
Abstract
Background
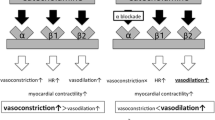
Clonidine is an imidazoline sympatholytic, acting on both α2-adrenergic and imidazoline receptors in the brainstem to induce antihypertensive and negative chronotropic effects in the vasculature and heart respectively.
Case presentation
A 69-year-old gentleman with hypertension presented to the emergency department after multiple syncopal episodes over the past 12 months. Electrocardiogram demonstrated sinus bradycardia with a heart rate of 42 beats per minute. It was hypothesized that the antihypertensive agent clonidine was responsible for inducing symptomatic bradycardia. Clonidine was thus gradually tapered and then discontinued over five days restoring normal sinus rhythm rates while avoiding hypertensive rebound related to sympathetic surge. His heart rate and blood pressure remained within normal limits after the clonidine taper and subsequent adjustments to his other hypertensive medications and he was discharged.
Conclusions
While clonidine has fallen out of favor for its indication as an antihypertensive, it remains a viable option for the use of opioid withdrawal, chronic pain, and smoking cessation, necessitating the appropriate clinical and pharmacological competencies for a physician to prescribe. A discussion of the clinical effects of clonidine brainstem receptor activation follows.
Similar content being viewed by others


Background
Cardiovascular drugs rank second to narcotics [1] as a source of adverse drug events and fatal poisonings in the U.S. Among adverse events, hypotension and dysrhythmias, including profound symptomatic bradycardia, represent significant risks for patients, especially the elderly (and is thus on the Beers criteria for potentially inappropriate medications to use in the elderly [2]). One cardiovascular drug, clonidine, is an imidazoline compound shown to have potent hypotensive actions in animals and humans [3]. Previously marketed as ‘Catapres,’ this sympatholytic drug has been clinically shown to reduce blood pressure and heart rate long before the discovery of G-protein coupled receptors such as the α2-adrenergic receptors were mechanistically linked to its phenotypic effect [4, 5]. While the initial reports of clonidine’s actions on the vasculature and heart provided ample evidence for its depressor and negative chronotropic actions respectively [6, 7], it remained unclear where in the body this agent exerted its pharmacological effects [8]. However, its robust hemodynamic properties including its potential for adverse outcomes including syncope [9] were evident early after its introduction into medical practice and remain a cautionary note to those continuing to employ it. Here, we present a case of clonidine adverse drug reaction and go on to discuss recent advances in the literature on clonidine pharmacology.
Case presentation
A 69-year-old male with a past medical history of insulin dependent diabetes (A1C = 6.3%), diabetic peripheral neuropathy, and no known coronary artery disease (CAD) or prior dysrhythmias was evaluated. He had a distant history of a transient ischemic attack, long standing hypertension, and no history of seizures or prior cerebrovascular accident (CVA). He presented to the emergency department (ED) shortly after a syncopal event while in the parking lot walking from his car to a grocery store. He had three prior syncopal events in the last year requiring hospitalization. He reported feeling dizzy prior to the most recent fall. As a consequence of the fall, he struck the right side of his face against the asphalt. He then regained consciousness and was able to resume an upright posture immediately. Upon arrival in the ED he was asymptomatic as he denied chest pain, shortness of breath, abdominal pain, headache, changes in vision, speech, strength, sensation, or gait. He noted that he often felt dizzy at least twice per week and that this persisted over the last several months. His current meds included simvastatin, clopidogrel, gabapentin, insulin, lisinopril, amlodipine and clonidine. He denied tobacco/illicit drug use and endorsed occasional alcohol use. He denied over the counter medications or therapies.
