
Abstract
Acute promyelocytic leukemia (APL) is characterized by the accumulation of promyelocytes in bone marrow. More than 95% of patients with this disease belong to typical APL, which express PML-RARA and are sensitive to differentiation induction therapy containing all-trans retinoic acid (ATRA) and arsenic trioxide (ATO), and they exhibit an excellent clinical outcome. Compared to typical APL, variant APL showed quite different aspects, and how to recognize, diagnose, and treat variant APL remained still challenged at present. Herein, we drew the genetic landscape of variant APL according to recent progresses, then discussed how they contributed to generate APL, and further shared our clinical experiences about variant APL treatment. In practice, when APL phenotype was exhibited but PML-RARA and t(15;17) were negative, variant APL needed to be considered, and fusion gene screen as well as RNA-sequencing should be displayed for making the diagnosis as soon as possible. Strikingly, we found that besides of RARA rearrangements, RARB or RARG rearrangements also generated the phenotype of APL. In addition, some MLL rearrangements, NPM1 rearrangements or others could also drove variant APL in absence of RARA/RARB/RARG rearrangements. These results indicated that one great heterogeneity existed in the genetics of variant APL. Among them, only NPM1-RARA, NUMA-RARA, FIP1L1-RARA, IRF2BP2-RARA, and TFG-RARA have been demonstrated to be sensitive to ATRA, so combined chemotherapy rather than differentiation induction therapy was the standard care for variant APL and these patients would benefit from the quick switch between them. If ATRA-sensitive RARA rearrangement was identified, ATRA could be added back for re-induction of differentiation. Through this review, we hoped to provide one integrated view on the genetic landscape of variant APL and helped to remove the barriers for managing this type of disease.
Similar content being viewed by others

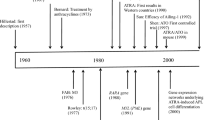

Background
APL is the most special subtype of AML, and it is characterized by the accumulation of promyelocytes in bone marrow, and mostly existence of PML-RARA. Before the application of ATRA and ATO, APL is the most dangerous AML subtype due to its high frequency of fatal bleeding when the chemotherapy is firstly conducted [1]. Subsequently, the differentiation induction therapy with ATRA and ATO has strikingly improved the clinical outcome of APL patients, and its long-term survival rate is ≥95%, but there are still 5% of patients dead of refractory/relapsed disease [2,3,4]. In detail, these refractory/relapsed APL patients are mainly consisted of high-risk typical APL and variant APL. It is widely accepted that high-risk typical APL can be improved by adding chemotherapy to ATRA and ATO, but how to recognize, diagnose and treat variant APL remains challenges.
Traditionally, variant APL mainly refers to RARA rearrangement-positive but PML-RARA-negative APL, but it is not comprehensive according to recent opinions. Besides of RARA rearrangements, RARB rearrangements, RARG rearrangements and other genetic events have been demonstrated to contribute to generate APL [5]. Therefore, we come up with the generalized criterion for variant APL, which is that all of PML-RARA-negative APL can be defined as variant APL whether variant RARA rearrangement exists or not. Up to now, how PML-RARA contributes to leukemogenesis has been well studied, and the mechanism of how ATRA and ATO target PML-RARA has been also uncovered. In contrast, the genetic heterogeneity, the mechanism of leukemogenesis, the choice of clinical treatment in variant APL remain largely unestablished. Herein, we drew the genetic landscape of variant APL according to recent progresses, then discussed how they contributed to generate APL, and further shared our clinical experiences about variant APL treatment.
Typical APL with PML-RARA
PML is located in chromosome 15q24, and PML together with its interacted partners formed one sub-nuclear structure through multimerization and organization, which is also known as NBs, to perform its function of tumor suppression and genomic stability maintenance [6]. RARA locates on chromosome band 17q21, and it shares one nearly 90% homology with RARB and RARG, which all belong to the RAR family. It is well known that RAR transcriptional pathway is critical for the development, maintenance, expansion, differentiation of HSCs [7]. In absent of ligand, RAR assembled one repressive-complex with its co-repressors NCoR/SMRT/HDAC, and bind to RARE on DNA to repress the RAR transcriptional pathway; in present of ligand, the co-repressors were dissociated from RAR, and it together with its cofactor RXR bind to RARE to activate the RAR transcriptional pathway [8]. PML-RARA is generated by t(15;17)(q24;q21), and it mainly contained three typical isoforms, such as bcr1, bcr2, and bcr3. The breakpoint of RARA is located on its intron-2, and it was the same for the three isoform, while the breakpoint of PML was on intron-6, exon-6, and intron-3, respectively. Therefore, bcr1 and bcr2 were also called the long isoform, while bcr3 was the short isoform. Besides of typical isoforms, more than 30 atypical isoforms have been identified, which shared large similarities with the typical ones [9]. PML-RARA generated APL phenotype mainly via acting as one repressor on RARA transcriptional network and disrupting the NBs. On the one hand, PML-RARA played one dominant-negative role on RARA/RXR-transcriptional network, and took control of RARE sites via recruiting the co-repressors to inhibit its transcription and activation, leading to the maturation arrested at the promyelocyte stage, enhanced self-renew, impairment of autophagy and apoptosis; On the other hand, NBs were disrupted and reformed by PML-RARA, and the self-renew was enhanced while DNA damage response, senescence, and apoptosis were inhibited [10]. Strikingly, the oncogenic fusion gene could be targeted by ATRA and ATO. ATRA directly bind to the RARA moiety of PML-RARA and induced its conformational change, leading it to dissociate the co-repressor complexes and recruit the co-activators, then RARA transcriptional network was re-activated. In addition, ATRA also led to PML-RARA degradation via ubiquitin-proteasome system [11, 12]. ATO directly bind to the specific cysteines residues located on zinc fingers in the B2 domains of PML, leading to the sumoylation of PML-RARA for its proteasomal degradation [13, 14]. ATRA and ATO targeted PML-RARA and induced its degradation via two totally different routes, so combination of ATRA and ATO cooperate in APL treatment, in which ATRA induced APL differentiation, and low-dose ATO induced differentiation while high-dose ATO induced apoptosis, and achieved excellent clinical responses in APL patients [15] (Fig. 1).

PML-RARA provided the therapeutic targets for ATRA and ATO in typical APL. B1 and 2: B box; CC: coiled-coil domain; DBD, DNA-binding domain; LBD, ligand binding domain; R: RING finger domain
Besides of PML-RARA, additional gene mutations have been identified in APL and they possibly cooperated with PML-RARA to participate in the leukemogenesis and therapeutic resistance. At diagnosis, several studies demonstrated that FLT3-ITD/TKD mutation was the most common additional gene mutation. WT1, NRAS/KRAS, and ARID1A/ARID1B mutations were also frequent [16,17,18,19,20]. Though epigenetic modifiers, such as DNMT3A, TET2, ASXL1, and IDH1/2, mutations were relatively rare in APL, they conferred APL one poor prognosis characterized by short overall survival duration and disease-free survival duration [17]. At relapse, the gene mutational landscape has skewed, and the most remarkable change was the emerged large amount of PML or RARA mutation [16, 18, 19, 21, 22]. Besides, the frequency of RUNX1 mutation also elevated compared to it at primary diagnosis [16, 18]. However, FLT3-ITD/TKD mutation was still the most common, and WT1, NRAS/KRAS, and ARID1A/ARID1B mutations followed [19]. Notably, the increased total frequency of additional genetic mutations was strongly associated with the high Sanz score at diagnosis [17]. Consistently, APL with these mutations preferred to fall in high-risk group and develop to relapsed disease during the clinical course. These results indicated that there were two different pathways for gene mutations to mediate therapeutic resistance and promote APL relapse at least. The first was that PML or RARA mutation impaired the binding of PML-RARA to ATO or ATRA, respectively; the second was that additional gene mutations, especially FLT3-ITD/TKD mutation, cooperated with PML-RARA to drive APL progression. Therefore, besides of PML-RARA, additional gene mutations were also important in the pathogenesis of APL.
RARA rearrangement in variant APL
Besides of PML-RARA, 16 additional RARA rearrangements have been identified up to now, which composed the most part of variant APL. Herein, each RARA rearrangement was described below according to the history of discovery.
PLZF-RARA
PLZF, also called ZBTB16, is a zinc finger transcription factor, which plays a critical role in the balance of self-renew and differentiation of HSCs and is involved in MLL-rearrangement-induced leukemogenesis. PLZF locates at chromosome 11q23, and t(11;17)(q23;q21) generated PLZF-RARA and RARA-PLZF. Notably, PLZF-RARA was the most common variant RARA rearrangement accounting 1% of APL, and various cases have been reported up to now, while its breakpoint was relatively conserved and only two isoforms were identified [23,24,25,26,27,28]. PLZF-RARA could block the myeloid differentiation and lead to leukemic transformation [29]. PLZF-RARA formed homodimers or heterodimers with RXRA to bind RARE, and recruited the co-repressor NCoR/SMRT/HDAC and NCoR/TBLR1 or PRC1 ploycomb group complex to play one dominate negative role on the RARA and RARG-signaling, whose target genes are critical for hematopoietic development [30,31,32]. The POZ domain on PLZF mediated the interaction between PLZF-RARA and PLZF or itself, and it was required for the activity of PLZF-RARA [33]. In contrast to PML-RARA, PLZF-RARA was resistant to ATRA treatment, which was reflected by its insensitive to ATRA-mediated degradation and its interaction with the corepressors not being dissociated with high-dose ATRA [34, 35]. Interestingly, HDAC inhibitor, which induced accumulation of acetylated histones to alter the PLZF-induced transcriptional repression, or 8-CPT-cAMP, which activated PKA to phosphorylate the Ser765 of PLZF/RARα and then disrupted its association with corepressors, could overcome the ATRA resistance of PLZF-RARA [36, 37]. Besides, the stability of PLZF-RARA was also regulated by the deubiquitinating enzyme USP37, which provided another target for PLZF-RARA treatment [38]. In addition to PLZF-RARA, RARA-PLZF also contributed to generate variant APL. For example, RARA-PLZF interacted with C/EBPα tethered to DNA, recruited HDAC1 to cause H3 deacetylation at C/EBPα target foci, and then decreased the expression of C/EBPα target genes to inhibit myeloid differentiation [39]. RARA-PLZF overcame PLZF-mediated repression of CRABPI via recruiting p300 and inducing its promoter hypomethylation [40]. In mouse model, PLZF-RARA generated CML-like phenotype, while NPM1-RARA more preferred to generate APL-like phenotype, in which some inconformity with clinical findings existed [41]. In clinic, all of PLZF-RARA-positive variant APL patients exhibited resistance to ATRA and ATO treatment, and combined chemotherapy functioned.
