
Abstract
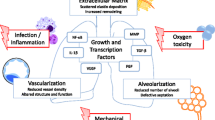
Bronchopulmonary dysplasia (BPD) is a multifactorial disease occurring as a consequence of premature birth, as well as antenatal and postnatal injury to the developing lung. BPD morbidity and severity depend on a complex interplay between prenatal and postnatal inflammation, mechanical ventilation, and oxygen therapy as well as associated prematurity-related complications. These initial hits result in ill-explored aberrant immune and reparative response, activation of pro-fibrotic and anti-angiogenic factors, which further perpetuate the injury. Histologically, the disease presents primarily by impaired lung development and an arrest in lung microvascular maturation. Consequently, BPD leads to respiratory complications beyond the neonatal period and may result in premature aging of the lung. While the numerous prenatal and postnatal stimuli contributing to BPD pathogenesis are relatively well known, the specific cell populations driving the injury, as well as underlying mechanisms are still not well understood. Recently, an effort to gain a more detailed insight into the cellular composition of the developing lung and its progenitor populations has unfold. Here, we provide an overview of the current knowledge regarding perinatal origin of BPD and discuss underlying mechanisms, as well as novel approaches to study the perturbed lung development.
Similar content being viewed by others

Introduction
Bronchopulmonary dysplasia (BPD) is the most common form of chronic lung disease in children and a leading cause of neonatal morbidity and mortality [1,2,3]. Since 1999, the disease has been defined by the need for supplemental oxygen at 36 weeks post-menstrual age, and is classified based on its severity, as well as oxygen and respiratory support requirements into three grades: mild, moderate, or severe [4, 5]. While advancements in neonatal care led to improvements in survival of premature infants, the incidence of BPD has not decreased. Not surprisingly, BPD is now less frequent in infants with birth weight > 1200 g, or born after 30 weeks of gestation, but affects extremely premature infants of lower gestational age [4,5,6].
BPD is a multifactorial disease which occurs predominantly as a consequence of prematurity leading to respiratory distress and consequent treatments in neonatal intensive care units (NICU), including mechanical ventilation (MV) and oxygen supplementation [2, 5]. In addition to the degree of prematurity, additional antenatal and perinatal risk factors include low body weight (LBW), infection, and maternal nutrition [6,7,8]. Moreover, preeclampsia and intrauterine growth restriction (IUGR) are identified as independent risks factors [6, 9,10,11]. Recent studies further indicate that prenatal smoke exposure might also contribute to the development of the disease [12, 13]. Finally, BPD may have some hereditary component [14, 15]. While increased prevalence of BPD is typically associated with male sex, in the long term, female patients with a history of BPD might be affected more severely [6, 16, 17].
In the past BPD was associated mostly with aggressive MV in more mature infants [18]. However, advances in ventilation technology, avoidance of MV, and more judicious use of oxygen result in a new histological phenotype characterized less by fibrosis and more by global arrest in alveolar and microvascular development, as well as impaired, and sometimes declining lung function [4, 5]. BPD is also associated with long-term sequelae, which often persist into adolescence or early adulthood, including neurodevelopmental and cognitive changes [19], impaired lung function [2, 20, 21], pulmonary vascular disease [2, 22], and cardiac dysfunction [2, 23]. Impaired immune development results in increased susceptibility to viral infections and higher risk of rehospitalization later in life [1, 4, 6]. The disease is further associated with increased incidence of asthma [21] and early-onset emphysema [24].
BPD constitutes a complex injury to the developing lung with heterogenous pathological features and outcomes, greatly depending on the degree of prematurity, as well as antenatal and postnatal exposures. In the following paragraphs we discuss underlying causes and mechanisms contributing to the development of BPD, as well as novel approaches to study BPD pathogenesis.
Intrauterine growth restriction, placental dysfunction, and preeclampsia
IUGR is defined as a failure of the fetus to reach its “biologically based potential” [25]. The condition can arise due to anatomical or functional disorders associated with maternal factors, maternal-placental-fetal unit, or genetic abnormalities [26,27,28,29]. IUGR often results in malnutrition, LBW, and permanent perturbations in metabolism and development [26, 28]. It is a known cause of prematurity and associated with increased morbidity and mortality [26, 30].
In experimental animal models, IUGR can be induced by various interventions, including uterine artery ligation [31], low protein diet (LPD) [32,33,34], or heat exposure [9]. Impaired alveolar and vascular formation during postnatal development and/or in adulthood were reported in various IUGR models in rats [32,33,34,35] and sheep [9, 36, 37]. IUGR in developing rat pups from LPD-fed mothers is associated with impaired lung development, as evidenced by increased alveolar septal thickness, Col1a1 expression and extracellular matrix (ECM) deposition at P23 [34]. These changes are preceded by an inhibition of GH/Stat5/IGF-1 signaling during the embryonic phase. Decreased IGF-1 levels were also reported in serum of BPD patients [38]. Notably, IGF-1 was shown to have anti-inflammatory properties, to preserve lung structure, and to prevent right ventricular hypertrophy (RVH) in a rat BPD model [39,40,41]. IUGR in rats further impaired embryonic VEGF and BMP signaling, and decreased microvascular and ECM formation postnatally [35]. Finally, microRNA microarray analysis revealed perturbations to “tissue repair” and “cellular communication” pathways [33]. In addition to structural changes, IUGR impairs lung function in developing rats [42, 43], while in clinical studies, IUGR and LBW are associated with poorer lung function in childhood [44, 45] and adulthood [46,47,48]. The impact on lung function may be directly related to a higher prevalence of BPD among the IUGR patients [11]. Finally, studies show that IUGR and LBW may contribute to development of chronical illness such as asthma [49] or chronic pulmonary obstructive disease (COPD) [50, 51].
Placental dysfunction and preeclampsia (PE), major causes of IUGR, also impact lung development. While the underlying molecular mechanisms remain unknown, PE is an independent risk factor for both, preterm delivery, and the development of BPD [6, 10, 52]. Pathological placental changes resulting from maternal vascular underperfusion (MVU) are associated with increased risk of BPD [53]. A recent meta-analysis of 211 studies show that placental vascular dysfunction in association with IUGR or being born small for gestational age (SGA) increases the risk of BPD and pulmonary hypertension (PH) [54]. Accordingly, decreased levels of cord blood angiogenic factors are strong predictors of BPD-associated PH [55]. Taken together, these data strongly indicates the strong association between the placental dysfunction of prematurity and vascular phenotype of BPD, supporting the so-called “vascular hypothesis” of BPD pathogenesis and the potential preventive use of proangiogenic agents in such patients [56].
While several preclinical models of PE have been designed, most differ from the human condition and replicate the condition to only a limited extent [57]. These include genetic models, such as the hypertensive BPH/5 mouse model [58], chronic hypoxia models [59, 60], or pharmacologically-induced models, such as nitric oxide inhibition [61, 62]. Widely used are also surgical models, such as the reduced uterine perfusion pressure (RUPP) [63], or selective RUPP rat model [64]. Interesting is also the CBA/J × DBA/2 J mouse model of recurrent miscarriage and spontaneous PE, recapitulating many features of the clinical condition, including renal damage, placental growth defects, restricted fetal growth, and increase sFLT-1 and leptin levels [65]. However, the animals do not become hypertensive, therefore not meeting the clinical criteria. Building on the notion of the importance of the above-mentioned sFLT-1 is another rat model, where PE is induced by intraamniotic sFLT-1 injections [66]. This model recapitulates retardation in lung growth, as well as findings of abnormal lung function, vessel density, and RVH. Importantly, both antenatal and postnatal treatment with selective anti-sFLT-1 antibody improved alveolarization, vessel formation, and lung function and decreased RVH in developing pups. Moreover, authors have shown similar result of anti-sFLT-1 treatment in the endotoxin-induced rat chorioamnionitis model [66].
