
Abstract
Background
Aidi Injection (ADI), a Chinese herbal preparation with anti-cancer activity, is used for the treatment of hepatocellular carcinoma (HCC). Several clinical studies have shown that co-administration of ADI with doxorubicin (DOX) is associated with reduced toxicity of chemotherapy, enhanced clinical efficacy and improved quality of life for patients. However, limited information is available about the herb-drug interactions between ADI and DOX. The study aimed to investigate the pharmacokinetic mechanism of herb-drug interactions between ADI and DOX in a rat model of HCC.
Methods
Experimental HCC was induced in rats by oral administration of diethylnitrosamine. The HCC rats were pretreated with ADI (10 mL/kg, intraperitoneal injection) for 14 consecutive days prior to administration of DOX (7 mg/kg, intravenous injection) to investigate pharmacokinetic interactions. Plasma concentrations of DOX and its major metabolite, doxorubicinol (DOXol), were determined using ultra-performance liquid chromatography-tandem mass spectrometry (UPLC-MS/MS).
Results
Preadministration of ADI significantly altered the pharmacokinetics of DOX in HCC rats, leading to increased plasma concentrations of both DOX and DOXol. The area under the plasma drug concentration-time curve (AUCs) of DOX and DOXol in rats pretreated with ADI were 3.79-fold and 2.92-fold higher, respectively, than those in control rats that did not receive ADI.
Conclusions
Increased levels of DOX and DOXol were found in the plasma of HCC rats pretreated with ADI.
Similar content being viewed by others

Background
Hepatocellular carcinoma (HCC) is one of the most common liver malignancies in regions where chronic hepatitis or liver diseases are prevalent, such as China [1]. Doxorubicin (DOX) is a key drug used in chemotherapy of HCC but its clinical utility is limited by both drug resistance and cardiotoxicity [2]. It has previously been suggested that the main active metabolite of DOX, doxorubicinol (DOXol), contributes to both the efficacy and toxicity of DOX [3].
HCC treatment is mainly inclined to comprehensive treatment. A large number of clinical trials have confirmed that natural products has a significant effect in the field of HCC, which not only improves the prognosis and quality of life of patients, but also improves the survival rate of patients [4,5,6]. Aidi Injection (ADI, Z52020236, China Food and Drug Administration), which contains extracts of Astragali Radix, A. senticosus, Ginseng Radix and Mylabris is widely used in China for the treatment of HCC. Several clinical studies have shown that combining ADI with chemotherapy reduces the toxicity of chemotherapy, enhances clinical efficacy and improves the quality of life of cancer patients [7,8,9]. Cantharidin, the major bioactive component of Cantharis, has potent antitumor activity, induces apoptosis in a variety of tumor cells, increases numbers of white blood cells and reduces the occurrence of bone marrow suppression [10,11,12,13]. Recent pharmacological studies have shown that astragalus polysaccharides have significant immunomodulatory activity [14], are hepatoprotective and antioxidant [15, 16], and have antitumor effects [17]. A. senticosus also has antitumor and immunomodulatory effects [18]. Ginsenosides, such as ginsenosides Rg3 and Rh2, show antitumor and antiangiogenic effects in various models using tumor cells and vascular endothelial cells [19, 20]. Many patients use ADI for HCC, before and after treatment with DOX, to reduce toxicity and improve the efficacy of chemotherapy [21, 22]. These effects are, however, insufficient to explain why ADI improves the clinical efficacy of chemotherapy drugs and the mechanism leading to increased efficacy needs to be explained. Plasma drug concentrations are generally believed to be proportional to the therapeutic effect and toxicity of a drug [23]. So far, there have been no reports describing research into pharmacokinetic herb-drug interactions between ADI and DOX and the nature of the interaction remains unknown.
There are potential risks in co-administering ADI with DOX in an outpatient setting and a pharmacokinetic study to evaluate potential interactions of ADI with DOX is needed. We hypothesized that ADI may alter the pharmacokinetics of DOX and used rats with experimental HCC to evaluate this hypothesis. HCC rats were pretreated with ADI (10 mL/kg, once a day, by intraperitoneal (i.p.) injection) for 14 consecutive days prior to administration of DOX (7 mg/kg by intravenous (i.v.) injection) to investigate pharmacokinetic interactions.
Methods
Chemicals and reagents
ADI (Product No. 20150627) was supplied by Guizhou Ebay Pharmaceutical Co., Ltd. (Guizhou, China). Diethylnitrosamine, doxorubicin (25316–40-9), doxorubicinol (54193–28-1), and the internal standard (IS) tropisetron hydrochloride (105826–92-4) were all purchased from Dalian Meilun Biotech Co., Ltd. (Liaoning, China). HPLC-grade acetonitrile, methanol and formic acid was supplied by Merck Company Inc. (Darmstadt, Germany). Distilled water was obtained from Watsons Group Co., Ltd. (Hong Kong, PRC). All chemicals and reagents used were of chromatographic or analytical grade.