NPM1-RARA
NPM1 is one nucleolar phosphoprotein and functions in various biological processes, including molecular chaperoning, ribosome biogenesis, DNA repair, and genome stability. NPM1 locates at chromosome 5q32, and t(5;17)(q32;q21) generated NPM1-RARA [42], while RARA-NPM1 was also found in some variant APL patients, but it lacked the ability of differentiation arrest [43]. The breakpoint of NPM1-RARA was variable, and the most common breakpoint located at NPM1 exon-4 and RARA exon-3, but additional 3 distinct transcripts have been identified at least [44]. Cytologically, NPM1-RARA-positive APL exhibited one different morphology from typical APL [44, 45], which was also found in the mouse model of NPM1-RARA [46], and clinically, it occasionally developed to aleukemic leukemia cutis with one relatively higher frequency compared to other variant APL [44, 47]. Interestingly, NPM1-RARA was not only limited in variant APL, but also identified in atypical acute myelomonocytic leukemia, CML, cutaneous mastocytosis, and myeloid sarcoma [45, 47,48,49]. In variant APL, NPM1-RARA bind to RARE as homodimers or as heterodimers with RXR, then recruited the co-repressor and exhibited one dominate negative effect on RARA to promote the leukemogenesis of variant APL, which was quite similar to PML-RARA [50]. In addition, NPM1-RARA could bind to TRADD to inhibit caspase activation and activate NF-κB as well as JNK signaling pathway [51, 52], while NPM1-RARA could decrease the TP53 expression and then impaired its transcriptional activity [53]. Similar to PML-RARA, NPM1-RARA generated one ATRA-sensitive variant APL [42, 44].
NUMA-RARA
NUMA is one microtubule-associated protein and exhibited some cell cycle-specific functions in different stages of mitosis. The most important one is the participation in spindle apparatus formation. NUMA locates at chromosome 11q13, and t(11;17)(q13;q21) generated NUMA-RARA, in which the breakpoint was at the exon-20 of NUMA and the intron-2 of RARA [54, 55]. In contrast to PML-RARA, PLZF-RARA, and NPM1-RARA, NUMA-RARA exhibited one dominate cytoplasmic localization with weak nucleus localization [56]. However, it also could form either homodimers or heterodimers with RXRA to bind RARE, and inhibited the RARA transcriptional activity, and enhanced the STAT3 transcriptional activity, which was similar to other RARA fusions [57]. The co-repressor SMRT was recruited by NUMA-RARA, and it could be released by ATRA, while the co-activator TRAM-1 was then recruited [58]. In depth, the α-helical coiled-coil domain of NUMA was critical for the activity of NUMA-RARA [58]. Consistent with it, the clinical case was also sensitive to ATRA treatment. Besides, the inducible neutrophil-specific NUMA-RARA expression mouse model, hCG-NUMA-RARA transgenic mouse, was also constructed, and it definitely reproduced the human APL-like phenotype in mouse, in which its RXRA interaction was required for the transformative activity of NUMA-RARA [59]. Interestingly, the copy number of NUMA-RARA was inversely correlated with the latency of disease onset [60]. Therefore, both of clinical and experimental study demonstrated that NUMA-RARA-positive variant APL was sensitive to ATRA treatment.
STAT5B-RARA
STAT5B belongs to the family of latent cytosolic transcription factors activated by Janus kinases, including STAT1, 2, 3, 4, 5A, 5B and 6, and it mediates the cellular response to activation of multiple cytokine receptors to regulate the proliferation and differentiation in hematopoiesis. The same as RARA, STAT5B also locates at chromosome 17p21. STAT5B-RARA is mostly caused by the cooperation of 17p11.2p11.1 inversion and 17q21.2 interstitial micro-deletion, and totally 3 different types of transcripts has been identified, in which the breakpoints were at the exon-14, exon-15, or exon-16 of STAT5B, and the exon-3 of RARA, respectively [61,62,63,64,65,66,67]. STAT5B-RARA formed either homodimers or heterodimers with RXRA to bind RARE, and then recruited the co-repressor SMRT, to inhibit transcriptional activity of RARA/RXRA [68]. In depth, the coiled-coil domain of STAT5B was required for this process. In addition, STAT5B-RARA could also activate the STAT3 oncogene pathway, which was similar to PML-RARA or PLZF-RARA [69]. STAT5B-RARA was demonstrated as one ATRA-resistant RARA rearrangement in experimental studies, and consistently, STAT5B-RARA-positive patients only responded to chemotherapy rather than ATO or ATRA [69,70,71,72,73].
PRKAR1A-RARA
PRKAR1A encodes the regulatory subunit type I-α of cyclic adenosine monophosphate-dependent protein kinase. The same as RARA, PRKAR1A also locates at chromosome 17, while it is on chromosome 17q24, and RARA is on chromosome 17q21. Interestingly, one cytogenetically cryptic recombination on chromosome 17 between these two gene locus leads to PRKAR1A-RARA. Alternative splicing PRKAR1A generated two different transcripts, the breakpoint for the longer in-frame fusion transcript located at the exon-3 of PRKAR1A and the exon-3 of RARA, while it for the shorter out-frame fusion transcript was at the exon-2 of PRKAR1A and the exon-3 of RARA [74]. Furthermore, PRKAR1A-RARA formed homodimers or heterodimers with RXRA to bind to RARE, but its DNA-binding characteristics was quite different from PLZF-RARA or PML-RARA. Undoubtedly, PRKAR1A-RARA could transform the primary HSPCs. In depth, the RIIa domain on PRKAR1A portion was required for forming homodimers and its binding to DNA, but not for the transformative activity. In contrast, RXRA interaction, which bind to the RARA portion of this fusion, played a critical role in transformation [75]. In clinic, the PRKAR1A-RARA-positive patient was induced to CR with the regimen containing ATRA, ATO and IDA, so whether it was sensitive to ATRA remained to be investigated.
BCOR-RARA
BCOR is one ubiquitously expressed nuclear protein, which functions as the corepressor of proto-oncoprotein BCL6. It plays a critical role in the development and differentiation of multiple hematopoietic lineages. Frequent BCOR mutation has been found in MDS and AML, which also indicated its tumor-suppressor role in hematopoiesis. Interestingly, BCOR, locating at chromosome Xp11, was involved by RARA rearrangement, and BCOR-RARA was generated by t(X;17)(p11;q12) [76, 77]. In BCOR-RARA, its breakpoint located at the exon-12 of BCOR and the exon-3 of RARA, and it exhibited a specific nuclear localization distinct from BCOR and BCL6. BCOR-RARA interacted with itself and BCL6 via the N-terminal BCOR portion, while associated with RXRE via forming BCOR-RARA/RXRA. Furthermore, BCOR-RARA displayed a dominant negative role in inhibiting ATRA-induced RARA transcriptional activation. In clinic, one BCOR-RARA-positive APL patient achieved CR via accepting ATRA plus IA regimen, but ATO or tamibarotene did not function in the induction at the first relapse; the other patient directly accepted IA regimen and reached to CR. Therefore, ATRA possibly was not sufficient to induce BCOR-RARA-positive APL to CR.
FIP1L1-RARA
FIP1L1 is an integral subunit of cleavage and polyadenylation specificity factor, and it interacts with poly(A)polymerase to stimulate polyadenylation. Previously, FIP1L1 has been found frequently fused with PDGFRA in hypereosinophilic syndrome/chronic eosinophilic leukemia. Up to now, it was also reported at one partner of RARA rearrangement. FIP1L1 located at chromosome 4q12, and t(4;17)(q12;q21) generated FIP1L1-RARA. The breakpoint was at exon-15 of FIP1L1 and the exon-3 of RARA for three FIP1L1-RARA transcripts caused by alternative splice and one RARA-FIP1L1 transcript, while at the intron-13 of FIP1L1 and the exon-3 of RARA for another FIP1L1-RARA transcript [78, 79]. FIP1L1-RARA formed the homodimers via the FIP1L1 portion and suppressed the RA-dependent transcriptional activity, but it could be reversed by high-dose ATRA. Therefore, FIP1L1-RARA was one ATRA-sensitive RARA rearrangement. Regretfully, this result could not be verified in clinic patients due to early death of retinoic acid syndrome. Interestingly, the same FIP1L1-RARA transcript, whose breakpoint was at exon-15 of FIP1L1 and the exon-3 of RARA, generated not only APL but also juvenile myelomonocytic leukemia [80], indicating the plasticity of FIP1L1-RARA-positive leukemic initial cells. Compared to FIP1L1-PDGFRA, in which the transformative activity relied on the C-terminal PDGFRA portion, FIP1L1-RARA functioned as the transcriptional repressor via the FIP1 motif at the FIP1L1 portion [81]. Though both of these two fusions involved FIP1L1, the critical domains for proto-ontogenesis were quite different, and this phenomenon was possibly attributed to different breakpoints as well as different partners for FIP1L1.