Finally, maternal malnutrition or overnutrition are also associated with metabolic changes leading to increased risk of developing diabetes, obesity, and metabolic syndrome in later life [67,68,69]. Combined insult of IUGR and maternal high-fat diet (HFD) increase the risk of early cardiovascular pathology in rats [31]. Furthermore, HFD in rat dams prior to conception, or HFD diet from pre-conception until lactation increases airway resistance in the offspring [70]. Taken together, these findings highlight the impact of perinatal nutrition on lung development and the origin of adult pulmonary disease.
Patent ductus arteriosus, pulmonary hypertension, and microbiome
Patent ductus arteriosus (PDA) is a frequent complication in very preterm infants, with up to 70% of infants born before 28th week of gestation requiring pharmacological or surgical treatment [71, 72]. The condition is associated with increased lung blood flow, impaired lung mechanics, oxidative stress, and increased need for MV. PDA is clinically often associated with RDS and BPD and is historically considered a risk factor for BPD [73,74,75,76,77]. However, whether PDA plays a causal role in BPD pathogenesis is not known [72, 78, 79]. In fact, multiple randomized control trials have failed to find a direct relationship between PDA and the development of BPD [78, 80,81,82,83]. PDA closure is performed either pharmacologically or surgically. Pharmacological closure, typically achieved with indomethacin or ibuprofen, was shown to improve alveolarization and lung mechanics, as well as the need for ventilator support [84,85,86]. Whether the improvement is due to PDA closure or the pharmacological agents themselves however is not fully understood. These improvements have not been observed after surgical closure, with some reports indicating that early surgical ligation itself may contribute to impaired alveolarization [78, 87, 88]. Additionally, studies indicate differences in outcome dependent on timing of the pharmacological closure [89, 90]. Consequently, recent studies and review literature recommend avoidance of surgical ligation and further investigations into timing of PDA closure [78, 91, 92].
During the fetal development, gas exchange is provided by placenta and fetal pulmonary vascular resistance (PVR) is high. Perinatal transition is normally marked by a significant decrease in PVR, resulting in up to tenfold increase in pulmonary blood flow [93,94,95]. However, in some instances this change in vascular resistance does not occur, resulting in PH. Neonatal PH is a frequent complication in premature infants, particularly those with extremely LBW [96]. While not fully understood, among the known risk factors contributing to neonatal PH are low birth weight, SGA status, oligohydramnios, PE, severity of BPD, and prolonged MV [94, 97]. A consensus approach to better classify pediatric PH recognizes 10 categories of pulmonary hypertensive vascular disease, including the BPD-PH category [98, 99]. Up to 25% of patients with moderate to severe BPD also develop PH [99,100,101]. It can present itself as primary PH, acute PH associated with RDS or chronic PH associated with BPD (BPD-PH). Additionally, neonatal PH can be exacerbated by a PDA. BPD-PH is characterized by aberrant pulmonary vascular growth and remodeling, RV failure, increased mortality, and increased risk of PH and right ventricular dysfunction in adulthood [99, 102]. Initial diagnosis of neonatal PH is typically based on echocardiography and clinical representation. Late onset of PH in BPD patients (3–4 months of age) is now well described and justifies continuous screening for PH in premature infants with BPD [96]. Current therapies are based on the underlying pathophysiology of neonatal PH and include maintaining adequate oxygen saturation, correction of acidosis, surfactant therapy, and the use of pulmonary vasodilators, such as inhaled nitric oxide and sildenafil [94, 95].
Another intriguing, but understudied factor impacting the perinatal period is the microbiome. Studies have revealed alterations in airway and lung microbiome of prematurely born infants and suggest a link between the microbiome and BPD severity. A recent systematic reviews showed that most studies indicate decreased bacterial diversity, higher levels of Ureoplasma, and lower levels of Staphylococcus in the tracheal aspirates (TAs) of preterm infants who went on to develop severe BPD [103,104,105,106,107]. BPD progression was further associated with microbial turnover and relative abundance of different bacterial strains. It is important to note that majority of the infants included in these studies have received prenatal or postnatal antibiotics, which has previously also been associated with increased risk and severity of BPD [103, 105, 108,109,110]. Finally, a recent study has explored the relationship between perinatal microbiome and metabolome. The authors observed an increase in Proteobacteria, a reduction in Lactobacilli, as well as reduction in fatty acid β-oxidation pathway in infants with BPD [111]. While this data suggests a role for airway microbiome in the regulation of inflammation, further studies are needed to identify the mechanisms by which microbiome at birth modulates and primes the pulmonary metabolome.
Prenatal inflammation
Increasing evidence suggests that pre- and postnatal inflammation play critical roles in the development of BPD. BPD is associated with an increase in pro-inflammatory and decrease in anti-inflammatory cytokine levels, lung neutrophil and monocyte infiltration, and macrophage activation. Whether pre- or postnatal inflammation contribute more to the development and severity of BPD is not known.
Multiple types of prenatal inflammation have been suggested as BPD risk factors, including chorioamnionitis [112], fetal inflammatory response syndrome [113], and neonatal leukemoid reaction [114]. Most widely studied in the context of BPD is chorioamnionitis, a complex syndrome associated with preterm delivery. Chorioamnionitis is an inflammation of chorion and amnion membranes, often caused by bacterial infection, and usually classified as either histological or clinical [115]. The histological form is characterized microscopically by inflammatory cells infiltration, while the clinical form is defined by abdominal pain, presence of uterine tenderness, fever, increased white blood count, and maternal and fetal tachycardia [115, 116]. Studies in experimental animals suggest that antenatal inflammation can affect the expression of grow factors, modulate the immune system, and contribute to structural changes in the lung, resembling the BPD phenotype [117]. Intrauterine inflammation in animal models is often induced by bacterial lipopolysaccharide (LPS). Pre- or postnatal LPS in rodents and sheep results in inflammation, alveolar hypoplasia, impaired surfactant production, and impaired pulmonary vascular function [118,119,120,121]. Importantly, amniotic concentrations of IL-6, IL-8, IL-1β, and TNF-α are increased in mothers of infants who develop BPD [122]. However, the clinical data on relationship between chorioamnionitis and BPD remain inconclusive [123,124,125]. This is partly due to an inconsistent diagnosis and lack of correlation between clinical and histological chorioamnionitis [116, 126]. In fact, some studies indicate that the increased risk of BPD is rather associated with the postnatal consequences of chorioamnionitis and prematurity, such as surfactant deficiency, neonatal sepsis, and need for MV [112, 126,127,128]. Additionally, postnatal sepsis directly increases BPD incidence and interrupts lung development by various mechanisms, including inflammation, oxidative stress, and endothelial injury [126, 129].
