
Abstract
Hepatocellular carcinoma (HCC) is one of the most common and lethal malignant tumors in the world. Therapeutic options for advanced HCC are limited. Systemic treatment, especially with conventional cytotoxic drugs, is usually ineffective. For more than a decade, sorafenib has been the only systemic drug that has been proven to be clinically effective for treating advanced HCC. However, over the past three years, the rapid progress of molecular targeted therapies has dramatically changed the treatment landscape for advanced HCC. Immune checkpoint therapies are now being incorporated into HCC therapies, and their combination with molecular targeted therapy is emerging as a tool to enhance the immune response. In this review, we summarize the development and progress of molecular targeted agents and immunotherapies in HCC.
Similar content being viewed by others


Background
Worldwide, liver cancers are predicted to rank sixth for incident cases and be the third leading cause of cancer-related death in 2020, with approximately 905,677 new cases and 830,180 deaths each year (Fig. 1A-C) [1]. Based on annual estimates, the World Health Organization predicts that more than 1.3 million people will die from liver cancer by 2040 (Fig. 1D) [2]. HCC is the most frequently occurring tumor among all primary liver cancers, accounting for 75%-85% of cases [3]. HCC is usually diagnosed at an advanced stage, for which there remain limited effective treatment options [4]. When applying palliative treatment at an advanced stage, the median post-diagnostic survival ranged from 6 to 12 months [5]. Systemic treatments may benefit patients with advanced-stage HCC [6].

Worldwide Epidemiology of Liver Cancer in 2020. Data source: GLOBOCAN 2020 (http://gco.iarc.fr/). (A) The estimated age-standardized incidences of liver cancer worldwide in 2020. (B) Bar charts of the estimated number of incident cases worldwide. (C) Bar charts of the estimated number of deaths worldwide. (D) WHO estimated the number of deaths from liver cancer from 2020 to 2040
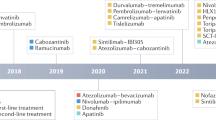
Until 2007, there were no effective treatment options for patients diagnosed with advanced-stage disease or patients who transitioned into advanced-stage disease after other treatments failed [7]. Sorafenib is the first and only systemic drug approved by the U.S. Food and Drug Administration (FDA) as standard treatment for advanced HCC between 2007 and 2016. However, rapid advances during the last 3 years have led to the approval of other molecular targeted drugs and several immune checkpoint inhibitors (ICIs), which have been added as therapeutic tools for advanced HCC (Tables 1, 2 and 3). In addition, the recent successful combination of atezolizumab and bevacizumab marks an important change in the first-line treatment of HCC (GO30140, NCT NCT02715531) [8]. To date, systemic therapy for advanced HCC includes molecular targeted therapy, immune checkpoint inhibitors, or a combination of both (Fig. 2).

Currently approved drugs for advanced HCC and timeline of pivotal clinical trials. The lines along the timeline indicate the time from the actual study start to FDA approval. The red boxes represent first-line therapies, and the green boxes represent second-line therapies
In this review, we present a summary of systemic drugs already approved for use in advanced HCC and those that have shown positive results in preclinical or clinical trials, providing a perspective on the future of advanced HCC therapies.
Agents approved by the FDA
Molecular targeted therapies
Over the last few decades, advances in molecular cell biology have greatly contributed to our knowledge of the molecular mechanisms of tumorigenesis and its progression, which in turn provides opportunities for the development of novel molecular targeted agents that inhibit molecular abnormalities as promising cancer treatments [9]. At present, molecular targeted therapy mainly includes tyrosine kinase inhibitors (TKIs) and/or monoclonal antibodies.
Sorafenib
Sorafenib is an oral TKI that targets the angiogenesis and proliferation pathways of tumors by blocking vascular endothelial growth factor receptor (VEGFR) 1–3, platelet-derived growth factor receptor (PDGFR) β-pathways, and Raf-MEK-ERK signaling, all of which participate in the pathogenesis of HCC (Fig. 4) [11, 12].
At ASCO 2007, the results of the SHARP (Sorafenib Hepatocellular Carcinoma Assessment Randomized Protocol; NCT00105443) were reported, which demonstrated the effectiveness of sorafenib against HCC. The primary endpoints were overall survival (OS) and time to symptomatic progression. Compared to the placebo group, the sorafenib treatment group had significantly prolonged OS (median OS 10.7 vs. 7.9 months, hazard ratio [HR]=0.69; 95% confidence interval [CI], 0.55–0.87; p<0.0001), while the time to symptomatic progression was not significantly different between the two groups (median time 4.1 vs. 4.9 months, HR=1.08, p=0.77). Statistically significant differences were found for secondary endpoints: the median time to radiologic progression improved by 3 months (5.5 vs. 2.8 months, P<0.001), and the sorafenib group had a higher disease-control rate than the placebo group (43% vs. 32%, P = 0.002). Common adverse effects observed in the sorafenib group included diarrhea, weight loss, hand-foot skin reactions, alopecia, anorexia, and voice changes, which were more frequent than those in the placebo group (P<0.001). Notably, 97% of the patients included in this trial were assessed as Child-Pugh liver function class A [13]. Based on these data, sorafenib was approved by the U.S. FDA in November 2007 for advanced HCC as first-line standard treatment.
Although sorafenib has a notable survival benefit in patients with advanced HCC, it has yet to be maintained. Owing to adverse events (AEs), many patients receiving sorafenib suffer from disease progression after dosage reductions or discontinuing treatment [14, 15]. In addition, 27% of patients in the SHARP trial had no initial response to sorafenib [13]. This reveals that resistance to sorafenib (both primary and acquired) greatly limits its survival benefit. In a study predicting survival in advanced HCC patients who permanently discontinued sorafenib, the median post-sorafenib survival was only 4.1 (95% CI 3.3-4.9) months. Liver decompensation, performance status (PS), tumor progression, and/or extrahepatic tumor spread were significantly associated with post-sorafenib survival [16].
Lenvatinib
As first-line treatment, most drugs subsequently tested in phase III trials failed to improve the prognosis compared to sorafenib; they also did not improve survival compared with placebo as second-line therapy. These drugs include erlotinib [17], brivanib [18], sunitinib [19], linifanib [20], everolimus [21], and tivantinib [22] (Fig. 4, Table 1). Insufficient antitumor activity, toxicity in a cirrhosis state, and inadequate patient selection were cited as reasons for these failures. Sorafenib has remained the sole effective first-line treatment option for over a decade until lenvatinib was finally proven to be non-inferior to sorafenib in terms of OS [23].
Lenvatinib is a small molecule inhibitor of VEGFR 1–3, fibroblast growth factor receptor (FGFR) 1–4, PDGFRα, KIT, and RET (Fig. 4) [24, 25]. Targeting the FGF signaling pathway in HCC differentiates lenvatinib from sorafenib.
In a multicenter, randomized, phase III, noninferiority trial (REFLECT; NCT01761266), lenvatinib exhibited noninferiority in overall survival compared with sorafenib (mOS, 13.6 vs. 12.3 months, HR=0.92, 95% CI, 0.79–1.06). All secondary efficacy endpoints in REFLECT were statistically significantly improved: the median progression-free survival (mPFS) was longer in the lenvatinib group than in the sorafenib group (mPFS, 7.3 vs. 3.6 months, HR=0.65, p< 0.0001); the median time to progression (mTTP) was 7.4 months in the lenvatinib group compared to 3.7 months in the sorafenib group (HR=0.61, p<0.0001); and the objective response rate was better (ORR, 18.8% vs. 6.5%, p<0.0001) based on the Response Evaluation Criteria in Solid Tumors (RECIST) version 1.1 after masked independent imaging review [23]. Based on the REFLECT results, the U.S. FDA approved lenvatinib for the first-line treatment of patients with advanced HCC on August 16, 2018 [26].
Regorafenib
Regorafenib is an oral inhibitor that inhibits many angiogenic and tumorigenic kinases, such as VEGFR1–3, tyrosine kinase with immunoglobulin and epidermal growth factor homology domain 2 (TIE2), PDGFRβ, FGFR1, B-RAF, RET, and KIT (Fig. 4) [27]. Regorafenib may exert a more far-reaching antiangiogenic activity owing to a combined blockade of the VEGFR2 and TIE2 pathways, which is supported by the results from a preclinical study by Tsai and Lee [28] showing a cooperative effect when combining anti-TIE2 and sorafenib, leading to an observed increase in overall survival in a melanoma model.
On April 27, 2017, the U.S. FDA expanded the indication for regorafenib as a second-line treatment for advanced HCC patients previously treated with sorafenib [29]. Approval was based on a randomized, placebo-controlled international phase III trial designed to assess the safety and efficacy of regorafenib in patients with HCC progressing during sorafenib treatment (RESORCE; NCT01774344) [30]. Regorafenib had a longer OS than placebo (mOS, 10.6 vs. 7.8 months, HR=0.63, p<0.0001) and prolonged progression-free survival and time to progression by mRECIST (mPFS 3.1 vs. 1.5 months, HR=0.46, p<0.0001; mTTP was 3.2 vs. 1.5 months, HR=0.44, p<0.0001)). The regorafenib group had an ORR of 11% versus 4% compared to the placebo group (p=0.0047), and the disease control rate (DCR) was 65% versus 35% (p<0.0001). Of note, the RESORCE trial was performed in patients who had progressed on prior sorafenib treatment; thus, the efficacy of regorafenib among sorafenib-intolerant patients has not yet been determined.
Additional analyses from the RESORCE trial suggested that regorafenib might offer clinical benefits independent of the latest sorafenib dose or the time to progression after previous sorafenib treatment and that the occurrence of AEs was independent of the last sorafenib dose [31]. Multiple clinical trials are currently evaluating the efficacy of regorafenib together with ICIs (Table 3).
Cabozantinib
Cabozantinib is a small-molecule tyrosine kinase inhibitor of MET and VEGFR 1-3, RET, KIT, AXL, and FLT3, all of which are associated with tumor pathogenesis (Fig. 4) [32, 33]. Cabozantinib has remarkable antitumor activity in HCC through dual inhibition of MET and VEGFR2 [33].
In an international, phase III double-blinded trial, 773 patients who progressed on no more than two prior systemic treatments including sorafenib were randomized to cabozantinib or placebo (CELESTIAL; NCT01908426). The primary endpoint of the CELESTIAL trial was that OS was significantly improved (mOS 10.2 vs. 8.0 months, HR=0.76, p=0.005). The secondary efficacy endpoints of the trial showed that cabozantinib significantly improved the PFS and ORR according to RECIST v1.1 (mPFS was 5.2 vs. 1.9 months, HR=0.44, p<0.001; ORR was 4% vs. <1%, p=0.009). Sixty-eight percent of patients in the cabozantinib group and 36% of patients in the placebo group experienced grade 3 or 4 AEs [34]. Based on these results, on January 14, 2019, the U.S. FDA approved cabozantinib for patients with HCC who had previously received sorafenib treatment [35]. Several trials evaluating the combinations of cabozantinib and ICIs in patients with advanced HCC are ongoing, including in combination with nivolumab and atezolizumab (Table 3).
Of note, in the CELESTIAL trial, 192 (27%) patients had received two previous systemic anticancer drugs, meaning that cabozantinib was used as third-line therapy. Within this subgroup, the OS was not significantly different (mOS 8.6 months vs. 8.6 months). However, cabozantinib improved the PFS (mPFS 3.7 vs. 1.9 months, HR=0.58, 95% CI, 0.41-0.83), suggesting that advanced HCC patients could use cabozantinib as a possible third-line option [34].
Ramucirumab
Ramucirumab, a humanized recombinant IgG1 monoclonal antibody, selectively binds to VEGFR-2, preventing activation of the VEGF pathway (Fig. 4) [36], and it showed anti-tumor activity in advanced HCC in a phase II trial [37]. A prospective study focusing on ramucirumab applied to treat the progression of advanced HCC while on sorafenib (REACH; NCT01140347) showed that ramucirumab did not lead to a significant improvement in OS compared to placebo (mOS 9.2 vs. 7.6 months, HR =0.87, p=0.14). However, in a subgroup with high baseline α-fetoprotein (AFP) concentrations (≥ 400 ng/mL), ramucirumab significantly prolonged the OS with a median OS of 7.8 months vs. 4.2 months (HR=0.674, p=0.006) [38].