OBFC2A-RARA
OBFC2A, locating at chromosome 2q32.3, encodes the single-stranded DNA binding protein 2, and it is required for DNA damage response and genomic stability. OBFC2A-RARA was generated by der(2)t(2;17)(q32;q21), and the breakpoint locates at the exon-5 of OBFC2A and exon-3 of RARA [82]. OBFC2A-RARA-positive APL was demonstrated sensitive to ATRA in vitro, but the case achieved CR with DA plus ATRA, so whether its sensitivity to ATRA in vivo was also the same as in vitro remained unknown.
TBLR1-RARA
TBLR1 encodes an F-box/beta-transducing repeat-containing protein, which is enriched in HSCs. TBLR1 locates at chromosome 3q26.32, and t(3;17)(q26;q21) generated TBLR1-RARA [83]. In addition to this translocation, one cryptic insertion of RARA into TBLR1 also generated TBLR1-RARA. The breakpoint of this fusion involved the exon-5 of TBLR1 and the exon-3 of RARA. TBLR1-RARA could form homodimers with itself and heterodimers with RXRA, and it exhibited a specific nuclear and cytoplasm localization. Due to its recruitment of transcriptional corepressors NCoR/SMRT/SIN3A/HDAC, the transcriptional activation function of TBLR1-RARA was diminished compared to RARA. Though TBLR1-RARA was sensitive to ATRA-mediated degradation in experimental studies, one TBLR1-RARA patient exhibited resistance to ATRA plus MA regimen, but showed responses to ATO plus mitoxantrone regimen, while the other patient responded to daunorubicin rather than ATRA. Therefore, TBLR1-RARA was definitely resistant to ATRA, but the mechanism of TBLR1-RARA sensitive to ATO remained to be further investigated.
GTF2I-RARA
GTF2I, locating at chromosome 7p11, is one phosphoprotein participating in transcription and signal transduction including growth factor signaling, cell cycle regulation, and TGF-β1 signaling. T(7;17)(q11;q21) generated GTF2I-RARA, and its breakpoint was at the exon-6 of GTF2I and the exon-3 of RARA [84]. GTF2I-RARA exhibited the diffuse nuclear distribution with a micropunctate pattern as well as the aggregation in the cytoplasm as macrogranules, and it could form homodimers and heterodimers with GTF2I. Consistent with the clinical case, GTF2I-RARA played a dominant negative role on the RARA/RXR transcriptional network and showed resistance to ATRA. Mechanically, GTF2I-RARA recruited NCoR/SMRT/HDAC3 transcriptional corepressors, but ATRA treatment could not dissociate its association with HDAC3. Besides, GTF2I-RARA directly up-regulated RNF8, while RNF8 interacted with RARA, then promoted its Lys48-linkage ubiquitylation and degradation to attenuate RARA transcriptional activation. Therefore, GTF2I-RARA exhibited ATRA resistance via multiple aspects, and the proteasome inhibitor, MG132, partially reversed ATRA resistance and synergistically induced GTF2I-RARA-positive APL differentiation with ATRA [85].
IRF2BP2-RARA
IRF2BP2 acts as a transcriptional corepressor and represses transactivation of NFAT to regulate cell cycle, apoptosis, and differentiation. It locates at chromosome 1q42.3, and t(1;17)(q42.3;q21.2) generates IRF2BP2-RARA. The breakpoint of this fusion was variable, and five different types of IRF2BP2-RARA transcripts has been identified at least [86,87,88,89]. IRF2BP2-RARA showed the association with itself and displayed repression on the RARE, but this inhibition could be overcome by ATRA. Furthermore, IRF2BP2-RARA also transformed murine HSPCs, while ATRA could abrogate this activity [90]. In clinic, there was one IRF2BP2-RARA-positive APL patient induced to CR by sole ATRA, though most of cases accepted ATRA plus chemotherapy-based regimen, so this result demonstrated that IRF2BP2-RARA was one ATRA-sensitive RARA rearrangement [89].
FNDC3B-RARA
FNDC3B, also called fibronectin type III domain containing 3B, is originally recognized to regulate adipocyte differentiation. FNDC3B locates at chromosome 3q26, which was the same as TBLR1, so FNDC3B-RARA was also generated by t(3;17)(q26;q21), and its breakpoint was at the exon-24 of FNDC3B and exon-3 of RARA [91]. In addition to FNDC3B-RARA, two reciprocal RARA-FNDC3B transcripts were also found in the same patient, and one was an in-frame fusion involving the exon-2 of RARA and exon-25 of FNDC3B, while the other was an out-of-frame fusion involving the same exon of RARA and exon-26 of FNDC3B. Furthermore, FNDC3B-RARA was conferred one dominate nuclear localization, and it could dimerize with itself, FNDC3B, and RXRA. Especially, it deregulated RARA transcriptional program by enhancing the repressor function of unliganded RARA to generate APL. Though FNDC3B-RARA was sensitive to ATRA-mediated degradation in vitro, the role of ATRA in this patient remained unestablished, and his CR mainly relied on DA regimen. In addition to fusion with RARA, FNDC3B exhibited relatively high expression in APL subtype according to FAB classification, and further progressively up-regulated in the process of ATRA-induced differentiation. When FNDC3B was knockdown, ATRA-induced differentiation could be partially impaired. Therefore, both FNDC3B and RARA played a critical role in ATRA-induced differentiation for APL, and FNDC3B-RARA possibly impaired both of their normal functions to contribute to block cell differentiation and generate APL.
STAT3-RARA
STAT3, similar to STAT5B, is also one of transducers in JAK-STAT signaling pathway, while STAT5B has been demonstrated as one partner of RARA rearrangement. STAT3 locates at chromosome 17p21.2, but cytogenetic analysis for two STAT3-RARA-positive APL patients did not identify the corresponding translocations. Totally, there were two transcripts of STAT3-RARA identifed, and their breakpoints located at the exon-3 of RARA and the exon-21/23 of STAT3, respectively [92]. Compared to the shorter one, the longer transcript reserved the phosphorylation site of STAT3. Fused to RARA, STAT3 was conferred one nuclear localization. Similar to PML-RARA, STAT3-RARA also formed homodimers. Though STAT3-RARA could be down-regulated by ATRA in vitro, these two patients exhibited resistance to ATRA or ATO. Besides, one of them achieved CR with HAG regimen, but the other one showed resistance to IA.
TFG-RARA
TFG, Trk-fused gene, functions in the endoplasmic reticulum and its associated microtubules. TFG locates at chromosome 3q12, and one t(3;14;17)(q12;q11;q21) generated TFG-RARA fusion [93]. The breakpoint was at the exon-7 of TFG as well as the exon-3 of RARA. Though IDA was added at the day 10 after ATRA initiation, TFG-RARA transcript has been significantly down-regulated and CR was finally achieved, indicating TFG-RARA was a ATRA-sensitive RARA rearrangement.
NUP98-RARA
NUP98 encodes a protein component of the nuclear pore complex, which is required for the nucleocytoplasmic transport of proteins and mRNA. Up to now, various NUP98 fusions were identified in AML or MDS. In addition, NUP98-RARA was found in one APL patient, and its breakpoint located at the exon-2 of NUP98 and the exon-3 of RARA [94]. NUP98-RARA exhibited an intracellular localization pattern, and the DBD but not LBD domain of RARA was required for its aberrant distribution. Similar to other RARA rearrangements, NUP98-RARA also dimerized with itself or RXR, and this association could be partially impaired by ATRA. NUP98-RARA exhibited responsive to ATRA-mediated degradation, and it was further enhanced by daunorubicin, which also was the same in differentiation induction. In clinic, this NUP98-RARA patient was sensitive to IA regimen, but whether he also was sensitive to ATRA remained unknown due to early discontinued this drug.
TNRC18-RARA
TNRC18 is encoded by trinucleotide repeat-containing gene 18, and in-frame fusion between the exon-5 of TNRC18 and the exon-3 of RARA generated TNRC18-RARA [95]. TNRC18-RARA exhibited one intranuclear distribution, which was similar to RARA, but it repressed RARE expression, and was resistant to ATRA induction. In addition, it formed homodimers or heterodimers with RXRA, and then activated its downstream signaling molecules, such as CDK9, AKT, and STAT3, but ATRA slightly repressed this TNRC18-RARA-mediated activation. Only one variant APL with TNRC18-RARA has been reported, and this patient exhibited resistance to APL therapy, which was consistent with experimental results, while AML therapy achieved his CR. Therefore, TNRC18-RARA was one ATRA-resistant RARA rearrangement.
Collectively, we found that all of variant RARA rearrangements shared some similar aspects with PML-RARA in molecular biology. First, the ability of forming homodimers or heretodimers with RXRA; Second, the recruitment of corepressors; Third, the dominate negative role on the RARA-transcriptional program. Though differences existed in the partners of RARA rearrangments and detailed functions of distinct fusions, these aspects determined to generate the similar phenotype of APL. However, variant RARA rearrangements lacked the target for ATO, while only a part of them was sensitive to ATRA-mediated differentiation induction and degradation. Therefore, it brought more difficulties to treat variant APL than typical APL (Fig. 2a).