Mechanical ventilation, hyperoxia, and postnatal inflammation
Prematurity in infants is furthermore directly associated with the need of respiratory support and ventilation. Ventilation constitutes a major risk factor for lung injury and BPD. Initial injury results from both, barotrauma and volutrauma, which initiate an influx of immune cells, particularly macrophages and neutrophils, and an increase in production of pro-inflammatory cytokines [130,131,132,133]. Elevated levels of pro-inflammatory IL-1β, IL-6, and IL-8 are found in TAs and blood of BPD patients [134]. Comparable cytokine profiles can be observed in ventilated rats, lambs, and baboons [135,136,137]. Similar to MV, hyperoxia exposure is as an independent mediator of lung inflammation [112, 131, 138,139,140]. Hyperoxia induces inflammation primarily via increase in IL-1β as evidenced by experiments with overexpression and blocking of IL-1β signaling [141,142,143,144]. Moreover, some evidence exists for the role of Csf1r+ monocytes and macrophages in hyperoxia-induced lung injury [138]. Finally, oxidative stress induced by hyperoxia impacts the expression of large number of genes implicated in cell cycle, signal transduction, ECM turnover, coagulation, and alveolar growth [145]. The initial inflammatory response disturbs the homeostasis and triggers additional mediators and growth factors which in turn impact alveolar and microvascular formation [146, 147]. Depending on the model used however, the inflammatory response induced by hyperoxia might be considered moderate. To better reflect the clinical situation, various double-hit models combining postnatal hyperoxia with either pre- or postnatal LPS administration were developed [148,149,150,151,152,153]. Results of these studies vary depending on species, strain, and LPS dosage. Overall, both pre- and postnatal endotoxin amplifies the effects of hyperoxia exposure in dose-dependent manner [148, 152]. Additionally, only animals exposed to both hits developed prominent PH [152], as well as intense local and systemic inflammatory response [149, 151].
Both, MV and hyperoxia exposure induce alveolar hypoplasia in mice [154, 155], rats [156, 157], lambs [130], and baboons [136]. Ventilation in premature infants is associated with underdeveloped terminal airspace epithelium and epithelial apoptosis [158,159,160]. Parallel observations of epithelial inflammation and shedding, as well as epithelial hyperplasia were made in ventilated lambs [161, 162] and baboons [163] respectively. MV and hyperoxia exposure are further associated with increased [164, 165] or decreased [166, 167] alveolar epithelial type 2 (AT2) cells proliferation. Finally, both hyperoxia [168] and cyclic stretch [169] disrupt epithelial permeability in vitro.
Reduction in lung microvascularization is seen in BPD patients [170, 171] and in various animal models [172, 173] alike. Both, hyperoxia and ventilation inhibit pro-angiogenic VEGF signaling in rodent [172,173,174], rabbits [175], and baboons [176], reminiscent of observations in the lungs, plasma, and TAs of BPD patients [177,178,179]. Decreased levels of additional pro-angiogenic factors have also been observed. Expression levels of eNOS are decreased in hyperoxia-exposed mice and ventilated lambs [174, 180]. Lowered Tie-2 and Ang-1 levels are found in ventilated preterm infants [181] and Ang-1 expression is decreased in hyperoxia-exposed mouse pups [182]. Similarly decreased in BPD patients and hyperoxia-exposed mice is the production of novel angiogenic markers such as Foxf1 and c-Kit [183].
The mesenchymal damage in the “new” BPD manifests in form of thickened alveolar septa, defective ECM deposition, and interstitial fibrosis [147, 184, 185]. Comparable findings were made in various animal models, including ventilated lambs and baboons [136, 186], as well as ventilated and hyperoxia-exposed rodents [172, 187,188,189]. Among the most studied in context of late lung development and BPD are the FGF, TGF-β, and PDGFA signaling pathways. Among the FGF family, pulmonary FGF10 expression is decreased [190], while FGF2 TA levels are increased in BPD patients [191]. Elevated FGF2 expression is also found during compensatory lung growth in hyperoxia-exposed rats [192]. PDGFA, as well as its receptor PDGFRA, are critical for secondary septation and their loss results in decreased myofibroblasts migration and proliferation [193,194,195]. Decreased PDGFRA expression is found in animal hyperoxia BPD models [151, 194, 196] and the low expression of PDGFRA is associated with increased BPD incidence in male patients [197]. TGF-β signaling is essential for fetal lung development and is dynamically regulated during alveolarization [198, 199]. Increase in TGF-β expression was reported in TAs of BPD patients [200]. Similarly, an increased TGF-β expression is associated with alveolar hypoplasia secondary to hyperoxia, which could be prevented by treatment with TGF-β neutralizing antibody [201, 202]. Perturbances in both, PDGFA and TGF-β signaling are associated with defects in secondary septation, during which elastin is deposited at the top of the protruding septa by myofibroblast. This ECM scaffold provides a base for further alveolar formation [147]. Impaired elastic fibres formation and an increased expression of elastin and elastin or collagen cross-linking enzymes were noted in BPD patients [203,204,205], as well as ventilated and hyperoxia-exposed rodents and lambs [186,187,188, 205, 206].
It is important to note that considerable efforts have been made to establish less invasive therapies, avoiding intubation and mechanical ventilation. These strategies include sustained inflation approaches, such as nasal continuous positive airways pressure (NCPAP) or synchronised nasal intermittent positive pressure ventilation (SNIPPV) [207, 208]. Particularly the combination of NCPAP and early surfactant-replacement therapy is more effective in preventing BPD than continuous MV and elective surfactant replacement [207, 209]. However, meta-analysis revealed that it is the avoidance of endotracheal MV, rather than sustained inflations strategies themselves, which decreases the risk of BPD and death [208]. As even a brief intubation early in life can have severe consequences, less invasive methods of surfactant delivery, such as less invasive surfactant administration (LISA) and minimally invasive surfactant therapy (MIST), have recently been developed. In combination with NCPAP these strategies currently represent very promising approaches to decrease the BPD occurrence [208, 210,211,212].
BPD phenotypes and endotypes
The multifactorial nature of BPD pathogenesis, combining various prenatal and postnatal insults results in several discrete endotypes and clinical phenotypes. As mentioned above, the biggest differences in clinical manifestation could be observed between the so-called “old”, profibrotic-like BPD phenotype and the “new” BPD characterized mainly by parenchymal and vascular damage [4, 5, 213]. Various classifications of BPD phenotypes have been proposed, including the categorization based on (i) severity [214], (ii) lung function (obstructive vs. restrictive phenotype) [215], or (iii) most effected tissue compartment [213, 216, 217]. Perhaps the most detailed classification, proposed in a recent review by Pierro et al., includes seven categories of BPD phenotypes: (i) parenchymal (characterized by alveolar simplification), (ii) peripheral airway (defined by bronchial hyperreactivity), (iii) central airway (stenosis, bronchomalacia, and tracheomalacia), (iv) interstitial (interstitial fibrosis and inflammation), (v) congestive (with pulmonary edema), (vi) vascular (dysmorphic vascularization and PH), and (vii) mixed phenotype [213].
Individual phenotypes do not only require different treatments but have different disease development and consequences in later life. The parenchymal phenotype, which is defined by arrest in alveolarization and decreased alveolar surface area, largely resembles emphysema. In fact, about two-thirds of BPD patients develop obstructive disease [215]. While the lung has capacity to continue alveolarization postnatally and parenchymal disease may improve over time, parenchymal damage in BPD has been previously associated with early onset chronic obstructive lung disease (COPD) in later life [213, 218]. The second obstructive phenotype—the peripheral airway phenotype, characterized by bronchoconstriction and hyperreactivity manifests similarly to asthma. However, studies have shown that at school age, these patients respond differently to treatments with β2-agonists than asthma patients and that the disease might be additionally characterized by structural changes in small airways [213, 216, 219].