To further investigate this result, the biomarker-driven REACH-2 trial (NCT02435433) was designed, evaluating ramucirumab versus placebo in advanced HCC patients with elevated AFP levels (≥400 ng/mL) who received prior sorafenib treatment. The primary endpoint of the REACH-2 trial showed that compared to the placebo group, OS was longer in the ramucirumab group (mOS 8.5 vs. 7.3 months, HR=0.71, p=0.0199). Ramucirumab also increased the PFS (mPFS 2.8 vs. 1.6 months, HR=0.452, p<0.0001), time to radiographic progression (mTTP 3.0 vs. 1.6 months, HR=0.427, p<0.0001), and disease control rate (59.9% vs. 38.9%, p=0.0006). REACH-2 was the first phase III trial to successfully screen for therapeutic candidates with biomarkers among patients with advanced HCC [39].
Based on these results, on May 10, 2019, the U.S. FDA authorized ramucirumab as a second-line treatment for HCC patients whose AFP was no less than 400 ng/mL [40].
Monotherapy with immune checkpoint inhibitors
The success of anti-cytotoxic T lymphocyte associated antigen 4 (CTLA-4) antibodies in blocking immune checkpoints in advanced melanoma patients offers hope for immunotherapy of tumors. HCC carcinogenesis is typically accompanied by chronic liver inflammation, and HCC could be immunogenic [41]. In general, immune tolerance occurs in HCC mainly due to myeloid-derived suppressor cells (MDSCs), alterations of immune checkpoint molecules (such as CTLA-4 and programmed cell death protein-1 [PD-1]), and enrichment of T-regulatory cells (Tregs) [42]. The first evidence showing that ICIs could make a significant difference in HCC treatment came from a phase II trial in which the safety and antitumor activity of tremelimumab (CTLA-4 blockade) supported further studies in patients with advanced HCC who developed HCV-induced cirrhosis [43] (Table 2). In addition, other clinical trials of ICI monotherapy for HCC have been performed (Table 2).
Nivolumab
Nivolumab, a monoclonal antibody, blocks the PD-1 signaling pathway (Figs. 3 and 4) and restores the anti-tumor immune activity. It has been authorized by the U.S. FDA for application in several tumors (e.g., melanoma, non-small cell lung cancer [NSCLC], and kidney cancer) [44]. CheckMate 040 (NCT01658878), a noncomparative phase I/II trial, was designed to investigate the safety and efficacy of nivolumab for advanced HCC patients by escalating and expanding the dose of the medication. Prior sorafenib was allowed. Safety and tolerability represented the primary endpoints for the dose-escalation phase (n=48), while ORR was the primary endpoint for the dose-expansion phase (n=214). During the dose-escalation phase, the safety profile of nivolumab was evaluated, and the patients’ tolerability of the medication was acceptable: 25% of the patients (12 of 48) had grade 3 or 4 treatment-related adverse events (TRAEs), and the incidence of TRAEs appeared to be dose-independent. In the dose-expansion phase, the ORR was 20% (42 of 214 patients, 95% CI, 15-26), with thirty-nine partial responses and three complete responses. The median duration of response was 9.9 months, and the mPFS was 4.0 months (95% CI, 2.9-5.4). Seventy-nine of 138 patients (57%) had their disease under control, with most disease stabilizations lasting for at least 6 months. Among uninfected untreated patients, the ORR was 23%, and the nine-month overall survival rate was 82%, which supported that nivolumab deserved further investigation as first-line therapy for advanced HCC patients [45]. Based on the ORR and the durable objective responses, on September 22, 2017, the U.S. FDA accelerated the approval of nivolumab to treat HCC patients who were first treated with sorafenib [46].

Mechanism of combination therapies. (A) Complementary mechanisms of PD-1/PD-L1 and CTLA-4 inhibitors. Presentation of tumor-associated antigen by the major histocompatibility complex (MHC) expressed by APCs results in the release of an activation signal in combination with a co-stimulatory signal via the B7-CD28 pathway, leading to activation of T cells in the lymph node; B7 also binds to CTLA-4 with a higher affinity than that of CD28, in which case T cells cannot be activated. PD-1 on T cells inhibits antigen-specific T cell activation by interacting with its ligands PD-L1 and PD-L2. Immune escape is induced through the PD-1/PD-L1 axis, as well as the B7/CTLA-4 axis. This figure was adapted from Kudo, et al. [44]. (B) VEGF modulates the immunosuppressive TME, and TKIs restore this suppressive effect. Red arrows represent promotion effects. APCs, antigen presenting cells; CTL, cytotoxic T lymphocyte; CTLA-4, cytotoxic T-lymphocyte antigen 4; iDC, immature dendritic cell; matDC, mature dendritic cell; MDSCs, myeloid-derived stem cells; PD-1, programmed cell death protein 1; PD-L1, programmed cell death-ligand 1; TAMs, tumor-associated macrophages; TME, tumor microenvironment; Tregs, regulatory T cells. Note: This is an open access article distributed under the Creative Commons Attribution License that permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited (CC BY 4.0)

Signaling pathways and molecular targeted therapies for HCC. * represents monotherapies approved by the FDA, ** represents agents as a component of combination therapy approved by the FDA. This figure was modified from Mossenta, et al. [10]. Note: This is an open access article distributed under the Creative Commons Attribution License that permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited (CC BY 4.0)
Another research team studied first-line treatment (nivolumab vs. sorafenib) for advanced HCC patients through a randomized and multicenter phase III study (CheckMate 459; NCT02576509) and reported its data at the 2019 Congress of European Society for Medical Oncology (ESMO). The primary endpoint, OS, was not statistically significant, as the mOS was 16.4 and 14.7 months in the nivolumab group and sorafenib group, respectively (HR=0.85, p=0.0752). Interestingly, patients receiving nivolumab had a higher complete response rate (ORR, 15% vs. 7%, respectively). Eighty-one patients (22%) taking nivolumab and 179 patients (46%) taking sorafenib had grade 3/4 TRAEs [47]. Although the above findings may not influence the current standard treatment, they give us insight that immunotherapy may also have a position in the first-line treatment for advanced HCC.
Pembrolizumab
Pembrolizumab, a monoclonal antibody blocking PD-1 (Figs. 3 and 4), has demonstrated potent anti-tumor activity and tolerable safety across many cancers [48], and its efficacy and safety were evaluated in advanced HCC patients via a nonrandomized, multicenter phase II trial (KEYNOTE-224; NCT02702414). KEYNOTE-224 revealed that 18 of 104 patients displayed an objective response (17%, 95% CI, 11–26) as per RECIST v1.1, with 1 (1%) achieving a complete response and 17 (16%) achieving a partial response. The median response duration was not reached (range 3.1–14.6+ months), the mTTP and mPFS were both 4.9 months, mOS was 12.9 months (95% CI, 9.7–15.5), and 54% of responding patients had response durations over 12 months [49]. Based on the above data, on November 9, 2018, pembrolizumab received accelerated approval by the U.S. FDA for patients with HCC progressing on sorafenib [50]. At the 2021 ASCO Gastrointestinal Cancer conference, the results of the KEYNOTE-224 cohort 2, which enrolled patients with advanced HCC without prior systemic therapy, were reported: the ORR was 16% (95% CI, 7-29), DCR was 57% (0 CR, 16% PR, 41% SD), mPFS was 4 months (95% CI, 2-6), and mOS was 17 months (95% CI, 8-NA). Based on these results, pembrolizumab monotherapy exhibited durable anti-tumor activity and promising overall survival, supporting further evaluation of pembrolizumab as a first-line treatment for advanced HCC [51].
Following KEYNOTE-224, a double-blind, randomized, phase III study (KEYNOTE-240; NCT02702401) was designed to further assess the safety and efficacy of pembrolizumab treatment. The results of KEYNOTE-240 were updated at the 2021 ASCO GI Conference: the mOS was 13.9 vs. 10.6 months compared to placebo (HR=0.771, 95% CI 0.617–0.964), mPFS was 3.3 vs. 2.8 months compared to placebo (HR=0.703, 95% CI 0.559–0.885), ORR was 18.3% for the pembrolizumab group and 4.4% for the placebo group, DCR was 61.9% vs. 53.3% compared to placebo, and the median TTP was 4.0 vs. 2.8 months [52]. The clinical activity of pembrolizumab was again demonstrated by KEYNOTE-240, confirming the findings from KEYNOTE-224, thus supporting its accelerated approval by the FDA. Two other phase III trials evaluating pembrolizumab as second-line therapy are currently ongoing (KEYNOTE-394 and KEYNOTE-937, Table 2). Pembrolizumab combined with molecular targeted drugs are now being evaluated through several phase I/II trials (Table 3).
Combination therapy
To achieve a more satisfactory therapeutic response rate than ICI monotherapy, combination therapies based on ICIs are being developed to treat cancer. In 2018, Larkin et al. discovered through the CheckMate067 trial that the use of combined nivolumab and ipilimumab (CTLA-4 inhibitor) was more effective than monotherapy in patients with advanced melanoma [53]. Possible mechanisms for this combination therapy are as follows: 1) blocking the CTLA-4 pathway in lymph nodes increases the activation and proliferation of T cells, leading to CD8+ T cell infiltration; 2) blocking the PD-1/PD-L1 pathway not only maintains the killing capacity of cytotoxic T lymphocytes (CTLs) [44] but also improves the ability of antigen-presenting cells to present tumor-associated antigens; and 3) given the high expression of CTLA-4 and PD-1 on Tregs, the combination of these two inhibitors indirectly reduces the immunosuppressive tumor microenvironment (TME) (Fig. 3A) [54].
Another area of interest is the combination of TKIs with ICIs. It is worth noting that molecular targeted agents (sorafenib, lenvatinib, regorafenib, cabozantinib, and ramucirumab) with beneficial effects on survival in patients with HCC all share the same characteristics, that is, anti-angiogenesis, highlighting the importance of this hallmark in cancer treatment [55]. The restoration of tumor vascular function may help to enhance the ability of other drugs (e.g., ICIs) that can be used in combination with anti-angiogenic molecules to kill tumor cells [56, 57]. TKIs with antiangiogenic activity exert immunomodulatory effects on the TME [58], which include promoting dendritic cell (DC) maturation [59], enhancing T cell trafficking and function [10], and reversing the hypoxia-induced immunosuppressive effect in tissue [60] (Fig. 3B).
Nivolumab + Ipilimumab
As discussed above, PD-1 plus CTLA-4 inhibitors promote the anti-tumor immune response through different complementary mechanisms that affect different signaling pathways. Nivolumab in combination with ipilimumab has been shown to be effective in many tumors (e.g., melanoma, NSCLC, renal cell carcinoma, mismatch repair-deficient/microsatellite instability-high metastatic colorectal cancer) [61,62,63,64]. In cohort 4 of CheckMate 040 (NCT01658878), 50 patients received 4 doses of nivolumab (1 mg/kg) with ipilimumab (3 mg/kg) every 3 weeks and then 240 mg of nivolumab every 2 weeks (arm A). In this randomized clinical trial, the dosage regimen of arm A showed a good safety profile, a promising objective response rate, and a durable response: the ORR was 32% per RECIST v1.1, with 4 complete responses and 12 partial responses; the median response duration was 17.5 months (4.6–30.5+ months), and 31% of the responses lasted over 24 months [45]. Based on these data, on March 10th, 2020, the dosage regimen of arm A received accelerated approval for advanced HCC patients who progressed on sorafenib treatment [65] (Table 3).
Recently, the 44-month follow-up results from CheckMate 040 were presented at the 2021 ASCO GI Conference. The mOS was 22.2 months in arm A. The 24-month OS rate improved to 46% (95% CI, 32–59), and the 36-month OS rate was 42% (95% CI, 28–55). The ORR remained at 32%, and the median response duration remained at 17.5 months (5–47+ months) in arm A. The DCR was higher in arm A than in arms B and C (54%, 43%, 49%, respectively). Nivolumab + ipilimumab as a second-line treatment continued to show clinical responses and long-term survival benefits [66].
Atezolizumab + Bevacizumab
Atezolizumab is an IgG1 monoclonal antibody that specifically binds to PD-L1 and interrupts its interaction with PD-1, thus reversing T cell suppression [67]. Bevacizumab, a humanized anti-VEGF monoclonal antibody, suppresses angiogenesis and tumor development in HCC [68], showing ORRs of 13% [69] and 14% [70] in single-agent phase II trials for advanced HCC. Atezolizumab + bevacizumab showed a tolerable safety profile and efficacy for treating unresectable HCC patients in GO30140 (NCT02715531) [8].
