
Abstract
Chemotherapy is one of the most important treatments for cancer therapy. However, chemotherapy resistance is a big challenge in cancer treatment. Due to chemotherapy resistance, drugs become less effective or no longer effective at all. In recent years, long non-coding RNA metastasis-associated lung adenocarcinoma transcript 1 (MALAT1) has been found to be associated with the development of chemotherapy resistance, suggesting that MALAT1 may be an important target to overcome chemotherapy resistance. In this review, we introduced the main mechanisms of chemotherapy resistance associated with MALAT1, which may provide new approaches for cancer treatment.
Similar content being viewed by others

Introduction
Over the past few decades, chemotherapy, as a supplement to surgical treatment, has become the treatment of choice for patients with advanced cancer, reducing tumor residue and preventing tumor recurrence. Numerous studies have shown that combined chemotherapy can significantly improve the prognosis of tumor patients [1, 2]. According to its cytotoxic mechanism, chemotherapy can be divided into the following six categories: (1) alkylation agents, such as cyclophosphamide; (2) anti-metabolites, such as 5-fluorouracil (5-FU) and methotrexate, which can inhibit DNA synthesis; (3) antibiotics, such as doxorubicin and mitomycin C; (4) DNA topoisomerase inhibitors, such as etoposide, which can prohibit transcription and replication; (5) mitosis inhibitors, such as paclitaxel and vincristine (VCR); (6) platinum-based drugs, such as cisplatin (DDP) and oxaliplatin (OXA), which can induce DNA-platinum adducts to block DNA repair [3,4,5,6]. Chemotherapy affects the cell cycle by inducing DNA breakdown and blocking DNA repair, which in turn leads to tumor cell death. Although chemotherapy is effective, intrinsic and acquired chemotherapy resistance remains a huge challenge in cancer therapy.
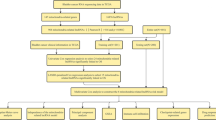
Chemotherapy resistance is a condition in which a disease becomes resistant to chemotherapy drugs. Once chemotherapeutic resistance occurs, the drug becomes less effective or no longer effective at all. For example, the recurrence rate of ovarian cancer (OC) increased to 70% due to DDP resistance, resulting in a 5-year survival rate of less than 50% [7]. The mechanism of chemotherapy resistance in cancer can be explained as follows: (1) increasing the ability of DNA damage repair; (2) influencing drug transport and metabolism, and affecting drug kinetics; (3) evading cell cycle checkpoint; (4) inhibiting cells apoptosis, and protecting cells from death; (5) promoting epithelial-mesenchymal transition (EMT); (6) altering the autophagy system of tumor cells; (7) modulating properties of cancer stem cells (Fig. 1). However, the exact mechanism remains to be studied.

Molecular mechanisms of MALAT1 in chemotherapy resistance. Molecular mechanisms of (A) DNA repair pathway, (B) drug efflux pump regulation, (C) cell cycle regulation, (D) apoptosis regulation, (E) EMT promoting, (F) autophagy regulation, and (G) stemness
Previous studies have focused on the mechanism of protein-coding genes in cancer chemotherapy resistance [8, 9]. However, protein-coding genes account for only about 2% of the human genome and most of the rest are untranslated genes, which has attracted wide attention in recent years. The transcription products of untranslated genes are called non-coding RNAs (ncRNAs), and long ncRNAs (lncRNAs) are one of them. LncRNAs are greater than 200 nucleotides in length and are initially thought to be transcriptional noise. However, emerging evidence suggests that lncRNAs play important roles in chemotherapy resistance of cancer [10,11,12,13].
MALAT1 was first identified in non-small cell lung cancer (NSCLC) patients and was upregulated in tumors with a high metastatic tendency [14]. MALAT1 gene is encoded on human chromosome 11q13.1, which has a high evolutionary conservation [15]. MALAT1 is approximately 8.7 knt and is transcribed by RNA polymerase II (Pol II) (Fig. 2) [15]. Abnormal expression of MALAT1 has been reported to be associated with the occurrence, progression, metastasis and chemotherapy resistance of cancers [16,17,18,19]. MALAT1 has been shown to be associated with various human diseases, including neoplastic and non-neoplastic diseases, such as oral squamous cell carcinoma, psoriasis, recurrent miscarriage, major adverse cardiac and cervical events [20,21,22,23,24]. Moreover, MALAT1 has been found to be a potential therapeutic target for cancer. For example, a recent study revealed that targeting the MALAT1/PARP1 (poly ADP-ribose polymerase 1) / LIG3 (DNA Ligase 3) complex resulted in DNA damage and apoptosis in multiple myeloma [25]. In addition, it has been proved that targeting MALAT1 also played a significant role in the treatment of gynecological cancer [26]. Previous studies have shown that MALAT1 affected cancer through multiple mechanisms. Therefore, we suspect that MALAT1 also plays a crucial role in chemotherapy resistance.

Schematic location of MALAT1. MALAT1 is located at 11q13.1, between LINC02736 and SNRPGP19. Its transcription products include MALAT1, mascRNA, and TALAM1 (the natural antisense RNA of MALAT1)
A large number of studies have found that there exists a high correlation between MALAT1 and chemotherapy resistance. However, there are few reviews on MALAT1 and chemotherapy resistance. Therefore, it is necessary to investigate the mechanism of MALAT1 in chemotherapy resistance to explore new tumor therapeutic targets.
In this review, we discussed the functions and mechanisms of MALAT1 in cancer chemotherapy resistance.