Animals
All experimental procedures were conducted according to the Institutional Animal Care guidelines and approved ethically by the Administration Committee of Experimental Animals, Guizhou Province, China. Male, pathogen-free, Sprague-Dawley rats (180–200 g) were purchased from Changsha Tianqing Biological Technology Co., Ltd. (Changsha, China, Certificate No. SCXK2016–0015) and acclimated for at least 1 week in their environmentally controlled quarters (25 °C ± 2 °C and 12/12 light/dark cycle), with free access to standard chow and water.
Induction of HCC in rats by diethylnitrosamine
Experimental HCC was induced in rats by oral administration of diethylnitrosamine (DEN), as previously described [24, 25]. In brief, DEN (95 μg/mL) was administered in drinking water for 4 consecutive weeks, administration was interrupted for 4 weeks, and then resumed for 8 weeks.
Animal treatment
On the last day of DEN administration, 24 HCC rats were randomly divided into two groups of 12 animals, a control group and an ADI group. The control group received saline (10 mL/kg, i.p.) once a day for 14 consecutive days and the ADI group received ADI (10 mL/kg, i.p.) once a day for 14 consecutive days. The rats were allowed free access to standard chow and water during these 14 days. Access to food was then prohibited for 12 h, with continued free access to water. Six rats from each group were then treated with DOX (7 mg/kg, i.v.). Blood samples (~ 250 μL) were collected from the tail vein into heparinized centrifuge tubes 0.033, 0.083, 0.167, 0.25, 0.333, 0.5, 1, 2, 4 and 8 h after DOX administration. Each blood sample was centrifuged for 5 min at 3306×g and an aliquot of the supernatant (100 μL) was transferred to a labeled plastic vial and stored at − 20 °C before analysis. At the end of study, all animals were euthanized by our veterinary staff in the animal care facility by carbon dioxide asphyxiation.
Pharmacokinetic studies
UPLC-ESI-MS conditions
Chromatographic conditions were based on preliminary research work carried out in our laboratory [26]. A Waters ACQUITY UPLC system (Waters Corp., Milford, MA, USA), coupled with a Waters TQD Quantum triple-quadrupole mass spectrometer equipped with an electrospray ionization (ESI) source, was used for determination of the chromatographic analytes. Waters MassLynx software v.4.1 was used for acquisition and data processing. Separation and quantification were performed using a BEH C18 column (50 mm × 2.1 mm × 1.7 μm, Waters, Wexford, Ireland). The column temperature was 45 °C and the flow rate was 0.35 mL/min. The eluent was a mixture of mobile phase A (acetonitrile containing 0.1% formic acid) and mobile phase B (water containing 0.1% formic acid), with a gradient program as follows: 0–0.5 min, 10–30% A; 0.5–1.5 min, 30–60% A; 1.5–2.0 min, 60–90% A; 2.0–3.0 min, 90–10% A. The samples were kept at 25 °C in the sample manager. The injection volume was 1.0 μL (partial loop with needle overfill mode). A strong needle wash solution (90:10, methanol-water, v/v) and a weak needle wash solution (10:90, acetonitrile-water, v/v) were used. The mass spectrometer was operated in positive ion mode, with optimized parameters set as follows: nitrogen gas flow, 650 L/h; capillary voltage, 3 kV; ion source temperature, 120 °C; desolvation temperature, 350 °C. Cone voltages were optimized and set at 20 V, 20 V, and 32 V for DOX, DOXol, and IS, respectively. Quantification was performed using selected or single ion recording mode by monitoring the parent ions (m/z 544.3 for DOX, m/z 546.3 for DOXol and m/z 285.3 for IS).
Sample preparation
Samples were thawed to room temperature before analysis. IS solution [50 μL, 50 ng/mL IS dissolved in water/acetonitrile (50:60, v/v)] was added to rat plasma (100 μL). After vortexing for 5 min, methanol containing 5% formic acid (450 μL) was added to precipitate the proteins. After vortexing, mixing and sonication for 5 min, the sample was centrifuged at 13,000×g for 10 min. The supernatant was then transferred to another tube and evaporated to dryness under a gentle stream of nitrogen. The residue was dissolved in mobile phase (mobile phase A: mobile phase B, 10/90; 400 μL), centrifuged at 13,000×g for 10 min, and an aliquot (1 μL) of the solution was injected into the UPLC-MS/MS.
