
Abstract
Polycythemia vera (PV) and essential thrombocythemia (ET) are both driven by JAK-STAT pathway activation and consequently much of the recent research efforts to improve the management and outcomes of patients with these neoplasms have centered around inhibition of this pathway. In addition to newer JAK inhibitors and improved interferons, promising novel agents exploiting a growing understanding of PV and ET pathogenesis and disease evolution mechanisms are being developed. These agents may modify the disease course in addition to cytoreduction. Histone deacetylase, MDM2 and telomerase inhibitors in patients with PV/ET have demonstrated clinically efficacy and serve as chief examples. Hepcidin mimetics, limiting iron availability to red blood cell precursors, offer an exciting alternative to therapeutic phlebotomy and have the potential to revolutionize management for patients with PV. Many of these newer agents are found to improve hematologic parameters and symptom burden, but their role in thrombotic risk reduction and disease progression control is currently unknown. The results of larger, randomized studies to confirm the early efficacy signals observed in phase 1/2 trials are eagerly awaited.
Similar content being viewed by others

Introduction
Polycythemia vera (PV) and essential thrombocythemia (ET) are the most common Philadelphia chromosome-negative myeloproliferative neoplasms (MPNs). This observation is not solely due to their incidence of 1.0–2.0 per 100,000 person-years, but also patients’ near-normal life expectancy, culminating in a prevalence of approximately 25–50 per 100,000 people [1]. Although biologically-distinct diseases, ET and PV share a pathogenesis generally rooted in JAK-STAT activation, which prompts the unregulated proliferation of hematopoietic stem/progenitor cells (HSCs). Ninety nine percent of PV is driven by acquired mutations in the Janus kinase 2 (JAK2) gene, almost always JAK2-V617F [2,3,4]. A JAK2-V617F mutation is found in approximately 50% of patients with ET [5,6,7,8]. In contrast to PV, about 20–25 and 5% of ET is characterized by a CALR and MPL mutation, respectively [9,10,11,12]. Up to 10% of ET has no detectable canonical driver mutation and is typically regarded as being “triple negative,” but rare non-canonical CALR, MPL and JAK2 variants have been detected in “triple negative” disease [3, 13].
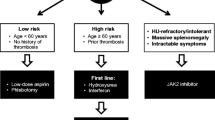
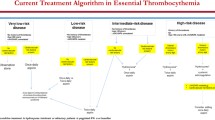
Although predicting a near-normal life expectancy, true to their categorization as myeloid malignancies, PV and ET are associated with an increased risk of the development of thrombosis, secondary myelofibrosis, myelodysplastic syndrome (MDS) and transformation to acute myeloid leukemia (AML) with these entities predicting a shorter life expectancy than otherwise expected with a PV or ET diagnosis itself [14,15,16,17]. In addition to these tangible outcomes, patients with PV and ET are also frequently burdened with symptoms including fatigue, night sweats, headaches, pruritus, early satiety/weight loss, and symptomatic splenomegaly [18]. It is for these reasons as well as to reduce the risk of arterial/venous thrombosis and bleeding that therapy for PV or ET is initiated. However, despite this need and a firm understanding of pathogenesis, hydroxyurea, anagrelide, pegylated interferon alfa 2a (PEG-IFN alfa-2a), ruxolitinib and busulfan constitute the bulk of cytoreductive therapies available to providers charged with the care of patients with PV and ET, but most have little to no evidence of disease-modifying activity. Inteferons have been used for the management of MPN for > 30 years and are favored by some providers due to their immunomodulatory, anti-inflammatory, antiangiogenic and anti-proliferative properties [19]. The introduction of pegylated interferon preparations improved tolerability, but are still associated with flu-like, autoimmune and psychiatric side effects [19]. A recent meta-analysis of 44 studies including 1359 patients treated with interferon (730 ET, 629 PV) showed high response rates among patients with PV (overall response rate [ORR] of 81%, complete hematologic response [CHR] of 59%) and ET (ORR of 77%, CHR of 49%) without significant differences observed between pegylated and non-pegylated formulations [20].
In addition to the high rates of CHR, symptomatic improvement and improvement in or resolution of need for therapeutic phlebotomy, many patients with PV and ET may experience a sustained decrease in JAK2-V617F allelic burden with a potential for disease modification due to malignant clone targeting by interferon [21]. A recent study evaluated interferon discontinuation among patients with PV and ET who accomplished CHR and demonstrated this practice to be safe with persistence of CHR particularly in patients with a driver mutation variant allele frequency < 10% at time of discontinuation; however disease transformation could not be evaluated due to the very low incidence of events [21]. Due to the overall favorable prognosis of patients with PV and ET, studies of survival and disease transformation may be challenging to conduct. The reduction of driver mutation allelic burden may be a feasible surrogate, but its relation to long-term outcomes is uncertain. Lifelong treatment represents a major burden for these chronically-ill patients and have to be well-tolerated without significant toxicities. Interferons may be especially attractive for the management of younger patients with PV or ET due to their lack of teratogenicity. The only unequivocal disease-modifying therapy is allogeneic hematopoietic stem cell transplantation (alloHCT) for which many patients with long life expectancy are not reasonable candidates.