Biogenesis and functions of MALAT1
Biogenesis of MALAT1
MALAT1, a ubiquitously expressed gene, is located on human chromosome 11q13 and mouse chromosome 19qA [27, 28]. Furthermore, the expression level of MALAT1 is quite high, even comparable to several protein-coding genes such as GADPH [28]. MALAT1 is similar to mRNA that encodes the protein and is synthesized by RNA Pol II. The transcript of MALAT1 is about 7 kb in human and 6.7 kb in mice [29, 30]. MALAT1 lacks a poly(A) tail at the 3' end, which differs from the typical cleavage and polyadenylation mechanism [31]. Instead, ribonuclease (RNase) P cuts the original transcript of MALAT1 into a mature 7 kb transcript, and into a much smaller transcription fragment at the 3’ end [29,30,31,32]. The RNase P cleaves MALAT1 at nt7518 to produce the 5' end of small RNA and the 3' end of mature MALAT1. The 3' end of mature MALAT1 is a highly conserved triple helix structure, distinct from the conventional poly (A) tail. It is composed of one A-rich tract and two U-rich motifs encoded by the genome [33]. Due to the unique triple helix structure at its 3’ end, MALAT1 has high stability and is not easy to cut [34]. Moreover, the natural antisense transcript TALAM1 also promotes the stability of MALAT1 through a feedforward positive regulatory loop [35]. Unlike other Pol II-produced RNAs, which are transported to the cytoplasm for further processing immediately after transcription, mature MALAT1 transcripts are enriched in nuclear speckles in human and mice [30, 36]. The location of MALAT1 suggests that MALAT1 is involved in physiological and pathological processes [37, 38]. However, knockdown of MALAT1 in mice does not cause phenotypic changes [34]. The possible reason is that MALAT1 has no obvious effect under normal conditions. Short fragments are cleaved and processed by RNase Z and CCA-adding enzymes to form a 61-nt-lncRNA, called MALAT1-associated small cytoplasmic RNA (mascRNA), which is then folded into a tRNA-like cloverleaf structure and transported into the cytoplasm (Fig. 3) [31]. However, the functions and effects of mascRNA remain to be further studied.

Biogenesis of MALAT1. The original MALAT1 is transcribed by RNA Pol II, and then cleaved by RNase P to form mature MALAT1 and a smaller RNA. The mature MALAT1 localizes to the nuclear speckles, while the smaller RNA is further processed into mascRNA by RNase Z and CCA-adding enzymes and transported into the cytoplasm
Biological functions of MALAT1
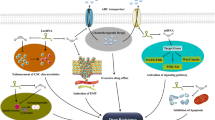
Collecting studies have found that MALAT1 can regulate biological function through intermolecular interactions. (1) Regulating gene transcription. In multiple myeloma (MM), the transcription factor Sp1 can be recruited by MALAT1 to promote the secretion of TGF-β by binding to latent transforming growth factor-β binding protein-3 [39]. (2) Regulating RNA splicing. MALAT1 can interact with the splicing factors of serine- and arginine-rich splicing factor (SRSF) 1, SRSF2, SRSF3 and other SR proteins, affect the distribution of splicing factors in the nuclear speckle domains, and regulate the alternative splicing of pre-mRNAs [40]. (3) Regulating protein activity. MALAT1, as a splicing factor proline-and glutamine-rich (SFPQ) bound competitor, will accelerate the dissociation of PTBP2 from the SFPQ/PTBP2 complex, enhance the function of PTBP2, and promote the proliferation and migration of tumor cells [41]. (4) Regulating epigenetic change. MALAT1 can recruit the suppressor of variegation 3–9 homolog 1 to MyoD-binding loci and cause the trimethylation of histone 3 lysine 9, which suppresses the transcriptional activity of MyoD [42]. (5) Regulating the nuclear and cytoplasmic transport of proteins. During cell division, MALAT1 alters the transport of the nucleus to the cytoplasm by binding to an abundant nuclear factor heterogeneous nuclear ribonucleoprotein C protein [43]. (6) Acting as a competitive endogenous RNA (ceRNA). Collecting studies have found that MALAT1 alters a series of life activities by acting as a ceRNA [44,45,46]. MALAT1 competitively sequestered miR-23b-3p and attenuated the inhibitory effect of miR-23b-3p on ATG12, thereby increasing the expression of ATG12 and promoting autophagy associated chemotherapy resistance of gastric cancer (GC) cells [44]. Moreover, MALAT1 can act as a ceRNA to inhibit miR-181c-5p, leading to berberine mediated inhibition of HMGB1 in poststroke inflammation [45]. MALAT1 regulated mTOR-mediated Tau hyperphosphorylation by acting as a ceRNA sponge of miR-144 in hippocampal cells [46]. Because MALAT1 extensively affects life activities and cell phenotypes through the above multiple pathways, it is possible that MALAT1 affects tumor chemotherapy resistance through the above mechanisms.
Mechanisms mediating chemotherapy resistance related to MALAT1 in cancers
MALAT1 associated with DNA damage repair pathway
To maintain genomic stability, normal cells can repair DNA damage caused by internal oxidative stress, external radiation, and cytotoxic drugs. If normal processes are blocked, the genome of cells may become unstable, leading to the development of tumors. There are two main molecular pathways to repair damaged DNA: homologous recombination (HR) pathway and non-homologous end junction (NHEJ) pathway [47, 48]. However, some cancer cells evade chemotherapy by enhancing their DNA repair abilities, leading to chemotherapy resistance [49].