Upon arrival, he was afebrile, pulse 42 BPM, respirations 16/min, blood pressure 164/62 mmHg, and O2 saturation was 97% on room air. His physical exam revealed an alert, oriented male with normal cognition. The remainder of his exam including a detailed neurological exam was unremarkable. Chest x-ray and head CT were normal. Blood work revealed normal electrolytes and glucose, with a negative troponin. Electrocardiogram (EKG) (Fig. 1) revealed marked sinus bradycardia. The patient was admitted and placed on telemetry for recurrent symptomatic sinus bradycardia. There were no ST or T wave or other EKG changes and the QT and QTc were normal (496 and 419 ms respectively). Further questioning revealed the patient was started on the clonidine for hypertension about 18 months prior. Due to a high degree of clinical suspicion early on in the hospital course, clonidine was discontinued from 0.3 mg BID over the course of five days to avoid severe rebound hypertension from sympathetic surge [10] (Fig. 2). His bradycardia resolved by day 2-3 of the taper and recurrent hypertension was alternatively treated. The patient was discharged with a pulse of 78 BPM and BP of 160/82 mmHg.

Electrocardiogram at admission demonstrates sinus bradycardia

SBP, systolic blood pressure; DBP, diastolic blood pressure; HR, heart rate; BPM, beats per minute
Over the course of his hospitalization, other potential etiologies were considered, including polypharmacy leading to pharmacodynamic and pharmacokinetic interactions as well as complications due to diabetes including autonomic neuropathy [11]. Diabetic autonomic neuropathy was less likely given that the cessation of clonidine relieved the bradycardia, although a synergistic effect of the drug and diabetes certainly cannot be ruled out.
Discussion and conclusions
Clonidine is an imidazoline sympatholytic, rendering it highly efficacious as a vascular depressor and negative chronotropic agent. The patient described presented with multiple episodes of syncope thought to be most likely due to bradycardia. This is a foreseeable outcome given the mechanism of this agent, an α2-adrenergic receptor agonist, coupled to lowering serum noradrenaline levels [12]. While the cardiovascular phenotypic consequences of clonidine are obvious, the incidence of adverse drug reactions to clonidine has not been sufficiently investigated [13]. Our focus is the enhanced understanding of clonidine’s basic molecular pharmacology that has occurred over the past several decades.
It has long been hypothesized that clonidine acts at critical foci within the the medulla of the brainstem. Here it exerts its effects on the autonomic nervous system to attenuate sympathetic outflow [14]. The rostral ventrolateral medulla (RVLM) is a nucleus considered by many to be the site of sympathoexcitatory generation, with efferents contributing powerfully to both vasomotor and chronotropic targets [15]. Within this specific brainstem nucleus exist barosensitive neurons synchronized to the cardiac cycle [16]. Foreseeably, when the baroreceptors detect a drop in vascular pressure, the efferent catecholaminergic response is then potentiated. Clonidine and other imidazoline derivatives likely exert their effects in this region to terminate presynaptic release of norepinephrine, and it is proposed they do so via their pharmacological actions on α2 or the controversial imidazoline receptors [17,18,19]. Furthermore, the cardio-inhibitory effect of clonidine is functionally antagonized by the local application of N-methyl-D-aspartate receptor (NMDA) blockers, highlighting the importance of glutamate signaling in cardiovascular depression in the RVLM [20, 21].
But it is only recently that evidence has been provided that clonidine is mechanistically coupled to both reducing sympathetic but also increasing parasympathetic outflow to the cardiovascular system. The primary parasympathetic driver to the heart comes from the premotor cardioinhibitory vagal neurons in the nucleus ambiguus of the medulla, which receives input from multiple brainstem nuclei including the nucleus tractus solitarius (NTS) [22, 23]. Inhibitory GABAergic input into the nucleus ambiguus prevents activation of the parasympathetic nuclei [24, 25]. Clonidine prevents the release of GABA in the nucleus ambiguus, thus disinhibition of that cluster of neurons potentiates parasympathetic outflow to the heart [26].
In addition to the central actions of clonidine, there is new additional evidence of a direct peripheral tissue effect. At the level of target cardiac and vascular structures, substantial expression of α2-adrenergic receptors exists where clonidine prevents the vesicular release of norepinephrine, thus functionally antagonizing local myocyte β-adrenergic activation.