Newer therapies with disease-modifying potential are needed for patients with PV and ET, particularly those patients deemed to have disease that is “higher risk” or resistant to currently available therapies. In this review we discuss some of the emerging and more promising therapies that are being explored and may soon become available to patients with PV or ET as well as some of the prospects for future investigation.
Optimizing the current treatment options
JAK inhibitors
Perhaps the greatest asset to the development of better or novel therapies for PV was the 2005 discovery of the gain-of-function JAK2 V617F exon 14 mutation and the recognition that it drives 95% of PV cases; most of the remaining cases of PV are explained by the presence of a JAK2 exon 12 mutation that was discovered 2 years later in 2007 [7, 22, 23]. The majority of contemporary therapies, including hydroxyurea, busulfan and interferons do not target the principal upstream element of JAK-STAT pathway activation. The JAK 1/2 tyrosine kinase inhibitor ruxolitinib is found to downregulate malignant MPN clone expression of pro-inflammatory cytokines like TNF-α and IL-6, which appear to be heavily dependent on JAK1 and JAK2-mediated activation of STAT3 [24,25,26]. In the RESPONSE trial, when compared with “standard therapy” (including hydroxyurea, interferon, anagrelide, lenalidomide/thalidomide, pipobroman, but excluding 32P, busulfan and chlorambucil), ruxolitinib demonstrated superior benefit with regards to hematocrit control, spleen volume reduction, and symptom burden reduction in patients intolerant of or with inadequate response to hydroxyurea [27]. Later follow-up and meta-analyses also supported its benefit in reducing the incidence of thrombosis compared to these standard therapies [28, 29]. Based on the RESPONSE trial results, ruxoltinib in 2014 became the first Food and Drug Administration (FDA)-approved therapy for PV, specifically for patients with PV who are resistant to or intolerant of hydroxyurea. The randomized, phase 3 RESPONSE-2 trial confirmed these findings in patients specifically without palpable splenomegaly [30]. Other JAK inhibitors, have been studied in PV. Gandotinib, a pan-JAK inhibitor with dose-dependent selectivity for JAK2, demonstrated a 95% ORR in patients with PV (including those previously receiving ruxolitinib) [31, 32] (Table 1).
Given the understanding of the role of JAK-STAT pathway activation in ET, JAK inhibition has been studied in this MPN as well. The first study of ruxolitinib in ET evaluated 39 patients who were refractory to or intolerant of hydroxyurea reported a favorable safety profile and that a majority of patients experienced white blood cell count, platelet count, spleen size and symptom burden reductions [38]. The MAJIC-ET trial randomized 110 patients with ET who were refractory to or intolerant of hydroxyurea to either ruxolitinib or best available therapy with most patients in the latter arm receiving anagrelide [39]. No differences in hematologic responses, thrombosis/bleeding or disease progression were observed between arms; however, a greater and more rapid symptom burden reduction was noted for patients receiving ruxolitinib [39]. RESET 272 is a phase 2 trial currently evaluating ruxolitinib in comparison to anagrelide in patients with ET who were refractory to or intolerant of hydroxyurea (NCT03123588). RUXBETA (NCT02962388) and Ruxobeat (NCT02577926) are ongoing phase 3 trials in the same population comparing ruxolitinib with best available therapy (including anagrelide and interferons). With regards to other JAK inhibitors, the same studies that evaluated gandotinib in PV reported a 90% ORR in ET, including 44% of patients with ET and no detectable JAK mutation [31, 32] (Table 2).
Limiting the enthusiasm for frontline ruxolitinib or other JAK inhibitor therapy for PV or ET is the lack of evidence for disease course modification including long-term risk of secondary (post-PV and post-ET myelofibrosis). Attempts to build upon the practical use of a JAK inhibitor for disease driven by JAK-STAT activation have included studies of combination therapy. The final results of a phase 2 trial of the combination of ruxolitinib with PEG-IFN alfa-2a showed the combination to be reasonably well-tolerated, to decrease both JAK2 V617F allelic burden and symptoms as well as ultimately lead to remission in 10 of 32 patients (31%) with PV who were almost entirely previously-intolerant of or refractory to PEG-IFN alfa-2a [33]. Randomized studies are needed to confirm the independent benefit of the addition of ruxolitinib to interferons for this population of patients.
