
Abstract
Background
Genomic instability and clonal evolution are hallmarks of progressing chronic myeloid leukemia (CML). Recently, we have shown that clonal evolution and blast crisis correlate with altered expression and activity of Separase, a cysteine endopeptidase that is a mitotic key player in chromosomal segregation and centriole duplication. Hyperactivation of Separase in human hematopoietic cells has been linked to a feedback mechanism that posttranslationally stimulates Separase proteolytic activity after imatinib therapy-induced reduction of Separase protein levels.
Methods and Results
In search for potential therapy-responsive transcriptional mechanisms we have investigated the role of the transcription factor c-MYB for Separase expression in CML cell lines (LAMA-84, K562, BV-173) and in clinical samples. Quantitative RT-PCR and Western blot immunostaining experiments revealed that c-MYB expression levels are decreased in an imatinib-dependent manner and positively correlate with Separase expression levels in cell lines and in clinical CML samples. RNA silencing of c-MYB expression in CML cell lines resulted in reduced Separase protein levels. Gelshift and ChIP assays confirmed that c-MYB binds to a putative c-MYB binding sequence located within the ESPL1 promoter.
Conclusions
Our data suggest that ESPL1/Separase is a regulatory target of c-MYB. Therefore, c-MYB, known to be required for BCR-ABL-dependent transformation of hematopoietic progenitors and leukemogenesis, may also control the Separase-dependent fidelity of mitotic chromosomal segregation and centriole duplication essential for maintenance of genomic stability.
Similar content being viewed by others


Background
ESPL1/Separase, a cysteine endopeptidase, is a key player of chromosomal segregation and centrosome duplication. In mitotic anaphase, it accomplishes proteolytic cleavage of Cohesin, a “glue” multi-protein complex that is responsible for cohesion of sister chromatids and of mother and daughter centrioles, the perpendicular oriented core structures of centrosomes [1–3]. Proper temporal and spatial activation of Separase proteolytic activity warrants chromosomal fidelity by establishing an accurate chromosomal segregation [4]. Furthermore, it is an essential prerequisite for semiconservative centriole duplication as the disengagement of mother and daughter centrioles licenses the cell cycle-associated duplication of centrosomes [5]. Failure to do so will result in premature segregation of chromatids and/or formation of anaphase bridges from lagging chromosomes [6]. Moreover, unscheduled (cell cycle uncoupled) activation of Separase can lead to aberrant high numbers of centrosomes (i.e. centrosome amplification) and subsequently to a defective mitotic spindle apparatus [7]. Both defects cause the emergence of aberrant karyotypes (aneuploidy), a hallmark of most advanced human malignancies [8–10]. In non-malignant cells where chromosome segregation and centrosomal duplication are tightly coupled to the cell cycle, Separase is temporary activated on metaphase to anaphase transition [11, 12]. Separase is tightly regulated on both translational and posttranslational levels. The latter includes multiple inhibitory mechanisms combining Securin binding, specific serine residue phosphorylation (pSer1126) by CyclinB1/CDK1, autocatalytic cleavage, and PP2A-dependent stabilization of Separase-bound Securin. All these mechanisms work together to prevent ectopic and unscheduled activation of intracellular Separase molecules [13–16].
In human cancer, ESPL1/Separase is frequently overexpressed and the resulting deregulated proteolytic activity is associated with the occurrence of supernumerary centrosomes, chromosomal missegregation and aneuploidy [6, 9, 13, 17]. Overexpression of Separase in the mammary gland of a MMTV-ESPL1 mouse model led to the development of highly aneuploid mammary carcinomas with high levels of chromosomal instability and aggressive disease phenotypes [18]. Consequently, Separase has been identified as an aneuploidy promoter that, when overexpressed and hyperactive, functions as an oncogene and renders cells susceptible not only for chromosomal missegregation-induced aneuploidy but also for DNA damage and loss of key tumor suppressor gene loci associated with tumorigenesis and disease progression [18–20].
This holds true for chronic myeloid leukemia (CML) as well, a clonal neoplastic disorder of hematopoietic stem cells caused by the genomic reciprocal translocation t(9;22)(q34;q11), which results in the formation of the Philadelphia chromosome. The fusion product, a BCR-ABL tyrosine kinase (TK) with deregulated TK activity, is the key player in CML pathogenesis. It affects various downstream signaling pathways by reprogramming the prior lineage commitment of hematopoietic stem and early progenitor cells [21]. Compromising multiple aspects of cellular behavior, including proliferation, apoptosis, cell to cell signaling and differentiation, the BCR-ABL oncoprotein triggers aberrant clonal hematopoiesis and drives disease progression from chronic phase (CP) toward the fully transformed phenotype of blast crisis (BC) [22]. Imatinib (IM) is a selective TK inhibitor (TKI) and presents one of the current first line treatments for CML [23, 24]. Despite significant decreases in BCR-ABL mRNA levels in the bone marrow compartment under IM long-term therapy, persistence of residual CML clones with low BCR-ABL expression and insensitivity to IM treatment makes disease eradication by TKI treatment unlikely [13, 25, 26]. Recent evidence suggests that kinase activity of BCR-ABL oncoprotein in CML stem cells is inhibited by TKI treatment without affecting CML stem cell survival [27, 28]. Obviously, additional cellular mechanisms promote CML stem cell survival and maintenance, rendering these cells TKI resistant and eventually promote relapse [29, 30]. About 35 % of patients in CP develop resistance or intolerance to IM and frequently undergo clonal evolution [31]. Clonal evolution denotes a heterogeneous entity of clonal molecular changes in BCR-ABL-positive hematopoietic stem/progenitor cells and has been described in about 30 % and 80 % of patients in accelerated phase (AP) and BC, respectively [32]. Emergence of altered chromosome numbers, collectively termed aneuploidy, involves an additional derivative chromosome 22, chromosome 17 abnormalities, trisomy 8, and are associated with poor prognosis [33, 34].