Recent studies have shown that MALAT1 affected many factors involved in DNA repair (Fig. 1A, Table 1). In MM, MALAT1 enhanced the NHEJ pathway by binding to PARP1 and LIG3, two key molecules in this pathway. Researchers found that anti-MALAT1 induced DNA damage and reduced drug resistance to bortezomib, melphalan, and doxorubicin, suggesting that MALAT1 may induce the development of resistance by enhancing DNA damage repair [25]. MALAT1 is not only involved in the NHEJ pathway, but also the HR pathway. In NSCLC, researchers found that targeting MALAT1 could induce DNA damage even when the NHEJ pathway was blocked. MALAT1 sponged miR-146a and miR-216b to protect BRCA1, functioning as a ceRNA. As BRCA1 was a key upregulation factor in the HR pathway, upregulation of BRCA1 expression led to enhancement of the HR pathway. Finally, MALAT1 overexpression resulted in DDP resistance in NSCLC cells [50].
In all, abnormally high expression of MALAT1 enhances the DNA repair ability of cancer cells through the above pathways, leading to drug resistance.
MALAT1 involved in drug efflux pump function
Reducing intracellular drug concentration is a major and direct way to obtain chemotherapy resistance in tumor cells, which is associated with drug efflux system, such as ATP-binding cassette (ABC) membrane transporter proteins [19, 20]. ABC proteins can affect pharmacokinetics by altering the transport of various drugs. Therefore, the overexpression of ABC protein is the main cause of multidrug resistance. Multidrug resistant protein 1 (MDR1), multidrug resistance like protein 1 (ABCC1 or MRP1), multidrug resistance associated protein 2, and ABC subfamily G2 are relatively common and well-known ABC proteins [51,52,53].
A large number of studies have shown that MALAT1 altered drug distribution by regulating the ABC proteins (Fig. 1B, Table 2). In glioblastoma, si-MALAT1 downregulated the expression of ZEB1, MDR1, MRP5 and LRP1, enhancing the sensitivity to Temozolomide (TMZ) [54, 55]. Conversely, overexpression of MALAT1 led to resistance to TMZ. However, the exact mechanism by which MALAT1 affects the expression of these proteins is still being investigated. It was found that overexpression of MALAT1 upregulated the expression of MRP1 and MDR1 by activating STAT3, thus promoting DDP resistance in NSCLC [56]. Another study pointed out that MALAT1 upregulated ABCC1 expression by activating Notch1, enhancing DDP resistance in OC cells [57]. In addition, MALAT1 was found to be involved in ABCA3 regulation by downregulating miR-335-3p in the multidrug resistant group of childhood acute lymphoblastic leukemia, which may lead to chemotherapy resistance [58]. In colorectal cancer (CRC), suppression of MALAT1 restrained cell progression by downregulation of the expression of ABC, MDR1, MRP1, and breast cancer drug resistant proteins through targeting miR-20b-5p, resulting in drug resistance of cancer cells to 5-FU [59]. In oral squamous cell carcinoma (OSCC), MALAT1 developed the DDP resistance of OSCC by upregulating P-glycoprotein expression [60].
Through these pathways, MALAT1 enhances the function of drug efflux pump in tumor cells, reduces intracellular drug concentration, and leads to drug resistance of tumors.
MALAT1 involved in cell cycle regulation
To ensure the quality and integrity of DNA replication, there is a mechanism in the cell cycle to solve the abnormal events that occur during DNA replication, which is known as the cell cycle checkpoint. The cell cycle checkpoint system consists of many molecules, including upstream protein kinases such as Ataxia telangiectasia-mutated and ataxia telangiectasis and Rad3 related, and downstream checkpoint proteins such as CHK1, CHK2, BRCA, and p53 [61, 62]. Cancer cell resistance to chemotherapy may be due to defects in factors associated with cell cycle checkpoints [63].
There is growing evidence that MALAT1 has a regulatory role in the cell cycle that reduces the sensitivity to chemotherapy (Fig. 1C, Table 3). For instance, it was found that MALAT1 promoted TMZ resistance in glioblastoma multiforme (GBM) cells. MALAT1 promoting the thymidylate synthase by downregulating miR-203, resulting in TMZ resistance, which was demonstrated by an increased percentage of cell population in G0/G1 phase [64]. Moreover, suppression of MALAT1 downregulated the expression of Cyclin D1 and CDK, and upregulated the expression of p53, p21, and p27, resulting in an increase of sensitivity of hepatocellular carcinoma (HCC) cells to 5-FU [65]. In addition, MALAT1 silencing greatly blocked the cell cycle of chronic myeloid leukemia (CML) cells and inhibited cell proliferation by releasing the sponging effect on miR-328, which attenuated the chemotherapy resistance of CML cells to imatinib [66]. Another study pointed out that MALAT1 was overexpressed in head and neck squamous cell carcinoma (HNSCC) cells. Downregulation of MALAT1 resulted in cell cycle arrest in G(2)/M phase and enhanced sensitivity of HNSCC cells to DDP [67].
In conclusion, the abnormal increase of MALAT1 can lead to the dysregulation of cell cycle, and then lead to the generation of drug resistance.
Apoptosis-related MALAT1 regulating chemosensitivity
Apoptosis is a complex programmed cell death process that can be triggered by caspase-mediated external or internal pathways, involving a variety of signaling pathways. Besides, the caspase-cascade system plays a crucial role in the induction, transduction, and amplification of intracellular apoptotic signals [68]. Moreover, caspases are also regulated by numerous molecules and pathways, influencing the apoptosis. Through the action of these regulators, apoptosis will be affected, and the cell phenotypes (such as chemotherapy resistance) will be altered accordingly [69].