This combination became a promising treatment and is being compared with sorafenib via a phase III trial. In a global, open-label, phase III trial, 501 patients randomly received either atezolizumab + bevacizumab or sorafenib (IMbrave 150; NCT03434379). The efficacy phase showed that the atezolizumab + bevacizumab group had a significantly longer OS and PFS: the estimated 6-month and 12-month survival rates were 84.8% (95% CI, 80.9–88.7) and 67.2% (95% CI, 61.3–73.1), corresponding to 72.2% (95% CI, 65.1–79.4) and 54.6% (95% CI, 45.2–64) for the sorafenib group; the mOS could not be evaluated (NE) in the atezolizumab + bevacizumab group versus 13.2 months (95% CI, 10.4–NE) in the sorafenib group (HR=0.58, p<0.001); and the estimated mPFS was 6.8 months and 4.3 months, respectively (HR=0.59, p<0.001). The objective response rates were also statistically significant: the ORR by RECIST v1.1 was 27.3% (95% CI, 22.5–32.5) in the combination group compared to 11.9% (95% CI, 7.4–18.0) in the sorafenib group (p<0.001) and 33.2% (95% CI, 28.1 to 38.6) versus 13.3% (95% CI, 8.4 to 19.6) per mRECIST (P<0.001). No serious adverse events with significant differences were observed between the two groups. The most common AEs, affecting more than 20% of patients in the atezolizumab + bevacizumab group, were hypertension (29.8%), fatigue (20.4%), and proteinuria (20.1%) [71]. On May 29, 2020, the U.S. FDA approved atezolizumab in combination with bevacizumab as a first-line treatment for advanced HCC patients based on the safety and efficacy revealed in IMbrave 150 [72] (Table 3).
At the 2021 ASCO Gastrointestinal Cancer Symposium, Finn et al. [73] updated the OS analysis for IMbrave 150. After an additional 12 months of follow-up, a sustained clinical efficacy benefit was achieved with atezolizumab + bevacizumab versus sorafenib: the mOS was 19.2 months in the combination group versus 13.4 months with sorafenib (HR=0.66 [95% CI, 0.52–0.85], P=0.0009). The updated data showed an ORR of 29.8% (95% CI, 24.8-35) by RECIST v1.1 in the combination group compared to 11.3% (95% CI, 6.9-17.3) in the sorafenib group, and more patients in the combination group achieved a complete response than previously reported (CR=7.7%). This combination regimen has the longest OS ever seen in first-line phase III studies, further confirming that this regimen can serve as a standard of care for patients with advanced HCC who have not received any systemic therapy before.
Novel therapies in development
Targeted therapies currently in clinical trials
Currently, five targeted therapies have been approved for the treatment of advanced HCC. Of these five therapies, four are small-molecule kinase inhibitors, and one is a monoclonal antibody against VEGFR2 (Fig. 2). In addition to these approved targeted therapies, a variety of targeted therapies are in clinical development.
VEGFR
There are many growth factors involved in tumor angiogenesis, the most important of which is the vascular endothelial growth factor (VEGF) family [74]. In addition to the mentioned inhibitors targeting VEGFR that have been approved by the FDA (sorafenib, lenvatinib, regorafenib, cabozantinib, and ramucirumab), there are several anti-VEGFR agents currently in clinical trials. Tivozanib is a potent VEGFR 1–3 TKI with relatively lesser effects on KIT and PDGFRβ and it has dose-dependent activity against HCC in vivo [75]. According to the existing NCT01835223 data (Table 1) [76], tivozanib showed an ORR (per RECIST v1.1) of 21%, mOS of 9 months (90% CI, 5.4–27.8), mPFS of 24 weeks, and a 1-year OS rate of 40%. Unfortunately, the study did not proceed to stage 2 due to not meeting the prespecified statistical plan. However, tivozanib had better anti-tumor efficacy than sorafenib and lenvatinib as first-line therapy, with ORRs of 21% vs. 6.5% (sorafenib) and 18.8% (lenvatinib), respectively. Since small molecular inhibitors can at least partially reverse immune dysfunction, the combination of tivozanib and ICIs is a promising option. As previously mentioned, a trial of tivozanib in combination with durvalumab is currently ongoing (Table 3).
In addition, at the 2020 ASCO Virtual Scientific Program, two other TKIs designed by Chinese companies reported exciting phase III results. In a first-line treatment setting, donafenib (ZGDH3, NCT02645981, Table 1) had a significantly longer overall survival than sorafenib: the mOS was 12.1 vs. 10.3 months (HR=0.831, p=0.0363) (Table 1). Additionally, donafenib exhibited favorable safety and tolerability compared to sorafenib: drug-related AEs of grade ≥ 3 occurred in 37.5% of patients in the donafenib group versus 49.7% in the sorafenib group (p=0.0018), and the incidence of treatment interruptions due to drug-related AEs was 30.3% in the donafenib group compared to 42.5% in the sorafenib group (P=0.0013) [77].
Apatinib has been investigated as second-line therapy in Chinese advanced HCC patients through a phase III study AHELP (NCT02329860, Table 1), which showed significantly longer OS and PFS compared to placebo and exhibited a tolerable safety profile. The mOS was 8.7 vs. 6.8 months (HR=0.785, p=0.0476), the mPFS was 4.5 vs. 1.9 months (HR=0.471, p<0.0001), the ORR was 10.7% vs. 1.5% per RECIST v1.1, and no new safety signals were noted [78].
c-MET
c-Met is a receptor tyrosine kinase for hepatocyte growth factor (HGF) [79], and abnormalities in c-Met are found in approximately 50% of HCC patients [80]. Aberrant c-MET activity is involved in HCC progression [81] and may lead to treatment resistance toward sorafenib [82]. c-MET inhibitors can be categorized into selective and nonselective. Nonselective c-MET inhibitors (e.g., tivantinib and cabozantinib) may exert anti-tumor effects, mainly owing to the inhibition of non-c-MET targets, and an “off-target” effect is associated with increased toxicity [83]. Conversely, selective c-MET inhibitors (e.g., tepotinib and capmatinib) are being tested in HCC and are thought to have reduced toxicity. Both selective and nonselective agents are being investigated in different trials for potential use in HCC patients as first- or second-line therapy (NCT01988493, NCT02115373; NCT01737827, NCT01964235, Table 1). Decaens et al. reported data of NCT02115373, showing that tepotinib was well tolerated and that the recommended phase 2 dose (500 mg) reached its primary endpoint in the phase 2 study: the 12-week PFS was 63.3% (one-sided binomial exact test: P < 0.0001) [84]. Selective c-MET inhibitors are more promising drug candidates. There are also ongoing studies evaluating the safety and tolerability of c-MET inhibitors combined with ICIs (NCT02795429, NCT02082210, Table 3).
TGFβ
The TGFβ pathway has dual anti- and pro-tumoral activities in cancer cells: in the early stage, it is beneficial for promoting cell cycle arrest and apoptosis [85], while in the advanced stage, it promotes tumor progression and metastasis by enhancing cell motility, epithelial-to-mesenchymal transition (EMT), invasiveness and stemness [86]. In addition, TGFβ signaling is a major modulator of immune cell proliferation, differentiation, development, and survival [87], which suppresses CD8+ T cells, NK cells, and DCs and increases CD4+CD25+ Tregs by promoting the differentiation of M2-type macrophages [88], leading to immunosuppression in HCC. Microenvironmental remodeling by TGFβ creates a beneficial microenvironment for tumor development and metastasis [86]. These findings provide a rationale for blocking TGFβ signaling in HCC treatment, especially in immunotherapy. Galunisertib (LY2157299) is a TGFβ receptor 1 inhibitor [89] that has been studied as monotherapy and combined with sorafenib in extensive phase I/II trials for HCC [90].
Endoglin
Endoglin (also known as CD105), a type 1 intact transmembrane glycoprotein, is a co-receptor for TGF-β ligands and plays an important role in fibrogenesis and angiogenesis [91]. Endoglin is mainly found in immature tumor vascular endothelial cells and contributes to tumor angiogenesis [92, 93]. Endoglin microvascular density has an essential and independent prognostic role in recurrent or metastatic HCC patients [94], and circulating serum endoglin could be detected in HCC patients [93, 95, 96], suggesting that endoglin may be a prognostic marker for HCC patients; however, it still needs further study. TRC105 (carotuximab) is an anti-endoglin antibody, yet it did not show adequate monotherapy efficacy in the phase II trial (NCT01375569) of TRC105 for post-sorafenib HCC patients [97]. The combination of TRC105 and sorafenib (NCT01306058) showed an ORR of 21% per RECIST at all four dose levels, with a mOS of 15.5 months and a mTTP of 3.8 months [98]. At the 2019 ASCO GI, Raghav et al. reported that TRC105 + sorafenib had encouraging signs of activity, with durable partial responses in 1 of 5 patients thus far in phase II (NCT01806064) [99].
FGF19/FGFR4
Among the family of tyrosine kinase receptors, fibroblast growth factor receptor 4 (FGFR4) is mostly expressed in the liver [100]. FGF19, one of the three endogenous FGFs, binds with the highest affinity to FGFR4 [101], and the FGF19 expression level is frequently reported to be associated with HCC prognosis [102]. After activation of the FGFR4 signaling pathway, HCC cells undergo proliferation, EMT, anti-apoptosis, angiogenesis, invasion, and drug resistance through activating four downstream pathways: PI3K/AKT/mTOR, RAS/RAF/MAPK, PLCγ/DAG/PKC, and GSK3β/β-catenin [103].. Abnormal expression of FGF19/FGFR4 promotes the development of HCC [102], making it a promising therapeutic target. Despite the relatively low selectivity of pan-FGFR inhibitors against FGFR4 [104], some TKIs specifically targeting FGF19/FGFR4 are being tested in different phases of clinical trials (Table 1) [105], among which the most promising candidate is BLU-554.
In a phase I trial (NCT02508467) to escalate and expand doses, fisogatinib (BLU-554) produced a clinical response in FGF19-positive patients with advanced HCC: the ORR per RECIST v1.1 was 17% (11 of 66 patients) in FGF19-positive patients and 0% in the FGF19-negative group; 53% of TRAEs in this study were grade 1/2, which were tolerable. This trial validates the effectiveness of blocking the FGF19/FGFR4 axis and the biomarker potential of FGF19 to screen HCC patients [106]. Another FGFR4 inhibitor, FGF401, has been investigated as monotherapy or in combination with PDR001 (NCT02325739).
CSF-1/CSF-1R
Colony-stimulating factor 1 (CSF-1) is a cytokine mainly produced by tumor cells that recruits macrophages under pathological conditions [107]. When CSF-1/CSF-1R is activated, tumor-associated macrophages (TAMs) secrete growth factors that contribute to tumor growth or metastasis, leading to a higher rate of recurrence [108, 109]. Ao et al. found that PLX3387 (CSF-1R inhibitor, also called pexidartinib) exhibited antitumor activity in both xenograft and allograft HCC models [110], and depletion of TAMs by drugs enhanced the antitumor effects of sorafenib [111]. Several TKIs targeting the CSF-1/CSF-1R axis are now being studied for treating advanced solid tumors, including HCC, such as pexidartinib (NCT02452424), chiauranib (NCT03245190), TPX-0022 (NCT03993873), and BLZ945 (NCT02829723).
Immune checkpoint therapies in clinical trials
Currently, two immune checkpoint inhibitors have been approved by FDA for the HCC treatment, both of which are PD-1 monoclonal antibodies. In addition to these approved therapies, there are a variety of promising immune checkpoints therapies in clinical trials.
PD-1 and PD-L1 inhibitors
In addition to nivolumab and pembrolizumab, there are a number of PD-1 inhibitors and PD-L1 inhibitors in clinical trials, including: (1) PD-1 inhibitors, such as tislezumab versus sorafenib as first-line treatment (RATIONALE 301, NCT03412773), camrelizumab (SHR-1210) as second-line treatment (NCT02989922, 2) PD-L1 inhibitors, namely durvalumab (NCT01693562, NCT03847428), and avelumab (NCT03389126). According to Qin et al. [112], the ORR of camrelizumab was 14.7%, and the 6-month OS was 74.4%. Lee et al. reported the results of a phase II trial of avelumab monotherapy, which demonstrated moderate efficacy and good tolerance with an ORR of 10%, DCR of 73.3%, mTTP of 4.4 months, and mOS of 14.2 months (Table 2) [113]. Except for these two trials, no other trials have reported any results yet.
