignancies. NPJ Precis Oncol. 2022;6(1):28.
Kastritis E, Terpos E, Roussou M, Gavriatopoulou M, Migkou M, Eleutherakis-Papaiakovou E, et al. Evaluation of the Revised International Staging System in an independent cohort of unselected patients with multiple myeloma. Haematologica. 2017;102(3):593–9.
Bygrave C, Pawlyn C, Davies F, Craig Z, Cairns D, Hockaday A, et al. Early relapse after high-dose melphalan autologous stem cell transplant predicts inferior survival and is associated with high disease burden and genetically high-risk disease in multiple myeloma. Br J Haematol. 2021;193(3):551–5.
Schavgoulidze A, Lauwers-Cances V, Perrot A, Cazaubiel T, Chretien ML, Moreau P, et al. Heterogeneity in long term outcomes for R-ISS stage II in newly diagnosed multiple myeloma patients. Haematol. 2022. https://doi.org/10.3324/haematol.2021.280566.
Dhodapkar MV. MGUS to myeloma: a mysterious gammopathy of underexplored significance. Blood. 2016;128(23):2599–606.
Oben B, Froyen G, Maclachlan KH, Leongamornlert D, Abascal F, Zheng-Lin B, et al. Whole-genome sequencing reveals progressive versus stable myeloma precursor conditions as two distinct entities. Nat Commun. 2021;12(1):1861.
Kumar S, Paiva B, Anderson KC, Durie B, Landgren O, Moreau P, et al. International Myeloma Working Group consensus criteria for response and minimal residual disease assessment in multiple myeloma. Lancet Oncol. 2016;17(8):e328–46.
Pantel K, Alix-Panabieres C. Liquid biopsy and minimal residual disease - latest advances and implications for cure. Nat Rev Clin Oncol. 2019;16(7):409–24.
Costa LJ, Derman BA, Bal S, Sidana S, Chhabra S, Silbermann R, et al. International harmonization in performing and reporting minimal residual disease assessment in multiple myeloma trials. Leukemia. 2021;35(1):18–30.
Rasche L, Chavan SS, Stephens OW, Patel PH, Tytarenko R, Ashby C, et al. Spatial genomic heterogeneity in multiple myeloma revealed by multi-region sequencing. Nat Commun. 2017;8(1):268.
Mithraprabhu S, Sirdesai S, Chen M, Khong T, Spencer A. Circulating Tumour DNA Analysis for Tumour Genome Characterisation and Monitoring Disease Burden in Extramedullary Multiple Myeloma. Int J Mol Sci. 2018;19(7):1858.
Mishima Y, Paiva B, Shi J, Park J, Manier S, Takagi S, et al. The Mutational Landscape of Circulating Tumor Cells in Multiple Myeloma. Cell Rep. 2017;19(1):218–24.
Raspadori A, Forcato C, Edoardo P, Papadopulos FM, Ferrarini A, Del Monaco V, et al. A High-Throughput Workflow for the Detection, Isolation and Genomic Analysis of Single Circulating Multiple Myeloma Cells. Blood. 2018;132(Supplement 1):5574.
Forcato C, Raspadori A, Ferrarini A, Terracciano M, Monaco VD, Garonzi M, et al. Abstract 2911: Genome-wide copy number profiling of single circulating multiple myeloma cells (CMMCs) reveals intra-patient convergent copy-number alterations (CNAs). Cancer Research. 2019;79(13_Supplement):2911.
Blombery PA, Ryland GL, Markham J, Guinto J, Wall M, McBean M, et al. Detection of clinically relevant early genomic lesions in B-cell malignancies from circulating tumour DNA using a single hybridisation-based next generation sequencing assay. Br J Haematol. 2018;183(1):146–9.
Foltz SM, Gao Q, Yoon CJ, Sun H, Yao L, Li Y, et al. Evolution and structure of clinically relevant gene fusions in multiple myeloma. Nat Commun. 2020;11(1):2666.
Chen M, Mithraprabhu S, Ramachandran M, Choi K, Khong T, Spencer A. Utility of Circulating Cell-Free RNA Analysis for the Characterization of Global Transcriptome Profiles of Multiple Myeloma Patients. Cancers (Basel). 2019;11(6):887.
Federico C, Sacco A, Belotti A, Ribolla R, Cancelli V, Giacomini A, et al. Circulating microRNAs and Their Role in Multiple Myeloma. Noncoding RNA. 2019;5(2):37.