Recent studies have fully revealed that MALAT1 was related to apoptosis and may lead to chemotherapy resistance (Fig. 1D, Table 4) [70]. For example, in NSCLC, polyphyllin I inhibited the expression of MALAT1, resulting in the inactivation of STAT3 signaling pathway and apoptosis in gefitinib-resistant cancer cells [71]. Another research group reported that inhibition of MALAT1 had also been found to alter apoptosis through the IKKα/NF-κB pathway, thereby boosting the sensitivity of cancer cells to 5-FU in HCC [65]. In cervical cancer, overexpression of MALAT1 was found to upregulate the expression of p-PI3K, p-AKT, and BRWD1, promoting DDP resistance [72]. Similarly, in GC cells, overexpression of MALAT1 can upregulate p-PI3K, p-AKT and p-STAT3, which also contributed to DDP resistance by altering apoptosis [73].
In general, the high expression of MALAT1 regulates apoptosis-related genes and molecules, affects cancer cell apoptosis, and triggers cancer drug resistance.
MALAT1 associated with EMT-related chemosensitivity
EMT is a process that alters the transform of polarized epithelial cells to motile mesenchymal cells characterized by the loss of E-cadherin through the activation of one or several factors such as SNAIL, SLUG, ZEBs, and TWIST [74, 75]. During EMT, epithelial cells lost their epithelial phenotype such as cell polarity and connection to the basement membrane and acquired a mesenchymal phenotype such as higher migration and invasion, resistance to apoptosis and degradation of the extracellular matrix. EMT is therefore a mechanism for tumor metastasis. In addition, recent studies have shown that multiple factors in EMT play an important role in the development of chemotherapy resistance.
Studies have shown that MALAT1 regulated EMT, thereby promoting EMT-induced chemotherapeutic resistance (Fig. 1E, Table 5). In CRC, MALAT1 knockdown enhanced E-cadherin expression and inhibited OXA-induced EMT, which may be a promising therapeutic target for CRC patients [76]. Li et al. [54] demonstrated that EMT and MALAT1 overexpression were associated with TMZ in drug-resistant GBM cells, suggesting that MALAT1 was involved in EMT-induced chemotherapy resistance. MALAT1 regulated EMT by upregulating ZEB1, making GBM cells resistant to TMZ. In OSCC, MALAT1 expression was higher in DDP-resistant cells. Further evidence suggested that MALAT1 was involved in EMT process through upregulation of P-gp and activation of PI3K/AKT/m-TOR signaling pathway, leading to DDP resistance [60]. In HCC, MALAT1 interacted with miR-140-5p to enhance the expression of Aurora-A, leading to EMT and the formation of chemotherapy resistance to sorafenib [77].
Therefore, MALAT1 can also induce drug resistance of cancer cells by promoting the EMT.
Autophagy-related MALAT1 associated with chemosensitivity
Autophagy is a basic process of degradation and reuse of cell components that is highly conserved in all eukaryotes. Autophagy is not only a "recycling" biological function, but also affects the response to infection, embryonic development and cellular variation, and directly affects the occurrence and development of tumors, as well as drug response and drug resistance [78,79,80]. The primary purpose of autophagy is to stabilize the intracellular environment. In normal cells, autophagy can reduce the risk of cancer. Paradoxically, autophagy is an important cause of chemotherapy failure in cancer cells [81].
In recent years, more and more studies have found that MALAT1 was strongly related to the regulation of autophagy in cancer cells (Fig. 1F, Table 6). In HCC, MALAT1 played a key role in the development of chemotherapy resistance by regulating autophagy. HIF-2α upregulated MALAT1 expression, which can act as a ceRNA of miR-216b to regulate autophagy, leading to 5-FU resistance [82]. Similarly, MALAT1 affected autophagy of GC cells through a variety of pathways, leading to tumor drug resistance. Hu et al. [44] revealed that MALAT1 targeted miR-23b-3p and reduced its inhibition of autophagy related gene (ATG) 12, leading to autophagy and chemotherapy resistance to 5-FU, DDP and VCR in GC. Another study pointed out that MALAT1 induced chemotherapy resistance of GC cells to DDP by inhibiting miR-30b and promoting ATG5 expression [83]. Moreover, it has been reported that propofol promotes DDP sensitivity by inhibiting autophagy in GC through MALAT1/miR-30e/ATG5 axis, suggesting that MALAT1 induced autophagy-associated chemotherapy resistance of GC cells to DDP [84].
In summary, MALAT1 overexpression affects the autophagy of cancer cells and leads to drug resistance.
MALAT1 involved in the stemness-related chemosensitivity
Cancer stemness is the phenotype like normal stem cells such as plasticity and self-renewal ability, and the cancer cells with these properties are known as cancer stem cells, which is recognized to be one of the important causes of chemotherapy resistance [85, 86].
Recent studies have shown that MALAT1 was involved in the cancer stemness (Fig. 1G, Table 7). In GC cells, MALAT1 acted as a stabilizer of SOX2 mRNA by binding to it directly, resulting in stemness and chemotherapy resistance to DDP [87]. In esophageal squamous cell carcinoma, MALAT1 directly bound to Yes-associated protein (YAP) and enhanced the transcription and expression of YAP, developing the stemness and resistance to DDP [88]. In OC, MALAT1 acted as a co-activator of YAP, inhibiting its translocation, promoting the expression of YAP and thereby enhancing the effect of YAP, which contributes to the stemness and chemotherapy resistance in OC to DDP [89].
As described above, MALAT1 can also contribute to cancer resistance to chemotherapeutic drugs by enhancing the stemness of cancer cells.