The genetic landscape of RAR-rearranged variant APL. (a) RARA-rearranged variant APL; (b) RARB-rearranged variant APL; (c) RARG-rearranged variant APL. ANK, ankyrin repeats; BBD, BCOR BCL6-binding domain; B1 and 2: B box; CC: coiled-coil domain; DBD, DNA-binding domain; DDD, dimerization/docking domain of the Type I alpha Regulatory subunit of cAMP-dependent protein kinase; FIP1: FIP1 binding domain for polymerase; FN3, fibronectin type 3 domain; GLEBS: Gle2/ Rae1-binding sequence; GLFG: Gly-Leu-Phe-Gly repeats; LBD, ligand binding domain; LisH: lissencephaly type-1-like homology motif; LZ, leucine zipper; ND, nucleoplasmin/nucleophosmin domain; NRD, Nuclear reassembly domain; PB1, Phox and Bem1 domain; PHD, plant homedomain finger transcription factor domain; PI, protein interaction domain; POZ: BTB/POZ domain; PQ-rich, proline-glutamine-enriched domain; Pro: proline-rich region; R: RING finger domain; RRM: RNA recognition motif; R1, I-repeat domains; SAD, Spindle association domain; SH2, Src homology 2 (SH2) domain; ZF, zinc finger domain
RARB rearrangement in variant APL
RARB located at chromosome 3p24, and up to now, only one type of RARB rearrangement, TBL1XR1-RARB, has been identified in variant APL [20, 96, 97]. TBL1XR1 has been reported as one partner of RARA in variant APL [83]. As to TBL1XR1 locating at chromosome 3q26, a t(3;3) or an inv. (3) could be found in these patients. In clinic, TBL1XR1-RARB-positive variant APL patients were all resistant to ATRA therapy, and the combined chemotherapy was more effective, but most of them experienced the refractory/relapsed disease. Molecularly, TBL1XR1-RARB could form the homo-dimers and exhibit a dominant negative effect against both RARA and RARB to block neutrophil differentiation. Functionally, it enhanced the replanting capacity and inhibited myeloid maturation of HSPCs. Consistent with clinical findings, TBL1XR1-RARB and its positive cells were also resistant to ATRA or ATO or Tamibarotene treatment. Therefore, TBL1XR1-RARB conferred a variant APL phenotype and showed resistance to ATRA (Fig. 2b).
RARG rearrangement in variant APL
Compared to RARB rearrangement, RARG rearrangement was relatively abundant. Recently, five different RARG rearrangements, including NUP98-RARG, PML-RARG, CPSF6-RARG, NPM1-RARG-NPM1 and HNRNPC-RARG, have been identified. NUP98-RARG was the first RARG rearrangement identified in variant APL, which was caused by t(11;12)(p15;q13) [98]. NUP98-RARG exhibited one unique nuclear localization, recruited RARA as well as NUP98, and showed similar transcriptional properties as PML-RARA [99]. In detail, the C-terminal GLFG domain of NUP98 and DNA binding domain of RARG were required for the transformation ability of NUP98-RARG in murine HSPCs. In contrast to clinical finding, NUP98-RARG-postive APL was sensitive to ATRA treatment in murine system, which possibly was attributed to the different genetic backgrounds. Up to now, additional NUP98-RARG-positive APL patients have been reported by different groups including our group, and the breakpoint for NUP98-RARG was conserved, which located at NUP98 exon-12 and RARG exon-4 [100,101,102,103,104]. Though NUP98-RARG transformed murine HSPCs was sensitive to ATRA treatment ex vivo, all of NUP98-RARG-positive APL patients showed resistance to ATRA in clinic, and chemotherapy was required for their CR achievement. This result indicated that the difference genetic background existed in different species influenced the phenotype of NUP98-RARG. Subsequently, PML-RARG, which was corresponding with t(12;15)(q13;q22), was identified in one variant APL patient, and was sensitive to AML therapy plus ATRA [105]. Two different PML-RARG transcripts were found, and both of them involved PML exon-3 and RARG 5′-UTR, in which the breakpoint for RARG located at exon-1 and exon-2, respectively. CPSF6-RARG has been reported by multiple groups [20, 106,107,108,109,110,111], and six different transcripts have been found at least, including one RARG-CPSF6 transcript. Consistently, all of CPSF6-RARG-positive patients exhibited resistance to APL therapy, but some of them has been demonstrated sensitive to DA or HA regimen. NPM1-RARG-NPM1 and HNRNPC-RARG were the latest RARG rearrangements in variant APL, and they also showed resistance to ATO and ATRA [112, 113]. Undoubtedly, RARG rearrangement-positive APL appropriately accepted AML therapy (Fig. 2c).
MLL rearrangement in variant APL
Besides of RAR rearrangements, MLL rearrangement could also be found in variant APL, especially in those RAR rearrangement-negative patients. In Jie Zhao et al.’ report, MLL-ELL combined with RPRD2-MLL and MLL-AF1Q were identified in two PML-RARA-negative patients respectively, while MLL-SEPT6 was found in one PML-RARA-positive patient, indicating MLL rearrangement could resemble APL [20]. In another study, t(11;17)(q23;q25) has been reported, and MLL rearrangement was involved, but the detailed partner for this fusion gene was not further analyzed [114]. Furthermore, we have also diagnosed one ELL-MLL positive variant APL patient, in which the RARA expression was significantly down-regulated when compared to other AML, typical APL or normal bone marrow [115]. Therefore, MLL rearrangement possibly resembled APL via repressing the RAR expression to further block cell differentiation at the promyelocytic stage. However, the detailed mechanism still needed to be investigated. There was no doubt that MLL rearrangement positive variant APL was more preferably sensitive to AML therapy but not APL therapy.
NPM1 rearrangement in variant APL
NPM1 mutation is the most common gene mutation in AML, but it exhibits the mutual exclusion with PML-RARA, so it is absent in APL [116]. Besides of NPM1 mutation, NPM1 fusions, such as NPM1-MLF1 [117], NPM1-HAUS1 [118], NPM1-RARA [44], NPM1-RARG-NPM1 [112], were identified in AML. Among them, NPM1-RARA and NPM1-RARG-NPM1 were associated with variant APL, and they have been described in previous section of our review. In addition, NPM1-CCDC28A was identified, and it was one brand new NPM1 rearrangement for variant APL [20]. CR was achieved by conventional chemotherapy plus ATRA in this patient. Except from NPM1-rearranged with RAR, sole NPM1 rearrangement also could generate variant APL, but it exhibited resistance to typical APL therapy.
Others
Besides of recurrent rearrangements mentioned above, one TBC1D15-RAB21 was identified in PML-RARA-negative APL, while one ARID1B-WASH4P was identified in PML-RARA-positive APL [20]. In clinic, the APL patient with PML-RARA/ARID1B-WASH4P exhibited sensitive to APL therapy, indicating ARID1B-WASH4P was not dominate in its pathogenesis, while the APL patient with TBC1D15-RAB21 received AML therapy plus ATRA and achieved CR, so whether TBC1D15-RAB21 was sensitive to APL therapy remained unknown.
It has been recognized that APL was generated by fusion genes, whether typical or variant APL. However, fusion gene-negative variant APL has also been reported up to now. One EZH2D185H-positive but fusion gene-negative patient exhibited APL phenotype by down-regulating RARA and RARG expression [119]. In this patient, AML therapy, like DA and FLAG regimens, was conducted but refractory disease was found. This specific case indicated that down-regulation of RARA and RARG possibly block cell differentiation at the promyelocytic stage and then contributed to generate APL phenotype.
Clinical practice for variant APL
Variant APL showed one great heterogeneous genetic feature. According to literature reports and our clinical experience, most of variant APL exhibited resistance to typical APL therapy, including ATRA and ATO. Though chemotherapy was effective as the alternative therapy, the prognosis of variant APL was still much inferior to typical APL, and it was closed to this of AML (Table 1). Besides, relatively high incidence of early death due to hemorrhage, and some aggressive molecular subtype, such as PLZF-RARA, STAT5B-RARA and GTF2I-RARA, also contributed to its inferior prognosis [28, 84, 110, 121]. Therefore, early identification and quick switch from ATRA to standard chemotherapy is very important for variant APL, and these patients could benefit from this strategy [102, 115]. So, we raised this schedule for APL patients. When APL was diagnosed in morphology, and ATRA should be applied as initial treatment. Immunophenotype analysis was also need to be done to confirm the diagnosis of APL. Then, according to European Leukemia Net recommends, RT-PCR/RQ-PCR/RT-QLAMP for PML-RARA, as well as FISH for t(15;17) should be displayed at the same time, and immunostaining with anti-PML antibody could also be helpful if possibly displayed [120]. If either of them was positive, the diagnosis of typical APL was definitely made. At this time, ATO should be immediately added, and chemotherapy was also needed to be considered if it was the high-risk typical APL. If both of them were negative, conventional PCR screen for variant RARA rearrangement and RNA-sequencing should be conducted to detect the pathogenic fusion gene for variant APL. Besides, ATRA should be immediately discontinued and then switched to standard 3 + 7 therapy for AML. If ATRA-sensitive RARA rearrangement, such as NPM1-RARA, NUMA-RARA, FIP1L1-RARA, IRF2BP2-RARA, or TFG-RARA was identified [42, 50, 54, 78, 79, 89, 93], ATRA could be considered to be added back. As to consolidation therapy adopted in typical or variant APL, it was the same as induction therapy. Furthermore, variant APL patients could be candidates for hematopoietic stem cell transplantation [28, 110], but this treatment could be only considered for typical APL when refractory/relapsed disease occurred (Fig. 3).

The suggested protocol for diagnosing and treating typical as well as variant APL
Conclusions
Though typical APL and variant APL shared various common features in clinical and genetic aspects, their pathogenesis and treatment were quite different. One great heterogeneity of genetics existed in variant APL. To our knowledge, RAR rearrangement-mediated variant APL possibly exhibited partially similar mechanisms with typical APL, but the details were truly different for each fusion due to the distinct partner of RAR. Furthermore, the leukemogenesis of some variant APLs even did not rely on the RAR rearrangement, while MLL rearrangement, NPM1 rearrangement, and some specific fusion or gene mutation could generate variant APL partially via down-regulating RAR expression. However, their detailed mechanism still remained largely unknown. In treatment, only a small part of RARA rearrangement-positive variant APL showed responsive to ATRA, and chemotherapy was the true backbone for the treatment of variant APL, which was contrast to typical APL. Therefore, quick distinguishing between variant APL and typical APL was important in clinical practice, and variant APL patients could benefit from immediate switch from APL therapy to APL therapy. In the future, there were also some questions needed to be answered. First, the detailed mechanism of leukemogenesis for PML-RARA has been well studied, but it for other RAR rearrangements was less investigated, so how they generated APL phenotype should be devoted to; Second, whether APL phenotype could be generated by non-RAR rearrangement and what was the detailed mechanism needed to be explored; Third, if additional new RAR rearrangements would be identified exhibited one great interest; Fourth, up to now, we mainly focused on the rearrangements in variant APL, but what was the landscape of additional mutations for variant APL should be further paid more attention to; Fifth, whether targeted therapy except of ATRA and ATO was available for variant APL called for investigations.