Pharmacokinetic analysis
Pharmacokinetic parameters were calculated using Drug and Statistic (DAS) pharmacokinetic software version 2.0 (Mathematical Pharmacology Professional Committee of China, Shanghai, China). All data were presented as the mean ± standard deviation (SD). A two tailed Student’s t-test was used to determine the significance of differences in pharmacokinetic parameters between the control group and the ADI group. P < 0.05 was considered to be statistically significant.
Results
Method validation
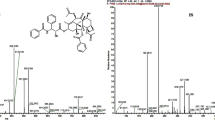
Plasma concentrations of DOX and DOXol were quantified using a validated UPLC-MS method previously developed in our laboratory [26]. Briefly, the retention times of tropisetron (IS), DOXol and DOX were 1.57, 1.60 and 1.74 min, respectively. Chromatograms of DOX, DOXol and IS in rat plasma is shown in Fig. 1. The lower limits of quantification (LLOQ) were 100 ng/mL for DOX and 10 ng/mL for DOXol. The mean recoveries of DOX and DOXol were 83.25–96.58% and the intra- and inter-day precisions were < 10%. DOX and DOXol in the analytical samples were stable for 12 h in the autosampler, for 72 h at − 20 °C and over three freeze-thaw cycles. Linearity, sensitivity, selectivity, accuracy, intra- and inter-day precision and stability of the method were validated according to the requirements for bioanalytical methods laid out in the Guidance for Industry Bioanalytical Method Validation Document from the American Food and Drug Administration.

Chromatograms of DOX, DOXol and tropisetron (I.S.) in rat plasma. Blank plasma sample (A); a plasma sample spiked with DOX, DOXol, and tropisetron (I.S.), respectively (B); and a plasma sample obtained from a rat 10 min after intravenous administration of doxorubicin (C). 1.Tropisetron 2. Doxorubicinol 3. Doxorubicin
Pharmacokinetic study
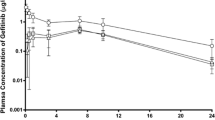
The effects of ADI on the pharmacokinetics of DOX in HCC rats were examined by administering a single dose of DOX (7 mg/kg, p.o.) to the rats. Pharmacokinetic investigation showed that the plasma concentration-time data for DOX were best fitted to a two-compartment intravenous open model. A one-compartment model was used to describe the pharmacokinetics of DOXol. Plasma concentrations of DOX were found to be significantly higher in the ADI group than in the control group (Fig. 2 and Table 1). The area under the plasma drug concentration-time curve (AUC) of DOX in the ADI group was 3.79-fold higher than that in the control group (P < 0.01). The half-life of distribution (t1/2α), apparent volume of distribution in the central compartment (V1) in the ADI group were also significantly higher than in the control group (P < 0.01). Meanwhile, compared with the control group, clearance (CL) of DOX in the ADI group was significantly lower (P < 0.05). There were no statistically significant differences in elimination half-life (t1/2β), elimination rate constant of drug from compartment 1 (K10), rate constant for movement of drug from compartment 1 to compartment 2 (K12) or rate constant for movement of drug from compartment 2 to compartment 1 (K21) between the two groups (P > 0.05). The AUC of DOXol, the main metabolite of DOX, was 2.92-fold higher in the ADI group than in the control group (Fig. 3 and Table 2). The mean residence time (MRT0-t), elimination half-life time (t1/2z) and peak concentration (Cmax) of DOXol were all significantly increased in the ADI group compared with the control group (2.22-, 2.39- and 3.46-fold, respectively) (P < 0.05). Pharmacokinetic analysis thus showed that preadministration of ADI significantly altered the pharmacokinetics of DOX in HCC rats, leading to elevation of plasma concentrations of both DOX and DOXol.

Plasma concentration-time profiles (Mean ± SD, n = 6) of DOX in control and ADI groups

Plasma concentration-time profiles (Mean ± SD, n = 6) of DOXol in control and ADI groups
Discussion
Despite improved diagnostic tools for HCC and much better survival rates of patients, the outcomes and prognoses of HCC patients remain poor because of poor liver function and advanced cancer stage. Transcatheter arterial chemoembolization (TACE) is the main treatment for unresectable HCC, and DOX is one of the commonly used drugs in TACE [27, 28]. TACE is not, however, ideal as a long-term cure since it often reduces immunity, aggravates the impairment of liver function and reduces life quality of HCC patients [29]. Finding a way to reduce liver injury and improve clinical efficacy and quality of life has thus become a key issue and many patients in Asia are seeking help from traditional herbal medicines [30, 31].