Ferreira B, Caetano J, Barahona F, Lopes R, Carneiro E, Costa-Silva B, et al. Liquid biopsies for multiple myeloma in a time of precision medicine. J Mol Med (Berl). 2020;98(4):513–25.
Hashemi M, Roshanzamir SM, Paskeh MDA, Karimian SS, Mahdavi MS, Kheirabad SK, et al. Non-coding RNAs and exosomal ncRNAs in multiple myeloma: An emphasis on molecular pathways. Eur J Pharmacol. 2023;941:175380.
Lo YMD, Han DSC, Jiang P, Chiu RWK. Epigenetics, fragmentomics, and topology of cell-free DNA in liquid biopsies. Sci. 2021;372(6538):eaaw3616.
Luo H, Wei W, Ye Z, Zheng J, Xu RH. Liquid Biopsy of Methylation Biomarkers in Cell-Free DNA. Trends Mol Med. 2021;27(5):482–500.
Liu MC, Oxnard GR, Klein EA, Swanton C, Seiden MV, Liu MC, et al. Sensitive and specific multi-cancer detection and localization using methylation signatures in cell-free DNA. Ann Oncol. 2020;31(6):745–59.
Klein EA, Hubbell E, Maddala T, Aravanis A, Beausang JF, Filippova D, et al. Development of a comprehensive cell-free DNA (cfDNA) assay for early detection of multiple tumor types: The Circulating Cell-free Genome Atlas (CCGA) study. Journal of Clinical Oncology. 2018;36(15_suppl):12021.
Oxnard GR, Klein EA, Seiden MV, Hubbell E, Venn O, Jamshidi A, et al. LBA77 - Simultaneous multi-cancer detection and tissue of origin (TOO) localization using targeted bisulfite sequencing of plasma cell-free DNA (cfDNA). Ann Oncol. 2019;30: v912.
Chiu BC, Zhang Z, Derman BA, Karpus J, Luo L, Zhang S, et al. Genome-wide profiling of 5-hydroxymethylcytosines in circulating cell-free DNA reveals population-specific pathways in the development of multiple myeloma. J Hematol Oncol. 2022;15(1):106.
Chiu BC, Zhang Z, Derman BA, Karpus J, Langerman S, Zeng C, et al. 5-Hydroxymethylcytosines in circulating cell-free DNA and overall survival in patients with multiple myeloma. Journal of Clinical Oncology. 2021;39(15_suppl):8032.
Yasui H, Kobayashi M, Sato K, Kondoh K, Ishida T, Kaito Y, et al. Circulating cell-free DNA in the peripheral blood plasma of patients is an informative biomarker for multiple myeloma relapse. Int J Clin Oncol. 2021;26(11):2142–50.
Mills JR, Barnidge DR, Dispenzieri A, Murray DL. High sensitivity blood-based M-protein detection in sCR patients with multiple myeloma. Blood cancer journal. 2017;7(8):e590-e.
Khier S, Lohan L. Kinetics of circulating cell-free DNA for biomedical applications: critical appraisal of the literature. Future Sci OA. 2018;4(4):Fso295.
Dutta AK, Hewett DR, Fink JL, Grady JP, Zannettino ACW. Cutting edge genomics reveal new insights into tumour development, disease progression and therapeutic impacts in multiple myeloma. Br J Haematol. 2017;178(2):196–208.
Kurtz DM, Soo J, Co Ting Keh L, Alig S, Chabon JJ, Sworder BJ, et al. Enhanced detection of minimal residual disease by targeted sequencing of phased variants in circulating tumor DNA. Nat Biotechnol. 2021;39(12):1537–47.
Acknowledgements
Not applicable.
Funding
This work was supported by National Natural Science Foundation of China (81800201, 81800202 and 81872322), Zhejiang Key Research and Development Project (2020C03014).
Author information
Authors and Affiliations
Contributions
LSC had the idea for the manuscript, performed the literature search, drafted the article, ZEF gave suggestions for this manuscript, CZ gave suggestion for the manuscript, and critically revised the work. The author(s) read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Li, S., Zhang, E. & Cai, Z. Liquid biopsy by analysis of circulating myeloma cells and cell-free nucleic acids: a novel noninvasive approach of disease evaluation in multiple myeloma. Biomark Res 11, 27 (2023). https://doi.org/10.1186/s40364-023-00469-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40364-023-00469-6