Availability of data and materials
Not applicable.
Abbreviations
- AML:
-
acute myeloid leukemia
- AMML:
-
acute myelomonocytic leukemia
- APL:
-
acute promyelocytic leukemia
- ATO:
-
arsenic trioxide
- ATRA:
-
all-trans retinoic acid
- BM:
-
bone marrow
- CML:
-
chronic myeloid leukemia
- CR:
-
complete remission
- DA:
-
doxorubicin plus cytarabine regimen
- DFS:
-
disease-free survival duration
- FAB:
-
French-American-British
- FISH:
-
fluorescence in situ hybridization
- HAG:
-
low-dose of homoharringtonine and cytarabine plus granulocyte colony-stimulating factor regimen
- HES/CEL:
-
hypereosinophilic syndrome/chronic eosinophilic leukemia
- HSCs:
-
hematopoietic stem cells
- HSPCs:
-
hematopoietic stem/progenitor cells
- IA:
-
idarubicin plus cytarabine regimen
- IDA:
-
idarubicin
- JMML:
-
juvenile myelomonocytic leukemia
- MDS:
-
myelodysplastic syndrome
- NBs:
-
nuclear bodies
- MA:
-
mitoxantrone plus cytarabine regimen
- MS:
-
myeloid sarcoma
- MTZ:
-
mitoxantrone
- OS:
-
Overall survival duration
- RA:
-
Retinoic acid
- RAR:
-
Retinoic acid receptor
- RARE:
-
Retinoic acid response element
- RQ-PCR:
-
Real-time quantitative polymerase chain reaction
- RT-QLAMP:
-
Reverse transcription–quenching loop-mediated isothermal amplification
- RT-PCR:
-
Reverse transcriptase polymerase chain reaction
References
Wang ZY, Chen Z. Acute promyelocytic leukemia: from highly fatal to highly curable. Blood. 2008;111(5):2505–15. https://doi.org/10.1182/blood-2007-07-102798.
Lo-Coco F, Avvisati G, Vignetti M, Thiede C, Orlando SM, Iacobelli S, et al. Retinoic acid and arsenic trioxide for acute promyelocytic leukemia. N Engl J Med. 2013;369:111–21.
Zhu HH, Wu DP, Du X, Zhang X, Liu L, Ma J, et al. Oral arsenic plus retinoic acid versus intravenous arsenic plus retinoic acid for non-high-risk acute promyelocytic leukaemia: a non-inferiority, randomised phase 3 trial. Lancet Oncol. 2018;19(7):871–9. https://doi.org/10.1016/S1470-2045(18)30295-X.
Zhu H, Hu J, Chen L, Zhou W, Li X, Wang L, et al. The 12-year follow-up of survival, chronic adverse effects, and retention of arsenic in patients with acute promyelocytic leukemia. Blood. 2016;128:1525–8.
Geoffroy MC, de The H. Classic and variants APLs, as viewed from a therapy response. Cancers (Basel). 2020;12(4). https://doi.org/10.3390/cancers12040967.
Li Y, Ma X, Wu W, Chen Z, Meng G. PML nuclear body biogenesis, carcinogenesis, and targeted therapy. Trends Cancer. 2020;6(10):889–906.
Altucci L, Leibowitz MD, Ogilvie KM, de Lera AR, Gronemeyer H. RAR and RXR modulation in cancer and metabolic disease. Nat Rev Drug Discov. 2007;6(10):793–810. https://doi.org/10.1038/nrd2397.
Martino OD, Welch JS. Retinoic acid receptors in acute myeloid leukemia therapy. Cancers (Basel). 2019;11(12). https://doi.org/10.3390/cancers11121915.
Liquori A, Ibanez M, Sargas C, Sanz MA, Barragan E, Cervera J. Acute Promyelocytic leukemia: a constellation of molecular events around a single PML-RARA fusion gene. Cancers (Basel). 2020;12(3). https://doi.org/10.3390/cancers12030624.
Noguera NI, Catalano G, Banella C, Divona M, Faraoni I, Ottone T, et al. Acute Promyelocytic leukemia: update on the mechanisms of Leukemogenesis, resistance and on innovative treatment strategies. Cancers (Basel). 2019;11(10). https://doi.org/10.3390/cancers11101591.
Chen GQ, Shen ZX, Wu F, Han JY, Miao JM, Zhong HJ, et al. Pharmacokinetics and efficacy of low-dose all-trans retinoic acid in the treatment of acute promyelocytic leukemia. Leukemia. 1996;10:825–8.
Huang ME, Ye YC, Chen SR, Chai JR, Lu JX, Zhoa L, et al. Use of all-trans retinoic acid in the treatment of acute promyelocytic leukemia. Blood. 1988;72(2):567–72. https://doi.org/10.1182/blood.V72.2.567.567.
Chen SJ, Zhou GB, Zhang XW, Mao JH, de The H, Chen Z. From an old remedy to a magic bullet: molecular mechanisms underlying the therapeutic effects of arsenic in fighting leukemia. Blood. 2011;117(24):6425–37. https://doi.org/10.1182/blood-2010-11-283598.
Zhang XW, Yan XJ, Zhou ZR, Yang FF, Wu ZY, Sun HB, et al. Arsenic trioxide controls the fate of the PML-RARalpha oncoprotein by directly binding PML. Science. 2010;328(5975):240–3. https://doi.org/10.1126/science.1183424.
Zhou GB, Zhao WL, Wang ZY, Chen SJ, Chen Z. Retinoic acid and arsenic for treating acute promyelocytic leukemia. PLoS Med. 2005;2:e12.
Iaccarino L, Ottone T, Alfonso V, Cicconi L, Divona M, Lavorgna S, et al. Mutational landscape of patients with acute promyelocytic leukemia at diagnosis and relapse. Am J Hematol. 2019;94(10):1091–7. https://doi.org/10.1002/ajh.25573.
Shen Y, Fu YK, Zhu YM, Lou YJ, Gu ZH, Shi JY, et al. Mutations of epigenetic modifier genes as a poor prognostic factor in acute Promyelocytic leukemia under treatment with all-trans retinoic acid and arsenic trioxide. EBioMedicine. 2015;2(6):563–71. https://doi.org/10.1016/j.ebiom.2015.04.006.
Fasan A, Haferlach C, Perglerova K, Kern W, Haferlach T. Molecular landscape of acute promyelocytic leukemia at diagnosis and relapse. Haematologica. 2017;102(6):e222–4. https://doi.org/10.3324/haematol.2016.162206.
Madan V, Shyamsunder P, Han L, Mayakonda A, Nagata Y, Sundaresan J, et al. Comprehensive mutational analysis of primary and relapse acute promyelocytic leukemia. Leukemia. 2016;30:1672–81.
Zhao J, Liang JW, Xue HL, Shen SH, Chen J, Tang YJ, et al. The genetics and clinical characteristics of children morphologically diagnosed as acute promyelocytic leukemia. Leukemia. 2019;33(6):1387–99. https://doi.org/10.1038/s41375-018-0338-z.
Alfonso V, Iaccarino L, Ottone T, Cicconi L, Lavorgna S, Divona M, et al. Early and sensitive detection of PML-A216V mutation by droplet digital PCR in ATO-resistant acute promyelocytic leukemia. Leukemia. 2019;33(6):1527–30. https://doi.org/10.1038/s41375-018-0298-3.
Zhu HH, Qin YZ, Huang XJ. Resistance to arsenic therapy in acute promyelocytic leukemia. N Engl J Med. 2014;370(19):1864–6. https://doi.org/10.1056/NEJMc1316382.
Han SB, Lim J, Kim Y, Kim HJ, Han K. A variant acute promyelocytic leukemia with t(11;17) (q23;q12); ZBTB16-RARA showing typical morphology of classical acute promyelocytic leukemia. Korean J Hematol. 2010;45(2):133–5. https://doi.org/10.5045/kjh.2010.45.2.133.
Langabeer SE, Preston L, Kelly J, Goodyer M, Elhassadi E, Hayat A. Molecular profiling: a case of ZBTB16-RARA acute Promyelocytic leukemia. Case Rep Hematol. 2017;2017:7657393.
Lechevalier N, Dulucq S, Bidet A. A case of acute promyelocytic leukaemia with unusual cytological features and a ZBTB16-RARA fusion gene. Br J Haematol. 2016;174:502.
Rohr SS, Pelloso LA, Borgo A, De Nadai LC, Yamamoto M, Rego EM, et al. Acute promyelocytic leukemia associated with the PLZF-RARA fusion gene: two additional cases with clinical and laboratorial peculiar presentations. Med Oncol. 2012;29(4):2345–7. https://doi.org/10.1007/s12032-011-0147-y.
Sainty D, Liso V, Cantu-Rajnoldi A, Head D, Mozziconacci MJ, Arnoulet C, et al. A new morphologic classification system for acute promyelocytic leukemia distinguishes cases with underlying PLZF/RARA gene rearrangements. Blood. 2000;96(4):1287–96.
Licht JD, Chomienne C, Goy A, Chen A, Scott AA, Head DR, et al. Clinical and molecular characterization of a rare syndrome of acute promyelocytic leukemia associated with translocation (11;17). Blood. 1995;85:1083–94.
Chen Z, Guidez F, Rousselot P, Agadir A, Chen SJ, Wang ZY, et al. PLZF-RAR alpha fusion proteins generated from the variant t(11;17)(q23;q21) translocation in acute promyelocytic leukemia inhibit ligand-dependent transactivation of wild-type retinoic acid receptors. Proc Natl Acad Sci U S A. 1994;91(3):1178–82. https://doi.org/10.1073/pnas.91.3.1178.