ADI(Z52020236) is the exclusive product of Guizhou Ebay Pharmaceutical Co. LTD, which was approved for clinical use since 2002. ADI, combined with TACE, is now widely used in the treatment of unresectable HCC in China. It has been reported that this combination can, to some extent, enhance the clinical effect, improve overall survival, increase quality of life for patients and reduce adverse events, including leukopenia, gastrointestinal side effects and liver damage [32]. In a previous study, we found that ADI reduced serum levels of alanine aminotransferase, aspartate aminotransferase, total bilirubinand alkaline phosphatasein rats with DEN-induced HCC, confirming its protective effect on liver function [26]. The clinical use of ADI is intravenous drip, 50 to 100 ml of ADI for adults, mixed with 0.9% sodium chloride injection or glucose injection, once a day. When combined with radiotherapy and chemotherapy, the course of treatment is synchronized with radiotherapy and chemotherapy. ADI is used for 10 days before and after surgery. For patients with advanced cachexia, ADI is used for 30 days or depending on the condition. According to human and rat dose conversion and the convenience of practical operation, rats were injected intraperitoneally with 10 mL/kg of ADI for 14 consecutive days [33].
Animal models of HCC are essential for understanding the cellular and molecular aspects of tumor development, as well as pharmacological and pharmacokinetic testing. Although the genetic engineering model of HCC can be used to prove the carcinogenic and tumor suppressive effects of cell and viral genes [34], chemically induced HCC models can cause both cirrhosis and HCC. The DEN-induced liver cancer model produces HCC in the context of fibrosis, similar to the liver microenvironment of almost all HCC patients [35]. Therefore, this method was adopted for experiments. The pharmacokinetic interaction between ADI and DOX was investigated in the 14th day after the end of DEN administration, we believe that DEN should disappear in the body, as the time between DEN and ADI is long enough. The main limitation of this experiment is that only one liver cancer model is used. To eliminate DEN interference, in the future, we will use other models to verify the results of this experiment, such as transplanted tumor models or spontaneous liver cancer models. In addition, we will investigate the distribution of DOX and DOXol in the tumor tissue when DOX is co-administered with ADI in subsequent experiments.
Our pharmacokinetic results show that plasma concentrations of DOX and DOXol were significantly higher in the ADI group. AUC of DOX and DOXol in the ADI group was 3.79-fold and 2.92-fold higher than that in the control group (both P < 0.01). It means there is herb-drug interactions between ADI and DOX. ADI can change the pharmacokinetics of DOX in HCC rats. To the best of our knowledge, the plasma protein binding rate of DOX is very low. Therefore, ADI is unlikely to change the plasma concentration of DOX and DOXol by competing with plasma protein binding. ADI alters DOX’s drug metabolism enzymes and transporters is a possible cause. DOX is mainly metabolized in the liver and excreted in bile, 50% of which are parent drugs, and 23% are active metabolites such as DOXol [36]. DOX can be converted into a semiquinone structure through single-electron reduction, and it can also form DOXol through C-13 hydroxylation in the cytoplasm by carbonyl reductase 1 (CBR1), which generally expressed in liver, heart and other tissues [37]. Various transporters, particularly P-gp (ABCB1, MDR1) and ABCC1 (MRP1), are thought to be play a role in resistance to DOX [2]. Generally, increased expression of P-gp results in increased DOX efflux. Many studies have shown that the resistance of DOX can be overcome by inhibiting P-gp [38,39,40]. The import transporter SLC22A16 has also been shown to be involved in intracellular transport of DOX [41]. Therefore, if ADI changes the activity or expression of CBR1, SLC22A16 and P-gp, it would change the pharmacokinetic behavior of DOX.
In our previous study, ADI also reduced mRNA levels and enzymatic activity of glutathione transferases (GSTs), and decreased protein expression of GST-π in the livers of HCC rats [24]. High expression of GST-π is known to accelerate the transformation and metabolism of anti-tumor drugs, shorten the duration of effective drug concentrations in cells and rapidly reduce the accumulation of drugs in target sites, thus reducing efficacy [42]. GSTs are considered to be potential targets to overcome chemoresistance in solid tumors [43], and reduction of GSTs activity may be the one of underlying mechanisms for the synergistic effect of ADI. Inhibition of GSTs activity cannot, however, explain why administration of ADI leads to elevated levels of DOX and DOXol, since GSTs are not involved in DOX metabolism. In summary, to explain why ADI changed pharmacokinetics of DOX, more experiments with rigorous design are needed.
DOX is an effective chemotherapeutic drug. DOXol is the most important component of DOX-induced cardiotoxicity. Hence, increased blood concentrations of DOX and DOXol, in addition to implying that it may increase the therapeutic effect of DOX and may lead to stronger toxic and side effects. However, many studies have shown that the related ingredients of Astragalus, A. senticosus, Ginseng can play a synergistic effect, protect the heart, and reduce the toxic and side effects of chemotherapy drugs, such as ginsenoside Rg1 [44], ginsenoside Rg3 [45,46,47], astragalus polysaccharide [48, 49], acanthopanax senticosides B [50]. Therefore, how the combination of ADI and DOX can enhance the efficacy of DOX and reduce its myocardial toxicity requires more experiments to verify.