Interferons
Inteferons remain a promising class of drugs with the potential for disease modification among patients with MPN [20]. Alternative interferon preparations for PV and ET patients are being studied and have largely been developed to improve upon the burdensome weekly administration schedule of currently-used pegylated interferons (e.g. PEG-IFN alfa-2a). Ropeginterferon alfa-2b is produced by Escherichia coli transformed with a human IFN plasmid and specifically modified by an additional N-terminus proline residue and single isomer that leads to an extended half-life allowing for dosing every 2 weeks [41, 42]. The PROUD-PV and CONTINUATION-PV phase 3 trials randomized 257 patients with PV and ≤ 3 years of cytoreductive therapy exposure 1:1 to receive ropeginterferon alfa-2b every 2 weeks or hydroxyurea [34, 41]. While PROUD-PV demonstrated no clear difference between treatment arms, patients treated with ropeginterferon alfa-2b on CONTINUATION-PV had better rate of composite complete hematological response with reduced symptom burden when compared with hydroxyurea (53% vs. 38%, p = 0.044) at 36 months follow-up [34]. In addition, the molecular response, defined as reductions in JAK2-V617F allelic burden, was significantly improved in the ropeginterferon alfa-2b arm when compared with standard therapy (66% vs. 27%; p < 0.0001); these results are durable based on 5 years of follow-up [43]. These data demonstrated that ropeginterferon alfa-2b was at least non-inferior to hydroxyurea for PV patients and led to the European Medical Agency approval of ropeginterferon alfa-2b for the frontline treatment of polycythemia without significant splenomegaly in 2019; the FDA has accepted a Biologics License Application for ropeginterferon alfa-2b for this indication within the United States and a decision is expected during 2021.
Interferons are also among the cytoreductive options for patients with ET. Similar to endeavors dedicated to PV, ropeginterferon alfa-2b is being currently studied in a phase 3 trial randomizing patients who are refractory to or intolerant of hydroxyurea to receive either anagrelide or ropeginterferon alfa-2b (NCT04285086).
Novel agents
In addition to efforts to optimize the use of current standard of care classes of agents, several novel compounds are being studied as therapy for PV. Targeting of epigenetic pathways, particularly the varied status of post-translational histone acetylation/de-acetylation which contribute to the regulation of gene transcription critical to cell cycling, offer a promising therapeautic opportunity [44]. The histone deacetylase inhibitors (HDACis) are theorized to reverse histone/DNA complex compaction and thus open the chromatin structure to restore pro-apoptotic gene transcription [44]. Although an oversimplified view, the HDACis are hypothesized to have a role in the treatment of MPN [45]. The HDACis vorinostat, panobinostat and givinostat have demonstrated that ability to inhibit proliferation and induce apoptosis of JAK2-mutated MPN cells in ex vivo studies [46,47,48]. Givinostat is a novel and potent histone deacetylase inhibitor that in ex vivo experiments was shown to suppress JAK2-V617F as well as STAT3 and STAT5 protein activation, ultimately leading to the inhibition of JAK2-V617F-mutated cell proliferation without affecting JAK2 wild-type cells [48]. Release of pro-inflammatory cytokines like IL-1, IL-6 and TNF-α by malignant cells is also shown to be inhibited with exposure to givinostat [49, 50]. A pilot phase 2 study of givinostat in patients with MPN (including 12 with PV) demonstrated significant reduction of splenomegaly and symptom burden, appreciable hematologic responses and a safe toxicity profile [51]. A subsequent phase 2 trial restricted to patients with PV unresponsive to hydroxyurea studied the addition of givinostat and reported impressive rate of pruritus resolution, good tolerance and a 50 and 55% complete (CR) and partial response (PR) rate, respectively [52]. More recently, the dose expansion phase of another phase 2 trial of givinostat monotherapy in 35 patients with previously-treated PV reported an 81% response rate, although grade ≤ 3 diarrhea, thrombocytopenia and creatinine elevations were observed in 35–50% of patients [35]. A phase 3 study comparing frontline givinostat to standard-of-care hydroxyurea in high-risk patients with PV is being planned [53] (Table 1). Similarly, IMG-7289 (bomedemstat) is a first-in-class, small molecule inhibitor of lysine-specific demethylase 1, which in in vitro and in vivo studies have shown to influence myeloid hematopoietic progenitor differentiation and self-renewal in JAK2-mutated cells [54]. A phase 2 study of IMG-7289 of patients with PV or ET refractory to prior cytoreductive therapy is underway (NCT04262141).
Another class of drugs being investigated for PV treatment is the MDM2 inhibitors. MDM2 acts a chief negative regulator of TP53, which is a pleiotropic and critical mediator of pro-apoptotic responses to cellular stress and damage [55]. MDM2-mediated TP53 regulation is multifactorial and includes direct inhibition of transcriptional activity as well as functioning as an E3 ubiquitin ligase modifier thus leading to TP53 proteasome-mediated degradation [55]. Overexpression of MDM2 downregulates TP53 activity in JAK2-mutated CD34+ MPN cells [56, 57]; conversely, in vitro treatment with an MDM2 inhibitor was shown to increase cellular TP53 levels/activity and consequently selective CD34+ PV cell death [56]. A phase 1 trial of idasanutlin, an oral MDM2 antagonist, in 13 patients with high-risk, previously-treated PV and ET (PV = 12, ET = 1) demonstrated a favorable toxicity profile with 7 out of 12 patients (58%) achieving CR or PR, including both symptomatic and molecular responses. The lone patient ET unfortunately developed deep venous thrombosis 1 week into therapy and was removed from the trial [58]. Interim analyses from an ongoing phase 2 study of idasanutlin for hydroxyurea-resistant/intolerant PV demonstrated that 9 of 16 (56%) patients achieved hematocrit control by 32 weeks of therapy [36]. Unfortunately, 63% of patients required dose modification with most prompted by significant nausea/vomiting and diarrhea [36] (Table 1). Recent data also suggest that idasanutlin stimulates the expansion of TP53-mutated clones, although these clones regressed with discontinuation of treatment [59]. Due to its side effect profile, the development of idasanutlin for PV management was discontinued. Novel and more potent MDM2 inhibitors have also been developed. KRT-232 was investigated in a phase 2 trial of patients with PV and hydroxyurea resistance/intolerance (NCT03669965), but the study was halted due to low accrual.