Conclusion and future perspectives
With the use of chemotherapy, the prognosis of cancer patients has improved dramatically, which is a huge advancement in cancer treatment. However, the development of chemotherapy resistance is becoming an important reason for chemotherapy failure. At present, lncRNAs have been proved to be important regulatory factors involved in numerous life activities. LncRNAs are specific in many respects and are equally expressed in terms of chemotherapy resistance. MALAT1, which has been studied the most, plays an important regulatory role in tumor development. MALAT1 has been a molecule of interest since its discovery as a predictive biomarker for lung cancer metastasis [90]. Current studies show that MALAT1 is a potential target not only for cancer therapy, but also for overcoming cancer resistance. Therefore, targeting MALAT1 treatment may not only be effective against tumor therapy itself, but also make chemotherapy more effective, which may contribute to the complex therapy of cancers and improve the prognosis of cancers. As an important adjuvant to chemotherapy, immunotherapy has developed rapidly in recent years. Chemotherapy combined with immunotherapy has been widely used, and MALAT1 has been found to play an important regulatory role in immunotherapy. For instance, MALAT1 inhibited the immune response to cancer by enhancing immune escape and immunosuppressive effects, potentially leading to failure of immunotherapy and poor prognosis [91, 92]. Therefore, it is urgent to explore the mechanism and influence of MALAT1 in immunotherapy, which may provide new targets and approaches for cancer therapy. In addition, some lncRNAs can mediate chemoresistance through immune pathways. For example, lncRNA PCAT-1 can induce KRAS-related chemoresistance through immunosuppression [93]. Although there is no conclusive evidence, based on the widespread expression of MALAT1 in the immune system and its multiple effects, we speculate that MALAT1 may also influence chemotherapy resistance by affecting the immune system. However, research on the effect of MALAT1 in the chemotherapy resistance and immunotherapy of cancers is still at the nascent stage. Substantial basic research and clinical trials are needed before these molecular approaches can be applied to the clinic. Due to the extensive regulatory roles of MALAT1, it will provide new targets for cancer prevention and treatment in the future.
Availability of data and materials
Not applicable.
Abbreviations
- MALAT1:
-
Metastasis-associated lung adenocarcinoma transcript 1
- 5-FU:
-
5-Fluorouracil
- VCR:
-
Vincristine
- DDP:
-
Cisplatin
- OXA:
-
Oxaliplatin
- OC:
-
Ovarian cancer
- EMT:
-
Epithelial-mesenchymal transition
- NcRNAs:
-
Non-coding RNAs
- LncRNAs:
-
Long non-coding RNAs
- NSCLC:
-
Non-small cell lung cancer
- Pol II:
-
Polymerase II
- RNase:
-
Ribonuclease
- PARP1:
-
Poly ADP-ribose polymerase 1
- LIG3:
-
DNA Ligase 3
- HMGB1:
-
High-mobility group box 1
- MascRNA:
-
MALAT1-associated small cytoplasmic RNA
- MM:
-
Multiple myeloma
- SRSF:
-
Serine- and arginine-rich splicing factor
- SFPQ:
-
Splicing factor proline-and glutamine-rich
- HR:
-
Homologous recombination
- NHEJ:
-
Non-homologous end junction
- CeRNA:
-
Competing endogenous RNA
- ABC:
-
ATP-binding cassette
- MDR1:
-
Multidrug resistant protein 1
- ABCC1 or MRP1:
-
Multidrug resistance like protein 1
- TMZ:
-
Temozolomide
- CRC:
-
Colorectal cancer
- OSCC:
-
Oral squamous cell carcinoma
- GBM:
-
Glioblastoma multiforme
- HCC:
-
Hepatocellular carcinoma
- CML:
-
Chronic myeloid leukemia
- HNSCC:
-
Head and neck squamous cell carcinoma
- GC:
-
Gastric cancer
- ATG:
-
Autophagy related gene
- YAP:
-
Yes-associated protein
References
Ikeda M, et al. Chemotherapy for hepatocellular carcinoma: current status and future perspectives. Jpn J Clin Oncol. 2018;48(2):103–14.
Aigner J, et al. The role of neoadjuvant chemotherapy in the management of primary breast cancer. Minerva Ginecol. 2011;63(3):261–74.
Falzone L, Salomone S, Libra M. Evolution of cancer pharmacological treatments at the turn of the third millennium. Front Pharmacol. 2018;9:1300.
Housman G, et al. Drug resistance in cancer: an overview. Cancers (Basel). 2014;6(3):1769–92.
Shewach DS, Kuchta RD. Introduction to cancer chemotherapeutics. Chem Rev. 2009;109(7):2859–61.
Swain SM. Chemotherapy: updates and new perspectives. Oncologist. 2011;16(Suppl 1):30–9.
Colombo N, Lorusso D, Scollo P. Impact of recurrence of ovarian cancer on quality of life and outlook for the future. Int J Gynecol Cancer. 2017;27(6):1134–40.
Yang D, et al. Association of BRCA1 and BRCA2 mutations with survival, chemotherapy sensitivity, and gene mutator phenotype in patients with ovarian cancer. JAMA. 2011;306(14):1557–65.
Sun F, et al. Causative role of PDLIM2 epigenetic repression in lung cancer and therapeutic resistance. Nat Commun. 2019;10(1):5324.
An J, Lv W, Zhang Y. LncRNA NEAT1 contributes to paclitaxel resistance of ovarian cancer cells by regulating ZEB1 expression via miR-194. Onco Targets Ther. 2017;10:5377–90.
Jia J, et al. The contrary functions of lncRNA HOTAIR/miR-17-5p/PTEN axis and Shenqifuzheng injection on chemosensitivity of gastric cancer cells. J Cell Mol Med. 2019;23(1):656–69.