Conclusions
In this study, an accurate and validated UPLC-MS/MS method was developed to determine DOX and DOXol concentrations in rat plasma and then used to investigate DOX pharmacokinetics. Using this method, we identified potential herb-drug interactions between ADI and DOX. The AUCs of DOX and DOXol in rats pretreated with ADI were 3.79-fold and 2.92-fold higher, respectively, than AUCs in the control group. Further studies are needed to better understand the synergistic effect of ADI and DOX. Since both DOX and DOXol are implicated in the cardiotoxicity, in future studies we will investigate the cardiotoxicity of DOX and the distribution of DOX and DOXol in the heart and tumor tissue when DOX is co-administered with ADI.
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Abbreviations
- ADI:
-
Aidi Injection
- DOX:
-
Doxorubicin
- DOXol:
-
Doxorubicinol
- UPLC-MS/MS:
-
Ultra-performance Liquid Chromatography-Tandem Mass Spectrometry
- DEN:
-
Diethylnitrosamine
- IS:
-
Internal standard
- AUC:
-
The area under the plasma drug concentration-time curve
- t1/2α :
-
The half-life of distribution
- V1 :
-
The apparent volume of distribution in the central compartment
- CL:
-
The clearance
- t1/2β :
-
The elimination half-life
- K10 :
-
The elimination rate constant of drug from compartment 1
- K12 :
-
The rate constant for movement of drug from compartment 1 to compartment 2
- K21 :
-
The rate constant for movement of drug from compartment 2 to compartment 1
- MRT0-t :
-
The mean residence time
- t1/2z :
-
The elimination half-life time
- Cmax :
-
The peak concentration
References
Omata M, Cheng AL, Kokudo N, Kudo M, Lee JM, Jia J, et al. Asia-Pacific clinical practice guidelines on the management of hepatocellular carcinoma: a 2017 update. Hepatol Int. 2017;11(4):317–70. https://doi.org/10.1007/s12072-017-9799-9.
Cox J, Weinman S. Mechanisms of doxorubicin resistance in hepatocellular carcinoma. Hepat Oncol. 2016;3(1):57–9. https://doi.org/10.2217/hep.15.41.
Thorn CF, Oshiro C, Marsh S, Hernandez-Boussard T, McLeod H, Klein TE, et al. Doxorubicin pathways: pharmacodynamics and adverse effects. Pharmacogenet Genomics. 2011;21(7):440–6. https://doi.org/10.1097/FPC.0b013e32833ffb56.
Hamza AA, Ahmed MM, Elwey HM, Amin A. Melissa officinalis protects against doxorubicin-induced cardiotoxicity in rats and potentiates its anticancer activity on MCF-7 cells. PLoS One. 2016;11(11):e0167049. https://doi.org/10.1371/journal.pone.0167049.
Amin A, Mahmoud-Ghoneim D, Syam MI, Daoud S. Neural network assessment of herbal protection against chemotherapeutic-induced reproductive toxicity. Theor Biol Med Model. 2012;9(1):1. https://doi.org/10.1186/1742-4682-9-1.
Korga A, Ostrowska M, Jozefczyk A, Iwan M, Wojcik R, Zgorka G, et al. Apigenin and hesperidin augment the toxic effect of doxorubicin against HepG2 cells. BMC Pharmacol Toxicol. 2019;20(1):22. https://doi.org/10.1186/s40360-019-0301-2.
Xiao Z, Wang C, Li L, Tang X, Li N, Li J, et al. Clinical efficacy and safety of Aidi injection plus docetaxel-based chemotherapy in advanced nonsmall cell lung Cancer: a Meta-analysis of 36 randomized controlled trials. Evid Based Complement Alternat Med. 2018;2018:7918258.
Xie G, Cui Z, Peng K, Zhou X, Xia Q, Xu D. Aidi injection, a traditional Chinese medicine injection, Could Be Used as an Adjuvant Drug to Improve Quality of Life of Cancer Patients Receiving Chemotherapy: A Propensity Score Matching Analysis Integr Cancer Ther. 2019;18:1534735418810799.
Xiao Z, Wang C, Zhou M, Hu S, Jiang Y, Huang X, et al. Clinical efficacy and safety of Aidi injection plus paclitaxel-based chemotherapy for advanced non-small cell lung cancer: a meta-analysis of 31 randomized controlled trials following the PRISMA guidelines. J Ethnopharmacol. 2019;228:110–22. https://doi.org/10.1016/j.jep.2018.09.024.