Other immune checkpoint inhibitors
In addition to the well-known PD-1/PD-L1 and CTLA-4, a series of inhibitory immune checkpoint molecules involved in the immune tolerance of HCC have been reported, including lymphocyte activation gene 3 (LAG-3) [114], T cell immunoglobulin mucin-3 (TIM-3)/galectin-9 (GLA-9) [115], T cell immunoglobulin and ITIM domain (TIGHT) [116], and adenosine A2a receptor. Zhou et al. [114] found that a PD-L1 inhibitor in combination with a TIM-3, LAG-3, or CTLA-4 inhibitor results in a revitalization of in vitro tumor-infiltrating lymphocyte (TIL) responses in most patients and further enhances its effect compared to PD-L1 monotherapy. The use of drugs targeting TIM-3/GLA-9 is well summarized in Ref [117]. Next-generation ICIs targeting LAG-3, TIM-3, and TIGHT are expected to provide survival benefits for HCC patients.
Agonists of co-stimulatory checkpoint pathways
Immune checkpoints include inhibitory pathways and co-stimulatory pathways. Agonists of co-stimulatory checkpoint pathways such as OX40, GITR, ICOS, CD27, and CD28 are under investigation for treating solid tumors, which have also been summarized in Ref [118]. PF-04518600 is an OX4 agonist antibody. In 2020, the results of a phase I trial (NCT02315066) investigating its safety and tolerability in patients with advanced HCC were published, showing that PF-04518600 was well-tolerated overall and provided meaningful disease control [119]. Glucocorticoid-induced tumor necrosis factor receptor family-related protein (GITR) is believed to promote the function of effector T cells and inhibit the function of Tregs [120]. Beek et al. found that agonistic targeting GITR enhances the function of tumor-infiltrating T cells in HCC, suggesting that it could be a promising target for immunotherapy [121]. Several antibodies targeting GITR including INCAGN01876 (NCT03126110), BMS-986156 (NCT04021043), and TRX518 (NCT02628574, NCT03861403) are being studied for solid tumors involving HCC.
Combination therapies in clinical trials
Regorafenib enhances the killing effect of T cells by modulating various immunoreactive molecules, helping to present antigens, and triggering T cell differentiation and accumulation [122]. Tumor-associated macrophages associated with angiogenesis in the TME are characterized by TIE2 expression, which is strongly associated with cancer cell intravasation into the circulatory system [123]. This evidence provided a rationale to combine regorafenib and pembrolizumab as first-line treatment for advanced HCC, which is being studied in a multicenter dose-escalation phase IIb trial (NCT03347292, Table 3).
Meanwhile, lenvatinib combined with pembrolizumab has shown promising antitumor activity and manageable toxicity in unresectable HCC through a phase Ib clinical trial (KEYNOTE-524/NCT03006926, Table 3), which supported a Breakthrough Therapy designation granted by the FDA in July 2019 [124]. Efficacy data from KEYNOTE-524 showed an ORR of 46% (95% CI, 36–56.3%) per mRECIST, the mPFS was 9.3 months (95% CI, 5.6–9.7) and mOS was 22 months (95% CI, 20.4–NE) [125]. As a single-arm study, lenvatinib + pembrolizumab from KEYNOTE-524 did not show evidence of meaningful improvement over approved therapies; therefore, a phase III trial is currently assessing the efficacy and safety of this combination compared to lenvatinib + placebo as a first-line option (LEAP-002/NCT03713593, Table 3).
Cabozantinib not only modulates peripheral and intratumor immune landscapes but also induces changes in the phenotype of MC38-CEA tumor cells, thereby increasing their vulnerability to cytotoxic effects mediated by T cells [126]. Immunogenic and immune subset modulations of cabozantinib support its clinical investigation in combination with immunotherapy. Multiple active trials are evaluating the efficacy of cabozantinib in combination with nivolumab/atezolizumab, including the COSMIC-312 phase III trial (NCT03755791, Table 3).
To improve the ORR over single-use nivolumab, a series of different combinations are being tested in clinical trials (Table 3), including sorafenib (NCT03439891), lenvatinib (NCT03418922), BMS-986253 (IL-8 inhibitor, NCT04050462), mogamulizumab (CCR4 inhibitor, NCT02705105), galunisertib (TGFβ inhibitor, NCT02423343) and Pexa-Vec (antitumor vaccine, NCT03071094).
Avelumab (a PD-L1 inhibitor) is being investigated in combination with axitinib/regorafenib via phase 1/2 trials (NCT03289533, NCT03475953, Table 3). VEGF Liver 100 (NCT03289533) is a phase Ib trial to evaluate the safety and efficacy of avelumab + axitinib (VEGFR inhibitor) as a first-line treatment in advanced HCC patients. At the 2019 ASCO GI Cancer, Kudo et al. reported the interim results of VEGF Liver 100: the ORR per mRECIST was 31.8% (95% CI, 13.9–54.9%), and the mPFS was 3.8 months (95% CI, 1.9–7.3) [127].
Camrelizumab was tested in combination with apatinib (VEGFR-2, RET, c-kit inhibitor) for treating advanced HCC patients in NCT02942329 [128]. At the data cutoff (June 15, 2018), 8 of 16 evaluable HCC patients achieved a partial response, with an ORR of 50% (95% CI, 24.7–75.4%). This combination has been evaluated in a phase III trial as a first-line treatment (NCT03764293) versus sorafenib and a phase II trial as a second-line treatment (NCT03463876) (Table 3).
A study investigating tivozanib (selective VEGFR 1–3 TKI) in combination with durvalumab for untreated advanced HCC updated its phase Ib data at the 2021 ASCO GI Conference. NCT03970616 (Table 3) enrolled 7 patients and found that untreated advanced HCC patients tolerated this combination well, with two of them achieving a partial response [129]. The phase II portion of the study is ongoing.
The results of NCT02519348 (Table 3), which evaluated durvalumab (D) combined with tremelimumab (T) in advanced HCC patients, have been reported at the 2020 ASCO Virtual Scientific Program, showing that T300+D (T 300 mg + D 1500 mg 1 dose followed by D Q4 weekly) provided the best benefit-risk profiles. The confirmed ORR by RECIST v1.1 in T300 + D was 24% (95% CI, 14.9–35.3%), mOS was 18.7 months (95% CI, 10.8–NR), and grade 3/4 TRAEs were observed in 26 patients (35.1%) in T300 +D [130]. T300 + D and durvalumab monotherapy are being investigated in the HIMALAYA trial (NCT03298451) as first-line treatment for advanced HCC patients. Besides, a phase III trial evaluating the efficacy and safety of IBI310 (CTLA-4 inhibitor) plus sintilimab (PD-1 inhibitor) versus sorafenib as first-line treatment for advanced HCC is currently recruiting (NCT04720716, Table 3).
Antibody-drug conjugates (ADCs)
Antibody-drug conjugates (ADCs) consist of a monoclonal antibody against a tumor associated antigen (TAA) on the surface of cancer cells that is conjugated to a cytotoxic drug (known as the payload) through a chemical linker [131]. This directed delivery strategy combines the precision of antibody targeting to antigens with the effective cell-killing activity of the payload, resulting in reduced systemic exposure and thus reduced toxicity [132]. ADCs are one of the rapidly growing areas in cancer drug development, with five ADCs having been approved by the FDA for clinical use (Adcetris, Kadcyla, Besponsa, Mylotarg, and Polivy) and over 100 ADCs being evaluated in clinical trials [133]. The discovery of a series of HCC-specific TAAs provides the rationale for applying ADCs in HCC treatment: glypican-3 (GPC3) [134], AFP, epithelial cell adhesion molecule (EpCAM) [135], tumor endothelial marker 1 (TEM1, or endosialin) [136], MAGE-A, NY-ESO-1 [137], and claudin-6 (CLDN6) [138]. In the past, there were pre-clinical studies of the anti-tumor effects of Anti-CD147 ILs-DOX [139], G7mAb-DOX [140], and MetFab-DOX [141] in HCC, as well as phase I/II trial (NCT01631552) of IMMU-132 [142] in solid tumors including HCC. Recently, Fu et al. designed and synthesized two anti-GPC3 ADCs with duocarmycin SA and pyrrolobenzodiazepine, which were named hYP7-DC and hYP7-PC, and demonstrated that these two ADCs had antitumor activity against both GPC3-positive HCC cell lines and xenograft models [143]. The hYP7-DC was also found to have synergistic effects with gemcitabine, and hYP7-PC induced tumor remission in a mouse model. Kong et al. developed a novel ADC drug, CLDN6-DM1 (DM1 being referred to as mertansine), and the preclinical data of CLDN6-DM1 showed robust antitumor effects against HCC cell lines and a xenograft mouse model, both as monotherapy and combined with sorafenib [144] (Table 4). All these efforts provide a new strategy for the treatment of HCC. Clinical trials of ADCs for HCC treatment in the near future are being anticipated.
Chimeric Antigen Receptor T Cells (CAR-T)
CAR is a fusion protein consisting of an antigen-binding domain (usually referring to the extracellular portion of the antibody), the signaling domains of the T cell receptor (TCR) chain, and additional co-stimulatory domains (e.g., CD28, OX40, CD137); therefore, CAR recognition is independent of major histocompatibility complex (MHC) restriction. Loss of MHC-associated antigen presentation by tumor cells is a key mechanism of cancer immune escape [148], and one strategy to overcome this escape is to genetically engineer autologous T cells with CAR, expand CAR T cells in vitro and infuse them back into patients to attack cancer cells [149]. In 2017, tisagenlecleucel (KYMRIAH) [150, 151] and axicabtagene ciloleucel (Yescarta) [152, 153] received successive FDA approval as CAR-T therapy for the treatment of acute lymphoblastic leukemia and diffuse large B-cell lymphoma. Breyanzi (lisocabtagene maraleucel) became the third FDA-approved CAR-T therapy in 2021 for certain types of non-Hodgkin lymphoma, including diffuse large B-cell lymphoma [154, 155]. Positive results of CAR-T therapy in hematologic malignancies raised hope for CAR-T therapy against solid tumors [156]. In preclinical studies, engineered GPC3-CAR-T cells could effectively eliminate GPC3-positive HCC cells in HCC xenograft mouse models, demonstrating their clinical potential for treatment [157,158,159]. There are currently 20 clinical trials in progress testing CAR-T therapy for HCC, 12 of which target glypican-3 (GPC-3) (Table 5). In 2020, Shi et al. [160] reported two sequential phase I studies, NCT02395250 and NCT03146234, which showed that GPC3-CAR-T cells have an initial safety profile and anti-tumor activity in advanced HCC patients. There were two confirmed partial responses of 13 patients, and one patient with sustained stable disease exhibited long-term survival (44.2 months). The 6-month, 1-year, and 3-year OS rates were 50.3%, 42.0%, and 10.5%, respectively, with a mOS of 278 days (95% CI, 48–615 days). There were no grade 3/4 neurotoxicity or CAR-T-related infusion reactions. In addition, CAR-T cells against other antigens are under investigation: anti-c-MET/PD-L1 (NCT03672305 and NCT03638206), anti-DR5 (NCT03941626 and NCT03638206), anti-EGFR (NCT03941626 and NCT03638206), anti-CD147 (NCT03993743), anti-ET1402 (NCT03349255), anti-NKG2D (NCT04550663), anti-mucin1 (NCT02587689), and anti-EpCAM (NCT03013712).
Bispecific T cell Engager (BiTE)
BiTEs are generated by two different single-chain variable fragments (scFvs) covalently linked by small peptides which are derived from the antigen-binding domains of anti-CD3 and anti-TAA antibodies [161]. BiTEs are believed to possess remarkable efficacy for tumor immunotherapy because of their mini size, high flexibility, and high-affinity binding between effector and target cells [162]. Blinatumomab, an anti-CD19/CD3-BiTE, has been successful in treating hematologic malignancies and was approved by the FDA for treating acute lymphocytic leukemia [163]. Zhang et al. reported that the anti-EpCAM BiTE 1H8/CD3 had effective antitumor activity against HCC cells both in vitro and in vivo [145]. Kebenko et al. [146] reported a phase I study of solitomab (MT110, targeted EpCAM and CD3, NCT00635596) for advanced solid tumors. The study was associated with dose-limiting toxicities, based on which it failed to escalate the dose to potentially therapeutic levels. ERY974 [147], an anti-GPC3/CD3 BiTE, had been evaluated for safety and efficacy in GPC3+ HCC patients in a phase I trial (NCT02748837), with no results yet (Table 4). To improve the efficiency of BiTEs and reduce on-target off-tumor toxicities, the identification of target molecules abundantly expressed by tumor cells compared to nonmalignant cells is necessary. In addition, BiTEs face the complex TME of solid tumors and drug resistance due to the loss of the target antigen expression. These are all key issues to be addressed in the future.