Zhang W, et al. Long non-coding RNA LINC00160 functions as a decoy of microRNA-132 to mediate autophagy and drug resistance in hepatocellular carcinoma via inhibition of PIK3R3. Cancer Lett. 2020;478:22–33.
Cai Q, et al. Long non-coding RNA GBCDRlnc1 induces chemoresistance of gallbladder cancer cells by activating autophagy. Mol Cancer. 2019;18(1):82.
Ji P, et al. MALAT-1, a novel noncoding RNA, and thymosin beta4 predict metastasis and survival in early-stage non-small cell lung cancer. Oncogene. 2003;22(39):8031–41.
Wilusz JE, Freier SM, Spector DL. 3′ End processing of a long nuclear-Retained Noncoding RNA Yields a tRNA-like Cytoplasmic RNA. Cell. 2008;135(5):919–32.
Xue D, et al. Long noncoding RNA MALAT1 enhances the docetaxel resistance of prostate cancer cells via miR-145-5p-mediated regulation of AKAP12. J Cell Mol Med. 2018;22(6):3223–37.
Chen Q, et al. Plasma long non-coding RNA MALAT1 is associated with distant metastasis in patients with epithelial ovarian cancer. Oncol Lett. 2016;12(2):1361–6.
Li Z, et al. Application of Long Noncoding RNAs in Osteosarcoma: Biomarkers and Therapeutic Targets. Cell Physiol Biochem. 2017;42(4):1407–19.
Yang C, et al. MALAT1 Promotes Tumorigenesis and Increases Cellular Sensitivity to Herceptin in HER2-positive Breast Cancer. Curr Cancer Drug Targets. 2021;21:860–9.
Zhang T, et al. Long noncoding RNA MALAT1 polymorphism predicts MACCEs in patients with myocardial infarction. BMC Cardiovasc Disord. 2022;22(1):152.
Hakimi P, et al. Association of seven fundamental genetic polymorphisms in long noncoding RNA MALAT1, SOX2OT and H19 with recurrent miscarriage in Turkish-Azeri Iranian population. Human Gene. 2022;33:201063.
Ghafouri-Fard S, et al. Association analysis of MALAT1 polymorphisms and risk of psoriasis among Iranian patients. Int J Immunogenet. 2022;49(2):83–7.
Ni W, et al. Meta-analysis of the association between MALAT1 rs619586 A>G polymorphism and cancer risk. J Int Med Res. 2020;48(7):300060520941969.
Ding YF, et al. Combined Impacts of Genetic Variants of Long Non-Coding RNA MALAT1 and the Environmental Carcinogen on the Susceptibility to and Progression of Oral Squamous Cell Carcinoma. Front Oncol. 2021;11:684941.
Hu Y, et al. Targeting the MALAT1/PARP1/LIG3 complex induces DNA damage and apoptosis in multiple myeloma. Leukemia. 2018;32(10):2250–62.
Qiao FH, Tu M, Liu HY. Role of MALAT1 in gynecological cancers: Pathologic and therapeutic aspects. Oncol Lett. 2021;21(4):333.
Wilusz JE. Long noncoding RNAs: Re-writing dogmas of RNA processing and stability. Biochim Biophys Acta. 2016;1859(1):128–38.
Zhang B, et al. The lncRNA Malat1 is dispensable for mouse development but its transcription plays a cis-regulatory role in the adult. Cell Rep. 2012;2(1):111–23.
Bernard D, et al. A long nuclear-retained non-coding RNA regulates synaptogenesis by modulating gene expression. Embo j. 2010;29(18):3082–93.
Hutchinson JN, et al. A screen for nuclear transcripts identifies two linked noncoding RNAs associated with SC35 splicing domains. BMC Genomics. 2007;8:39.
Torabi SF, DeGregorio SJ, Steitz JA. tRNA-like leader-trailer interaction promotes 3’-end maturation of MALAT1. RNA. 2021;27(10):1140–7.
Clemson CM, et al. An architectural role for a nuclear noncoding RNA: NEAT1 RNA is essential for the structure of paraspeckles. Mol Cell. 2009;33(6):717–26.
Wu Y, et al. Long Noncoding RNA MALAT1: Insights into its Biogenesis and Implications in Human Disease. Curr Pharm Des. 2015;21(34):5017–28.
Eißmann M, et al. Loss of the abundant nuclear non-coding RNA MALAT1 is compatible with life and development. RNA Biol. 2012;9(8):1076–87.
Zong X, et al. Natural antisense RNA promotes 3’ end processing and maturation of MALAT1 lncRNA. Nucleic Acids Res. 2016;44(6):2898–908.
Sone M, et al. The mRNA-like noncoding RNA Gomafu constitutes a novel nuclear domain in a subset of neurons. J Cell Sci. 2007;120(Pt 15):2498–506.
Liu JY, et al. Pathogenic role of lncRNA-MALAT1 in endothelial cell dysfunction in diabetes mellitus. Cell Death Dis. 2014;5(10): e1506.
Michalik KM, et al. Long noncoding RNA MALAT1 regulates endothelial cell function and vessel growth. Circ Res. 2014;114(9):1389–97.
Li B, et al. Activation of LTBP3 gene by a long noncoding RNA (lncRNA) MALAT1 transcript in mesenchymal stem cells from multiple myeloma. J Biol Chem. 2014;289(42):29365–75.
Tripathi V, et al. The nuclear-retained noncoding RNA MALAT1 regulates alternative splicing by modulating SR splicing factor phosphorylation. Mol Cell. 2010;39(6):925–38.