Liu D, Chen Z. The effects of cantharidin and cantharidin derivates on tumour cells. Anti Cancer Agents Med Chem. 2009;9(4):392–6. https://doi.org/10.2174/1871520610909040392.
Han W, Wang S, Liang R, Wang L, Chen M, Li H, et al. Non-ionic surfactant vesicles simultaneously enhance antitumor activity and reduce the toxicity of cantharidin. Int J Nanomedicine. 2013;8:2187–96. https://doi.org/10.2147/IJN.S43568.
Kim JA, Kim Y, Kwon BM, Han DC. The natural compound cantharidin induces cancer cell death through inhibition of heat shock protein 70 (HSP70) and Bcl-2-associated athanogene domain 3 (BAG3) expression by blocking heat shock factor 1 (HSF1) binding to promoters. J Biol Chem. 2013;288(40):28713–26. https://doi.org/10.1074/jbc.M113.488346.
Zhang W, Ma YZ, Song L, Wang CH, Qi TG, Shao GR. Effect of cantharidins in chemotherapy for hepatoma: a retrospective cohort study. Am J Chin Med. 2014;42(3):561–7. https://doi.org/10.1142/S0192415X14500360.
Zhang YM, Liu YQ, Liu D, Zhang L, Qin J, Zhang Z, et al. The effects of Astragalus polysaccharide on bone marrow-derived mesenchymal stem cell proliferation and morphology induced by A549 lung Cancer cells. Med Sci Monit. 2019;25:4110–21. https://doi.org/10.12659/MSM.914219.
Zheng Y, Ren W, Zhang L, Zhang Y, Liu D, Liu Y. A review of the pharmacological action of Astragalus polysaccharide. Front Pharmacol. 2020;11:349. https://doi.org/10.3389/fphar.2020.00349.
Kondeva-Burdina M, Shkondrov A, Simeonova R, Vitcheva V, Krasteva I, Ionkova I. In vitro/in vivo antioxidant and hepatoprotective potential of defatted extract and flavonoids isolated from Astragalus spruneri Boiss. (Fabaceae). Food Chem. Toxicol. 2018;111:631–40.
Li LK, Kuang WJ, Huang YF, Xie HH, Chen G, Zhou QC, et al. Anti-tumor effects of Astragalus on hepatocellular carcinoma in vivo. Indian J Pharmacol. 2012;44(1):78–81. https://doi.org/10.4103/0253-7613.91872.
Yoon TJ, Yoo YC, Lee SW, Shin KS, Choi WH, Hwang SH, et al. Anti-metastatic activity of Acanthopanax senticosus extract and its possible immunological mechanism of action. J Ethnopharmacol. 2004;93(2–3):247–53. https://doi.org/10.1016/j.jep.2004.03.052.
Yang X, Zou J, Cai H, Huang X, Yang X, Guo D, et al. Ginsenoside Rg3 inhibits colorectal tumor growth via down-regulation of C/EBPβ/NF-κB signaling. Biomed Pharmacother. 2017;96:1240–5. https://doi.org/10.1016/j.biopha.2017.11.092.
Dai G, Sun B, Gong T, Pan Z, Meng Q, Ju W. Ginsenoside Rb2 inhibits epithelial-mesenchymal transition of colorectal cancer cells by suppressing TGF-β/Smad signaling. Phytomedicine. 2019;56:126–35. https://doi.org/10.1016/j.phymed.2018.10.025.
Xiao Z, Jiang Y, Chen XF, Wang CQ, Xu WH, Liu Y, et. al. The Hepatorenal Toxicity and Tumor Response of Chemotherapy With or Without Aidi Injection in Advanced Lung Cancer: A Meta-Analysis of 80 Randomized Controlled Trials. Clin Ther. 2020;42(3):515–43.e31.
Xiao Z, Liang R, Wang CQ, Xu S, Li N, He Y, et al. Can Aidi injection alleviate the toxicity and improve the clinical efficacy of radiotherapy in lung cancer?: a meta-analysis of 16 randomized controlled trials following the PRISMA guidelines. Medicine (Baltimore). 2016;95(35):e4517. https://doi.org/10.1097/MD.0000000000004517.
Fan J, de Lannoy IA. Pharmacokinetics. Biochem Pharmacol. 2014;87(1):93–120. https://doi.org/10.1016/j.bcp.2013.09.007.
Lu Y, Pan J, Yang ST, Wu Q, Liu CH, Li YJ, et al. Effect of Aidi injection on glutathione-S-transferase of diethynitrosamine-induced hepatocellular carcinoma model rats. Chin Pharma Bull. 2018;34(8):1170–4.