In addition to BPD phenotypes, two main BPD endotypes are often recognized: (i) infection/inflammation (including chorioamnionitis) and (ii) placental dysfunction (including preeclampsia and IUGR) [213, 220]. While chorioamnionitis and infection represent the more common endotype, placental dysfunction in combination with IUGR constitute a more prominent background for vascular BPD phenotype [54, 221]. Ideally, in accordance with emerging personalized medicine approaches, distinct BPD phenotypes will be considered when assigning therapies and identifying possible complications in later life. However, details regarding underlying mechanisms and cellular integrations pertaining to individual phenotypes have not been yet deciphered.
Novel approaches to study the preterm lung
BPD in single-cell resolution
Understanding the lung composition on single-cell level has been a focus of substantial scientific research for more than a decade. During this period, more than 300 single-cell RNA sequencing (scRNA-seq) or single-nuclei RNA sequencing (snRNA-seq) human or animal lung datasets have been published. However, only a small fraction of these is dedicated to late lung development, and even fewer to the pathogenesis of BPD.
First exploratory scRNA-seq analysis of postnatal developing lung in mice were performed by Cohen et al., constructing a detailed single-cell map of the developing lung and lung progenitor cell populations covering the period from embryonic day (E)12.5 to postnatal day (P)7 [222]. The study identified 10 non-immune and 12 immune cell populations, revealing dynamic changes in population sizes, particularly during the pseudoglandular (E12.5) and canalicular stage (E18/19) of lung development. This data were further expanded in another study, characterizing lung immune (CD45+) cells between E18.5 and P21 [223]. When comparing the prenatal and postnatal immune cells authors identified a gradual increase in macrophage heterogeneity, as well as rapid increase in the proportion of lymphoid populations (2% vs 60% of all immune cells, respectively). Additional studies have focused on the developmental changes and postnatal adaptation in lung epithelial, endothelial, and stromal populations [224,225,226,227].
Several scRNA-seq studies have contributed to establishing a cell atlas of human fetal lung development [228,229,230,231]. No study to date has analysed lungs of BPD patients, although the scRNA-seq analysis of TA-derived cells to establish novel biomarkers and aid in stratifying BPD into endotypes has recently been proposed [232]. In contrast to the lack of studies in humans, few studies have explored the BPD pathogenesis at the single-cell level in the neonatal mouse hyperoxia model [151, 183, 226]. As mentioned above, the expression levels of angiogenic markers FOXF1 and c-KIT are decreased in the lungs of BPD patients [183]. This was confirmed by scRNA-seq in adult mouse lungs where hyperoxia exposure for first 7 days of life decreased the number of c-Kit+ endothelial cells (ECs) progenitors. Importantly, authors showed that adoptive transfer of c-Kit+ ECs improved lung angiogenesis and alveolarization in developing hyperoxia-exposed mice [183]. Substantial expression changes in all cell compartments were observed in the largest to date study of hyperoxia-exposed developing mice by Hurskainen et al., profiling over 66.000 lung cells at P3, P7, and P14 [151]. In this study, hyperoxia caused gradual changes in cell composition and expression patterns, particularly after 7 days of exposure. Within the stroma, authors identified transcriptomic shifts in myofibroblasts, pericytes, and Col13a1+ fibroblast, which were also among the most active signal senders and receivers in the hyperoxic lung. The study further revealed a substantial depletion of gCap (general capillary) cells and an increase in number of Car4+ aCap cells (aerocytes) after hyperoxia exposure [151]. The gCap cells were previously identified as putative distal lung vascular progenitors and regulators of capillary homeostasis, vasomotor tone, and repair [233]. The depletion of gCap ECs may contribute not only to the developmental injury, but also to the lack of repair capacity and an increased susceptibility to lung injury later in life, implying potential benefits of EC-derived cell therapies in BPD [151, 234]. In parallel, the Car4+ aCap cells showed pathological gene expression characterized by pro-inflammatory and anti-angiogenic markers. This is in agreement with recent reports, that aCap cells might contribute to septation [227] and revascularization following injury [235]. Moreover, the study highlighted the importance of inflammation in hyperoxia injury, with majority of the impacted transcriptional programs related to inflammatory response [151]. Finally, a recent study explored the long-term implication of neonatal hyperoxia [236]. Authors mapped lung cell populations in developing (P7) and adult (P60) mice previously exposed to hyperoxia for the first 3 days of life and identified persistent changes to AT2 subpopulations, predicting lasting perturbations to lung architecture and function [236].
While scRNA-seq studies so far have provided us with a somewhat complete map of the postnatally developing mouse lung, presently only a small portion of data derived from these studies have contributed to our understanding of BPD pathogenesis. As such, parallel studies in humans are still needed and further studies of BPD lungs are necessary to improve our interpretation of data obtained from animal studies. Finally, additional techniques, such as lineage tracing and spatial transcriptomics should be used to complement scRNA-seq to further investigate the role of newly identified cell populations, particularly rare cell subtypes and potential putative progenitors.
Lung stem/progenitor cells—opportunities to regenerate the preterm lung
Lung constitutes a quiescent organ, with the turnover time gradually decreasing along the proximal–distal axis [237, 238]. Following injury, lung cells are typically activated by their microenvironment and directed to participate in remodelling or repair [237, 239, 240]. The same injurious stimuli can damage or inhibit stem cells' ability to differentiate, leading to decreased, incorrect, or inappropriately timed production of particular cell populations, contributing to the development of lung disease [241]. The role of stem cells in the dysplastic pulmonary growth, premature lung aging, and pathogenesis of BPD has previously been proposed [241]. However, while multiple populations of lung endothelial, epithelial, and stromal stem or progenitor cells have been described, relatively little is known about their role in late lung development or BPD [237]. Particularly of interest are questions why resident stem cells lose their function and whether it is more feasible to restore their progenitor potential, or rather supplement the injured lung with undamaged, exogenous, therapeutic stem cells.
Epithelial stem cells are the most studied putative progenitors in the lung [242,243,244]. These include proximal airways basal cells, secretory cells, bronchial alveolar stem cells (BASC), and distal AT2 cells. The fate of the progenitor basal cells, characterized by the expression of luminal cytokeratin KRT8, seems to be largely guided by the NOTCH signaling. Low levels of NOTCH expression predispose basal cells toward the secretory phenotype, while high levels lead to differentiation into goblet cells, and the absence of NOTCH results in the ciliated phenotype [245,246,247,248,249]. Although the progenitor capacity of BASC cells has been demonstrated in mice, their exact role in postnatal lung growth and even their existence in human lungs still remain controversial [237, 244, 250]. On the other hand, the role of distal AT2 cells in lung repair is well-established and has been broadly studied. Lineage-tracing and scRNA-seq studies shown, that (alveolar type) AT1 and AT2 cells originate from a common bipotent progenitor. In humans, the AT1/AT2 progenitors were reported in developing lungs at gestational week 15 [230]. Studies in mice suggest that the bipotent AT1/AT2 population splits into independent cell lines by E18.5 [251, 252]. AT2 cells were repeatedly shown to self-renew, differentiate into AT1 cells, and exhibit repair capacities even in matured lungs [253,254,255,256]. In regard to BPD pathogenesis, increased compensatory AT2-to-AT1 trans-differentiation was shown in the developing hyperoxia-exposed rats [257]. Early postnatal hyperoxia-exposure in mice resulted in reduced AT2 proliferation which persisted for up to 2 months [167]. Contradicting observations were however made in premature ventilated baboons, where AT2 hyperproliferation was observed [164], perhaps indicating that the nature, intensity, and timing of the injurious stimulus are critical in determining the way progenitor populations respond. Indeed, the molecular mechanisms involved in the AT2 progenitor capacity are largely unknown. Among the proposed pathways are the WTN, EGFR, and KRAS signaling pathways [256, 258]. Further, reports of progenitor-like AT1 cells [259] and the AT1-to-AT2 trans-differentiation also exist [260], and a specific Hopx+ AT1 population was shown to generate new AT2 cells in an adult mice post-pneumonectomy [261]. Finally, the progenitor role of so-called respiratory airway secretory cells (RAS) was also recently revealed [262]. RAS, which are located in human, but not mice proximal airways, differentiate exclusively into AT2 cells, a process regulated by NOTCH and WNT signalling. While this study explored the potential role of RAS in adult lung disease, future studies are needed to reveal their role in the neonatal lung.