Suggested systemic treatment strategies
Recently, approved systemic agents do offer more options for the treatment of advanced HCC patients; however, the lack of head-to-head comparative studies between first- and second-line regimens for systemic treatment has led to confusion when making clinical decisions. Thus, systematic review and network meta-analysis are particularly important at this stage. A meta-analysis of 14 clinical trials conducted by Sonbol et al. showed that atezolizumab + bevacizumab was superior in the first-line setting compared to sorafenib (HR=0.58), lenvatinib (HR=0.63), and nivolumab (HR=0.68). In the second-line setting, all drugs showed a PFS benefit compared with placebo, with only regorafenib and cabozantinib converting to OS benefit. In addition, cabozantinib and regorafenib showed a PFS advantage over ramucirumab and pembrolizumab, but only regorafenib had a statistically significant OS benefit compared to ramucirumab (HR=0.71). For patients with advanced HCC whose AFP levels were at or above 400 ng/mL, there was no significant difference in PFS or OS among regorafenib, cabozantinib, and ramucirumab [164]. Park et al. incorporated 13 first-line studies and 11 second-line studies to obtain similar results. In first-line therapies, atezolizumab + bevacizumab had the greatest OS benefit, while lenvatinib had the greatest ORR benefit. In second-line therapies, cabozantinib had the greatest PFS benefit and ORR benefit compared to placebo [165]. In 2020, ASCO assembled an expert panel to perform a systematic review of published phase III trials and to develop evidence-based clinical practice guidelines for systemic therapy of advanced HCC [166]. According to these guidelines, atezolizumab + bevacizumab is the standard treatment for most patients with advanced HCC in Child-Pugh grade A and Eastern Cooperative Oncology Group Performance Status (ECOG PS) 0–1, the details of which are shown in Fig. 5. For patients with contraindications to immunotherapy, sorafenib or lenvatinib is preferred for first-line treatment.

Suggested systemic treatment strategies for advanced HCC. This algorithm is derived from recommendations of “Systemic Therapy for Advanced Hepatocellular Carcinoma: ASCO Guideline” [166]
Conclusions
Undoubtedly, the systemic treatments of advanced HCC have ushered in prime time (Fig. 4). Angiogenesis and immune evasion are two common features of cancers [167]. As a pioneer in the era of immune combination therapy, the atezolizumab + bevacizumab regimen is the first combination proven to be more effective than sorafenib in the first-line treatment of advanced HCC, which is regarded as a milestone and an encouraging breakthrough in the treatment of advanced HCC. Notably, with atezolizumab + bevacizumab being promoted as a preferred first-line treatment [164,165,166], the following questions arise: are the existing second-line treatment options still valid? Can the existing first-line TKIs become the second-line treatment after immunotherapy? These points remain to be addressed by future studies. As mentioned earlier, several phase III trials in the first-line setting are underway (e.g., CheckMate-9DW, LEAP2, COSMIC-312, HIMALAYA, Table 3), and the potential first-line treatment options are likely to continue to increase, making clinical decisions more challenging.
Our review not only lists the new drugs approved by FDA for HCC treatment, but also summarizes novel drug candidates under investigation. Selective inhibitors of VEGFR, c-MET, TGFβ, endoglin, and FGFR4 have shown good tolerability and efficacy in phase I/II or phase III studies, making these compounds promising for future treatment. In the field of immunotherapy, in addition to targeting PD-1/PD-L1 and CTLA-4, an increasing number of ICIs, such as LAG-3, TIM-3, and GITR, are gradually demonstrating their efficacy. Meanwhile, as more and more HCC-specific TAAs are identified, other immunotherapeutic approaches, including ADCs, CAR-T, and BiTEs, are being further developed.
The development of new drugs for HCC treatment may not be as impressive as for some other tumors, and few predictive biomarkers have yet been identified (except AFP for ramucirumab and perhaps FGF19 for FGFR4 inhibitors). Based on the results of recent clinical trials, it seems that a single drug may not be sufficient for the treatment of HCC [168], therefore combination therapy represents a major research direction for the systemic treatment of advanced HCC. In addition, it is important to find biomarkers that can predict the treatment response to guide systemic therapy strategy. With the continued development of new therapeutic strategies, it is expected that the treatment outcomes for HCC will be greatly improved in the future.
Availability of data and materials
The materials supporting the conclusion of this review have been included within the article.
Abbreviations
- ADCs:
-
Antibody-drug conjugates
- AEs:
-
Adverse events
- ASCO:
-
American society of clinical oncology
- BiTE:
-
Bispecific T cell engager
- CAR-T:
-
Chimeric antigen receptor T Cells
- CI:
-
Confidence interval
- CTL:
-
Cytotoxic T lymphocyte
- CTLA-4:
-
Cytotoxic T lymphocyte associated antigen 4
- DCR:
-
Disease control rate
- FGF:
-
Fibroblast growth factor
- FGFR:
-
Fibroblast growth factor receptor
- HCC:
-
Hepatocellular carcinoma
- HR:
-
Hazard ratio
- ICIs:
-
Immune checkpoint inhibitors
- MDSCs:
-
Myeloid-derived suppressor cells
- ORR:
-
Objective response rate
- OS:
-
Overall survival
- PDGFR:
-
Platelet-derived growth factor receptor
- PFS:
-
Progression-free survival
- RECIST:
-
Response evaluation criteria in solid tumors
- TAMs:
-
Tumor-associated macrophages
- TKIs:
-
Tyrosine kinase inhibitors
- TME:
-
Tumor microenvironment
- TRAEs:
-
Treatment-related adverse events
- Tregs:
-
T-regulatory cells
- TTP:
-
Time to progression
- VEGFR:
-
Vascular endothelial growth factor receptor
References
Cancer Today [https://gco.iarc.fr/today/home]. Accessed 20 Feb 2021.
Cancer Tomorrow [https://gco.iarc.fr/tomorrow]. Accessed 20 Feb 2021.
Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68(6):394–424.
Sim HW, Knox J. Hepatocellular carcinoma in the era of immunotherapy. Curr Probl Cancer. 2018;42(1):40–8.
Alqahtani A, Khan Z, Alloghbi A, Said Ahmed TS, Ashraf M, Hammouda DM. Hepatocellular Carcinoma: Molecular Mechanisms and Targeted Therapies. Medicina (Kaunas). 2019;55:9.
Llovet JM, Zucman-Rossi J, Pikarsky E, Sangro B, Schwartz M, Sherman M, et al. Hepatocellular carcinoma. Nat Rev Dis Primers. 2016;2:16018.
Forner A, Reig M, Bruix J. Hepatocellular carcinoma. Lancet. 2018;391(10127):1301–14.
Lee MS, Ryoo BY, Hsu CH, Numata K, Stein S, Verret W, et al. Atezolizumab with or without bevacizumab in unresectable hepatocellular carcinoma (GO30140): an open-label, multicentre, phase 1b study. Lancet Oncol. 2020;21(6):808–20.
Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–74.
Mossenta M, Busato D, Baboci L, Cintio FD, Toffoli G, Bo MD. New Insight into Therapies Targeting Angiogenesis in Hepatocellular Carcinoma. Cancers (Basel). 2019;11:8.
Wilhelm SM, Carter C, Tang L, Wilkie D, McNabola A, Rong H, et al. BAY 43-9006 exhibits broad spectrum oral antitumor activity and targets the RAF/MEK/ERK pathway and receptor tyrosine kinases involved in tumor progression and angiogenesis. Cancer Res. 2004;64(19):7099–109.
Chang YS, Adnane J, Trail PA, Levy J, Henderson A, Xue D, et al. Sorafenib (BAY 43-9006) inhibits tumor growth and vascularization and induces tumor apoptosis and hypoxia in RCC xenograft models. Cancer Chemother Pharmacol. 2007;59(5):561–74.
Llovet JM, Ricci S, Mazzaferro V, Hilgard P, Gane E, Blanc JF, et al. Sorafenib in advanced hepatocellular carcinoma. N Engl J Med. 2008;359(4):378–90.
Cheng AL, Kang YK, Chen Z, Tsao CJ, Qin S, Kim JS, et al. Efficacy and safety of sorafenib in patients in the Asia-Pacific region with advanced hepatocellular carcinoma: a phase III randomised, double-blind, placebo-controlled trial. Lancet Oncol. 2009;10(1):25–34.
Iavarone M, Cabibbo G, Piscaglia F, Zavaglia C, Grieco A, Villa E, et al. Field-practice study of sorafenib therapy for hepatocellular carcinoma: a prospective multicenter study in Italy. Hepatology. 2011;54(6):2055–63.
Iavarone M, Cabibbo G, Biolato M, Della Corte C, Maida M, Barbara M, et al. Predictors of survival in patients with advanced hepatocellular carcinoma who permanently discontinued sorafenib. Hepatology. 2015;62(3):784–91.
Zhu AX, Rosmorduc O, Evans TR, Ross PJ, Santoro A, Carrilho FJ, et al. SEARCH: a phase III, randomized, double-blind, placebo-controlled trial of sorafenib plus erlotinib in patients with advanced hepatocellular carcinoma. J Clin Oncol. 2015;33(6):559–66.
Johnson PJ, Qin S, Park JW, Poon RT, Raoul JL, Philip PA, et al. Brivanib versus sorafenib as first-line therapy in patients with unresectable, advanced hepatocellular carcinoma: results from the randomized phase III BRISK-FL study. J Clin Oncol. 2013;31(28):3517–24.
Cheng AL, Kang YK, Lin DY, Park JW, Kudo M, Qin S, et al. Sunitinib versus sorafenib in advanced hepatocellular cancer: results of a randomized phase III trial. J Clin Oncol. 2013;31(32):4067–75.
Cainap C, Qin S, Huang WT, Chung IJ, Pan H, Cheng Y, et al. Linifanib versus Sorafenib in patients with advanced hepatocellular carcinoma: results of a randomized phase III trial. J Clin Oncol. 2015;33(2):172–9.
Zhu AX, Kudo M, Assenat E, Cattan S, Kang YK, Lim HY, et al. Effect of everolimus on survival in advanced hepatocellular carcinoma after failure of sorafenib: the EVOLVE-1 randomized clinical trial. Jama. 2014;312(1):57–67.
Rimassa L, Assenat E, Peck-Radosavljevic M, Pracht M, Zagonel V, Mathurin P, et al. Tivantinib for second-line treatment of MET-high, advanced hepatocellular carcinoma (METIV-HCC): a final analysis of a phase 3, randomised, placebo-controlled study. Lancet Oncol. 2018;19(5):682–93.
Kudo M, Finn RS, Qin S, Han KH, Ikeda K, Piscaglia F, et al. Lenvatinib versus sorafenib in first-line treatment of patients with unresectable hepatocellular carcinoma: a randomised phase 3 non-inferiority trial. Lancet. 2018;391(10126):1163–73.
Matsui J, Funahashi Y, Uenaka T, Watanabe T, Tsuruoka A, Asada M. Multi-kinase inhibitor E7080 suppresses lymph node and lung metastases of human mammary breast tumor MDA-MB-231 via inhibition of vascular endothelial growth factor-receptor (VEGF-R) 2 and VEGF-R3 kinase. Clin Cancer Res. 2008;14(17):5459–65.
Yamamoto Y, Matsui J, Matsushima T, Obaishi H, Miyazaki K, Nakamura K, et al. Lenvatinib, an angiogenesis inhibitor targeting VEGFR/FGFR, shows broad antitumor activity in human tumor xenograft models associated with microvessel density and pericyte coverage. Vasc Cell. 2014;6:18.
FDA approves lenvatinib for unresectable hepatocellular carcinoma [https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-lenvatinib-unresectable-hepatocellular-carcinoma]
Wilhelm SM, Dumas J, Adnane L, Lynch M, Carter CA, Schütz G, et al. Regorafenib (BAY 73-4506): a new oral multikinase inhibitor of angiogenic, stromal and oncogenic receptor tyrosine kinases with potent preclinical antitumor activity. Int J Cancer. 2011;129(1):245–55.