Pan J, Lu Y, Zhang S, Li YT, Sun J, Liu CH, et al. Differential changes in the pharmacokinetics of doxorubicin in diethylnitrosamine-induced hepatocarcinoma model rats. Xenobiotica. 2020;50(10):1251–7. https://doi.org/10.1080/00498254.2020.1765049.
Lu Y, Pan J, Yang ST, Liu CH, Li YJ, Liu T. Effect of Aidi injection on in vivo pharmacokinetics of doxorubicin in Sprague-Dawley rats by UPLC-MS method. Chin. Pharma. Bull. 2018;34(3):423–7.
Gomes AS, Monteleone PA, Sayre JW, Finn RS, Sadeghi S, Tong MJ, et al. Comparison of triple-drug transcatheter arterial chemoembolization (TACE) with single-drug TACE using doxorubicin-eluting beads: long-term survival in 313 patients. AJR Am J Roentgenol. 2017;209(4):722–32. https://doi.org/10.2214/AJR.17.18219.
Sun JH, Zhou GH, Zhang YL, Nie CH, Zhou TY, Ai J, et al. Chemoembolization of liver cancer with drug-loading microsphere 50–100 μm. Oncotarget. 2017;8(3):5392–9. https://doi.org/10.18632/oncotarget.14281.
Cao W, Li J, Hu C, Shen J, Liu X, Xu Y, et al. Symptom clusters and symptom interference of HCC patients undergoing TACE: a cross-sectional study in China. Support Care Cancer. 2013;21(2):475–83. https://doi.org/10.1007/s00520-012-1541-5.
Wan YM, Li YH, Xu ZY, Wu HM, Xu Y, Yang M, et al. The effect of Transarterial chemoembolization in combination with Kang'ai injection on patients with intermediate stage hepatocellular carcinoma: a prospective study. Integr Cancer Ther. 2018;17(2):477–85. https://doi.org/10.1177/1534735417734913.
Hu B, Wang SS, Du Q. Traditional Chinese medicine for prevention and treatment of hepatocarcinoma: from bench to bedside. World J Hepatol. 2015;7(9):1209–32. https://doi.org/10.4254/wjh.v7.i9.1209.
Dai Y, Gao S, Liu X, Gao Q, Zhang L, Fan X, et al. Effect of Aidi injection plus TACE on hepatocellular carcinoma: a meta-analysis of randomized controlled trials. Evid Based Complement Alternat Med. 2018;9196409.
Sun XF, Pei YT, Yin QW, Wu MS, Yang GT. Application of Aidi injection (艾迪注射液) in the bronchial artery infused neo-adjuvant chemotherapy for stage III a non-small cell lung cancer before surgical operation. Chin J Integr Med. 2010;16(6):537–41. https://doi.org/10.1007/s11655-010-0569-y.
Wu L, Tang ZY, Li Y. Experimental models of hepatocellular carcinoma: developments and evolution. J Cancer Res Clin Oncol. 2009;135(8):969–81. https://doi.org/10.1007/s00432-009-0591-7.
Santos NP, Colaço AA, Oliveira PA. Animal models as a tool in hepatocellular carcinoma research: a review. Tumour Biol. 2017;39(3):1010428317695923. https://doi.org/10.1177/1010428317695923.
Lal S, Mahajan A, Chen WN, Chowbay B. Pharmacogenetics of target genes across doxorubicin disposition pathway: a review. Curr Drug Metab. 2010;11(1):115–28. https://doi.org/10.2174/138920010791110890.
Bains OS, Karkling MJ, Grigliatti TA, Reid RE, Riggs KW. Two nonsynonymous single nucleotide polymorphisms of human carbonyl reductase 1 demonstrate reduced in vitro metabolism of daunorubicin and doxorubicin. Drug Metab Dispos. 2009;37(5):1107–14. https://doi.org/10.1124/dmd.108.024711.
Chen T, Wang C, Liu Q, Meng Q, Sun H, Huo X, et al. Dasatinib reverses the multidrug resistance of breast cancer MCF-7 cells to doxorubicin by downregulating P-gp expression via inhibiting the activation of ERK signaling pathway. Cancer Biol Ther. 2015;16(1):106–14. https://doi.org/10.4161/15384047.2014.987062.
Zhao YY, Yu L, Liu BL, He XJ, Zhang BY. Downregulation of P-gp, Ras and p-ERK1/2 contributes to the arsenic trioxide-induced reduction in drug resistance towards doxorubicin in gastric cancer cell lines. Mol Med Rep. 2015;12(5):7335–43. https://doi.org/10.3892/mmr.2015.4367.
Nanayakkara AK, Follit CA, Chen G, Williams NS, Vogel PD, Wise JG. Targeted inhibitors of P-glycoprotein increase chemotherapeutic-induced mortality of multidrug resistant tumor cells. Sci Rep. 2018;8(1):967. https://doi.org/10.1038/s41598-018-19325-x.