Boukarabila H, Saurin AJ, Batsche E, Mossadegh N, van Lohuizen M, Otte AP, et al. The PRC1 Polycomb group complex interacts with PLZF/RARA to mediate leukemic transformation. Genes Dev. 2009;23(10):1195–206. https://doi.org/10.1101/gad.512009.
Tomita A, Buchholz DR, Obata K, Shi YB. Fusion protein of retinoic acid receptor alpha with promyelocytic leukemia protein or promyelocytic leukemia zinc finger protein recruits N-CoR-TBLR1 corepressor complex to repress transcription in vivo. J Biol Chem. 2003;278(33):30788–95. https://doi.org/10.1074/jbc.M303309200.
Hong SH, David G, Wong CW, Dejean A, Privalsky ML. SMRT corepressor interacts with PLZF and with the PML-retinoic acid receptor alpha (RARalpha) and PLZF-RARalpha oncoproteins associated with acute promyelocytic leukemia. Proc Natl Acad Sci U S A. 1997;94(17):9028–33. https://doi.org/10.1073/pnas.94.17.9028.
Dong S, Zhu J, Reid A, Strutt P, Guidez F, Zhong HJ, et al. Amino-terminal protein-protein interaction motif (POZ-domain) is responsible for activities of the promyelocytic leukemia zinc finger-retinoic acid receptor-alpha fusion protein. Proc Natl Acad Sci U S A. 1996;93(8):3624–9. https://doi.org/10.1073/pnas.93.8.3624.
Ruthardt M, Testa U, Nervi C, Ferrucci PF, Grignani F, Puccetti E, et al. Opposite effects of the acute promyelocytic leukemia PML-retinoic acid receptor alpha (RAR alpha) and PLZF-RAR alpha fusion proteins on retinoic acid signalling. Mol Cell Biol. 1997;17(8):4859–69. https://doi.org/10.1128/MCB.17.8.4859.
Koken MH, Daniel MT, Gianni M, Zelent A, Licht J, Buzyn A, et al. Retinoic acid, but not arsenic trioxide, degrades the PLZF/RARalpha fusion protein, without inducing terminal differentiation or apoptosis, in a RA-therapy resistant t(11;17)(q23;q21) APL patient. Oncogene. 1999;18:1113–8.
He LZ, Tolentino T, Grayson P, Zhong S, Warrell RP Jr, Rifkind RA, et al. Histone deacetylase inhibitors induce remission in transgenic models of therapy-resistant acute promyelocytic leukemia. J Clin Invest. 2001;108:1321–30.
Jiao B, Ren ZH, Liu P, Chen LJ, Shi JY, Dong Y, et al. 8-CPT-cAMP/all-trans retinoic acid targets t(11;17) acute promyelocytic leukemia through enhanced cell differentiation and PLZF/RARalpha degradation. Proc Natl Acad Sci U S A. 2013;110(9):3495–500. https://doi.org/10.1073/pnas.1222863110.
Yang WC, Shih HM. The deubiquitinating enzyme USP37 regulates the oncogenic fusion protein PLZF/RARA stability. Oncogene. 2013;32(43):5167–75. https://doi.org/10.1038/onc.2012.537.
Girard N, Tremblay M, Humbert M, Grondin B, Haman A, Labrecque J, et al. RARalpha-PLZF oncogene inhibits C/EBPalpha function in myeloid cells. Proc Natl Acad Sci U S A. 2013;110(33):13522–7. https://doi.org/10.1073/pnas.1310067110.
Guidez F, Parks S, Wong H, Jovanovic JV, Mays A, Gilkes AF, et al. RARalpha-PLZF overcomes PLZF-mediated repression of CRABPI, contributing to retinoid resistance in t(11;17) acute promyelocytic leukemia. Proc Natl Acad Sci U S A. 2007;104(47):18694–9. https://doi.org/10.1073/pnas.0704433104.
Cheng GX, Zhu XH, Men XQ, Wang L, Huang QH, Jin XL, et al. Distinct leukemia phenotypes in transgenic mice and different corepressor interactions generated by promyelocytic leukemia variant fusion genes PLZF-RARalpha and NPM-RARalpha. Proc Natl Acad Sci U S A. 1999;96(11):6318–23. https://doi.org/10.1073/pnas.96.11.6318.
Redner RL, Rush EA, Faas S, Rudert WA, Corey SJ. The t(5;17) variant of acute promyelocytic leukemia expresses a nucleophosmin-retinoic acid receptor fusion. Blood. 1996;87:882–6.
Pollock SL, Rush EA, Redner RL. NPM-RAR, not the RAR-NPM reciprocal t(5;17)(q35;q21) acute promyelocytic leukemia fusion protein, inhibits myeloid differentiation. Leuk Lymphoma. 2014;55:1383–7.
Kikuma T, Nakamachi Y, Noguchi Y, Okazaki Y, Shimomura D, Yakushijin K, et al. A new transcriptional variant and small azurophilic granules in an acute promyelocytic leukemia case with NPM1/RARA fusion gene. Int J Hematol. 2015;102(6):713–8. https://doi.org/10.1007/s12185-015-1857-2.
Yanagisawa R, Ogiso Y, Yoshikawa K, Tanaka M, Matsuda K, Ishii E. Myelomonocytic differentiation associated with NPM1-RARA rearrangement. Br J Haematol. 2017;179:183.
Rego EM, Ruggero D, Tribioli C, Cattoretti G, Kogan S, Redner RL, et al. Leukemia with distinct phenotypes in transgenic mice expressing PML/RAR alpha, PLZF/RAR alpha or NPM/RAR alpha. Oncogene. 2006;25(13):1974–9. https://doi.org/10.1038/sj.onc.1209216.
Otsubo K, Horie S, Nomura K, Miyawaki T, Abe A, Kanegane H. Acute promyelocytic leukemia following aleukemic leukemia cutis harboring NPM/RARA fusion gene. Pediatr Blood Cancer. 2012;59(5):959–60. https://doi.org/10.1002/pbc.24199.
Kanegane H, Nomura K, Abe A, Makino T, Ishizawa S, Shimizu T, et al. Spontaneous regression of aleukemic leukemia cutis harboring a NPM/RARA fusion gene in an infant with cutaneous mastocytosis. Int J Hematol. 2009;89(1):86–90. https://doi.org/10.1007/s12185-008-0216-y.
Li Y, Shao H, Fu B. Coexistence of t(5;17)/NPM1-RARA and t(9;22)/BCR-ABL1 in chronic myeloid leukemia at initial diagnosis. Ann Hematol. 2019;98:1319–21.
Redner RL, Chen JD, Rush EA, Li H, Pollock SL. The t(5;17) acute promyelocytic leukemia fusion protein NPM-RAR interacts with co-repressor and co-activator proteins and exhibits both positive and negative transcriptional properties. Blood. 2000;95(8):2683–90. https://doi.org/10.1182/blood.V95.8.2683.
Chattopadhyay A, Abecassis I, Redner RL. NPM-RAR binding to TRADD selectively inhibits caspase activation, while allowing activation of NFkappaB and JNK. Leuk Lymphoma. 2015;56(12):3401–6. https://doi.org/10.3109/10428194.2015.1023799.
Chattopadhyay A, Hood BL, Conrads TP, Redner RL. Extrinsic apoptosis is impeded by direct binding of the APL fusion protein NPM-RAR to TRADD. Mol Cancer Res. 2014;12:1283–91.
Swaney EM, Chattopadhyay A, Abecassis I, Rush EA, Redner RL. The leukemic oncoprotein NPM1-RARA inhibits TP53 activity. Leuk Lymphoma. 2016;57(8):1933–7. https://doi.org/10.3109/10428194.2015.1124992.
Wells RA, Catzavelos C, Kamel-Reid S. Fusion of retinoic acid receptor alpha to NuMA, the nuclear mitotic apparatus protein, by a variant translocation in acute promyelocytic leukaemia. Nat Genet. 1997;17:109–13.
Wells RA, Hummel JL, De Koven A, Zipursky A, Kirby M, Dube I, et al. A new variant translocation in acute promyelocytic leukaemia: molecular characterization and clinical correlation. Leukemia. 1996;10(4):735–40.
Hummel JL, Zhang T, Wells RA, Kamel-Reid S. The retinoic acid receptor alpha (RARalpha) chimeric proteins PML-, PLZF-, NPM-, and NuMA-RARalpha have distinct intracellular localization patterns. Cell Growth Differ. 2002;13:173–83.
Sukhai MA, Wu X, Xuan Y, Zhang T, Reis PP, Dube K, et al. Myeloid leukemia with promyelocytic features in transgenic mice expressing hCG-NuMA-RARalpha. Oncogene. 2004;23(3):665–78. https://doi.org/10.1038/sj.onc.1207073.
Dong S, Qiu J, Stenoien DL, Brinkley WR, Mancini MA, Tweardy DJ. Essential role for the dimerization domain of NuMA-RARalpha in its oncogenic activities and localization to NuMA sites within the nucleus. Oncogene. 2003;22:858–68.
Sukhai MA, Thomas M, Xuan Y, Chan LS, Hamadanizadeh SA, Zhang T, et al. Evidence of functional interaction between NuMA-RARalpha and RXRalpha in an in vivo model of acute promyelocytic leukemia. Oncogene. 2008;27(34):4666–77. https://doi.org/10.1038/onc.2008.106.
Sukhai MA, Thomas M, Hamadanizadeh SA, Xuan Y, Wells RA, Kamel-Reid S. Correlation among nuclear localization of NuMA-RARalpha, deregulation of gene expression and leukemic phenotype of hCG-NuMA-RARalpha transgenic mice. Leuk Res. 2011;35:670–6.
Arnould C, Philippe C, Bourdon V, Gr goire MJ, Berger R, Jonveaux P: The signal transducer and activator of transcription STAT5b gene is a new partner of retinoic acid receptor alpha in acute promyelocytic-like leukaemia. Hum Mol Genet. 1999, 8:1741–1749, 9, DOI: https://doi.org/10.1093/hmg/8.9.1741.