Tsai JH, Lee WM. Tie2 in tumor endothelial signaling and survival: implications for antiangiogenic therapy. Mol Cancer Res. 2009;7(3):300–10.
Regorafenib [https://www.fda.gov/drugs/resources-information-approved-drugs/regorafenib]
Bruix J, Qin S, Merle P, Granito A, Huang Y-H, Bodoky G, et al. Regorafenib for patients with hepatocellular carcinoma who progressed on sorafenib treatment (RESORCE): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet. 2017;389(10064):56–66.
Finn RS, Merle P, Granito A, Huang YH, Bodoky G, Pracht M, et al. Outcomes of sequential treatment with sorafenib followed by regorafenib for HCC: Additional analyses from the phase III RESORCE trial. J Hepatol. 2018;69(2):353–8.
Yakes FM, Chen J, Tan J, Yamaguchi K, Shi Y, Yu P, et al. Cabozantinib (XL184), a novel MET and VEGFR2 inhibitor, simultaneously suppresses metastasis, angiogenesis, and tumor growth. Mol Cancer Ther. 2011;10(12):2298–308.
Xiang Q, Chen W, Ren M, Wang J, Zhang H, Deng DY, et al. Cabozantinib suppresses tumor growth and metastasis in hepatocellular carcinoma by a dual blockade of VEGFR2 and MET. Clin Cancer Res. 2014;20(11):2959–70.
Abou-Alfa GK, Meyer T, Cheng AL, El-Khoueiry AB, Rimassa L, Ryoo BY, et al. Cabozantinib in Patients with Advanced and Progressing Hepatocellular Carcinoma. N Engl J Med. 2018;379(1):54–63.
FDA approves cabozantinib for hepatocellular carcinoma [https://www.fda.gov/drugs/fda-approves-cabozantinib-hepatocellular-carcinoma]
Spratlin JL, Cohen RB, Eadens M, Gore L, Camidge DR, Diab S, et al. Phase I pharmacologic and biologic study of ramucirumab (IMC-1121B), a fully human immunoglobulin G1 monoclonal antibody targeting the vascular endothelial growth factor receptor-2. J Clin Oncol. 2010;28(5):780–7.
Zhu AX, Finn RS, Mulcahy M, Gurtler J, Sun W, Schwartz JD, et al. A phase II and biomarker study of ramucirumab, a human monoclonal antibody targeting the VEGF receptor-2, as first-line monotherapy in patients with advanced hepatocellular cancer. Clin Cancer Res. 2013;19(23):6614–23.
Zhu AX, Park JO, Ryoo BY, Yen CJ, Poon R, Pastorelli D, et al. Ramucirumab versus placebo as second-line treatment in patients with advanced hepatocellular carcinoma following first-line therapy with sorafenib (REACH): a randomised, double-blind, multicentre, phase 3 trial. Lancet Oncol. 2015;16(7):859–70.
Zhu AX, Kang YK, Yen CJ, Finn RS, Galle PR, Llovet JM, et al. Ramucirumab after sorafenib in patients with advanced hepatocellular carcinoma and increased α-fetoprotein concentrations (REACH-2): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2019;20(2):282–96.
FDA approves ramucirumab for hepatocellular carcinoma [FDA approves ramucirumab for hepatocellular carcinoma]
Hato T, Goyal L, Greten TF, Duda DG, Zhu AX. Immune checkpoint blockade in hepatocellular carcinoma: current progress and future directions. Hepatology. 2014;60(5):1776–82.
Lu C, Rong D, Zhang B, Zheng W, Wang X, Chen Z, et al. Current perspectives on the immunosuppressive tumor microenvironment in hepatocellular carcinoma: challenges and opportunities. Mol Cancer. 2019;18(1):130.
Sangro B, Gomez-Martin C, de la Mata M, Inarrairaegui M, Garralda E, Barrera P, et al. A clinical trial of CTLA-4 blockade with tremelimumab in patients with hepatocellular carcinoma and chronic hepatitis C. J Hepatol. 2013;59(1):81–8.
Kudo M. Immune Checkpoint Inhibition in Hepatocellular Carcinoma: Basics and Ongoing Clinical Trials. Oncology. 2017;92(Suppl 1):50–62.
El-Khoueiry AB, Sangro B, Yau T, Crocenzi TS, Kudo M, Hsu C, et al. Nivolumab in patients with advanced hepatocellular carcinoma (CheckMate 040): an open-label, non-comparative, phase 1/2 dose escalation and expansion trial. Lancet. 2017;389(10088):2492–502.
FDA grants accelerated approval to nivolumab for HCC previously treated with sorafenib [https://www.fda.gov/drugs/resources-information-approved-drugs/fda-grants-accelerated-approval-nivolumab-hcc-previously-treated-sorafenib]. Accessed 22 Sept 2017.
Yau TPJ, Finn RS, Cheng A-L, Mathurin P, Edeline J, et al. CheckMate 459: A randomized, multi-center phase III study of nivolumab (NIVO) vs sorafenib (SOR) as first-line (1L) treatment in patients (pts) with advanced hepatocellular carcinoma (aHCC) [abstract LBA38_PR]. Annals of Oncology. 2019;30:v874–5.
Sharma P, Allison JP. The future of immune checkpoint therapy. Science. 2015;348(6230):56–61.
Zhu AX, Finn RS, Edeline J, Cattan S, Ogasawara S, Palmer D, et al. Pembrolizumab in patients with advanced hepatocellular carcinoma previously treated with sorafenib (KEYNOTE-224): a non-randomised, open-label phase 2 trial. Lancet Oncol. 2018;19(7):940–52.
FDA grants accelerated approval to pembrolizumab for hepatocellular carcinoma [https://www.fda.gov/drugs/fda-grants-accelerated-approval-pembrolizumab-hepatocellular-carcinoma]. Accessed 9 Nov 2018.
Jean-Luc Van Laethem IB, Mark Karwal, Chris Verslype, Hans Van Vlierberghe, Adel Kardosh, Vittorina Zagonel, Per Stal, Debashis Sarker, Daniel H. Palmer, Arndt Vogel, Julien Edeline, Stéphane Cattan, Masatoshi Kudo, Ann-Lii Cheng, Sadahisa Ogasawara, Abby B. Siegel, Michael Jon Chisamore, Anran Wang, Andrew X. Zhu: Pembrolizumab (pembro) monotherapy for previously untreated advanced hepatocellular carcinoma (HCC): Phase II KEYNOTE-224 study. J Clin Oncol. 2021; 39 (suppl 33); abstr 297.
Philippe Merle JE, Bouattour M, Cheng A-L, Chan SL, Yau T, Garrido M, et al. Finn: Pembrolizumab (pembro) vs placebo (pbo) in patients (pts) with advanced hepatocellular carcinoma (aHCC) previously treated with sorafenib: Updated data from the randomized, phase III KEYNOTE-240 study. J Clin Oncol. 2021;39(suppl 33):abstr 268.
Wolchok JD, Chiarion-Sileni V, Gonzalez R, Rutkowski P, Grob JJ, Cowey CL, et al. Overall Survival with Combined Nivolumab and Ipilimumab in Advanced Melanoma. N Engl J Med. 2017;377(14):1345–56.
Zongyi Y, Xiaowu L. Immunotherapy for hepatocellular carcinoma. Cancer Lett. 2020;470:8–17.
Weis SM, Cheresh DA. Tumor angiogenesis: molecular pathways and therapeutic targets. Nat Med. 2011;17(11):1359–70.
Jain RK. Normalizing tumor vasculature with anti-angiogenic therapy: a new paradigm for combination therapy. Nat Med. 2001;7(9):987–9.
Liu K, Zhang X, Xu W, Chen J, Yu J, Gamble JR, et al. Targeting the vasculature in hepatocellular carcinoma treatment: Starving versus normalizing blood supply. Clin Transl Gastroenterol. 2017;8(6):e98.
Kwilas AR, Donahue RN, Tsang KY, Hodge JW. Immune consequences of tyrosine kinase inhibitors that synergize with cancer immunotherapy. Cancer Cell Microenviron. 2015:2(1).
Alfaro C, Suarez N, Gonzalez A, Solano S, Erro L, Dubrot J, et al. Influence of bevacizumab, sunitinib and sorafenib as single agents or in combination on the inhibitory effects of VEGF on human dendritic cell differentiation from monocytes. Br J Cancer. 2009;100(7):1111–9.
Pinter M, Jain RK, Duda DG. The Current Landscape of Immune Checkpoint Blockade in Hepatocellular Carcinoma: A Review. JAMA Oncol. 2021;7(1):113–23.
Hodi FS, Chesney J, Pavlick AC, Robert C, Grossmann KF, McDermott DF, et al. Combined nivolumab and ipilimumab versus ipilimumab alone in patients with advanced melanoma: 2-year overall survival outcomes in a multicentre, randomised, controlled, phase 2 trial. Lancet Oncol. 2016;17(11):1558–68.
Hellmann MD, Rizvi NA, Goldman JW, Gettinger SN, Borghaei H, Brahmer JR, et al. Nivolumab plus ipilimumab as first-line treatment for advanced non-small-cell lung cancer (CheckMate 012): results of an open-label, phase 1, multicohort study. Lancet Oncol. 2017;18(1):31–41.
Motzer RJ, Tannir NM, McDermott DF, Arén Frontera O, Melichar B, Choueiri TK, et al. Nivolumab plus Ipilimumab versus Sunitinib in Advanced Renal-Cell Carcinoma. N Engl J Med. 2018;378(14):1277–90.
Overman MJ, Lonardi S, Wong KYM, Lenz HJ, Gelsomino F, Aglietta M, et al. Durable Clinical Benefit With Nivolumab Plus Ipilimumab in DNA Mismatch Repair-Deficient/Microsatellite Instability-High Metastatic Colorectal Cancer. J Clin Oncol. 2018;36(8):773–9.
FDA grants accelerated approval to nivolumab and ipilimumab combination for hepatocellular carcinoma [https://www.fda.gov/drugs/resources-information-approved-drugs/fda-grants-accelerated-approval-nivolumab-and-ipilimumab-combination-hepatocellular-carcinoma]. Accessed 10 Mar 2020.
El-Khoueiry TY AB, Kang Y-K, Kim T-Y, Santoro A, Sangro B, Melero I, et al. Nivolumab (NIVO) plus ipilimumab (IPI) combination therapy in patients (Pts) with advanced hepatocellular carcinoma (aHCC): Long-term results from CheckMate 040. J Clin Oncol. 2021;39(suppl 33):abstr 269.
Herbst RS, Soria JC, Kowanetz M, Fine GD, Hamid O, Gordon MS, et al. Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature. 2014;515(7528):563–7.
Finn RS, Bentley G, Britten CD, Amado R, Busuttil RW. Targeting vascular endothelial growth factor with the monoclonal antibody bevacizumab inhibits human hepatocellular carcinoma cells growing in an orthotopic mouse model. Liver Int. 2009;29(2):284–90.
Siegel AB, Cohen EI, Ocean A, Lehrer D, Goldenberg A, Knox JJ, et al. Phase II trial evaluating the clinical and biologic effects of bevacizumab in unresectable hepatocellular carcinoma. J Clin Oncol. 2008;26(18):2992–8.
Boige V, Malka D, Bourredjem A, Dromain C, Baey C, Jacques N, et al. Efficacy, safety, and biomarkers of single-agent bevacizumab therapy in patients with advanced hepatocellular carcinoma. Oncologist. 2012;17(8):1063–72.
Finn RS, Qin S, Ikeda M, Galle PR, Ducreux M, Kim TY, et al. Atezolizumab plus Bevacizumab in Unresectable Hepatocellular Carcinoma. N Engl J Med. 2020;382(20):1894–905.
FDA approves atezolizumab plus bevacizumab for unresectable hepatocellular carcinoma [https://www.fda.gov/drugs/drug-approvals-and-databases/fda-approves-atezolizumab-plus-bevacizumab-unresectable-hepatocellular-carcinoma]. Accessed 29 May 2020.
Finn SQ RS, Ikeda M, Galle PR, Ducreux M, Kim T-Y, Lim HY, et al. IMbrave150: Updated overall survival (OS) data from a global, randomized, open-label phase III study of atezolizumab (atezo) + bevacizumab (bev) versus sorafenib (sor) in patients (pts) with unresectable hepatocellular carcinoma (HCC). J Clin Oncol. 2021;39(suppl 33):abstr 267.