Ji Q, et al. Long non-coding RNA MALAT1 promotes tumour growth and metastasis in colorectal cancer through binding to SFPQ and releasing oncogene PTBP2 from SFPQ/PTBP2 complex. Br J Cancer. 2014;111(4):736–48.
Chen X, et al. Malat1 regulates myogenic differentiation and muscle regeneration through modulating MyoD transcriptional activity. Cell Discov. 2017;3:17002.
Yang F, et al. MALAT-1 interacts with hnRNP C in cell cycle regulation. FEBS Lett. 2013;587(19):3175–81.
YiRen H, et al. Long noncoding RNA MALAT1 regulates autophagy associated chemoresistance via miR-23b-3p sequestration in gastric cancer. Mol Cancer. 2017;16(1):174.
Cao DW, et al. The lncRNA Malat1 functions as a ceRNA to contribute to berberine-mediated inhibition of HMGB1 by sponging miR-181c-5p in poststroke inflammation. Acta Pharmacol Sin. 2020;41(1):22–33.
Lu C, et al. MALAT1 Regulated mTOR-Mediated Tau Hyperphosphorylation by Acting as a ceRNA of miR144 in Hippocampus Cells Exposed to High Glucose. Clin Interv Aging. 2021;16:1185–91.
Li X, Heyer WD. Homologous recombination in DNA repair and DNA damage tolerance. Cell Res. 2008;18(1):99–113.
Chang HHY, et al. Non-homologous DNA end joining and alternative pathways to double-strand break repair. Nat Rev Mol Cell Biol. 2017;18(8):495–506.
Hosoya N, Miyagawa K. Targeting DNA damage response in cancer therapy. Cancer Sci. 2014;105(4):370–88.
Huang J, et al. Targeting MALAT1 induces DNA damage and sensitize non-small cell lung cancer cells to cisplatin by repressing BRCA1. Cancer Chemother Pharmacol. 2020;86(5):663–72.
Wang J, et al. Overexpression of ABCB1 Transporter Confers Resistance to mTOR Inhibitor WYE-354 in Cancer Cells. Int J Mol Sci. 2020;21(4):1387.
Lin H, et al. KDM5c inhibits multidrug resistance of colon cancer cell line by down-regulating ABCC1. Biomed Pharmacother. 2018;107:1205–9.
Hsu HH, et al. Oxaliplatin resistance in colorectal cancer cells is mediated via activation of ABCG2 to alleviate ER stress induced apoptosis. J Cell Physiol. 2018;233(7):5458–67.
Li H, et al. Long Non-Coding RNA MALAT1 Decreases the Sensitivity of Resistant Glioblastoma Cell Lines to Temozolomide. Cell Physiol Biochem. 2017;42(3):1192–201.
Kim SS, et al. Targeted nanocomplex carrying siRNA against MALAT1 sensitizes glioblastoma to temozolomide. Nucleic Acids Res. 2018;46(3):1424–40.
Fang Z, et al. LncRNA-MALAT1 contributes to the cisplatin-resistance of lung cancer by upregulating MRP1 and MDR1 via STAT3 activation. Biomed Pharmacother. 2018;101:536–42.
Bai L, et al. Knockdown of MALAT1 enhances chemosensitivity of ovarian cancer cells to cisplatin through inhibiting the Notch1 signaling pathway. Exp Cell Res. 2018;366(2):161–71.
Pouyanrad S, Rahgozar S, Ghodousi ES. Dysregulation of miR-335-3p, targeted by NEAT1 and MALAT1 long non-coding RNAs, is associated with poor prognosis in childhood acute lymphoblastic leukemia. Gene. 2019;692:35–43.
Tang D, et al. Inhibition of MALAT1 reduces tumor growth and metastasis and promotes drug sensitivity in colorectal cancer. Cell Signal. 2019;57:21–8.
Wang R, Lu X, Yu R. lncRNA MALAT1 Promotes EMT Process and Cisplatin Resistance of Oral Squamous Cell Carcinoma via PI3K/AKT/m-TOR Signal Pathway. Onco Targets Ther. 2020;13:4049–61.
Bartek J, Lukas J. Chk1 and Chk2 kinases in checkpoint control and cancer. Cancer Cell. 2003;3(5):421–9.
Abraham RT. Cell cycle checkpoint signaling through the ATM and ATR kinases. Genes Dev. 2001;15(17):2177–96.
Smith J, et al. The ATM-Chk2 and ATR-Chk1 pathways in DNA damage signaling and cancer. Adv Cancer Res. 2010;108:73–112.
Chen W, et al. MALAT1 is a prognostic factor in glioblastoma multiforme and induces chemoresistance to temozolomide through suppressing miR-203 and promoting thymidylate synthase expression. Oncotarget. 2017;8(14):22783–99.
Ji DG, et al. Inhibition of MALAT1 sensitizes liver cancer cells to 5-flurouracil by regulating apoptosis through IKKα/NF-κB pathway. Biochem Biophys Res Commun. 2018;501(1):33–40.
Wen F, et al. LncRNA MALAT1 promotes cell proliferation and imatinib resistance by sponging miR-328 in chronic myelogenous leukemia. Biochem Biophys Res Commun. 2018;507(1–4):1–8.
Kangboonruang K, et al. MALAT1 Decreases the Sensitivity of Head and Neck Squamous Cell Carcinoma Cells to Radiation and Cisplatin. Anticancer Res. 2020;40(5):2645–55.
Fan TJ, et al. Caspase family proteases and apoptosis. Acta Biochim Biophys Sin (Shanghai). 2005;37(11):719–27.
Tourneur L, et al. Absence or low expression of fas-associated protein with death domain in acute myeloid leukemia cells predicts resistance to chemotherapy and poor outcome. Cancer Res. 2004;64(21):8101–8.