Recently, we have reported that enhanced rates of acquired chromosomal aberrations, clonal evolution and fast disease progression (time to BC) in CML patients undergoing long-term IM treatment correlate with enhanced proteolytic activity of Separase in the respective in vitro models [35]. Mechanistically, this was linked to a BCR-ABL-dependent regulatory feedback mechanism that posttranslationally stimulates Separase proteolytic activity after IM-induced decreases of Separase expression in b3a2 BCR-ABL fusion type CML cell lines [35]. To date, it was unclear what underlying transcriptional or translational mechanism may be involved in the IM-dependent regulation of Separase expression in BCR-ABL-positive cells.
The transcription factor c-MYB is known to play an important role in BCR-ABL-dependent leukemogenesis. The expression of c-MYB is enhanced by BCR-ABL [36–38]. C-MYB is a 75 KDa nuclear protein and a leucine zipper transcription factor encoded by the proto-oncogene c-MYB. It plays a pivotal role in proliferation, survival and differentiation of normal myeloid progenitors [39]. Conditional knockout of c-MYB expression in adult hematopoietic stem cells causes loss of self-renewal due to impaired proliferation and accelerated differentiation [40]. On the other hand, overexpression of c-MYB in myeloid and erythroid cell lines has been demonstrated to block differentiation and prevents maturation-associated growth arrest [41]. Aberrant (enhanced) c-MYB expression has been found in various human malignancies including T-cell leukemia and acute and chronic myeloid leukemias [42]. Moreover, various genetic lesions affecting c-MYB activity in human leukemias, such as chromosomal translocation, gene duplication and truncations have been reported [39, 43, 44]. However, in CML cells, the c-MYB gene has been reported to be intact but protein levels are often increased, in part, due to enhanced protein stability via BCR-ABL-regulated activation of PI-3 K/Akt/GSKIIIß dependent pathways [36]. This altered regulatory mechanism has been considered to explain why leukemic blast cells appear to depend on high c-MYB expression levels more than their normal counterparts [45]. Functional studies of c-MYB by ChIP-Seq experiments revealed that c-MYB functions as a hematopoietic master regulator [46]. It binds directly near or within 793 genes thereby affecting more than 2300 genes that make up the gene signatures for normal and leukemic stem/progenitor cells and myeloid development. Despite being usually considered as a transactivator, c-MYB is also able to directly repress many target genes pointing to an important role for myelopoiesis and leukemogenesis through both positive and negative transcriptional regulation [47]. For example, c-MYB modulates the expression of CD34, c-Kit, c-Myc, Flt-3 and Bcl-2 all playing important roles for proliferation and survival of hematopoietic cells [38]. Moreover, c-MYB directly regulates CyclinB1 expression and contributes to the control of the G2/M cell cycle phase [48, 49], thereby directly controlling one of the key posttranslational inhibitors of Separase [13, 14].
In this study, we set out to investigate the role of c-MYB for the regulation of ESPL1/Separase expression in CML. We report that c-MYB binds to a c-MYB binding motif located within the ESPL1 promoter and functions as a positive regulator of ESPL1/Separase expression as demonstrated by c-MYB-directed siRNA silencing. Moreover, IM treatment led to equally decreased c-MYB and ESPL1/Separase expression levels in CML cell lines and primary cells.
Results
In search for transcriptional mechanisms that could explain the IM-associated downregulation of Separase protein levels we analyzed the conditional context between c-MYB expression, Separase and IM treatment. Confirming and expanding previously published work we performed cell culture experiments on four human cell lines [13, 35]. Of these, U937 cells served as model for leukemic but BCR-ABL-negative cells. All cell lines were treated with therapeutic doses of IM (1 to 5 μM) as performed in our previous studies [13, 50–52]. In accordance with data from extensive studies on the dose-dependent effects and time kinetics of IM we applied lower IM doses (range: 1 to 2.5 μM) for leukemia-derived p210BCR-ABL-positive cells (LAMA-84 and K562) than for p210BCR-ABL-negative cells (U937, 5 μM) [53, 54]. Treating CML cell lines with IM doses higher than 2.5 μM for a longer period than 24 h impeded the collection of sufficient viable cells for Western blot analysis (data not shown).
Concerted decrease of c-MYB and ESPL1 expression levels under IM treatment
Treatment of the CML cell lines LAMA-84 and K562 with therapeutic IM doses for 24 h revealed decreased expression levels for c-MYB and ESPL1/Separase on both transcriptional and protein levels as shown on representative Western blot composite images (Fig. 1, Table 1). It should be emphasized that the weak treatment schedule (≤2.5 μM IM for 24 h) still enabled CrkL phosphorylation (Fig. 1, lower panel). Thus, when compared to the respective untreated cells, the decreasing c-MYB protein levels in LAMA-84 (−25.7 ± 9.6 %) and K562 (−37.1 ± 9.6 %) cells concurred with decreased ESPL1 transcript levels of −90 ± 3.3 % and −25.0 ± 9.7 %, respectively. One might argue that the observed decline may be due to IM-related changes in the cell cycle. However, FACS analysis of tested cells revealed no differences neither in G2/M cell proportion nor in the apoptotic cell fraction (<12 %) that could clarify the observed decreases in c-MYB and ESPL1/Separase expression levels (compare Fig. 3b in Ref. [13]). Treatment of BCR-ABL-negative control cells (U937) with IM revealed no changes in c-MYB protein and ESPL1/Separase expression levels (Table 1, Fig. 1).