Yang J, Yan J, Liu B. Targeting VEGF/VEGFR to Modulate Antitumor Immunity. Front Immunol. 2018;9:978.
Nakamura K, Taguchi E, Miura T, Yamamoto A, Takahashi K, Bichat F, et al. KRN951, a highly potent inhibitor of vascular endothelial growth factor receptor tyrosine kinases, has antitumor activities and affects functional vascular properties. Cancer Res. 2006;66(18):9134–42.
Fountzilas C, Gupta M, Lee S, Krishnamurthi S, Estfan B, Wang K, et al. A multicentre phase 1b/2 study of tivozanib in patients with advanced inoperable hepatocellular carcinoma. Br J Cancer. 2020;122(7):963–70.
Feng Bi SQ, Shanzhi G, Bai Y, Chen Z, Wang Z, Ying J, et al. Donafenib versus sorafenib as first-line therapy in advanced hepatocellular carcinoma: An open-label, randomized, multicenter phase II/III trial. J Clin Oncol. 2020;38(suppl):abstr 4506.
Qin S, Li Q, Gu S, Chen X, Lin L, Wang Z, et al. Apatinib as second-line or later therapy in patients with advanced hepatocellular carcinoma (AHELP): a multicentre, double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Gastroenterol Hepatol. 2021.
Fasolo A, Sessa C, Gianni L, Broggini M. Seminars in clinical pharmacology: an introduction to MET inhibitors for the medical oncologist. Ann Oncol. 2013;24(1):14–20.
Xin Y, Jin D, Eppler S, Damico-Beyer LA, Joshi A, Davis JD, et al. Population pharmacokinetic analysis from phase I and phase II studies of the humanized monovalent antibody, onartuzumab (MetMAb), in patients with advanced solid tumors. J Clin Pharmacol. 2013;53(11):1103–11.
Boccaccio C, Comoglio PM. Invasive growth: a MET-driven genetic programme for cancer and stem cells. Nat Rev Cancer. 2006;6(8):637–45.
Chen J, Jin R, Zhao J, Liu J, Ying H, Yan H, et al. Potential molecular, cellular and microenvironmental mechanism of sorafenib resistance in hepatocellular carcinoma. Cancer Lett. 2015;367(1):1–11.
Bouattour M, Raymond E, Qin S, Cheng AL, Stammberger U, Locatelli G, et al. Recent developments of c-Met as a therapeutic target in hepatocellular carcinoma. Hepatology. 2018;67(3):1132–49.
Decaens T, Barone C, Assenat E, Wermke M, Fasolo A, Merle P, et al. Phase 1b/2 trial of tepotinib in sorafenibpretreated advanced hepatocellular carcinoma with MET overexpression. Br J Cancer. 2021.
Drabsch Y, ten Dijke P. TGF-β signalling and its role in cancer progression and metastasis. Cancer Metastasis Rev. 2012;31(3-4):553–68.
Neuzillet C, Tijeras-Raballand A, Cohen R, Cros J, Faivre S, Raymond E, et al. Targeting the TGFbeta pathway for cancer therapy. Pharmacol Ther. 2015;147:22–31.
Chen W, Ten Dijke P. Immunoregulation by members of the TGFβ superfamily. Nat Rev Immunol. 2016;16(12):723–40.
de Gramont A, Faivre S, Raymond E. Novel TGF-beta inhibitors ready for prime time in onco-immunology. Oncoimmunology. 2017;6(1):e1257453.
Yingling JM, McMillen WT, Yan L, Huang H, Sawyer JS, Graff J, et al. Preclinical assessment of galunisertib (LY2157299 monohydrate), a first-in-class transforming growth factor-β receptor type I inhibitor. Oncotarget. 2018;9(6):6659–77.
Kelley RK, Gane E, Assenat E, Siebler J, Galle PR, Merle P, et al. A Phase 2 Study of Galunisertib (TGF-β1 Receptor Type I Inhibitor) and Sorafenib in Patients With Advanced Hepatocellular Carcinoma. Clin Transl Gastroenterol. 2019;10(7):e00056.
Jeng KS, Sheen IS, Lin SS, Leu CM, Chang CF. The Role of Endoglin in Hepatocellular Carcinoma. Int J Mol Sci. 2021;22:6.
Wikström P, Lissbrant IF, Stattin P, Egevad L, Bergh A. Endoglin (CD105) is expressed on immature blood vessels and is a marker for survival in prostate cancer. Prostate. 2002;51(4):268–75.
Henry-Berger J, Mouzat K, Baron S, Bernabeu C, Marceau G, Saru JP, et al. Endoglin (CD105) expression is regulated by the liver X receptor alpha (NR1H3) in human trophoblast cell line JAR. Biol Reprod. 2008;78(6):968–75.
Yang LY, Lu WQ, Huang GW, Wang W. Correlation between CD105 expression and postoperative recurrence and metastasis of hepatocellular carcinoma. BMC Cancer. 2006;6:110.
Elnemr DM, Abdel-Azeez HA, Labib HA, Abo-Taleb FM. Clinical relevance of serum endoglin level in Egyptian hepatocellular carcinoma patients. Clin Lab. 2012;58(9-10):1023–8.
Teama S, Fawzy A, Teama S, Helal A, Drwish AD, Elbaz T, et al. Increased Serum Endoglin and Transforming Growth Factor β1 mRNA Expression and Risk of Hepatocellular Carcinoma in Cirrhotic Egyptian Patients. Asian Pac J Cancer Prev. 2016;17(5):2429–34.
Duffy AG, Ulahannan SV, Cao L, Rahma OE, Makarova-Rusher OV, Kleiner DE, et al. A phase II study of TRC105 in patients with hepatocellular carcinoma who have progressed on sorafenib. United European Gastroenterol J. 2015;3(5):453–61.
Duffy AG, Ma C, Ulahannan SV, Rahma OE, Makarova-Rusher O, Cao L, et al. Phase I and Preliminary Phase II Study of TRC105 in Combination with Sorafenib in Hepatocellular Carcinoma. Clin Cancer Res. 2017;23(16):4633–41.
RTL KPSR, Paluri RK, Mody K, Simpson B, Adams BJ, Theuer CP. Ahmed Omar Kaseb: An open-label phase Ib/2 trial of TRC105 plus sorafenib in patients with advanced/metastatic hepatocellular carcinoma (HCC) (NCT01806064). J Clin Oncol. 2019;37(suppl 34):abstr 268).
Hughes SE. Differential expression of the fibroblast growth factor receptor (FGFR) multigene family in normal human adult tissues. J Histochem Cytochem. 1997;45(7):1005–19.
Lee S, Choi J, Mohanty J, Sousa LP, Tome F, Pardon E, et al. Structures of β-klotho reveal a 'zip code'-like mechanism for endocrine FGF signalling. Nature. 2018;553(7689):501–5.
Wu X, Ge H, Lemon B, Vonderfecht S, Weiszmann J, Hecht R, et al. FGF19-induced hepatocyte proliferation is mediated through FGFR4 activation. J Biol Chem. 2010;285(8):5165–70.
Lu X, Chen H, Patterson AV, Smaill JB, Ding K. Fibroblast Growth Factor Receptor 4 (FGFR4) Selective Inhibitors as Hepatocellular Carcinoma Therapy: Advances and Prospects. J Med Chem. 2019;62(6):2905–15.
Dai S, Zhou Z, Chen Z, Xu G, Chen Y. Fibroblast Growth Factor Receptors (FGFRs): Structures and Small Molecule Inhibitors. Cells. 2019;8(6).
Yue S, Li Y, Chen X, Wang J, Li M, Chen Y, et al. FGFR-TKI resistance in cancer: current status and perspectives. J Hematol Oncol. 2021;14(1):23.
Kim RD, Sarker D, Meyer T, Yau T, Macarulla T, Park JW, et al. First-in-Human Phase I Study of Fisogatinib (BLU-554) Validates Aberrant FGF19 Signaling as a Driver Event in Hepatocellular Carcinoma. Cancer Discov. 2019;9(12):1696–707.
Jia JB, Wang WQ, Sun HC, Zhu XD, Liu L, Zhuang PY, et al. High expression of macrophage colony-stimulating factor-1 receptor in peritumoral liver tissue is associated with poor outcome in hepatocellular carcinoma after curative resection. Oncologist. 2010;15(7):732–43.
Pollard JW. Tumour-educated macrophages promote tumour progression and metastasis. Nat Rev Cancer. 2004;4(1):71–8.
Condeelis J, Pollard JW. Macrophages: obligate partners for tumor cell migration, invasion, and metastasis. Cell. 2006;124(2):263–6.
Ao JY, Zhu XD, Chai ZT, Cai H, Zhang YY, Zhang KZ, et al. Colony-Stimulating Factor 1 Receptor Blockade Inhibits Tumor Growth by Altering the Polarization of Tumor-Associated Macrophages in Hepatocellular Carcinoma. Mol Cancer Ther. 2017;16(8):1544–54.
Zhang W, Zhu XD, Sun HC, Xiong YQ, Zhuang PY, Xu HX, et al. Depletion of tumor-associated macrophages enhances the effect of sorafenib in metastatic liver cancer models by antimetastatic and antiangiogenic effects. Clin Cancer Res. 2010;16(13):3420–30.
Qin S, Ren Z, Meng Z, Chen Z, Chai X, Xiong J, et al. Camrelizumab in patients with previously treated advanced hepatocellular carcinoma: a multicentre, open-label, parallel-group, randomised, phase 2 trial. Lancet Oncol. 2020;21(4):571–80.
Lee DW, Cho EJ, Lee JH, Yu SJ, Kim YJ, Yoon JH, et al. Phase II Study of Avelumab in Patients with Advanced Hepatocellular Carcinoma Previously Treated with Sorafenib. Clin Cancer Res. 2021;27(3):713–8.
Zhou G, Sprengers D, Boor PPC, Doukas M, Schutz H, Mancham S, et al. Antibodies Against Immune Checkpoint Molecules Restore Functions of Tumor-Infiltrating T Cells in Hepatocellular Carcinomas. Gastroenterology. 2017;153(4):1107–1119.e1110.
Anderson AC, Joller N, Kuchroo VK. Lag-3, Tim-3, and TIGIT: Co-inhibitory Receptors with Specialized Functions in Immune Regulation. Immunity. 2016;44(5):989–1004.
Yu X, Harden K, Gonzalez LC, Francesco M, Chiang E, Irving B, et al. The surface protein TIGIT suppresses T cell activation by promoting the generation of mature immunoregulatory dendritic cells. Nat Immunol. 2009;10(1):48–57.
Liu F, Liu Y, Chen Z. Tim-3 expression and its role in hepatocellular carcinoma. J Hematol Oncol. 2018;11(1):126.
Marin-Acevedo JA, Dholaria B, Soyano AE, Knutson KL, Chumsri S, Lou Y. Next generation of immune checkpoint therapy in cancer: new developments and challenges. J Hematol Oncol. 2018;11(1):39.
El-Khoueiry AB, Spano J-P, Angevin E, Doi T, Bullock AJ, Harris WP, et al. Analysis of OX40 agonist antibody (PF-04518600) in patients with hepatocellular carcinoma. J Clin Oncol. 2020;38(4_suppl):523.
Buzzatti G, Dellepiane C, Del Mastro L. New emerging targets in cancer immunotherapy: the role of GITR. ESMO Open. 2020;4(Suppl 3):e000738.
van Beek AA, Zhou G, Doukas M, Boor PPC, Noordam L, Mancham S, et al. GITR ligation enhances functionality of tumor-infiltrating T cells in hepatocellular carcinoma. Int J Cancer. 2019;145(4):1111–24.
Tsai AK, Khan AY, Worgo CE, Wang LL, Liang Y, Davila E. A Multikinase and DNA-PK Inhibitor Combination Immunomodulates Melanomas, Suppresses Tumor Progression, and Enhances Immunotherapies. Cancer Immunol Res. 2017;5(9):790–803.
Noy R, Pollard JW. Tumor-associated macrophages: from mechanisms to therapy. Immunity. 2014;41(1):49–61.