In comparison to epithelial cells, less is known in regard to lung resident endothelial and mesenchymal stem cells. The rational for the search for endothelial progenitor cells (EPCs) is based in the hypothesis that the lung development is driven by pulmonary vessel formation [56, 263]. Numerous studies support the existence of resident EPCs in the postnatally developing lungs and a defective lung vascularization can be found in both, BPD patients and animal models of BPD [170, 172, 176, 177]. Moreover, inhibition of vessel formation in developing animals stunts lung development and results in alveolar hypoplasia [172, 264,265,266]. Importantly, pro-angiogenic interventions proved effective in improving lung alveolarization in animal BPD models [172, 267,268,269]. Reduction in number of resident and circulating EPCs was observed in murine BPD model [174], and the hyperoxia exposure decreased proliferation in human fetal lung endothelial colony-forming cells (ECFCs) in vitro [234]. Importantly, intravenously administered human cord blood-derived ECFCs were effective in restoring lung function, alveolar and vascular growth, and colony-formic capacity of resident ECFCs in hyperoxia-exposed developing mice [234]. Finally, some efforts have recently been made to identify markers of resident lung EPCs. Among the promising proposed candidates are above-mentioned markers FOXF1 and c-KIT, which expression is decreased in the lungs of hyperoxia-exposed rodents and BPD patients alike [151, 183].
The best described and most attractive among the somatic stem cells are mesenchymal stromal cells (MSCs), which can be easily isolated from bone marrow (BM-MSCs) or umbilical cord (UC-MSCs). Several studies have demonstrated the therapeutic properties of exogenous MSCs in experimental BPD, where UC- and BM-MSCs restored lung architecture and function, and attenuated inflammation and PH in developing rodents [270,271,272,273,274]. This evidence prompted further interest in MSC-based cell therapies for BPD and selected approaches are currently in early phase clinical trials [275,276,277,278]. Besides the cell-based therapies, MSC research further encompasses the study of cell-derived products, mainly extracellular vesicles (EVs). EVs represent a heterogenous population with smaller EVs (30–100 nm) also being referred to as exosomes [279]. Multiple studies have shown their role in cell communication during both, organ homeostasis and disease [280]. Human UC-MSC-derived EVs were shown to improve alveolar and vascular development, as well as lung function and RVH in hyperoxia-exposed developing mice [281,282,283]. Similar results were further observed in studies employing EVs from amniotic fluid-derived [284] and Wharton’s Jelly-derived MSCs [285]. Studies indicate that EVs exert their mostly anti-inflammatory effects by promoting an immunosuppressive CCR2-associated myeloid cell phenotype [283]. Similarly, antenatal delivery of BM-MSCs-derived EVs benefited rats with endotoxin-induced chorioamnionitis, resulting in reduced cytokine levels and improved lung growth and mechanics [286]. Finally, UC-MSCs-derived EVs protected lung architecture, vessel formation and inflammatory modulation in LPS-injected and mechanically ventilated developing mice [287].
In addition to exogenous MSCs, the notion of the resident lung MSC (L-MSC) population come from reports of MSCs in TAs from prematurely born infants, where their presence was identified as indicator of BPD morbidity and severity [288,289,290]. Resident L-MSCs were also described in human fetal lungs (gestational week 15–17) [291] and hyperoxia-exposed developing rodents [292, 293]. Hyperoxia exposure increased the number of L-MSCs and triggered expression of pro-inflammatory, pro-fibrotic, and anti-angiogenic genes [151, 293]. A scRNA-seq cell communication analysis revealed inflammatory signals from immune populations as main drivers of hyperoxia-induced changes in L-MSCs [293]. Importantly, hyperoxia-exposed human fetal L-MSCs exhibited decreased colony-forming capacity [291], while L-MSCs isolated from hyperoxia-exposed animals had decreased ability to support angiogenesis [292]. Although minimal criteria for MSCs characterization have been officially established [294], the definition remains rather crude and the identification of organ-specific MSCs, including the L-MSCs, lacks standardization. As a result, no L-MSC-specific marker has been accepted to date, although few markers have been proposed [292, 293, 295,296,297]. Notable among these is LY6A, also known as SCA-1 (stem cell antigen 1) [295, 297]. Recent studies have shown that L-MSCs may constitute a rather heterogenous population and their study might require more advanced methods, such as scRNA-seq or sc-proteomics [292, 293]. Another recently emerging candidate resident MSC population are the Gli-1+ repair-supportive mesenchymal cells [298, 299]. Progenitor properties of Gli-1+ cells were previously described in other organs, including bones [300, 301], teeth [300], and liver [302]. In the lung, Gli-1+ cells co-express Acta2, Fgf10 and Pdgfra, thus resembling alveolar fibroblasts [225, 298]. In mice Gli-1+ MSCs were shown to aid epithelial regeneration following naphthalene-induced airway injury [298], and were shown to be increased in bleomycin-induced lung fibrosis in mice [303]. A more detailed characterization of all types of lung resident stem cell populations will clearly be of essence in understanding their role in normal and impaired lung development and regeneration.
Conclusions
Great advancements in the understanding of BPD pathophysiology have been made since its first description almost 60 years ago. However, prevention and treatment of this multifactorial disease still pose major challenges. Moreover, most treatment strategies, however lifesaving, can contribute to the disease pathogenesis. Because BPD occurs in a still developing lung, there is a notable risk of life-long adverse effects. Additionally, due to the scarcity of human material, most of our current knowledge derives from experimental animal, mostly rodent models. Aberrant immune response, activation of pro-fibrotic and anti-angiogenic factors, as well as defects in alveolar and capillary formation are among the main features of BPD pathogenesis. More recent studies suggest additional roles in the development of BPD for maternal obesity, second-hand smoking, and pollution. However, no single animal model can fully replicate the complex nature of BPD. Therefore, how exactly do the prenatal and postnatal factors interrelate during the development of BPD, effect patients' recovery, and possibly contribute to the susceptibility to pulmonary diseases in later life remains unknown. Finally, the heterogenous nature of pulmonary cellular landscape represents a great challenge when identifying individual effector cells, particularly rare progenitors. Future novel multi-omics and interdisciplinary approaches will allow for more in-depth identification of rare cell types, cellular dynamics, and novel biomarkers. This knowledge will further enable development of more personalized therapeutics relevant to disease prevention, as well as acute and long-term organ repair.
Availability of data and materials
Not applicable.