Telomerase activity inhibition via targeting of the RNA template of the telomerase subunit hTERC can inhibit the growth of megakaryocytic colony-forming units obtained from patients with ET while sparing the normal megakaryocytic proliferation of samples from healthy controls [60, 61]. Imetelstat, the telomerase activity inhibitor used in these preclinical studies, demonstrated a rapid, durable and impressive (89%) complete hematologic response rate in 18 patients with ET refractory to prior therapy [40] (Table 2).
The iron metabolism pathway has been the subject of recent and unique targeting for the management of PV, which is essentially treated by either inducing or exacerbating iron deficiency with serial therapeutic phlebotomies targeting a hematocrit goal of < 45% based on the CYTO-PV study results [62]; however, iron deficiency can produce significant symptomatology. Both iron deficiency and expanded erythropoiesis in patients with PV may lead to suppression of hepcidin, the chief regulator of iron homeostasis [63]. Lack of hepcidin enhances the availability of iron for erythropoiesis in patients with PV [37, 64]. PTG-300, a first-in-class, subcutaneous hepcidin mimetic, is in phase 2 testing for patients with low- and high-risk PV requiring therapeutic phlebotomies (NCT04057040). Interim data on 13 patients showed that the drug is well-tolerated and led to phlebotomy independence in all treated patients [37]. Additionally, PTG-300 normalized iron stores in as short as 4 weeks of therapy and improved patient’s symptom burden [37] (Table 1).
Conclusion
Although most patients with PV or ET will enjoy a normal life expectancy, some higher-risk patients suffer complications and are at increased risk of disease transformation and early mortality. Endeavors aimed at improving the quality of life and outcomes of patients with PV and ET are focused on hematologic control and thrombotic risk reduction. Hydroxyurea or interferon therapy is the current standard of care for higher-risk patients with ruxolitinib or anagrelide being reserved for second-line management. Longer-acting interferons with more convenient administration schedules such as ropeginterferon alfa-2b, which appears to be disease-modifying and superior to hydroxyurea, offer more options for the patient with PV. Alternatively, combining current standard-of-care therapies may offer synergy and better patient outcomes, as demonstrated with the combination of ruxolitinib and PEG-IFN alfa-2a.
If available, clinical trials should be offered to patients who are failed by these therapies, which may soon be supplanted by agents with optimized JAK-STAT pathway inhibition. The novel agents exploiting mechanisms critical to MPN HSC proliferation, such as inhibitors of histone deacetylase, MDM2 and telomerase, are currently being studied. Hepcidin mimetics offer an opportunity to normalize the disrupted iron homeostasis observed in PV, while alleviating the need for therapeutic phlebotomies. Randomized trials of new agents in comparison to reference standards are necessary to confirm their value for patients with PV and ET. Correlative studies exploring molecular and metabolic changes may help to understand treatment potential to modify the course of disease. Continued high-impact research may soon foster the development of disease-modifying therapies for PV and ET and satisfy this need for the optimal management of patients with these MPNs.
Availability of data and materials
Not applicable.
Abbreviations
- alloHCT:
-
Allogeneic hematopoietic stem cell transplantation
- AML:
-
Acute myeloid leukemia
- CHR:
-
Complete hematologic response
- ET:
-
Essential thrombocythemia
- FDA:
-
Food and Drug Administration
- HDACi:
-
Histone deacetylase inhibitor
- HSC:
-
Hematopoietic stem/progenitor cells
- JAK2 :
-
Janus kinase 2
- MDS:
-
Myelodysplastic syndrome
- MPN:
-
Myeloproliferative neoplasm
- ORR:
-
Overall response rate
- PEG-IFN alfa-2a:
-
Pegylated interferon alfa 2a
- PV:
-
Polycythemia vera
References
Shallis RM, Wang R, Davidoff A, Ma X, Podoltsev NA, Zeidan AM. Epidemiology of the classical myeloproliferative neoplasms: the four corners of an expansive and complex map. Blood Rev. 2020;42:100706. https://doi.org/10.1016/j.blre.2020.100706.
Tefferi A, Vannucchi AM. Genetic risk assessment in myeloproliferative neoplasms. Mayo Clin Proc. 2017;92(8):1283–90. https://doi.org/10.1016/j.mayocp.2017.06.002.