It was clear from its first description in the 1960s that clonidine possessed excellent sympatholytic activity, reducing blood pressure and slowing the heart. Its aforementioned negative chronotropic effects can thus predictably lead to overt bradycardia and syncope [27]. While it has fallen out of favor as a first line antihypertensive agent, new reports have shown efficacy in chronic pain [28], Tourette’s Syndrome [29], opioid withdrawal [30], and smoking cessation [31]. It is still employed for resistant hypertension, revealing that it still has an important adjunctive role. But foreseeably, its less frequent use as a hypertensive agent has resulted in clinicians becoming increasingly unfamiliar with its pharmacology and therapeutic outcomes. And finally, the collective understanding of the molecular basis for clonidine’s mechanistic impact on cardiovascular hemodynamics continues to evolve. Understanding the molecular and cellular basis of inhibiting sympathetic tone may provide a crucial link to the clinical phenotypic expression of syncope.
Abbreviations
- CAD:
-
coronary artery disease
- CVA:
-
Cerebrovascular accident
- ED:
-
emergency department
- EKG:
-
electrocardiogram
- NMDA:
-
N-methyl-D-aspartate
- NTS:
-
nucleus tractus solitarius
- RVLM:
-
rostral ventrolateral medulla
References
Givens ML. Toxic bradycardias in the critically ill poisoned patient. Emerg Med Int. 2012;2012:852051.
By the American Geriatrics Society Beers Criteria Update Expert Panel. American Geriatrics Society 2015 updated beers criteria for potentially inappropriate medication use in older adults. J Am Geriatr Soc. 2015;63(11):2227–46.
Ng J, et al. Properties of catapres, a new hypotensive drug: a preliminary report. N Z Med J. 1967;66(425):864–70.
Simpson GM, Kunz-Bartholini E, Watts TP. A preliminary evaluation of the sedative effects of catapres, a new antihypertensive agent, in chronic schizophrenic patients. J Clin Pharmacol J New Drugs. 1967;7(4):221–5.
Barnett AJ, Cantor S. Observations on the hypotensive action of “Catapre” (ST 155) in man. Med J Aust. 1968;1(3):87–91.
Lowenstein J. Drugs five years later: clonidine. Ann Intern Med. 1980;92(1):74–7.
Scriabine A, et al. Cardiac slowing effects of clonidine (ST-155) in dogs. J Pharmacol Exp Ther. 1970;171(2):256–64.
Head GA, Gundlach AL, Musgrave IF. Recent advances in imidazoline receptor research: ligands--localization and isolation--signaling--functional and clinical studies. J Auton Nerv Syst. 1998;72(2-3):74–9.
Muir AL, Burton JL, Lawrie DM. Circulatory effects at rest and exercise of clonidine, an imidazoline derivative with hypotensive properties. Lancet. 1969;2(7613):181–4.
Shaw M, Matsa R. Clonidine withdrawal induced sympathetic surge. BMJ Case Rep. 2015;1-3.
Golusinski LL Jr. And B.W. Blount, Clonidine-induced bradycardia. J Fam Pract. 1995;41(4):399–401.
Kanagy NL. Alpha(2)-adrenergic receptor signalling in hypertension. Clin Sci (Lond). 2005;109(5):431–7.
Isbister GK, et al. Adult clonidine overdose: prolonged bradycardia and central nervous system depression, but not severe toxicity. Clin Toxicol (Phila). 2017;55(3):187–92.
Granata AR, Cohen MI. Rhythmic properties of neurons in the rostral ventrolateral medulla of the rat in vitro: effects of clonidine. J Neurophysiol. 2002;88(5):2262–79.
Granata AR, et al. A1 noradrenergic neurons tonically inhibit sympathoexcitatory neurons of C1 area in rat brainstem. Brain Res. 1986;377(1):127–46.