Analysis of c-MYB and ESPL1 transcript levels, c-MYB and Separase protein levels in LAMA-84 (a), K562 (b) and BCR-ABL-negative control cells (U937) (c) upon IM treatment. Treatment (dose, period) was performed as noted in Table 1. Level changes (Δ-values in %) are shown as calculated from comparison with the corresponding untreated cells. Upper panel. Transcript levels were analyzed by qRT-PCR, protein levels by Western blot immunostaining densitometry. Representative Western blot images are shown in the lower panel. In all qRT-PCR experiments the housekeeping gene Gus (beta-glucuronidase) served as internal standard. For Western blot immunostaining experiments Actin was used as loading control and reference parameter. Densitometric data are derived from at least triplicate experiments and are denoted in Table 1. P-values are given above the respective column. Abbreviations: ns, not significant; nd, not determined
Similar results were obtained when paired clinical samples (n = 5), each pair derived from the same CML patient before (at diagnosis) and during IM therapy were comparatively monitored for c-MYB and ESPL1 transcript levels (Fig. 2). Two female and three male patients, all with b3a2 BCR-ABL fusion type, were analyzed. The median age was 58 years (range, 47 to 78). Mean time between diagnosis and sampling of the second specimen from the same patient after achievement of MMR under IM treatment was 3.6 years (range, 2 to 6.5). These experiments suggest synchronous regulatory mechanisms for c-MYB and ESPL1/Separase expression upon IM treatment in vitro and in vivo.

Comparative analysis of c-MYB and ESPL1 transcript levels in paired cDNA samples of CML patients (n = 5) before and under IM therapy. Percent changes (Δ-values are differences between means) are shown corresponding to expression level changes within paired samples, each pair derived from the same patient at differing time points (sample at diagnosis (before IM treatment) vs. sample after major molecular response (MMR) achievement under IM therapy). All transcript levels were normalized to Gus and represent mean values of triplicate qRT-PCR assays
Decreased Separase expression levels after c-MYB silencing in CML cell lines
To test the direct regulatory influence of c-MYB on Separase expression we silenced c-Myb transcription in LAMA-84 and BV-173 cells and monitored the influence of the resulting decline in c-MYB protein levels on Separase expression (Fig. 3). The siRNA-induced decrease in available c-MYB molecules resulted in decreased c-MYB protein levels in BV-173 (−49 ± 13.6 %) and LAMA-84 cells (−47.6 ± 7.9 %). This suggests a direct regulatory relationship between c-MYB and ESPL1/Separase.

Separase expression after c-MYB silencing by RNAi in BV-173 and LAMA-84 cells. BV-173 (panel a) and LAMA-84 cells (panel b) were treated with negative control siRNA (nc) and c-MYB-specific siRNA (siRNA). C-MYB transcript levels are measured by qRT-PCR (left column). The house-keeping gene Gus (beta-glucuronidase) served as internal standard. C-MYB and Separase regulation on protein levels were determined by quantitative Western blot immunostaining experiments 48 h post transfection (middle and right columns, respectively). Corresponding representative Western blot images are shown in the very right panels of A and B. Actin served as loading control for Western blot immunostaining. All densitometric data are derived from at least triplicate experiments
c-MYB binds to a putative c-MYB recognition site located within the ESPL1 promoter
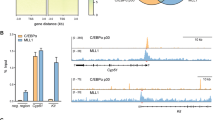
The ESPL1 promoter features two hormone responsive elements and two TP53 binding sites that are located 12,777 and 16,059 bases upstream of the predicted transcription start site (TSS) [20]. For details go to the Human Genome Browser (http://www.ncbi.nlm.nih.gov/gene?cmd=Retrieve&dopt=Graphics&list_uids=9700) and find the TSS at position 53,662,083 (+ strand) of the NC_000012.11 Chromosome 12 Reference GRCh37.913 Primary Assembly. The Champion ChIP Transcription Factor Search Portal based on SABiosciences’ proprietary database known as DECODE (DECipherment Of DNA Elements) was used to identify the putative c-MYB binding site within the Espl1 promoter (for more information see http://www.sabiosciences.com/chipqpcrsearch.php?app=TFBS). A hypothetical c-MYB binding site was found 15,623 bases upstream of the TSS (53,646,460 - 53,646,470) and thus, is located between both TP53 sites (see Fig. 4a). In order to determine whether the putative c-MYB binding site of the ESPL1 promoter can actually bind the c-MYB protein, we performed electrophoretic mobility shift assays (EMSA). Native nuclear extracts prepared from exponentially growing BV-173 cells were incubated with a FITC-labeled double stranded oligonucleotide (Fig. 4b lane 1) featuring the c-MYB binding site of the ESPL1 promoter. As shown in Fig. 4b left panel, addition of nuclear extract to the FITC-labeled oligonucleotide (lane 2) led to the formation of an oligo/protein complex with retarded electrophoretic migration (shift). Addition to the incubation mix of a 100fold molar excess of unlabeled c-MYB binding site oligonucleotide successfully competed with the formation of the shifted signal (lane 3). For corroboration of c-MYB protein binding, the native agarose gel was blotted under denaturing conditions followed by immunostaining with an anti-c-MYB antibody (right panel). The contribution of c-MYB to the shift signal (lane 2) was confirmed. In addition, the unlabeled competitor oligonucleotide (lane 3) appeared as shifted signal as well confirming specific abrogation of the FITC signal (left panel, lane 3) by the unlabeled competitor DNA. The second and lower band may be due altered electrophoretic mobility of at least some of the native oligo/c-MYB complexes after addition of 100fold excess of the unlabelled competitor. To demonstrate in vivo binding of c-MYB protein to the putative c-MYB recognition site located within the ESPL1 promoter we performed ChIP experiments on BV-173 cells. As shown in Fig. 4c a significant enrichment of ESPL1 promoter-related target DNA was observed when using a c-MYB-specific IgG antibody for chromatin immunoprecipitation when compared to the unspecific IgG antibody control. In summary, our in vitro and in vivo data suggest that c-MYB directly binds to the ESPL1 promoter and positively regulates ESPL1/Separase expression.