Vainchenker W, Kralovics R. Genetic basis and molecular pathophysiology of classical myeloproliferative neoplasms. Blood. 2017;129(6):667–79. https://doi.org/10.1182/blood-2016-10-695940.
Stein BL, Oh ST, Berenzon D, Hobbs GS, Kremyanskaya M, Rampal RK, et al. Polycythemia vera: an appraisal of the biology and management 10 years after the discovery of JAK2 V617F. J Clin Oncol. 2015;33(33):3953–60. https://doi.org/10.1200/JCO.2015.61.6474.
Baxter EJ, Scott LM, Campbell PJ, East C, Fourouclas N, Swanton S, et al. Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet. 2005;365(9464):1054–61. https://doi.org/10.1016/S0140-6736(05)71142-9.
Jones AV, Kreil S, Zoi K, Waghorn K, Curtis C, Zhang L, et al. Widespread occurrence of the JAK2 V617F mutation in chronic myeloproliferative disorders. Blood. 2005;106(6):2162–8. https://doi.org/10.1182/blood-2005-03-1320.
Kralovics R, Passamonti F, Buser AS, Teo SS, Tiedt R, Passweg JR, et al. A gain-of-function mutation of JAK2 in myeloproliferative disorders. N Engl J Med. 2005;352(17):1779–90. https://doi.org/10.1056/NEJMoa051113.
Passamonti F, Rumi E, Pietra D, Della Porta MG, Boveri E, Pascutto C, et al. Relation between JAK2 (V617F) mutation status, granulocyte activation, and constitutive mobilization of CD34+ cells into peripheral blood in myeloproliferative disorders. Blood. 2006;107(9):3676–82. https://doi.org/10.1182/blood-2005-09-3826.
Pardanani AD, Levine RL, Lasho T, Pikman Y, Mesa RA, Wadleigh M, et al. MPL515 mutations in myeloproliferative and other myeloid disorders: a study of 1182 patients. Blood. 2006;108(10):3472–6. https://doi.org/10.1182/blood-2006-04-018879.
Pikman Y, Lee BH, Mercher T, McDowell E, Ebert BL, Gozo M, et al. MPLW515L is a novel somatic activating mutation in myelofibrosis with myeloid metaplasia. PLoS Med. 2006;3(7):e270. https://doi.org/10.1371/journal.pmed.0030270.
Klampfl T, Gisslinger H, Harutyunyan AS, Nivarthi H, Rumi E, Milosevic JD, et al. Somatic mutations of calreticulin in myeloproliferative neoplasms. N Engl J Med. 2013;369(25):2379–90. https://doi.org/10.1056/NEJMoa1311347.
Nangalia J, Massie CE, Baxter EJ, Nice FL, Gundem G, Wedge DC, et al. Somatic CALR mutations in myeloproliferative neoplasms with nonmutated JAK2. N Engl J Med. 2013;369(25):2391–405. https://doi.org/10.1056/NEJMoa1312542.
Rumi E, Cazzola M. How I treat essential thrombocythemia. Blood. 2016;128(20):2403–14. https://doi.org/10.1182/blood-2016-05-643346.
Mills KI, McMullin MF. Mutational spectrum defines primary and secondary myelofibrosis. Haematologica. 2014;99(1):2–3. https://doi.org/10.3324/haematol.2013.101279.
Yogarajah M, Tefferi A. Leukemic transformation in myeloproliferative neoplasms: a literature review on risk, characteristics, and outcome. Mayo Clin Proc. 2017;92(7):1118–28. https://doi.org/10.1016/j.mayocp.2017.05.010.
Hultcrantz M, Bjorkholm M, Dickman PW, Landgren O, Derolf AR, Kristinsson SY, et al. Risk for arterial and venous thrombosis in patients with myeloproliferative neoplasms: a population-based cohort study. Ann Intern Med. 2018;168(5):317–25. https://doi.org/10.7326/M17-0028.
Landtblom AR, Bower H, Andersson TM, Dickman PW, Samuelsson J, Bjorkholm M, et al. Second malignancies in patients with myeloproliferative neoplasms: a population-based cohort study of 9379 patients. Leukemia. 2018;32(10):2203–10. https://doi.org/10.1038/s41375-018-0027-y.
Radia D, Geyer HL. Management of symptoms in polycythemia vera and essential thrombocythemia patients. Hematol Am Soc Hematol Educ Program. 2015;2015(1):340–8. https://doi.org/10.1182/asheducation-2015.1.340.
Silver RT, Kiladjian JJ, Hasselbalch HC. Interferon and the treatment of polycythemia vera, essential thrombocythemia and myelofibrosis. Expert Rev Hematol. 2013;6(1):49–58. https://doi.org/10.1586/ehm.12.69.
Bewersdorf JP, Giri S, Wang R, Podoltsev N, Williams RT, Tallman MS, et al. Interferon alpha therapy in essential thrombocythemia and polycythemia vera-a systematic review and meta-analysis. Leukemia. 2020. https://doi.org/10.1038/s41375-020-01020-4.