Abbreviations
- aCap:
-
Aerocytes capillary cell
- Ang-1:
-
Angiotensin 1
- AT1:
-
Alveolar type 1 cell
- AT2:
-
Alveolar type 2 cell
- BASCS:
-
Bronchial alveolar stem cells
- BM-MSC:
-
Bone marrow-derived mesenchymal stromal cell
- BPD:
-
Bronchopulmonary dysplasia
- Car4:
-
Carbonic anhydrase 4
- CD45:
-
PTPRC, protein tyrosine phosphatase receptor type C
- c-Kit:
-
KIT proto-oncogene, receptor tyrosine kinase
- Col1a1:
-
Collagen 1a1
- Col13a1:
-
Collagen 13a1
- COPD:
-
Chronic pulmonary obstructive disease
- Csf1r:
-
Colony stimulating factor 1 receptor
- E:
-
Embryonic day
- EC:
-
Endothelial cell
- ECM:
-
Extracellular matrix
- eNOS:
-
Endothelial Nitric oxide synthase 3
- EPC:
-
Endothelial progenitor cell
- EV:
-
Extracellular vesicle
- FGF:
-
Fibroblast growth factor
- FGF2:
-
Fibroblast growth factor 2
- FGF10:
-
Fibroblast growth factor 10
- FOXF1:
-
Forkhead box F1
- gCap:
-
General capillary cell
- GH:
-
Growth hormone
- HFD:
-
High fat diet
- Hopx:
-
HOP homeobox
- IGF-1:
-
Insulin-like growth factor 1
- IL-6:
-
Interleukin 6
- IL-8:
-
Interleukin 8
- IL-1β:
-
Interleukin 1 beta
- IUGR:
-
Intrauterine growth restriction
- KRT8:
-
Keratin 8
- LBW:
-
Low body weight
- LISA:
-
Less invasive surfactant administration
- L-MSC:
-
Lung mesenchymal stromal cell
- LPD:
-
Low protein dietactin
- LPS :
-
Lipopolysaccharide
- LY6A:
-
Lymphocyte antigen 6 complex, locus A
- MIST:
-
Minimally invasive surfactant therapy
- MSC:
-
Mesenchymal stromal cell
- MV:
-
Mechanical ventilation
- MVU:
-
Maternal vascular underperfusion
- NCPAP:
-
Nasal continuous positive airways pressure
- NICU:
-
Neonatal intensive care unit
- NOTCH:
-
NOTCH protein
- P:
-
Postnatal day
- PDA:
-
Patent ductus arteriosus
- PDGFA:
-
Plateled-derived growth factor alpha
- PDGFRA:
-
Plateled-derived growth factor receptor alpha
- PE:
-
Preeclampsia
- PH:
-
Pulmonary hypertension
- PVR:
-
Pulmonary vascular resistance
- RAS:
-
Respiratory airway secretory cells
- RVH:
-
Right vascular hypertrophy
- SCA1:
-
Stem cell antigen 1
- sc-proteomics:
-
Single cell proteomics
- scRNA-seq:
-
Single cell RNA sequencing
- sFlt-1:
-
Soluble fms-like tyrosine kinase 1
- SGA:
-
Small for gestational age
- SNIPPV:
-
Nasal intermittent positive pressure ventilation
- snRNA-seq:
-
Single nuclear RNA sequencing
- Stat5:
-
Signal transducer and activator of transcription 5A
- TA:
-
Tracheal aspirate
- TGF-β:
-
Transcription growth factor beta
- Tie-2:
-
TEK receptor tyrosine kinase
- TNF-α:
-
Tumor necrosis factor alpha
- UC-MSC:
-
Umbilical cord-derived mesenchymal stromal cell
- VEGF:
-
Vascular endothelial growth factor
References
Hilgendorff A, Oâ€TMReilly MA (2015) Bronchopulmonary dysplasia early changes leading to long-term consequences. Front Med. https://doi.org/10.3389/fmed.2015.00002
Thébaud B, Goss KN, Laughon M et al (2019) Bronchopulmonary dysplasia. Nat Rev Dis Primers 5:78
Warburton D (2017) Overview of lung development in the newborn human. Neonatology 111:398–401
Duijts L, van Meel ER, Moschino L et al (2020) European Respiratory Society guideline on long-term management of children with bronchopulmonary dysplasia. Eur Respir J 55:1900788
Jobe AH, Bancalari E (2001) Bronchopulmonary dysplasia. Am J Respir Crit Care Med 163:1723–1729
Sucre J, Haist L, Bolton CE, Hilgendorff A (2021) Early changes and indicators characterizing lung aging in neonatal chronic lung disease. Front Med 8:665152
Sahni M, Bhandari V (2020) Recent advances in understanding and management of bronchopulmonary dysplasia. F100Res. 9:703
Biniwale MA, Ehrenkranz RA (2006) The role of nutrition in the prevention and management of bronchopulmonary dysplasia. Semin Perinatol 30:200–208
Rozance PJ, Seedorf GJ, Brown A, Roe G, O’Meara MC, Gien J, Tang J-R, Abman SH (2011) Intrauterine growth restriction decreases pulmonary alveolar and vessel growth and causes pulmonary artery endothelial cell dysfunction in vitro in fetal sheep. Am J Physiology Lung Cell Mol Physiol 301:L860–L871
Collaborators of the Hypertensive Disorders of Pregnancy Study Group, Rocha G, de Lima FF, Machado AP, Guimarães H (2018) Preeclampsia predicts higher incidence of bronchopulmonary dysplasia. J Perinatol 38:1165–1173
Arigliani M, Stocco C, Valentini E et al (2021) Lung function between 8 and 15 years of age in very preterm infants with fetal growth restriction. Pediatr Res 90:657–663
Gilliland FD (2000) Maternal smoking during pregnancy, environmental tobacco smoke exposure and childhood lung function. Thorax 55:271–276
Isaksen CV, Austgulen R, Chedwick L, Romundstad P, Vatten L, Craven C (2004) Maternal Smoking, Intrauterine Growth Restriction, and Placental Apoptosis. Pediatr Dev Pathol 7:433–442
Bhandari V (2006) Familial and Genetic Susceptibility to Major Neonatal Morbidities in Preterm Twins. Pediatrics 117:1901–1906
Lal CV, Ambalavanan N (2015) Genetic predisposition to bronchopulmonary dysplasia. Semin Perinatol 39:584–591
Binet M-E, Bujold E, Lefebvre F, Tremblay Y, Piedboeuf B, for the Canadian Neonatal NetworkTM, (2012) Role of gender in morbidity and mortality of extremely premature neonates. Amer J Perinatol 29:159–166
Vrijlandt EJ, Gerritsen J, Boezen HM, Duiverman EJ, the Dutch POPS-19 Collaborative Study Group* (2005) Gender differences in respiratory symptoms in 19-year-old adults born preterm. Respir Res 6:117
Northway WH, Rosan RC, Porter DY (1967) Pulmonary disease following respirator therapy of hyaline-membrane disease: bronchopulmonary dysplasia. N Engl J Med 276:357–368
Cheong JLY, Doyle LW (2018) An update on pulmonary and neurodevelopmental outcomes of bronchopulmonary dysplasia. Semin Perinatol 42:478–484
Northway WH, Moss RB, Carlisle KB, Parker BR, Popp RL, Pitlick PT, Eichler I, Lamm RL, Brown BW (1990) Late pulmonary sequelae of bronchopulmonary dysplasia. N Engl J Med 323:1793–1799
Fawke J, Lum S, Kirkby J, Hennessy E, Marlow N, Rowell V, Thomas S, Stocks J (2010) Lung function and respiratory symptoms at 11 years in children born extremely preterm: The EPICure Study. Am J Respir Crit Care Med 182:237–245
Goss KN, Beshish AG, Barton GP et al (2018) Early pulmonary vascular disease in young adults born preterm. Am J Respir Crit Care Med 198:1549–1558