Electrophoretic mobility shift assay (EMSA) using synthetic ESPL1 promoter-derived c-MYB binding site probes and Chromatin immunoprecipitation (ChIP). a Schematic drawing depicting the ESPL1 Separase promoter and location of predicted regulatory DNA motifs (drawing not to scale) (Pati 2008). The arrow shows the predicted transcription start site (TSS). Abbreviations: TATA, TATA box; PRE, progesterone responsive element; ERE, estrogen responsive element; p53, p53 binding element; c-MYB, predicted c-MYB binding element. Numbers denote upstream distances with respect to the TSS. b A FITC-labeled double stranded DNA oligonucleotide corresponding to the putative c-MYB binding site of the ESPL1 promoter was incubated with native BV-173 nuclear extract. DNA/protein complexes were resolved on a 0.5 % TBE 1.0 % native LE/GTG agarose gel. The left panel (tonal inversion) shows FITC-related fluorescence signaling of the gel before blotting, the right panel depicts the corresponding anti-c-MYB Western blot immunostaining. The lanes represent: lane 1, DNA target (FITC-labeled oligonucleotide) without nuclear extract; lane 2, DNA target with nuclear extract; lane 3, DNA target with nuclear extract and with 100fold molar excess of analogous unlabeled oligonucleotide as binding competitor. c ChIP analysis of BV-173 cells. DNA fragments immunoprecipitated by anti-c-MYB IgG and a non-binding control IgG were amplified by qRT-PCR. Results are expressed as percentage of input (average). ChIP results are derived from at least triplicate qRT-PCR measurements
Discussion
In search for transcriptional mechanisms that may explain the previously observed IM-associated downregulation of Separase protein levels in CML [13] followed by posttranslational hyperactivation of Separase proteolytic activity, we have analyzed the conditional context between c-MYB expression, Separase and IM treatment. We found that the transcription factor c-MYB, known to play a pivotal role in proliferation, survival and differentiation of normal myeloid progenitors, is a direct positive regulator of Separase expression. Specifically, we demonstrated by EMSA (Fig. 4b) and ChIP that c-MYB interacts with a putative c-MYB binding motif located within the ESPL1 promoter. In fact, in all tested CML cell lines (K562, LAMA-84, BV-173) and in primary cells from CML patients coinciding c-MYB and ESPL1/Separase expression levels irrespective to therapeutic treatment (IM) and silencing (siRNA) conditions (Figs. 1, 2 and 3) were observed. This suggests that the ESPL1 promoter is under transcriptional control of the transcription factor c-MYB concurring with former results of Kohmura and coworkers who found a downregulation of c-MYB mRNA levels in K562 cells after incubation with IM [55].
Our results further coincide with previous studies reporting that p210BCR-ABL functions as enhancer of c-MYB expression and plays an important role in BCR-ABL-dependent leukemogenesis as the leukemic blast cells appear to rely on high levels of c-MYB protein more than normal progenitors [37, 45]. Moreover, c-MYB promotes centriole duplication and, when mutated, can lead to centrosome amplification as demonstrated in mouse and Drosophila model systems [56, 57]. Previous microarray data point to c-MYB as part of the regulatory network associated with CML progression including clonal evolution and genetic instability [58, 59].
The positive regulatory impact of c-MYB on ESPL1/Separase expression explains well the observations of Patel and Gordon who found abnormally high Separase expression levels in CML cells of chronic and blast phase when compared to normal CD34+ cells. It was then speculated that BCR-ABL expression may upregulate ESPL1/Separase and influence the occurrence of centrosomal aberrations [60].
Our data provide the first proof of direct regulatory relationship between BCR-ABL, c-MYB and ESPL1/Separase and give a plausible mechanistic explanation on how BCR-ABL may trigger dislocation between the centrosome-centriole cycle and the cell cycle in CML contributing to clonal evolution and genomic instability. The therapeutic administration of IM “normalizes” c-MYB protein levels - as shown in LAMA-84 and K562 cells (Fig. 1) - either by slowing down the transcriptional rate of c-MYB or by antagonizing enhanced c-MYB protein stability elicited by BCR-ABL via the PI-3 K/AKT pathway [36, 55]. The anticipated decrease of c-MYB protein levels after IM treatment is also in line with data of Flamant and coworkers who demonstrated that the drug results in a rapid increase in the expression of regulatory microRNAs such as miR-150 that is a strong negative regulator of c-MYB protein expression [61, 62].
Conclusions
In conclusion, our data suggest that ESPL1/Separase is a regulatory target of c-MYB and that c-MYB expression is modulated by IM treatment. Therefore, c-MYB, known to be required for BCR-ABL-dependent transformation of hematopoietic progenitors and leukemogenesis, may also influence Separase-related proteolytic events such as chromosomal segregation and centriole duplication, the ordered course of which is essential for maintenance of centrosomal and genomic stability.
Methods
Cell lines and culture conditions
Four human cell lines (K562, LAMA-84, BV-173, U937) were investigated. Of these, K562, LAMA-84 and BV-173 are BCR-ABL-positive IM responsive CML cell lines. U937 cells served as BCR-ABL-negative control cells. All cell lines were obtained from the DSMZ (German Collection of Microorganisms and Cell Cultures, Braunschweig, Germany) and were cultured in RPMI-1640 medium supplemented with 10 % fetal bovine serum and 1 % penicillin-streptomycin (Gibco/Invitrogen, Karlsruhe, Germany) at 37 °C in 5 % CO2 atmosphere. Exponentially growing cells were used in at least triplicate experiments.
Patients and ethics statement
Paired cDNA samples of randomly chosen CML patients (n = 5) from the German CML-Study IV (registered at www.clinicaltrials.gov as # NCT00055874) were investigated by qRT-PCR. Each sample pair was derived from peripheral blood of the same patient at the time point of diagnosis (before IM treatment) and after achievement of major molecular response (MMR) under IM therapy. Blood sampling was performed in the context of regular therapeutic monitoring. The procedure followed the declaration of Helsinki and was approved by the IRB/Medizinische Ethikkommision II der Medizinischen Fakultät Mannheim der Ruprecht-Karls-Universität Heidelberg. (http://www.umm.uni-heidelberg.de/inst/ethikkommission, # 2013-509 N-MA from 2013-02-21). Written informed consent was obtained from all patients. Two female and three male patients, all with b3a2 BCR-ABL fusion type, were analyzed. The median age was 58 years (range, 47 to 78). Mean time between diagnosis and sampling of the second specimen from the same patient after achievement of MMR under IM treatment was 3.6 years (range, 2 to 6.5).