Daltro De Oliveira R, Soret-Dulphy J, Zhao L-P, Marcault C, Gauthier N, Verger E, et al. Interferon-Alpha (IFN) therapy discontinuation is feasible in myeloproliferative neoplasm (MPN) patients with complete hematological remission. Blood. 2020;136(Supplement 1):35–6.
Passamonti F, Elena C, Schnittger S, Skoda RC, Green AR, Girodon F, et al. Molecular and clinical features of the myeloproliferative neoplasm associated with JAK2 exon 12 mutations. Blood. 2011;117(10):2813–6. https://doi.org/10.1182/blood-2010-11-316810.
Scott LM, Tong W, Levine RL, Scott MA, Beer PA, Stratton MR, et al. JAK2 exon 12 mutations in polycythemia vera and idiopathic erythrocytosis. N Engl J Med. 2007;356(5):459–68. https://doi.org/10.1056/NEJMoa065202.
Kleppe M, Kwak M, Koppikar P, Riester M, Keller M, Bastian L, et al. JAK-STAT pathway activation in malignant and nonmalignant cells contributes to MPN pathogenesis and therapeutic response. Cancer Discov. 2015;5(3):316–31. https://doi.org/10.1158/2159-8290.CD-14-0736.
Koschmieder S, Mughal TI, Hasselbalch HC, Barosi G, Valent P, Kiladjian JJ, et al. Myeloproliferative neoplasms and inflammation: whether to target the malignant clone or the inflammatory process or both. Leukemia. 2016;30(5):1018–24. https://doi.org/10.1038/leu.2016.12.
Verstovsek S, Kantarjian H, Mesa RA, Pardanani AD, Cortes-Franco J, Thomas DA, et al. Safety and efficacy of INCB018424, a JAK1 and JAK2 inhibitor, in myelofibrosis. N Engl J Med. 2010;363(12):1117–27. https://doi.org/10.1056/NEJMoa1002028.
Vannucchi AM, Kiladjian JJ, Griesshammer M, Masszi T, Durrant S, Passamonti F, et al. Ruxolitinib versus standard therapy for the treatment of polycythemia vera. N Engl J Med. 2015;372(5):426–35. https://doi.org/10.1056/NEJMoa1409002.
Verstovsek S, Vannucchi AM, Griesshammer M, Masszi T, Durrant S, Passamonti F, et al. Ruxolitinib versus best available therapy in patients with polycythemia vera: 80-week follow-up from the RESPONSE trial. Haematologica. 2016;101(7):821–9. https://doi.org/10.3324/haematol.2016.143644.
Masciulli A, Ferrari A, Carobbio A, Ghirardi A, Barbui T. Ruxolitinib for the prevention of thrombosis in polycythemia vera: a systematic review and meta-analysis. Blood Adv. 2020;4(2):380–6. https://doi.org/10.1182/bloodadvances.2019001158.
Passamonti F, Griesshammer M, Palandri F, Egyed M, Benevolo G, Devos T, et al. Ruxolitinib for the treatment of inadequately controlled polycythaemia vera without splenomegaly (RESPONSE-2): a randomised, open-label, phase 3b study. Lancet Oncol. 2017;18(1):88–99. https://doi.org/10.1016/S1470-2045(16)30558-7.
Verstovsek S, Mesa RA, Salama ME, Li L, Pitou C, Nunes FP, et al. A phase 1 study of the Janus kinase 2 (JAK2)(V617F) inhibitor, gandotinib (LY2784544), in patients with primary myelofibrosis, polycythemia vera, and essential thrombocythemia. Leuk Res. 2017;61:89–95. https://doi.org/10.1016/j.leukres.2017.08.010.
Berdeja J, Palandri F, Baer MR, Quick D, Kiladjian JJ, Martinelli G, et al. Phase 2 study of gandotinib (LY2784544) in patients with myeloproliferative neoplasms. Leuk Res. 2018;71:82–8. https://doi.org/10.1016/j.leukres.2018.06.014.
Sorensen AL, Mikkelsen SU, Knudsen TA, Bjorn ME, Andersen CL, Bjerrum OW, et al. Ruxolitinib and interferon-alpha2 combination therapy for patients with polycythemia vera or myelofibrosis: a phase II study. Haematologica. 2020;105(9):2262–72. https://doi.org/10.3324/haematol.2019.235648.
Gisslinger H, Klade C, Georgiev P, Krochmalczyk D, Gercheva-Kyuchukova L, Egyed M, et al. Ropeginterferon alfa-2b versus standard therapy for polycythaemia vera (PROUD-PV and CONTINUATION-PV): a randomised, non-inferiority, phase 3 trial and its extension study. Lancet Haematol. 2020;7(3):e196–208. https://doi.org/10.1016/S2352-3026(19)30236-4.
Rambaldi A, Iurlo A, Vannucchi AM, Noble R, von Bubnoff N, Guarini A, et al. Safety and efficacy of the maximum tolerated dose of givinostat in polycythemia vera: a two-part phase Ib/II study. Leukemia. 2020;34(8):2234–7. https://doi.org/10.1038/s41375-020-0735-y.