Lal S, Wong ZW, Jada SR, Xiang X, Chen Shu X, Ang PC, et al. Novel SLC22A16 polymorphisms and influence on doxorubicin pharmacokinetics in Asian breast cancer patients. Pharmacogenomics. 2007;8(6):567–75. https://doi.org/10.2217/14622416.8.6.567.
Dong SC, Sha HH, Xu XY, Hu TM, Lou R, Li H, et al. Glutathione S-transferase π: a potential role in antitumor therapy. Drug Des Devel Ther. 2018;12:3535–47. https://doi.org/10.2147/DDDT.S169833.
Pljesa-Ercegovac M, Savic-Radojevic A, Matic M, Coric V, Djukic T, Radic T, et al. Glutathione transferases: potential targets to overcome chemoresistance in solid tumors. Int J Mol Sci. 2018;19(12):3785. https://doi.org/10.3390/ijms19123785.
Zhu C, Wang Y, Liu H, Mu H, Lu Y, Zhang J, et al. Oral administration of Ginsenoside Rg1 prevents cardiac toxicity induced by doxorubicin in mice through anti-apoptosis. Oncotarget. 2017;8(48):83792–801. https://doi.org/10.18632/oncotarget.19698.
Wang X, Chen L, Wang T, Jiang X, Zhang H, Li P, et al. Ginsenoside Rg3 antagonizes adriamycin-induced cardiotoxicity by improving endothelial dysfunction from oxidative stress via upregulating the Nrf2-ARE pathway through the activation of akt. Phytomedicine. 2015;22(10):875–84. https://doi.org/10.1016/j.phymed.2015.06.010.
Zhou B, Yan Z, Liu R, Shi P, Qian S, Qu X, et al. Prospective study of transcatheter arterial chemoembolization (TACE) with ginsenoside Rg3 versus TACE alone for the treatment of patients with advanced hepatocellular carcinoma. Radiology. 2016;280(2):630–9. https://doi.org/10.1148/radiol.2016150719.
Li L, Ni J, Li M, Chen J, Han L, Zhu Y, et al. Ginsenoside Rg3 micelles mitigate doxorubicin-induced cardiotoxicity and enhance its anticancer efficacy. Drug Delivery. 2017;24(1):1617–30. https://doi.org/10.1080/10717544.2017.1391893.
Cao Y, Ruan Y, Shen T, Huang X, Li M, Yu W, et al. Astragalus polysaccharide suppresses doxorubicin-induced cardiotoxicity by regulating the PI3k/Akt and p38MAPK pathways. Oxidative Med Cell Longev. 2014;2014:674219.
Cao Y, Shen T, Huang X, Lin Y, Chen B, Pang J, et al. Astragalus polysaccharide restores autophagic flux and improves cardiomyocyte function in doxorubicin-induced cardiotoxicity. Oncotarget. 2017;8(3):4837–48. https://doi.org/10.18632/oncotarget.13596.
Liang Q, Yu X, Qu S, Xu H, Sui D. Acanthopanax senticosides B ameliorates oxidative damage induced by hydrogen peroxide in cultured neonatal rat cardiomyocytes. Eur J Pharmacol. 2010;627(1):209–15. https://doi.org/10.1016/j.ejphar.2009.10.055.
Acknowledgements
Not applicable.
Funding
This work was financially supported by the National Natural Science Foundation of China (No. 81860718), the Youth Science and Technology Talent Growth Project of Guizhou Provincial Department of Education (KY [2021]171), the Science and Technology Department of Guizhou Province ([2015]7361, [2017]5718), the Team Project of Guizhou Provincial Science and Technology Department ([2016]5613\5677), the Central Guidance for Local Science and Technology Projects ([2018]4006). These entities had no role in the design of the studies or the collection, analysis, or interpretation of data or in writing the manuscript.
Author information
Authors and Affiliations
Contributions
YL, YJL and TL designed research; YL, JP, XQZ, SZ, CL, JS, YTL, SC, JH, CC, YW, YJL, TL performed research and analyzed the data; YL and TL wrote the paper. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
In vivo experiments were performed in accordance with international guidelines and experimental procedures performed with due approval from the Ethical Committee on Animal Studies of Guizhou Medical University (Approval number 1801207).
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Lu, Y., Pan, J., Zhu, X. et al. Pharmacokinetic herb-drug interactions between Aidi injection and doxorubicin in rats with diethylnitrosamine-induced hepatocellular carcinoma. BMC Pharmacol Toxicol 22, 48 (2021). https://doi.org/10.1186/s40360-021-00515-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40360-021-00515-9