IM treatment
Cells were treated with IM (Biomol GmbH, Hamburg, Germany) in concentrations of 1 to 5 μM for 24 h (K562, LAMA-84) and 48 h (U937) according to the rationale pointed out in references [13, 35]. Untreated cells served as controls.
Western blot analysis, antibodies
Western blot immunostaining of Separase and Actin was performed as described previously [13]. For c-MYB detection, an anti-c-MYB monoclonal rabbit antibody (ab45150; Abcam, Cambridge, UK) at a dilution of 1:10,000 was used. Signals were visualized with a ChemiDoc™ XRS+ System (BIO-RAD, München, Germany) after secondary antibody staining (goat anti-rabbit IgG HRP conjugated antibody (1:10,000; Santa Cruz Biotechnology Inc., Heidelberg, Germany) utilizing SuperSignal West Femto Maximum Sensitivity Substrate (Thermo Fisher Scientific, Bonn, Germany). Image acquisition and densitometric analysis was performed using Image Lab Software (version 3.0.1, BIO-RAD). All values were normalized with Actin as loading control. Image cropping and tonal adjustments across the entire image were performed with Adobe Photoshop CS4 (Adobe Systems Inc., San Jose, CA, USA).
RNA extraction and quantification of ESPL1 and c-MYB transcripts by qRT-PCR
Total RNA was extracted using RNeasy kit (Qiagen, Hilden, Germany) and reverse transcribed using Superscript II kit (Gibco/Invitrogen). For quantification of ESPL1 and c-MYB transcript levels, the commercial Hs_MYB_1_SG and Hs_ESPL1_1_SG QuantiTect Primer Assays (Qiagen) were employed according to the instructions (two-step Light Cycler 480 protocol) of the manufacturer, respectively. For normalization, the housekeeping gene beta-glucuronidase (Gus, NM_000181, GUSB, primer set Hs_GUSB_1_SG, QuantiTect Primer Assay, Qiagen) was amplified. QRT-PCR was performed with the Roche LightCycler 480 System, using LC480 DNA Master SYBR Green and the standard LightCycler protocol (Roche Diagnostics, Mannheim, Germany). Relative transcript levels calculated from triplicate measurements were calculated by the 2-ΔΔCT method with values normalized to Gus and relative to transcription in untreated control cells [63].
C-MYB silencing by siRNA
C-MYB-specific siRNA (FlexiTube GeneSolution GS4602 for c-MYB) was purchased from Qiagen. As negative controls the same cells were transfected with AllStars Negative Control siRNA (Qiagen), a nonsilencing siRNA with no homology to any known mammalian gene. Transfection was accomplished using the Nucleofector manual (program T016, Lonza GmbH, Köln, Germany). For siRNA treatment, 1.5x106 cells were resuspended in 100 μl Cell Line Nucleofector Solution V (Lonza) containing 18 μl Supplement S Solution (Lonza). The siRNA was added to a final concentration of 0.01 nmol per 106 cells. 24 h after transfection c-MYB transcript levels were analyzed by qRT-PCR. Protein lysates for Western blot immunostaining experiments were prepared 48 h after transfection.
ESPL1 promoter electrophoretic mobility shift assay (EMSA)
Native nuclear extracts were prepared from BV-173 cells according to a rapid micropreparation method [64]. FITC-labeled oligonucleotides (32mers) corresponding to the c-MYB-binding site sequence in the human ESPL1 promoter (Human genome browser http://www.ncbi.nlm.nih.gov/gene?cmd=Retrieve&dopt=Graphics&list_uids=9700), NC_000012.11 chromosome 12 reference GRCh37.p13 primary assembly; c-MYB binding position: chr12: 53,646,460 - 53,646,470) were synthesized by Sigma GmbH (Rödermark, Germany). Oligonucleotide sequences (c-MYB binding sequence underlined) were: MYB_sense, FITC-CTCCCACCCACCCAACTGGTCCCTCCGGTCTG; MYB_antisense, FITC-CAGACCGGAGGGACCAGTTGGGTGGGTGGGAG. EMSA was performed using the EMSA kit (order no. E33075) of Life Technologies GmbH (Darmstadt, Germany) according to the instructions of the manufacturer. In brief, 2 μg of nuclear extract were mixed with binding buffer prior to addition of 30 ng of annealed double stranded oligonucleotide corresponding to the c-MYB binding site within the ESPL1 promoter. After incubation for 20 min at RT, 2 μl of EMSA loading dye was added and the DNA/protein complexes were resolved by electrophoresis (80 V for 90 min) on a 0.5 % TBE 1.0 % native LE/GTG agarose gel. Detection of FITC-labeled DNA oligonucleotides was performed using a ChemiDoc™ XRS+ System (BIO-RAD). Consecutively, the same agarose gel was blotted onto Immobilon-P membrane (Millipore, Bedford, USA) using a Trans Blot SD semi-dry electrophoretic transfer cell (BIO-RAD) at 15 V (current limit at 5 mA/cm2) for 30 min. C-MYB Protein detection was performed as described in the Western blot analysis section.
Chromatin immunoprecipitation (ChIP)
ChIP was performed using the EpiTect ChIP OneDay Kit (#334471, Qiagen, Hilden) according to the instructions of the manufacturer. In brief, 1.5x106 BV-173 cells per sample were lysed and sonicated (5 times 6 s, 0.5 W) using the Vibra-Cell™ system (Sonics, Newtown, USA). Immunoprecipitation was performed with an anti-c-MYB antibody (ab17851, Abcam, Cambridge, UK) and an unspecific control IgG1 control antibody (ab91353, Abcam) according to the manual. For quantification of the immunoprecipitated DNA target, the ChIP qPCR Primer Assay GPH1003144(+)02A (Sabioscience/Qiagen) was applied employing the Roche LightCycler 480 System, using LC480 DNA Master SYBR Green and the standard LightCycler protocol as already described (Roche Diagnostics, Mannheim, Germany). Results derived from triplicate experiments were expressed as percentage of input sample taken before immunoprecipitation during the ChIP procedure. Therefore, the Ct values of the IP fractions were normalized to the input fraction.