Allen AM, Guyenet PG. Alpha 2-adrenoceptor-mediated inhibition of bulbospinal barosensitive cells of rat rostral medulla. Am J Phys. 1993;265(5 Pt 2):R1065–75.
Ernsberger P, et al. Role of imidazole receptors in the vasodepressor response to clonidine analogs in the rostral ventrolateral medulla. J Pharmacol Exp Ther. 1990;253(1):408–18.
Schmitt H, Fenard S. Decrease in the sympatho-inhibitory action of clonidine after destruction of the sympatho-inhibitory area. Experientia. 1973;29(10):1247–9.
Ernsberger P, et al. Clonidine binds to imidazole binding sites as well as alpha 2-adrenoceptors in the ventrolateral medulla. Eur J Pharmacol. 1987;134(1):1–13.
Wang WZ, Yuan WJ, Su DF. Blockade of N-methyl-D-aspartate receptors within the rostral ventrolateral medulla antagonizes clonidine-induced cardiovascular effects. Auton Neurosci. 2003;109(1-2):21–8.
Wang WZ, et al. Interaction between clonidine and N-methyl-D-aspartate receptors in the caudal ventrolateral medulla of rats. Exp Brain Res. 2004;158(2):259–64.
Mendelowitz D. Advances in parasympathetic control of heart rate and cardiac function. News Physiol Sci. 1999;14:155–61.
Mendelowitz D, Kunze DL. Identification and dissociation of cardiovascular neurons from the medulla for patch clamp analysis. Neurosci Lett. 1991;132(2):217–21.
Wang X. Propofol and isoflurane enhancement of tonic gamma-aminobutyric acid type a current in cardiac vagal neurons in the nucleus ambiguus. Anesth Analg. 2009;108(1):142–8.
Jameson HS, Pinol RA, Mendelowitz D. Purinergic P2X receptors facilitate inhibitory GABAergic and glycinergic neurotransmission to cardiac vagal neurons in the nucleus ambiguus. Brain Res. 2008;1224:53–62.
Philbin KE, Bateman RJ, Mendelowitz D. Clonidine, an alpha2-receptor agonist, diminishes GABAergic neurotransmission to cardiac vagal neurons in the nucleus ambiguus. Brain Res. 2010;1347:65–70.
McComb MN, Chao JY, Ng TM. Direct vasodilators and sympatholytic agents. J Cardiovasc Pharmacol Ther. 2016;21(1):3–19.
Ginosar Y, Riley ET, Angst MS. Analgesic and sympatholytic effects of low-dose intrathecal clonidine compared with bupivacaine: a dose-response study in female volunteers. Br J Anaesth. 2013;111(2):256–63.
Jiao F, et al. Clinical observation on treatment of Tourette syndrome in Chinese children by clonidine adhesive patch. Eur J Paediatr Neurol. 2016;20(1):80–4.
Mannelli P, et al. The combination very low-dose naltrexone-clonidine in the management of opioid withdrawal. Am J Drug Alcohol Abuse. 2012;38(3):200–5.
Gourlay SG, Stead LF, Benowitz NL. Clonidine for smoking cessation. Cochrane Database Syst Rev. 2004;3:CD000058.
Acknowledgements
N/A
Funding
There is no funding source to report.
Availability of data and materials
The datasets used and/or analyzed during the current study available from the corresponding author on reasonable request.
Author information
Authors and Affiliations
Contributions
AJS, CM, AS, and JR analyzed and interpreted patient data and EKG. AJS and JR were the primary contributors in writing the manuscript. AJS made the figures based on patient data and EKG. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Written consent was obtained from the patient to report individual data within the text and images for publication.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Sandweiss, A.J., Morrison, C.M., Spichler, A. et al. A case report of clonidine induced syncope: a review of central actions of an old cardiovascular drug. BMC Pharmacol Toxicol 19, 6 (2018). https://doi.org/10.1186/s40360-018-0198-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40360-018-0198-1