Mascarenhas J, Higgins B, Anders D, Burbury K, El-Galaly TC, Gerds AT, et al. Safety and efficacy of idasanutlin in patients (pts) with hydroxyurea (HU)-resistant/intolerant polycythemia vera (PV): results of an international Phase II study. Blood. 2020;136(Supplement 1):29–31.
Ginzburg Y, Kremyanskaya M, Kuykendall AT, Yacoub A, Yang J, Gupta SK, et al. Hepcidin mimetic (PTG-300) reverses iron deficiency while controlling hematocrit in polycythemia vera patients. Blood. 2020;136(Supplement 1):40–1.
Verstovsek S, Passamonti F, Rambaldi A, Barosi G, Rumi E, Gattoni E, et al. Ruxolitinib for essential thrombocythemia refractory to or intolerant of hydroxyurea: long-term phase 2 study results. Blood. 2017;130(15):1768–71. https://doi.org/10.1182/blood-2017-02-765032.
Harrison CN, Mead AJ, Panchal A, Fox S, Yap C, Gbandi E, et al. Ruxolitinib vs best available therapy for ET intolerant or resistant to hydroxycarbamide. Blood. 2017;130(17):1889–97. https://doi.org/10.1182/blood-2017-05-785790.
Baerlocher GM, Oppliger Leibundgut E, Ottmann OG, Spitzer G, Odenike O, McDevitt MA, et al. Telomerase inhibitor Imetelstat in patients with essential thrombocythemia. N Engl J Med. 2015;373(10):920–8. https://doi.org/10.1056/NEJMoa1503479.
Gisslinger H, Zagrijtschuk O, Buxhofer-Ausch V, Thaler J, Schloegl E, Gastl GA, et al. Ropeginterferon alfa-2b, a novel IFNalpha-2b, induces high response rates with low toxicity in patients with polycythemia vera. Blood. 2015;126(15):1762–9. https://doi.org/10.1182/blood-2015-04-637280.
Illes A, Pinczes LI, Egyed M. A pharmacokinetic evaluation of ropeginterferon alfa-2b in the treatment of polycythemia vera. Expert Opin Drug Metab Toxicol. 2021;17(1):3–7.
Gisslinger H, Klade C, Georgiev P, Krochmalczyk D, Gercheva-Kyuchukova L, Egyed M, et al. Long-term use of ropeginterferon Alpha-2b in polycythemia vera: 5-year results from a randomized controlled study and its extension. Blood. 2020;136(Supplement 1):33.
Delcuve GP, Khan DH, Davie JR. Roles of histone deacetylases in epigenetic regulation: emerging paradigms from studies with inhibitors. Clin Epigenetics. 2012;4(1):5. https://doi.org/10.1186/1868-7083-4-5.
Bose P, Verstovsek S. Investigational histone deacetylase inhibitors (HDACi) in myeloproliferative neoplasms. Expert Opin Investig Drugs. 2016;25(12):1393–403. https://doi.org/10.1080/13543784.2016.1250882.
Akada H, Akada S, Gajra A, Bair A, Graziano S, Hutchison RE, et al. Efficacy of vorinostat in a murine model of polycythemia vera. Blood. 2012;119(16):3779–89. https://doi.org/10.1182/blood-2011-02-336743.
Wang Y, Fiskus W, Chong DG, Buckley KM, Natarajan K, Rao R, et al. Cotreatment with panobinostat and JAK2 inhibitor TG101209 attenuates JAK2V617F levels and signaling and exerts synergistic cytotoxic effects against human myeloproliferative neoplastic cells. Blood. 2009;114(24):5024–33. https://doi.org/10.1182/blood-2009-05-222133.
Guerini V, Barbui V, Spinelli O, Salvi A, Dellacasa C, Carobbio A, et al. The histone deacetylase inhibitor ITF2357 selectively targets cells bearing mutated JAK2(V617F). Leukemia. 2008;22(4):740–7. https://doi.org/10.1038/sj.leu.2405049.
Carta S, Tassi S, Semino C, Fossati G, Mascagni P, Dinarello CA, et al. Histone deacetylase inhibitors prevent exocytosis of interleukin-1beta-containing secretory lysosomes: role of microtubules. Blood. 2006;108(5):1618–26. https://doi.org/10.1182/blood-2006-03-014126.
Golay J, Cuppini L, Leoni F, Mico C, Barbui V, Domenghini M, et al. The histone deacetylase inhibitor ITF2357 has anti-leukemic activity in vitro and in vivo and inhibits IL-6 and VEGF production by stromal cells. Leukemia. 2007;21(9):1892–900. https://doi.org/10.1038/sj.leu.2404860.
Rambaldi A, Dellacasa CM, Finazzi G, Carobbio A, Ferrari ML, Guglielmelli P, et al. A pilot study of the histone-deacetylase inhibitor givinostat in patients with JAK2V617F positive chronic myeloproliferative neoplasms. Br J Haematol. 2010;150(4):446–55.