Lewandowski AJ, Bradlow WM, Augustine D, Davis EF, Francis J, Singhal A, Lucas A, Neubauer S, McCormick K, Leeson P (2013) Right ventricular systolic dysfunction in young adults born preterm. Circulation 128:713–720
Wong PM, Lees AN, Louw J, Lee FY, French N, Gain K, Murray CP, Wilson A, Chambers DC (2008) Emphysema in young adult survivors of moderate-to-severe bronchopulmonary dysplasia. Eur Respir J 32:321–328
Gordijn SJ, Beune IM, Ganzevoort W (2018) Building consensus and standards in fetal growth restriction studies. Best Pract Res Clin Obstet Gynaecol 49:117–126
Rosenberg A (2008) The IUGR Newborn. Semin Perinatol 32:219–224
Briana DD, Malamitsi-Puchner A (2020) Perinatal biomarkers implying ‘Developmental Origins of Health and Disease’ consequences in intrauterine growth restriction. Acta Paediatr 109:1317–1322
Kuiper-Makris C, Selle J, Nüsken E, Dötsch J, Alejandre Alcazar MA (2021) Perinatal nutritional and metabolic pathways: early origins of chronic lung diseases. Front Med 8:667315
Brodsky D, Christou H (2004) Current concepts in intrauterine growth restriction. J Intensive Care Med 19:307–319
Garite TJ, Clark R, Thorp JA (2004) Intrauterine growth restriction increases morbidity and mortality among premature neonates. Am J Obstet Gynecol 191:481–487
Miller TA, Dodson RB, Mankouski A, Powers KN, Yang Y, Yu B, Zinkhan EK (2019) Impact of diet on the persistence of early vascular remodeling and stiffening induced by intrauterine growth restriction and a maternal high-fat diet. Am J Physiol Heart Circ Physiol 317:H424–H433
Zana-Taieb E, Butruille L, Franco-Montoya M-L et al (2013) Effect of two models of intrauterine growth restriction on alveolarization in rat lungs: morphometric and gene expression analysis. PLoS ONE 8:e78326
Dravet-Gounot P, Morin C, Jacques S, Dumont F, Ely-Marius F, Vaiman D, Jarreau P-H, Méhats C, Zana-Taïeb E (2017) Lung microRNA deregulation associated with impaired alveolarization in rats after intrauterine growth restriction. PLoS ONE 12:e0190445
Nawabi J, Vohlen C, Dinger K et al (2018) Novel functional role of GH/IGF-I in neonatal lung myofibroblasts and in rat lung growth after intrauterine growth restriction. Am J Physiol Lung Cell Mol Physiol 315:L623–L637
Kuiper-Makris C, Zanetti D, Vohlen C, Fahle L, Müller M, Odenthal M, Felderhoff-Müser U, Dötsch J, Alejandre Alcazar MA (2020) Mendelian randomization and experimental IUGR reveal the adverse effect of low birth weight on lung structure and function. Sci Rep 10:22395
Maritz GS, Cock ML, Louey S, Joyce BJ, Albuquerque CA, Harding R (2001) Effects of fetal growth restriction on lung development before and after birth: a morphometric analysis. Pediatr Pulmonol 32:201–210
Lipsett J, Tamblyn M, Madigan K, Roberts P, Cool JC, Runciman SI, McMillen IC, Robinson J, Owens JA (2006) Restricted fetal growth and lung development: A morphometric analysis of pulmonary structure. Pediatr Pulmonol 41:1138–1145
Löfqvist C, Hellgren G, Niklasson A, Engström E, Ley D, Hansen-Pupp I, and the WINROP Consortium (2012) Low postnatal serum IGF-I levels are associated with bronchopulmonary dysplasia (BPD). Acta Paediatr 101:1211–1216
Seedorf G, Kim C, Wallace B, Mandell EW, Nowlin T, Shepherd D, Abman SH (2020) rhIGF-1/BP3 Preserves lung growth and prevents pulmonary hypertension in experimental bronchopulmonary dysplasia. Am J Respir Crit Care Med 201:1120–1134
Puzik A, Rupp J, Tröger B, Göpel W, Herting E, Härtel C (2012) Insulin-like growth factor-I regulates the neonatal immune response in infection and maturation by suppression of IFN-γ. Cytokine 60:369–376
Hellstrom A, Perruzzi C, Ju M et al (2001) Low IGF-I suppresses VEGF-survival signaling in retinal endothelial cells: direct correlation with clinical retinopathy of prematurity. Proc Natl Acad Sci 98:5804–5808
Khazaee R, McCaig LA, Yamashita C, Hardy DB, Veldhuizen RAW (2019) Maternal protein restriction during perinatal life affects lung mechanics and the surfactant system during early postnatal life in female rats. PLoS ONE 14:e0215611
Alejandre Alcazar MA, Östreicher I, Appel S, Rother E, Vohlen C, Plank C, Dötsch J (2012) Developmental regulation of inflammatory cytokine-mediated Stat3 signaling: the missing link between intrauterine growth restriction and pulmonary dysfunction? J Mol Med 90:945–957
Kotecha SJ, Watkins WJ, Heron J, Henderson J, Dunstan FD, Kotecha S (2010) Spirometric Lung Function in School-Age Children: Effect of Intrauterine Growth Retardation and Catch-up Growth. Am J Respir Crit Care Med 181:969–974
Ronkainen E, Dunder T, Kaukola T, Marttila R, Hallman M (2016) Intrauterine growth restriction predicts lower lung function at school age in children born very preterm. Arch Dis Child Fetal Neonatal Ed 101:F412–F417
Cai Y, Shaheen SO, Hardy R, Kuh D, Hansell AL (2016) Birth weight, early childhood growth and lung function in middle to early old age: 1946 British birth cohort. Thorax 71:916–922
Harris C, Lunt A, Bisquera A, Peacock J, Greenough A (2021) Intrauterine growth retardation and lung function of very prematurely born young people. Pediatr Pulmonol 56:2284–2291
Canoy D, Pekkanen J, Elliott P, Pouta A, Laitinen J, Hartikainen A-L, Zitting P, Patel S, Little MP, Jarvelin M-R (2007) Early growth and adult respiratory function in men and women followed from the fetal period to adulthood. Thorax 62:396–402
Svanes C, Omenaas E, Heuch JM, Irgens LM, Gulsvik A (1998) Birth characteristics and asthma symptoms in young adults: results from a population-based cohort study in Norway. Eur Respir J 12:1366–1370
Barker DJ, Godfrey KM, Fall C, Osmond C, Winter PD, Shaheen SO (1991) Relation of birth weight and childhood respiratory infection to adult lung function and death from chronic obstructive airways disease. BMJ 303:671–675
Stocks J, Sonnappa S (2013) Early life influences on the development of chronic obstructive pulmonary disease. Ther Adv Respir Dis 7:161–173
Hansen AR, Barnés CM, Folkman J, McElrath TF (2010) Maternal preeclampsia predicts the development of bronchopulmonary dysplasia. J Pediatr 156:532–536
Mestan KK, Check J, Minturn L, Yallapragada S, Farrow KN, Liu X, Su E, Porta N, Gotteiner N, Ernst LM (2014) Placental pathologic changes of maternal vascular underperfusion in bronchopulmonary dysplasia and pulmonary hypertension. Placenta 35:570–574