Chen H, Pan J, Yao L, Wu L, Zhu J, Wang W, et al. Acute promyelocytic leukemia with a STAT5b-RARalpha fusion transcript defined by array-CGH, FISH, and RT-PCR. Cancer Genet. 2012;205(6):327–31. https://doi.org/10.1016/j.cancergen.2012.02.007.
Iwanaga E, Nakamura M, Nanri T, Kawakita T, Horikawa K, Mitsuya H, et al. Acute promyelocytic leukemia harboring a STAT5B-RARA fusion gene and a G596V missense mutation in the STAT5B SH2 domain of the STAT5B-RARA. Eur J Haematol. 2009;83(5):499–501. https://doi.org/10.1111/j.1600-0609.2009.01324.x.
Kluk MJ, Abo RP, Brown RD, Kuo FC, Dal Cin P, Pozdnyakova O, et al. Myeloid neoplasm demonstrating a STAT5B-RARA rearrangement and genetic alterations associated with all-trans retinoic acid resistance identified by a custom next-generation sequencing assay. Cold Spring Harb Mol Case Stud. 2015;1(1):a000307. https://doi.org/10.1101/mcs.a000307.
Kusakabe M, Suzukawa K, Nanmoku T, Obara N, Okoshi Y, Mukai HY, et al. Detection of the STAT5B-RARA fusion transcript in acute promyelocytic leukemia with the normal chromosome 17 on G-banding. Eur J Haematol. 2008;80(5):444–7. https://doi.org/10.1111/j.1600-0609.2008.01042.x.
Pessina C, Basilico C, Genoni A, Meroni E, Elli L, Granata P, et al. A new acute myeloid leukemia case with STAT5B-RARA gene fusion due to 17q21.2 interstitial deletion. Leuk Lymphoma. 2017;58(8):1977–80. https://doi.org/10.1080/10428194.2016.1262952.
Peterson JF, He RR, Nayer H, Cuevo RS, Smadbeck JB, Vasmatzis G, et al. Characterization of a rarely reported STAT5B/RARA gene fusion in a young adult with newly diagnosed acute promyelocytic leukemia with resistance to ATRA therapy. Cancer Genet. 2019;237:51–4. https://doi.org/10.1016/j.cancergen.2019.06.007.
Maurer AB, Wichmann C, Gross A, Kunkel H, Heinzel T, Ruthardt M, et al. The Stat5-RARalpha fusion protein represses transcription and differentiation through interaction with a corepressor complex. Blood. 2002;99(8):2647–52. https://doi.org/10.1182/blood.v99.8.2647.
Dong S, Tweardy DJ. Interactions of STAT5b-RARalpha, a novel acute promyelocytic leukemia fusion protein, with retinoic acid receptor and STAT3 signaling pathways. Blood. 2002;99:2637–46.
Ciangola G, Gurnari C, Paterno G, Mirabile M, Angelini M, Lavorgna S, et al. STAT5b-RARa-positive acute myeloid leukemia: diagnostic and therapeutic challenges of a rare AML subtype. Leuk Res. 2019;78:21–3. https://doi.org/10.1016/j.leukres.2019.01.004.
Wang YY, Hao J, Liu ZY, Weng XQ, Sheng Y, Jiang CL, et al. Novel STAT5B-RARA fusion transcript in acute promyelocytic leukemia: identification and treatment response. Leuk Lymphoma. 2015;56(9):2731–4. https://doi.org/10.3109/10428194.2015.1007454.
Zhang C, Wang Y, Liu B, Gong B, Gong X, Liu Y, et al. Clinical characteristics of acute promyelocytic leukemia with the STAT5B-RARA fusion gene. Blood Cells Mol Dis. 2018;69:71–3. https://doi.org/10.1016/j.bcmd.2017.09.007.
Strehl S, Konig M, Boztug H, Cooper BW, Suzukawa K, Zhang SJ, et al. All-trans retinoic acid and arsenic trioxide resistance of acute promyelocytic leukemia with the variant STAT5B-RARA fusion gene. Leukemia. 2013;27:1606–10.
Catalano A, Dawson MA, Somana K, Opat S, Schwarer A, Campbell LJ, et al. The PRKAR1A gene is fused to RARA in a new variant acute promyelocytic leukemia. Blood. 2007;110(12):4073–6. https://doi.org/10.1182/blood-2007-06-095554.
Qiu JJ, Lu X, Zeisig BB, Ma Z, Cai X, Chen S, et al. Leukemic transformation by the APL fusion protein PRKAR1A-RAR{alpha} critically depends on recruitment of RXR{alpha}. Blood. 2010;115(3):643–52. https://doi.org/10.1182/blood-2009-07-232652.
Yamamoto Y, Tsuzuki S, Tsuzuki M, Handa K, Inaguma Y, Emi N. BCOR as a novel fusion partner of retinoic acid receptor alpha in a t(X;17)(p11;q12) variant of acute promyelocytic leukemia. Blood. 2010;116:4274–83.
Ichikawa S, Ichikawa S, Ishikawa I, Takahashi T, Fujiwara T, Harigae H. Successful treatment of acute promyelocytic leukemia with a t(X;17)(p11.4;q21) and BCOR-RARA fusion gene. Cancer Genet. 2015;208:162–3.
Kondo T, Mori A, Darmanin S, Hashino S, Tanaka J, Asaka M. The seventh pathogenic fusion gene FIP1L1-RARA was isolated from a t(4;17)-positive acute promyelocytic leukemia. Haematologica. 2008;93:1414–6.
Menezes J, Acquadro F, Perez-Pons de la Villa C, Garcia-Sanchez F, Alvarez S, Cigudosa JC. FIP1L1/RARA with breakpoint at FIP1L1 intron 13: a variant translocation in acute promyelocytic leukemia. Haematologica. 2011;96(10):1565–6. https://doi.org/10.3324/haematol.2011.047134.
Buijs A, Bruin M. Fusion of FIP1L1 and RARA as a result of a novel t(4;17)(q12;q21) in a case of juvenile myelomonocytic leukemia. Leukemia. 2007;21(5):1104–8. https://doi.org/10.1038/sj.leu.2404596.
Iwasaki J, Kondo T, Darmanin S, Ibata M, Onozawa M, Hashimoto D, et al. FIP1L1 presence in FIP1L1-RARA or FIP1L1-PDGFRA differentially contributes to the pathogenesis of distinct types of leukemia. Ann Hematol. 2014;93(9):1473–81. https://doi.org/10.1007/s00277-014-2085-1.
Won D, Shin SY, Park CJ, Jang S, Chi HS, Lee KH, et al. OBFC2A/RARA: a novel fusion gene in variant acute promyelocytic leukemia. Blood. 2013;121(8):1432–5. https://doi.org/10.1182/blood-2012-04-423129.
Chen Y, Li S, Zhou C, Li C, Ru K, Rao Q, et al. TBLR1 fuses to retinoid acid receptor alpha in a variant t(3;17)(q26;q21) translocation of acute promyelocytic leukemia. Blood. 2014;124(6):936–45. https://doi.org/10.1182/blood-2013-10-528596.
Li J, Zhong HY, Zhang Y, Xiao L, Bai LH, Liu SF, et al. GTF2I-RARA is a novel fusion transcript in a t(7;17) variant of acute promyelocytic leukaemia with clinical resistance to retinoic acid. Br J Haematol. 2015;168(6):904–8. https://doi.org/10.1111/bjh.13157.
Yan W, Li J, Zhang Y, Yin Y, Cheng Z, Wang J, et al. RNF8 is responsible for ATRA resistance in variant acute promyelocytic leukemia with GTF2I/RARA fusion, and inhibition of the ubiquitin-proteasome pathway contributes to the reversion of ATRA resistance. Cancer Cell Int. 2019;19(1):84. https://doi.org/10.1186/s12935-019-0803-4.
Shimomura Y, Mitsui H, Yamashita Y, Kamae T, Kanai A, Matsui H, et al. New variant of acute promyelocytic leukemia with IRF2BP2-RARA fusion. Cancer Sci. 2016;107(8):1165–8. https://doi.org/10.1111/cas.12970.
Yin CC, Jain N, Mehrotra M, Zhagn J, Protopopov A, Zuo Z, et al. Identification of a novel fusion gene, IRF2BP2-RARA, in acute promyelocytic leukemia. J Natl Compr Cancer Netw. 2015;13(1):19–22. https://doi.org/10.6004/jnccn.2015.0005.
Mazharuddin S, Chattopadhyay A, Levy MY, Redner RL. IRF2BP2-RARA t(1;17)(q42.3;q21.2) APL blasts differentiate in response to all-trans retinoic acid. Leuk Lymphoma. 2018;59(9):2246–9. https://doi.org/10.1080/10428194.2017.1421761.
Liu Y, Xu F, Hu H, Wen J, Su J, Zhou Q, et al. A rare case of acute promyelocytic leukemia with IRF2BP2-RARA fusion; and literature review. Onco Targets Ther. 2019;12:6157–63. https://doi.org/10.2147/OTT.S217622.
Jovanovic JV, Chillon MC, Vincent-Fabert C, Dillon R, Voisset E, Gutierrez NC, et al. The cryptic IRF2BP2-RARA fusion transforms hematopoietic stem/progenitor cells and induces retinoid-sensitive acute promyelocytic leukemia. Leukemia. 2017;31(3):747–51. https://doi.org/10.1038/leu.2016.338.
Cheng CK, Wang AZ, Wong THY, Wan TSK, Cheung JS, Raghupathy R, et al. FNDC3B is another novel partner fused to RARA in the t(3;17)(q26;q21) variant of acute promyelocytic leukemia. Blood. 2017;129(19):2705–9. https://doi.org/10.1182/blood-2017-02-767707.