Statistical analysis
Statistical significance of unpaired data was analyzed by the Student’s t-test using the GraphPad Prism software version 6.0 (GraphPad Inc., La Jolla, USA). Values of p < 0.05 were considered significant.
Abbreviations
- AP:
-
accelerated phase
- BC:
-
blast crisis
- ChIP:
-
chromatin immunoprecipitation
- CML:
-
chronic myeloid leukemia
- CP:
-
chronic phase
- EMSA:
-
electrophoretic mobility shift assay
- ERE:
-
estrogen responsive element
- IM:
-
imatinib
- PRE:
-
progesterone responsive element
- qRT-PCR:
-
quantitative reverse transcription-PCR
- RNAi:
-
RNA-interference
- TK:
-
tyrosine kinase
- TSS:
-
transcription start site
References
Chestukhin A, Pfeffer C, Milligan S, DeCaprio JA, Pellman D. Processing, localization, and requirement of human separase for normal anaphase progression. Proc Natl Acad Sci U S A. 2003;100(8):4574–9. doi:10.1073/pnas.0730733100.
Schockel L, Mockel M, Mayer B, Boos D, Stemmann O. Cleavage of cohesin rings coordinates the separation of centrioles and chromatids. Nat Cell Biol. 2011;13(8):966–72. doi:10.1038/ncb2280.
Simmons-Kovacs LA, Haase SB. Cohesin: it’s not just for chromosomes anymore. Cell Cycle. 2010;9(9):1750–3.
Uhlmann F. Secured cutting: controlling separase at the metaphase to anaphase transition. EMBO Rep. 2001;2(6):487–92. doi:10.1093/embo-reports/kve113.
Nigg EA. Centrosome duplication: of rules and licenses. Trends Cell Biol. 2007;17(5):215–21. doi:10.1016/j.tcb.2007.03.003.
Zhang N, Ge G, Meyer R, Sethi S, Basu D, Pradhan S, et al. Overexpression of Separase induces aneuploidy and mammary tumorigenesis. Proc Natl Acad Sci U S A. 2008;105(35):13033–8. doi:10.1073/pnas.0801610105.
Nigg EA, Raff JW. Centrioles, centrosomes, and cilia in health and disease. Cell. 2009;139(4):663–78. doi:10.1016/j.cell.2009.10.036.
Chan JY. A clinical overview of centrosome amplification in human cancers. Int J Biol Sci. 2011;7(8):1122–44.
Meyer R, Fofanov V, Panigrahi A, Merchant F, Zhang N, Pati D. Overexpression and mislocalization of the chromosomal segregation protein separase in multiple human cancers. Clin Cancer Res. 2009;15(8):2703–10. doi:10.1158/1078-0432.CCR-08-2454.
Mitelman F, Levan G, Nilsson PG, Brandt L. Non-random karyotypic evolution in chronic myeloid leukemia. Int J Cancer. 1976;18(1):24–30.
Agircan FG, Schiebel E. Sensors at centrosomes reveal determinants of local separase activity. PLoS Genet. 2014;10(10):e1004672. doi:10.1371/journal.pgen.1004672.
Shindo N, Kumada K, Hirota T. Separase sensor reveals dual roles for separase coordinating cohesin cleavage and cdk1 inhibition. Dev Cell. 2012;23(1):112–23. doi:10.1016/j.devcel.2012.06.015.
Haass W, Stehle M, Nittka S, Giehl M, Schrotz-King P, Fabarius A, et al. The proteolytic activity of separase in BCR-ABL-positive cells is increased by imatinib. PLoS One. 2012;7(8):e42863. doi:10.1371/journal.pone.0042863.
Holland AJ, Taylor SS. Cyclin-B1-mediated inhibition of excess separase is required for timely chromosome disjunction. J Cell Sci. 2006;119(Pt 16):3325–36. doi:10.1242/jcs.03083.
Waizenegger I, Gimenez-Abian JF, Wernic D, Peters JM. Regulation of human separase by securin binding and autocleavage. Curr Biol. 2002;12(16):1368–78.
Yim H, Erikson RL. Regulation of the final stage of mitosis by components of the pre-replicative complex and a polo kinase. Cell Cycle. 2011;10(9):1374–7.
Basu D, Zhang N, Panigrahi AK, Horton TM, Pati D. Development and validation of a fluorogenic assay to measure separase enzyme activity. Anal Biochem. 2009;392(2):133–8. doi:10.1016/j.ab.2009.05.046.
Mukherjee M, Byrd T, Brawley VS, Bielamowicz K, Li XN, Merchant F, et al. Overexpression and constitutive nuclear localization of cohesin protease Separase protein correlates with high incidence of relapse and reduced overall survival in glioblastoma multiforme. J Neurooncol. 2014. doi:10.1007/s11060-014-1458-6.
Mukherjee M, Ge G, Zhang N, Edwards DG, Sumazin P, Sharan SK, et al. MMTV-Espl1 transgenic mice develop aneuploid, estrogen receptor alpha (ERalpha)-positive mammary adenocarcinomas. Oncogene. 2013. doi:10.1038/onc.2013.493.
Pati D. Oncogenic activity of separase. Cell Cycle. 2008;7(22):3481–2.
Sawyers CL. Chronic myeloid leukemia. N Engl J Med. 1999;340(17):1330–40. doi:10.1056/NEJM199904293401706.
Calabretta B, Perrotti D. The biology of CML blast crisis. Blood. 2004;103(11):4010–22. doi:10.1182/blood-2003-12-4111.
Druker BJ, Guilhot F, O’Brien SG, Gathmann I, Kantarjian H, Gattermann N, et al. Five-year follow-up of patients receiving imatinib for chronic myeloid leukemia. N Engl J Med. 2006;355(23):2408–17. doi:10.1056/NEJMoa062867.
Hochhaus A, Schenk T, Erben P, Ernst T, La Rosee P, Muller MC. Cause and management of therapy resistance. Best Pract Res Clin Haematol. 2009;22(3):367–79. doi:10.1016/j.beha.2009.05.004.