Finazzi G, Vannucchi AM, Martinelli V, Ruggeri M, Nobile F, Specchia G, et al. A phase II study of Givinostat in combination with hydroxycarbamide in patients with polycythaemia vera unresponsive to hydroxycarbamide monotherapy. Br J Haematol. 2013;161(5):688–94. https://doi.org/10.1111/bjh.12332.
Chifotides HT, Bose P, Verstovsek S. Givinostat: an emerging treatment for polycythemia vera. Expert Opin Investig Drugs. 2020;29(6):525–36. https://doi.org/10.1080/13543784.2020.1761323.
Jutzi JS, Kleppe M, Dias J, Staehle HF, Shank K, Teruya-Feldstein J, et al. LSD1 inhibition prolongs survival in mouse models of MPN by selectively targeting the disease clone. Hemasphere. 2018;2(3):e54. https://doi.org/10.1097/HS9.0000000000000054.
Shangary S, Wang S. Targeting the MDM2-p53 interaction for cancer therapy. Clin Cancer Res. 2008;14(17):5318–24. https://doi.org/10.1158/1078-0432.CCR-07-5136.
Lu M, Wang X, Li Y, Tripodi J, Mosoyan G, Mascarenhas J, et al. Combination treatment in vitro with Nutlin, a small-molecule antagonist of MDM2, and pegylated interferon-alpha 2a specifically targets JAK2V617F-positive polycythemia vera cells. Blood. 2012;120(15):3098–105. https://doi.org/10.1182/blood-2012-02-410712.
Nakatake M, Monte-Mor B, Debili N, Casadevall N, Ribrag V, Solary E, et al. JAK2(V617F) negatively regulates p53 stabilization by enhancing MDM2 via La expression in myeloproliferative neoplasms. Oncogene. 2012;31(10):1323–33. https://doi.org/10.1038/onc.2011.313.
Mascarenhas J, Lu M, Kosiorek H, Virtgaym E, Xia L, Sandy L, et al. Oral idasanutlin in patients with polycythemia vera. Blood. 2019;134(6):525–33. https://doi.org/10.1182/blood.2018893545.
Marcellino BK, Farnoud N, Cassinat B, Lu M, Verger E, McGovern E, et al. Transient expansion of TP53 mutated clones in polycythemia vera patients treated with idasanutlin. Blood Adv. 2020;4(22):5735–44. https://doi.org/10.1182/bloodadvances.2020002379.
Mosoyan G, Kraus T, Ye F, Eng K, Crispino JD, Hoffman R, et al. Imetelstat, a telomerase inhibitor, differentially affects normal and malignant megakaryopoiesis. Leukemia. 2017;31(11):2458–67. https://doi.org/10.1038/leu.2017.78.
Baerlocher GM, Haubitz M, Braschler TR, Brunold C, Burington B, Oppliger Leibundgut E, et al. Imetelstat inhibits growth of megakaryocyte colony-forming units from patients with essential thrombocythemia. Blood Adv. 2019;3(22):3724–8. https://doi.org/10.1182/bloodadvances.2019000167.
Marchioli R, Finazzi G, Specchia G, Cacciola R, Cavazzina R, Cilloni D, et al. Cardiovascular events and intensity of treatment in polycythemia vera. N Engl J Med. 2013;368(1):22–33. https://doi.org/10.1056/NEJMoa1208500.
Nemeth E, Ganz T. The role of hepcidin in iron metabolism. Acta Haematol. 2009;122(2–3):78–86. https://doi.org/10.1159/000243791.
Ginzburg YZ, Feola M, Zimran E, Varkonyi J, Ganz T, Hoffman R. Dysregulated iron metabolism in polycythemia vera: etiology and consequences. Leukemia. 2018;32(10):2105–16. https://doi.org/10.1038/s41375-018-0207-9.
Acknowledgements
None.
Funding
None.
Author information
Authors and Affiliations
Contributions
R.M.S. and N.A.P. conceptualized and wrote the manuscript. The author(s) read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
All authors consent to publication.
Competing interests
N.A.P. consulted for and received honoraria from Alexion, Pfizer, Agios Pharmaceuticals, Blueprint Medicines, Incyte, Novartis, Celgene, Bristol-Myers Squib, CTI BioPharma and PharmaEssentia. N.A.P. received research funding (all to the institution) from Boehringer Ingelheim, Astellas Pharma, Daiichi Sankyo, Sunesis Pharmaceuticals, Jazz Pharmaceuticals, Pfizer, Astex Pharmaceuticals, CTI biopharma, Celgene, Genentech, AI Therapeutics, Samus Therapeutics, Arog Pharmaceuticals, Kartos Therapeutics. None of these relationships were related to the development of this manuscript. All other authors report no relevant disclosures/competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Shallis, R.M., Podoltsev, N.A. Emerging agents and regimens for polycythemia vera and essential thrombocythemia. Biomark Res 9, 40 (2021). https://doi.org/10.1186/s40364-021-00298-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40364-021-00298-5