Yao L, Wen L, Wang N, Liu T, Xu Y, Ruan C, et al. Identification of novel recurrent STAT3-RARA fusions in acute promyelocytic leukemia lacking t(15;17)(q22;q12)/PML-RARA. Blood. 2018;131(8):935–9. https://doi.org/10.1182/blood-2017-09-807370.
Chong ML, Cheng H, Xu P, You H, Wang M, Wang L, et al. TFG-RARA: a novel fusion gene in acute promyelocytic leukemia that is responsive to all-trans retinoic acid. Leuk Res. 2018;74:51–4. https://doi.org/10.1016/j.leukres.2018.09.012.
Zhu HH, Yang MC, Wang F, Lou YJ, Jin J, Li K, et al. Identification of a novel NUP98-RARA fusion transcript as the 14(th) variant of acute promyelocytic leukemia. Am J Hematol. 2020;95(7):E184–6. https://doi.org/10.1002/ajh.25807.
Wang Z, Wen L, Zhang L, Xu X, Chen X, Yao L, et al. Identification of a novel TNRC18-RARA fusion in acute promyelocytic leukemia lacking t(15;17)(q24;q12)/PML-RARA. Mol Carcinog. 2021;60(2). https://doi.org/10.1002/mc.23276.
Osumi T, Tsujimoto SI, Tamura M, Uchiyama M, Nakabayashi K, Okamura K, et al. Recurrent RARB translocations in acute Promyelocytic leukemia lacking RARA translocation. Cancer Res. 2018;78:4452–8.
Shiba N, Yoshida K, Hara Y, Yamato G, Shiraishi Y, Matsuo H, et al. Transcriptome analysis offers a comprehensive illustration of the genetic background of pediatric acute myeloid leukemia. Blood Adv. 2019;3(20):3157–69. https://doi.org/10.1182/bloodadvances.2019000404.
Such E, Cervera J, Valencia A, Barragan E, Ibanez M, Luna I, et al. A novel NUP98/RARG gene fusion in acute myeloid leukemia resembling acute promyelocytic leukemia. Blood. 2011;117(1):242–5. https://doi.org/10.1182/blood-2010-06-291658.
Qiu JJ, Zeisig BB, Li S, Liu W, Chu H, Song Y, et al. Critical role of retinoid/rexinoid signaling in mediating transformation and therapeutic response of NUP98-RARG leukemia. Leukemia. 2015;29(5):1153–62. https://doi.org/10.1038/leu.2014.334.
Such E, Cordon L, Sempere A, Villamon E, Ibanez M, Luna I, et al. In vitro all-trans retinoic acid sensitivity of acute myeloid leukemia blasts with NUP98/RARG fusion gene. Ann Hematol. 2014;93:1931–3.
Luo H, Zhang S, Li K, Chen XH, Li YC, Sun Y, et al. A novel entity of acute myeloid leukaemia with recurrent RARG-rearrangement resembling acute promyelocytic leukaemia. Leuk Res. 2019;77:14–6. https://doi.org/10.1016/j.leukres.2018.12.009.
Zhang X, Li F, Wang J, Suo S, Ling Q, Yu W, et al. RARgamma-rearrangements resemble acute promyelocytic leukemia and benefit from 3 + 7 regimen. Leuk Lymphoma. 2019;60(7):1831–4. https://doi.org/10.1080/10428194.2018.1553302.
Wei W, Liu Q, Song F, Cao H, Liu M, Jiang Y, et al. Alkaloid-based regimen is beneficial for acute myeloid leukemia resembling acute promyelocytic leukemia with NUP98/RARG fusion and RUNX1 mutation: a case report. Medicine (Baltimore). 2020;99(40):e22488. https://doi.org/10.1097/MD.0000000000022488.
Tao S, Song L, Deng Y, Chen Y, Shi Y, Gan Y, et al. Acute myeloid leukemia with NUP98-RARG gene fusion similar to acute Promyelocytic leukemia: case report and literature review. Onco Targets Ther. 2020;13:10559–66. https://doi.org/10.2147/OTT.S273172.
Ha JS, Do YR, Ki CS, Lee C, Kim DH, Lee W, et al. Identification of a novel PML-RARG fusion in acute promyelocytic leukemia. Leukemia. 2017;31(9):1992–5. https://doi.org/10.1038/leu.2017.167.
Zhang Z, Jiang M, Borthakur G, Luan S, Huang X, Tang G, et al. Acute myeloid leukemia with a novel CPSF6-RARG variant is sensitive to homoharringtonine and cytarabine chemotherapy. Am J Hematol. 2020;95(2):E48–51. https://doi.org/10.1002/ajh.25689.
Liu T, Wen L, Yuan H, Wang Y, Yao L, Xu Y, et al. Identification of novel recurrent CPSF6-RARG fusions in acute myeloid leukemia resembling acute promyelocytic leukemia. Blood. 2018;131(16):1870–3. https://doi.org/10.1182/blood-2017-11-818716.
Miller CA, Tricarico C, Skidmore ZL, Uy GL, Lee YS, Hassan A, et al. A case of acute myeloid leukemia with promyelocytic features characterized by expression of a novel RARG-CPSF6 fusion. Blood Adv. 2018;2(11):1295–9. https://doi.org/10.1182/bloodadvances.2017014183.
Qin YZ, Huang XJ, Zhu HH. Identification of a novel CPSF6-RARG fusion transcript in acute myeloid leukemia resembling acute promyelocytic leukemia. Leukemia. 2018;32(10):2285–7. https://doi.org/10.1038/s41375-018-0095-z.
Wen L, Xu Y, Yao L, Wang N, Wang Q, Liu T, et al. Clinical and molecular features of acute promyelocytic leukemia with variant retinoid acid receptor fusions. Haematologica. 2019;104:e195–9.
Han X, Jin C, Zheng G, Li Y, Wang Y, Zhang E, et al. Acute myeloid leukemia with CPSF6-RARG fusion resembling acute promyelocytic leukemia with extramedullary infiltration. Ther Adv Hematol. 2021;12:2040620720976984.
Chen X, Wang F, Zhang Y, Teng W, Cao P, Ma X, et al. A novel NPM1-RARG-NPM1 chimeric fusion in acute myeloid leukaemia resembling acute promyelocytic leukaemia but resistant to all-trans retinoic acid and arsenic trioxide. Br J Cancer. 2019;120(11):1023–5. https://doi.org/10.1038/s41416-019-0456-z.
Su Z, Liu X, Xu Y, Hu W, Zhao C, Zhao H, et al. Novel reciprocal fusion genes involving HNRNPC and RARG in acute promyelocytic leukemia lacking RARA rearrangement. Haematologica. 2020;105(7):e376–8. https://doi.org/10.3324/haematol.2019.244715.
Kang LC, Smith SV, Kaiser-Rogers K, Rao K, Dunphy CH. Two cases of acute myeloid leukemia with t(11;17) associated with varying morphology and immunophenotype: rearrangement of the MLL gene and a region proximal to the RARalpha gene. Cancer Genet Cytogenet. 2005;159(2):168–73. https://doi.org/10.1016/j.cancergencyto.2004.10.009.
Zhang X, Huang X, Xu H, Li J, Yu W. MLL-rearrangement can resemble acute promyelocytic leukemia. Leuk Lymphoma. 2019;60:2841–3.
Kunchala P, Kuravi S, Jensen R, McGuirk J, Balusu R. When the good go bad: mutant NPM1 in acute myeloid leukemia. Blood Rev. 2018;32(3):167–83. https://doi.org/10.1016/j.blre.2017.11.001.
Berger R, Busson M, Baranger L, Helias C, Lessard M, Dastugue N, et al. Loss of the NPM1 gene in myeloid disorders with chromosome 5 rearrangements. Leukemia. 2006;20(2):319–21. https://doi.org/10.1038/sj.leu.2404063.
Campregher PV, de Oliveira PW, Lisboa B, Puga R, Deolinda ER, Helman R, et al. A novel mechanism of NPM1 cytoplasmic localization in acute myeloid leukemia: the recurrent gene fusion NPM1-HAUS1. Haematologica. 2016;101(7):e287–90. https://doi.org/10.3324/haematol.2015.137364.
Coccaro N, Zagaria A, Orsini P, Anelli L, Tota G, Casieri P, et al. RARA and RARG gene downregulation associated with EZH2 mutation in acute promyelocytic-like morphology leukemia. Hum Pathol. 2018;80:82–6.
Sanz MA, Fenaux P, Tallman MS, Estey EH, Lowenberg B, Naoe T, et al. Management of acute promyelocytic leukemia: updated recommendations from an expert panel of the European LeukemiaNet. Blood. 2019;133(15):1630–43. https://doi.org/10.1182/blood-2019-01-894980.
Wang L, Yan X, He J. Does acute promyelocytic leukemia patient with the STAT5B/RARa fusion gene respond well to decitabine?: a case report and literature review. Medicine (Baltimore). 2020;99(43):e22923. https://doi.org/10.1097/MD.0000000000022923.
Acknowledgements
We thanked the supports from all members of Department of Hematology, The First Affiliated Hospital, Zhejiang University College of Medicine.
Funding
This study was funded by the National Natural Science Foundation of China (81800199, 81670124), the Natural Science Foundation of Zhejiang Province (LY21H080003, LQ21H160010), and Medical Science and Technology Foundation of Zhejiang Province (2020KY204).
Author information
Authors and Affiliations
Contributions
X.Z. designed the study. X.Z. and J.-W. S. reviewed literatures and made summary. X.Z. wrote this manuscript. W.-J. Y. and J.J. revised the manuscript. All authors approved the manuscript. X.Z. and J.-W. S. should be considered equal first authors.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
All authors have agreed to publish this manuscript.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Zhang, X., Sun, J., Yu, W. et al. Current views on the genetic landscape and management of variant acute promyelocytic leukemia. Biomark Res 9, 33 (2021). https://doi.org/10.1186/s40364-021-00284-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40364-021-00284-x