Bolton-Gillespie E, Schemionek M, Klein HU, Flis S, Hoser G, Lange T, et al. Genomic instability may originate from imatinib-refractory chronic myeloid leukemia stem cells. Blood. 2013;121(20):4175–83. doi:10.1182/blood-2012-11-466938.
Kumari A, Brendel C, Hochhaus A, Neubauer A, Burchert A. Low BCR-ABL expression levels in hematopoietic precursor cells enable persistence of chronic myeloid leukemia under imatinib. Blood. 2012;119(2):530–9. doi:10.1182/blood-2010-08-303495.
Hamilton A, Helgason GV, Schemionek M, Zhang B, Myssina S, Allan EK, et al. Chronic myeloid leukemia stem cells are not dependent on Bcr-Abl kinase activity for their survival. Blood. 2012;119(6):1501–10. doi:10.1182/blood-2010-12-326843.
Perl A, Carroll M. BCR-ABL kinase is dead; long live the CML stem cell. J Clin Invest. 2011;121(1):22–5. doi:10.1172/JCI43605.
Morotti A, Panuzzo C, Fava C, Saglio G. Kinase-inhibitor-insensitive cancer stem cells in chronic myeloid leukemia. Expert Opin Biol Ther. 2014;14(3):287–99. doi:10.1517/14712598.2014.867323.
Naka K, Hoshii T, Hirao A. Novel therapeutic approach to eradicate tyrosine kinase inhibitor resistant chronic myeloid leukemia stem cells. Cancer Sci. 2010;101(7):1577–81. doi:10.1111/j.1349-7006.2010.01584.x.
Schnittger S, Bacher U, Dicker F, Kern W, Alpermann T, Haferlach T, et al. Associations between imatinib resistance conferring mutations and Philadelphia positive clonal cytogenetic evolution in CML. Genes Chromosomes Cancer. 2010;49(10):910–8. doi:10.1002/gcc.20801.
Cortes J, O’Dwyer ME. Clonal evolution in chronic myelogenous leukemia. Hematol Oncol Clin North Am. 2004;18(3):671–84. doi:10.1016/j.hoc.2004.03.012.
O’Dwyer ME, Mauro MJ, Kurilik G, Mori M, Balleisen S, Olson S, et al. The impact of clonal evolution on response to imatinib mesylate (STI571) in accelerated phase CML. Blood. 2002;100(5):1628–33. doi:10.1182/blood-2002-03-0777.
Verma D, Kantarjian H, Shan J, O’Brien S, Estrov Z, Garcia-Manero G, et al. Survival outcomes for clonal evolution in chronic myeloid leukemia patients on second generation tyrosine kinase inhibitor therapy. Cancer. 2010;116(11):2673–81. doi:10.1002/cncr.25015.
Haass W, Kleiner H, Weiss C, Haferlach C, Schlegelberger B, Muller MC, et al. Clonal evolution and blast crisis correlate with enhanced proteolytic activity of separase in BCR-ABL b3a2 fusion type CML under imatinib therapy. PLoS One. 2015;10(6):e0129648. doi:10.1371/journal.pone.0129648.
Corradini F, Cesi V, Bartella V, Pani E, Bussolari R, Candini O, et al. Enhanced proliferative potential of hematopoietic cells expressing degradation-resistant c-Myb mutants. J Biol Chem. 2005;280(34):30254–62. doi:10.1074/jbc.M504703200.
Lidonnici MR, Corradini F, Waldron T, Bender TP, Calabretta B. Requirement of c-Myb for p210(BCR/ABL)-dependent transformation of hematopoietic progenitors and leukemogenesis. Blood. 2008;111(9):4771–9. doi:10.1182/blood-2007-08-105072.
Manzotti G, Mariani SA, Corradini F, Bussolari R, Cesi V, Vergalli J, et al. Expression of p89(c-Mybex9b), an alternatively spliced form of c-Myb, is required for proliferation and survival of p210BCR/ABL-expressing cells. Blood Cancer J. 2012;2(5):e71. doi:10.1038/bcj.2012.16.
Greig KT, Carotta S, Nutt SL. Critical roles for c-Myb in hematopoietic progenitor cells. Semin Immunol. 2008;20(4):247–56. doi:10.1016/j.smim.2008.05.003.
Lieu YK, Reddy EP. Conditional c-myb knockout in adult hematopoietic stem cells leads to loss of self-renewal due to impaired proliferation and accelerated differentiation. Proc Natl Acad Sci U S A. 2009;106(51):21689–94. doi:10.1073/pnas.0907623106.
Selvakumaran M, Liebermann DA, Hoffman-Liebermann B. Deregulated c-myb disrupts interleukin-6- or leukemia inhibitory factor-induced myeloid differentiation prior to c-myc: role in leukemogenesis. Mol Cell Biol. 1992;12(6):2493–500.
Pattabiraman DR, Gonda TJ. Role and potential for therapeutic targeting of MYB in leukemia. Leukemia. 2013;27(2):269–77. doi:10.1038/leu.2012.225.
Clappier E, Cuccuini W, Kalota A, Crinquette A, Cayuela JM, Dik WA, et al. The C-MYB locus is involved in chromosomal translocation and genomic duplications in human T-cell acute leukemia (T-ALL), the translocation defining a new T-ALL subtype in very young children. Blood. 2007;110(4):1251–61. doi:10.1182/blood-2006-12-064683.
Tomita A, Watanabe T, Kosugi H, Ohashi H, Uchida T, Kinoshita T, et al. Truncated c-Myb expression in the human leukemia cell line TK-6. Leukemia. 1998;12(9):1422–9.
Calabretta B, Sims RB, Valtieri M, Caracciolo D, Szczylik C, Venturelli D, et al. Normal and leukemic hematopoietic cells manifest differential sensitivity to inhibitory effects of c-myb antisense oligodeoxynucleotides: an in vitro study relevant to bone marrow purging. Proc Natl Acad Sci U S A. 1991;88(6):2351–5.