Mohammad RM, et al. Broad targeting of resistance to apoptosis in cancer. Semin Cancer Biol. 2015;35 Suppl(0):S78-s103.
Yang Q, et al. Polyphyllin I modulates MALAT1/STAT3 signaling to induce apoptosis in gefitinib-resistant non-small cell lung cancer. Toxicol Appl Pharmacol. 2018;356:1–7.
Wang N, et al. MALAT1 promotes cisplatin resistance in cervical cancer by activating the PI3K/AKT pathway. Eur Rev Med Pharmacol Sci. 2018;22(22):7653–9.
Dai Q, Zhang T, Li C. LncRNA MALAT1 Regulates the Cell Proliferation and Cisplatin Resistance in Gastric Cancer via PI3K/AKT Pathway. Cancer Manag Res. 2020;12:1929–39.
Buehler D, et al. Expression of epithelial-mesenchymal transition regulators SNAI2 and TWIST1 in thyroid carcinomas. Mod Pathol. 2013;26(1):54–61.
Montemayor-Garcia C, et al. The role of epithelial mesenchymal transition markers in thyroid carcinoma progression. Endocr Pathol. 2013;24(4):206–12.
Li P, et al. MALAT1 Is Associated with Poor Response to Oxaliplatin-Based Chemotherapy in Colorectal Cancer Patients and Promotes Chemoresistance through EZH2. Mol Cancer Ther. 2017;16(4):739–51.
Fan L, et al. Long Noncoding RNA MALAT1 Contributes to Sorafenib Resistance by Targeting miR-140-5p/Aurora-A Signaling in Hepatocellular Carcinoma. Mol Cancer Ther. 2020;19(5):1197–209.
Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell. 2008;132(1):27–42.
Vempati RK, Malla RR. Autophagy-Induced Drug Resistance in Liver Cancer. Crit Rev Oncog. 2020;25(1):21–30.
Sridhar S, et al. Autophagy and disease: always two sides to a problem. J Pathol. 2012;226(2):255–73.
Li YJ, et al. Autophagy and multidrug resistance in cancer. Chin J Cancer. 2017;36(1):52.
Yuan P, et al. The HIF-2α-MALAT1-miR-216b axis regulates multi-drug resistance of hepatocellular carcinoma cells via modulating autophagy. Biochem Biophys Res Commun. 2016;478(3):1067–73.
Xi Z, Si J, Nan J. LncRNA MALAT1 potentiates autophagy-associated cisplatin resistance by regulating the microRNA-30b/autophagy-related gene 5 axis in gastric cancer. Int J Oncol. 2019;54(1):239–48.
Zhang YF, et al. Propofol facilitates cisplatin sensitivity via lncRNA MALAT1/miR-30e/ATG5 axis through suppressing autophagy in gastric cancer. Life Sci. 2020;244:117280.
Atashzar MR, et al. Cancer stem cells: A review from origin to therapeutic implications. J Cell Physiol. 2020;235(2):790–803.
Kim Y, et al. Cancer stem cells and their mechanism of chemo-radiation resistance. Int J Stem Cells. 2009;2(2):109–14.
Xiao Y, et al. LncRNA MALAT1 increases the stemness of gastric cancer cells via enhancing SOX2 mRNA stability. FEBS Open Bio. 2019;9(7):1212–22.
Yao Q, et al. Long noncoding RNA MALAT1 promotes the stemness of esophageal squamous cell carcinoma by enhancing YAP transcriptional activity. FEBS Open Bio. 2019;9(8):1392–402.
Wu X, et al. The Long Non-Coding RNA MALAT1 Enhances Ovarian Cancer Cell Stemness by Inhibiting YAP Translocation from Nucleus to Cytoplasm. Med Sci Monit. 2020;26:e922012.
Goyal B, et al. Diagnostic, prognostic, and therapeutic significance of long non-coding RNA MALAT1 in cancer. Biochim Biophys Acta Rev Cancer. 2021;1875(2):188502.
Hou ZH, et al. Long non-coding RNA MALAT1 promotes angiogenesis and immunosuppressive properties of HCC cells by sponging miR-140. Am J Physiol Cell Physiol. 2020;318(3):C649-c663.
Wang QM, et al. LncRNA MALAT1 promotes tumorigenesis and immune escape of diffuse large B cell lymphoma by sponging miR-195. Life Sci. 2019;231:116335.
Domvri K, et al. Exosomal lncRNA PCAT-1 promotes Kras-associated chemoresistance via immunosuppressive miR-182/miR-217 signaling and p27/CDK6 regulation. Oncotarget. 2020;11(29):2847–62.
Acknowledgements
Not applicable.
Funding
This work was supported by National Natural Science Foundation of China (Grant No. 82072835) to K Wang, Key Research and Development Joint Program of Liaoning Province (Grant No. 2020JH 2/10300139) to K Wang, Natural Science Foundation of Liaoning Province (Grant No. 2019-MS-360) to K Wang, Shenyang Science and Technology Bureau Plan Projects (Grant No. 20–205-4–076) to K Wang, 345 Talent Project of Shengjing Hospital of China Medical University to K Wang, and Outstanding Scientific Fund of Shengjing Hospital to K Wang.
Author information
Authors and Affiliations
Contributions
KW, GZ and XW conceived the review; JH, GZ, YW, and KW wrote the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Hou, J., Zhang, G., Wang, X. et al. Functions and mechanisms of lncRNA MALAT1 in cancer chemotherapy resistance. Biomark Res 11, 23 (2023). https://doi.org/10.1186/s40364-023-00467-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40364-023-00467-8