MERCK AND EISAI RECEIVE THIRD BREAKTHROUGH THERAPY DESIGNATION FROM FDA FOR KEYTRUDA® (PEMBROLIZUMAB) PLUS LENVIMA® (LENVATINIB) COMBINATION TREATMENT [https://eisai.mediaroom.com/Merck-and-Eisai-Receive-Third-Breakthrough-Therapy-Designation-from-FDA-for-KEYTRUDA-R-pembrolizumab-plus-LENVIMA-R-lenvatinib-Combination-Treatment]
Finn RS, Ikeda M, Zhu AX, Sung MW, Baron AD, Kudo M, et al. Phase Ib Study of Lenvatinib Plus Pembrolizumab in Patients With Unresectable Hepatocellular Carcinoma. J Clin Oncol. 2020;38(26):2960–70.
Kwilas AR, Ardiani A, Donahue RN, Aftab DT, Hodge JW. Dual effects of a targeted small-molecule inhibitor (cabozantinib) on immune-mediated killing of tumor cells and immune tumor microenvironment permissiveness when combined with a cancer vaccine. J Transl Med. 2014;12:294.
Masatoshi Kudo KM, Wada Y, Inaba Y, Sakamoto Y, Kurosaki M, Umeyama Y, et al. First-line avelumab + axitinib in patients with advanced hepatocellular carcinoma: Results from a phase 1b trial (VEGF Liver 100). J Clin Oncol. 2019;37(suppl):abstr 4072.
Xu J, Zhang Y, Jia R, Yue C, Chang L, Liu R, et al. Anti-PD-1 Antibody SHR-1210 Combined with Apatinib for Advanced Hepatocellular Carcinoma, Gastric, or Esophagogastric Junction Cancer: An Open-label, Dose Escalation and Expansion Study. Clin Cancer Res. 2019;25(2):515–23.
Renuka V, Iyer DL, Dayyani F, Phan AT, Needle MN, Abrams TA. DEDUCTIVE: A study of tivozanib in combination with durvalumab in subjects with untreated advanced hepatocellular carcinoma—Phase Ib results. J Clin Oncol. 2021;39(suppl 33):abstr 294.
Robin Kate Kelley BS, Harris WP, Ikeda M, Okusaka T, Kang Y-K, Qin S, et al. Efficacy, tolerability, and biologic activity of a novel regimen of tremelimumab (T) in combination with durvalumab (D) for patients (pts) with advanced hepatocellular carcinoma (aHCC). J Clin Oncol. 2020;38(suppl):abstr 4508.
Birrer MJ, Moore KN, Betella I, Bates RC. Antibody-Drug Conjugate-Based Therapeutics: State of the Science. J Natl Cancer Inst. 2019;111(6):538–49.
Khongorzul P, Ling CJ, Khan FU, Ihsan AU, Zhang J. Antibody-Drug Conjugates: A Comprehensive Review. Mol Cancer Res. 2020;18(1):3–19.
Chau CH, Steeg PS, Figg WD. Antibody-drug conjugates for cancer. Lancet. 2019;394(10200):793–804.
Capurro M, Wanless IR, Sherman M, Deboer G, Shi W, Miyoshi E, et al. Glypican-3: a novel serum and histochemical marker for hepatocellular carcinoma. Gastroenterology. 2003;125(1):89–97.
Yamashita T, Forgues M, Wang W, Kim JW, Ye Q, Jia H, et al. EpCAM and alpha-fetoprotein expression defines novel prognostic subtypes of hepatocellular carcinoma. Cancer Res. 2008;68(5):1451–61.
Christian S, Winkler R, Helfrich I, Boos AM, Besemfelder E, Schadendorf D, et al. Endosialin (Tem1) is a marker of tumor-associated myofibroblasts and tumor vessel-associated mural cells. Am J Pathol. 2008;172(2):486–94.
Schmidt N, Flecken T, Thimme R. Tumor-associated antigen specific CD8(+) T cells in hepatocellular carcinoma - a promising target for immunotherapy. Oncoimmunology. 2014;3(9):e954919.
Huang L, Zhao C, Sun K, Yang D, Yan L, Luo D, et al. Downregulation of CLDN6 inhibits cell proliferation, migration, and invasion via regulating EGFR/AKT/mTOR signalling pathway in hepatocellular carcinoma. Cell Biochem Funct. 2020;38(5):541–8.
Wang J, Wu Z, Pan G, Ni J, Xie F, Jiang B, et al. Enhanced doxorubicin delivery to hepatocellular carcinoma cells via CD147 antibody-conjugated immunoliposomes. Nanomedicine. 2018;14(6):1949–61.
Ma Z, He H, Sun F, Xu Y, Huang X, Ma Y, et al. Selective targeted delivery of doxorubicin via conjugating to anti-CD24 antibody results in enhanced antitumor potency for hepatocellular carcinoma both in vitro and in vivo. J Cancer Res Clin Oncol. 2017;143(10):1929–40.
Chen X, Ding G, Gao Q, Sun J, Zhang Q, Du L, et al. A human anti-c-Met Fab fragment conjugated with doxorubicin as targeted chemotherapy for hepatocellular carcinoma. PLoS ONE. 2013;8(5):e63093.
Bardia A, Messersmith WA, Kio EA, Berlin JD, Vahdat L, Masters GA, et al. Sacituzumab govitecan, a Trop-2-directed antibody-drug conjugate, for patients with epithelial cancer: final safety and efficacy results from the phase I/II IMMU-132-01 basket trial. Ann Oncol. 2021;32(6):746–56.
Fu Y, Urban DJ, Nani RR, Zhang YF, Li N, Fu H, et al. Glypican-3-Specific Antibody Drug Conjugates Targeting Hepatocellular Carcinoma. Hepatology. 2019;70(2):563–76.
Kong FE, Li GM, Tang YQ, Xi SY, Loong JHC, Li MM, et al. Targeting tumor lineage plasticity in hepatocellular carcinoma using an anti-CLDN6 antibody-drug conjugate. Sci Transl Med. 2021;13(579).
Zhang P, Shi B, Gao H, Jiang H, Kong J, Yan J, et al. An EpCAM/CD3 bispecific antibody efficiently eliminates hepatocellular carcinoma cells with limited galectin-1 expression. Cancer Immunol Immunother. 2014;63(2):121–32.
Kebenko M, Goebeler ME, Wolf M, Hasenburg A, Seggewiss-Bernhardt R, Ritter B, et al. A multicenter phase 1 study of solitomab (MT110, AMG 110), a bispecific EpCAM/CD3 T-cell engager (BiTE®) antibody construct, in patients with refractory solid tumors. Oncoimmunology. 2018;7(8):e1450710.
Ishiguro T, Sano Y, Komatsu SI, Kamata-Sakurai M, Kaneko A, Kinoshita Y, et al. An anti-glypican 3/CD3 bispecific T cell-redirecting antibody for treatment of solid tumors. Sci Transl Med. 2017;9:410.
June CH, O'Connor RS, Kawalekar OU, Ghassemi S, Milone MC. CAR T cell immunotherapy for human cancer. Science. 2018;359(6382):1361–5.
Feins S, Kong W, Williams EF, Milone MC, Fraietta JA. An introduction to chimeric antigen receptor (CAR) T-cell immunotherapy for human cancer. Am J Hematol. 2019;94(S1):S3–9.
FDA approves tisagenlecleucel for B-cell ALL and tocilizumab for cytokine release syndrome [https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-tisagenlecleucel-b-cell-all-and-tocilizumab-cytokine-release-syndrome]. Accessed 7 Sept 2017.
Maude SL, Laetsch TW, Buechner J, Rives S, Boyer M, Bittencourt H, et al. Tisagenlecleucel in Children and Young Adults with B-Cell Lymphoblastic Leukemia. N Engl J Med. 2018;378(5):439–48.
Neelapu SS, Locke FL, Bartlett NL, Lekakis LJ, Miklos DB, Jacobson CA, et al. Axicabtagene Ciloleucel CAR T-Cell Therapy in Refractory Large B-Cell Lymphoma. N Engl J Med. 2017;377(26):2531–44.
FDA approves CAR-T cell therapy to treat adults with certain types of large B-cell lymphoma [https://www.fda.gov/news-events/press-announcements/fda-approves-car-t-cell-therapy-treat-adults-certain-types-large-b-cell-lymphoma]. Accessed 21 Mar 2018.
Abramson JS, Palomba ML, Gordon LI, Lunning MA, Wang M, Arnason J, et al. Lisocabtagene maraleucel for patients with relapsed or refractory large B-cell lymphomas (TRANSCEND NHL 001): a multicentre seamless design study. Lancet. 2020;396(10254):839–52.
FDA Approves New Treatment For Adults With Relapsed Or Refractory Large-B-Cell Lymphoma [https://www.fda.gov/news-events/press-announcements/fda-approves-new-treatment-adults-relapsed-or-refractory-large-b-cell-lymphoma]. Accessed 5 Feb 2021.
Dal Bo M, De Mattia E, Baboci L, Mezzalira S, Cecchin E, Assaraf YG, et al. New insights into the pharmacological, immunological, and CAR-T-cell approaches in the treatment of hepatocellular carcinoma. Drug Resist Updat. 2020;51:100702.
Batra SA, Rathi P, Guo L, Courtney AN, Fleurence J, Balzeau J, et al. Glypican-3-Specific CAR T Cells Coexpressing IL15 and IL21 Have Superior Expansion and Antitumor Activity against Hepatocellular Carcinoma. Cancer Immunol Res. 2020;8(3):309–20.
Gao H, Li K, Tu H, Pan X, Jiang H, Shi B, et al. Development of T cells redirected to glypican-3 for the treatment of hepatocellular carcinoma. Clin Cancer Res. 2014;20(24):6418–28.
Wu X, Luo H, Shi B, Di S, Sun R, Su J, et al. Combined Antitumor Effects of Sorafenib and GPC3-CAR T Cells in Mouse Models of Hepatocellular Carcinoma. Mol Ther. 2019;27(8):1483–94.
Shi D, Shi Y, Kaseb AO, Qi X, Zhang Y, Chi J, et al. Chimeric Antigen Receptor-Glypican-3 T-Cell Therapy for Advanced Hepatocellular Carcinoma: Results of Phase I Trials. Clin Cancer Res. 2020;26(15):3979–89.
Felices M, Lenvik TR, Davis ZB, Miller JS, Vallera DA. Generation of BiKEs and TriKEs to Improve NK Cell-Mediated Targeting of Tumor Cells. Methods Mol Biol. 2016;1441:333–46.
Goebeler ME, Bargou RC. T cell-engaging therapies - BiTEs and beyond. Nat Rev Clin Oncol. 2020;17(7):418–34.
Ma W, Wu L, Zhou F, Hong Z, Yuan Y, Liu Z. T Cell-Associated Immunotherapy for Hepatocellular Carcinoma. Cell Physiol Biochem. 2017;41(2):609–22.
Sonbol MB, Riaz IB, Naqvi SAA, Almquist DR, Mina S, Almasri J, et al. Systemic Therapy and Sequencing Options in Advanced Hepatocellular Carcinoma: A Systematic Review and Network Meta-analysis. JAMA Oncol. 2020;6(12):e204930.
Park R, Lopes da Silva L, Nissaisorakarn V, Riano I, Williamson S, Sun W, et al. Comparison of Efficacy of Systemic Therapies in Advanced Hepatocellular Carcinoma: Updated Systematic Review and Frequentist Network Meta-Analysis of Randomized Controlled Trials. J Hepatocell Carcinoma. 2021;8:145–54.
Gordan JD, Kennedy EB, Abou-Alfa GK, Beg MS, Brower ST, Gade TP, et al. Systemic Therapy for Advanced Hepatocellular Carcinoma: ASCO Guideline. J Clin Oncol. 2020;38(36):4317–45.
Chen DS, Hurwitz H. Combinations of Bevacizumab With Cancer Immunotherapy. Cancer J. 2018;24(4):193–204.
Faivre S, Rimassa L, Finn RS. Molecular therapies for HCC: Looking outside the box. J Hepatol. 2020;72(2):342–52.
Acknowledgments
Not applicable.
Funding
This study was supported by the National Natural Science Foundation of China (Grants No. 81570537 and No. 81974074 to Y.C.).
Author information
Authors and Affiliations
Contributions
H.Z. reviewed the relevant literature and completed the figures and tables, and wrote the manuscript. Y.C. offered main direction and significant guidance for this manuscript. H.Z., W.Z., and L.J. revised the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Zhang, H., Zhang, W., Jiang, L. et al. Recent advances in systemic therapy for hepatocellular carcinoma. Biomark Res 10, 3 (2022). https://doi.org/10.1186/s40364-021-00350-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40364-021-00350-4