Lorenzo PI, Brendeford EM, Gilfillan S, Gavrilov AA, Leedsak M, Razin SV, et al. Identification of c-Myb target genes in K562 cells reveals a role for c-Myb as a master regulator. Genes Cancer. 2011;2(8):805–17. doi:10.1177/1947601911428224.
Zhao L, Glazov EA, Pattabiraman DR, Al-Owaidi F, Zhang P, Brown MA, et al. Integrated genome-wide chromatin occupancy and expression analyses identify key myeloid pro-differentiation transcription factors repressed by Myb. Nucleic Acids Res. 2011;39(11):4664–79. doi:10.1093/nar/gkr024.
Deng QL, Ishii S, Sarai A. Binding site analysis of c-Myb: screening of potential binding sites by using the mutation matrix derived from systematic binding affinity measurements. Nucleic Acids Res. 1996;24(4):766–74.
Nakata Y, Shetzline S, Sakashita C, Kalota A, Rallapalli R, Rudnick SI, et al. c-Myb contributes to G2/M cell cycle transition in human hematopoietic cells by direct regulation of cyclin B1 expression. Mol Cell Biol. 2007;27(6):2048–58. doi:10.1128/MCB.01100-06.
Fabarius A, Giehl M, Rebacz B, Kramer A, Frank O, Haferlach C, et al. Centrosome aberrations and G1 phase arrest after in vitro and in vivo treatment with the SRC/ABL inhibitor dasatinib. Haematologica. 2008;93(8):1145–54. doi:10.3324/haematol.12793.
Giehl M, Fabarius A, Frank O, Erben P, Zheng C, Hafner M, et al. Expression of the p210BCR-ABL oncoprotein drives centrosomal hypertrophy and clonal evolution in human U937 cells. Leukemia. 2007;21(9):1971–6. doi:10.1038/sj.leu.2404834.
Fabarius A, Giehl M, Frank O, Duesberg P, Hochhaus A, Hehlmann R, et al. Induction of centrosome and chromosome aberrations by imatinib in vitro. Leukemia. 2005;19(9):1573–8. doi:10.1038/sj.leu.2403861.
Deininger M, Buchdunger E, Druker BJ. The development of imatinib as a therapeutic agent for chronic myeloid leukemia. Blood. 2005;105(7):2640–53. doi:10.1182/blood-2004-08-3097.
Di Gion P, Kanefendt F, Lindauer A, Scheffler M, Doroshyenko O, Fuhr U, et al. Clinical pharmacokinetics of tyrosine kinase inhibitors: focus on pyrimidines, pyridines and pyrroles. Clin Pharmacokinet. 2011;50(9):551–603. doi:10.2165/11593320-000000000-00000.
Kohmura K, Miyakawa Y, Kawai Y, Ikeda Y, Kizaki M. Different roles of p38 MAPK and ERK in STI571-induced multi-lineage differentiation of K562 cells. J Cell Physiol. 2004;198(3):370–6. doi:10.1002/jcp.10426.
Fung SM, Ramsay G, Katzen AL. Mutations in Drosophila myb lead to centrosome amplification and genomic instability. Development. 2002;129(2):347–59.
Tan FE, Vladar EK, Ma L, Fuentealba LC, Hoh R, Espinoza FH, et al. Myb promotes centriole amplification and later steps of the multiciliogenesis program. Development. 2013;140(20):4277–86. doi:10.1242/dev.094102.
Oehler VG, Yeung KY, Choi YE, Bumgarner RE, Raftery AE, Radich JP. The derivation of diagnostic markers of chronic myeloid leukemia progression from microarray data. Blood. 2009;114(15):3292–8. doi:10.1182/blood-2009-03-212969.
Zheng C, Li L, Haak M, Brors B, Frank O, Giehl M, et al. Gene expression profiling of CD34+ cells identifies a molecular signature of chronic myeloid leukemia blast crisis. Leukemia. 2006;20(6):1028–34. doi:10.1038/sj.leu.2404227.
Patel H, Gordon MY. Abnormal centrosome-centriole cycle in chronic myeloid leukaemia? Br J Haematol. 2009;146(4):408–17. doi:10.1111/j.1365-2141.2009.07772.x.
Flamant S, Ritchie W, Guilhot J, Holst J, Bonnet ML, Chomel JC, et al. Micro-RNA response to imatinib mesylate in patients with chronic myeloid leukemia. Haematologica. 2010;95(8):1325–33. doi:10.3324/haematol.2009.020636.
Lu J, Guo S, Ebert BL, Zhang H, Peng X, Bosco J, et al. MicroRNA-mediated control of cell fate in megakaryocyte-erythrocyte progenitors. Dev Cell. 2008;14(6):843–53. doi:10.1016/j.devcel.2008.03.012.
Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) Method. Methods. 2001;25(4):402–8. doi:10.1006/meth.2001.1262.
Andrews NC, Faller DV. A rapid micropreparation technique for extraction of DNA-binding proteins from limiting numbers of mammalian cells. Nucleic Acids Res. 1991;19(9):2499.
Acknowledgements
We thank Susanne Brendel for excellent technical assistance.
Support and funding
WS received a grant from the Deutsche Krebshilfe (www.krebshilfe.de, no. 110625) and from the Deutsche José Carreras Leukämie-Stiftung e.V. (www.carreras-stiftung.de/, no. DJCLS R 14/11). The funders had no role in study design, data collection and analysis, interpretation of data and in writing the manuscript.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
WP and WS contributed to the experimental design, analyzed and interpreted the data, and wrote the manuscript. WP HK MS performed the experiments. WP SR HK MS MCM WKH AF WS provided expertise and editing. All authors read and approved the final manuscript.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Prinzhorn, W., Stehle, M., Kleiner, H. et al. c-MYB is a transcriptional regulator of ESPL1/Separase in BCR-ABL-positive chronic myeloid leukemia. Biomark Res 4, 5 (2016). https://doi.org/10.1186/s40364-016-0059-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40364-016-0059-2