Pierro M, Villamor-Martinez E, van Westering-Kroon E, Alvarez-Fuente M, Abman SH, Villamor E (2021) Association of the dysfunctional placentation endotype of prematurity with bronchopulmonary dysplasia: a systematic review, meta-analysis and meta-regression. Thorax Thoraxjnl. 2020:216485
Mestan KK, Gotteiner N, Porta N, Grobman W, Su EJ, Ernst LM (2017) Cord blood biomarkers of placental maternal vascular underperfusion predict bronchopulmonary dysplasia-associated pulmonary hypertension. J Pediatr 185:33–41
Abman SH (2001) Bronchopulmonary dysplasia: “a vascular hypothesis.” Am J Respir Crit Care Med 164:1755–1756
Gatford KL, Andraweera PH, Roberts CT, Care AS (2020) Animal models of preeclampsia: causes, consequences, and interventions. Hypertension 75:1363–1381
Davisson RL, Hoffmann DS, Butz GM, Aldape G, Schlager G, Merrill DC, Sethi S, Weiss RM, Bates JN (2002) Discovery of a spontaneous genetic mouse model of preeclampsia. Hypertension 39:337–342
Zhou J, Xiao D, Hu Y, Wang Z, Paradis A, Mata-Greenwood E, Zhang L (2013) Gestational hypoxia induces preeclampsia-like symptoms via heightened endothelin-1 signaling in pregnant rats. Hypertension 62:599–607
Tong W, Allison BJ, Brain KL et al (2022) Chronic hypoxia in ovine pregnancy recapitulates physiological and molecular markers of preeclampsia in the mother, placenta, and offspring. Hypertension 79:1525–1535
Yallampalli C, Garfield RE (1993) Inhibition of nitric oxide synthesis in rats during pregnancy produces signs similar to those of preeclampsia. Am J Obstet Gynecol 169:1316–1320
Witlin AG, Gangula PRR, Thompson ML, Yallampalli C (2002) Growth and fertility rates in the offspring of pregnant rats treated with L-ω nitro-L-arginine methyl ester (L-NAME), a nitric oxide inhibitor. Am J Obstet Gynecol 186:89–93
Li J, LaMarca B, Reckelhoff JF (2012) A model of preeclampsia in rats: the reduced uterine perfusion pressure (RUPP) model. Am J Physiol Heart Circ Physiol 303:H1–H8
Schenone MH, Mari G, Schlabritz-Loutsevitch N, Ahokas R (2015) Effects of selective reduced uterine perfusion pressure in pregnant rats. Placenta 36:1450–1454
Ahmed A, Singh J, Khan Y, Seshan SV, Girardi G (2010) A new mouse model to explore therapies for preeclampsia. PLoS ONE 5:e13663
Wallace B, Peisl A, Seedorf G, Nowlin T, Kim C, Bosco J, Kenniston J, Keefe D, Abman SH (2018) Anti–sFlt-1 therapy preserves lung alveolar and vascular growth in antenatal models of bronchopulmonary dysplasia. Am J Respir Crit Care Med 197:776–787
Longo S, Bollani L, Decembrino L, Di Comite A, Angelini M, Stronati M (2013) Short-term and long-term sequelae in intrauterine growth retardation (IUGR). J Matern Fetal Neonatal Med 26:222–225
Ravelli G-P, Stein ZA, Susser MW (1976) Obesity in young men after famine exposure in utero and early infancy. N Engl J Med 295:349–353
Wallace JM, Bourke DA, Aitken RP, Palmer RM, Da Silva P, Cruickshank MA (2000) Relationship between nutritionally-mediated placental growth restriction and fetal growth, body composition and endocrine status during late gestation in adolescent sheep. Placenta 21:100–108
Dinger K, Koningsbruggen-Rietschel S, v., Dötsch J, Alejandre Alcazar MA, (2020) Identification of critical windows of metabolic programming of metabolism and lung function in male offspring of obese dams. Clin Transl Sci 13:1065–1070
Hamrick SEG, Hansmann G (2010) Patent ductus arteriosus of the preterm infant. Pediatrics 125:1020–1030
Dani C, Pratesi S (2020) Patent ductus arteriosus and oxidative stress in preterm infants: a narrative review. Transl Pediatr 9:835–839
Rojas MA, Gonzalez A, Bancalari E, Claure N, Poole C, Silva-Neto G (1995) Changing trends in the epidemiology and pathogenesis of neonatal chronic lung disease. J Pediatr 126:605–610
Schena F, Francescato G, Cappelleri A, Picciolli I, Mayer A, Mosca F, Fumagalli M (2015) Association between hemodynamically significant patent ductus arteriosus and bronchopulmonary dysplasia. J Pediatr 166:1488–1492
Gentle SJ, Travers CP, Clark M, Carlo WA, Ambalavanan N (2022) Patent ductus arteriosus and development of bronchopulmonary dysplasia with pulmonary hypertension. Am J Respir Crit Care Med 207(7):921-928
Marshall DD, Kotelchuck M, Young TE, Bose CL, Kruyer L, O’Shea TM, the North Carolina Neonatologists Association (1999) Risk factors for chronic lung disease in the surfactant era: a north carolina population-based study of very low birth weight infants. Pediatrics 104:1345–1350
Brown ER (1979) Increased risk of bronchopulmonary dysplasia in infants with patent ductus arteriosus. J Pediatr 95:865–866
Clyman RI (2018) Patent ductus arteriosus, its treatments, and the risks of pulmonary morbidity. Semin Perinatol 42:235–242
Hennelly M, Greenberg RG, Aleem S (2021) An update on the prevention and management of bronchopulmonary dysplasia. PHMT 12:405–419
Fowlie PW, Davis PG, McGuire W (2010) Prophylactic intravenous indomethacin for preventing mortality and morbidity in preterm infants. Cochrane Database Syst Rev. https://doi.org/10.1002/14651858.CD000174.pub2
Kluckow M, Jeffery M, Gill A, Evans N (2014) A randomised placebo-controlled trial of early treatment of the patent ductus arteriosus. Arch Dis Child Fetal Neonatal Ed 99:F99–F104
Ohlsson A, Shah SS (2020) Ibuprofen for the prevention of patent ductus arteriosus in preterm and/or low birth weight infants. Cochrane Database Syst Rev. https://doi.org/10.1002/14651858.CD004213.pub5
Ohlsson A, Walia R, Shah SS (2018) Ibuprofen for the treatment of patent ductus arteriosus in preterm or low birth weight (or both) infants. Cochrane Database Syst Rev. https://doi.org/10.1002/14651858.CD003481.pub7
Cooke L, Steer PA, Woodgate PG (2003) Indomethacin for asymptomatic patent ductus arteriosus in preterm infants. Cochrane Database Syst Rev. https://doi.org/10.1002/14651858.CD003745
Liebowitz M, Koo J, Wickremasinghe A, Allen IE, Clyman RI (2017) Effects of prophylactic indomethacin on vasopressor-dependent hypotension in extremely preterm infants. J Pediatr 182:21-27.e2
McCurnin D, Seidner S, Chang L-Y et al (2008) Ibuprofen-induced patent ductus arteriosus closure: physiologic, histologic, and biochemical effects on the premature lung. Pediatrics 121:945–956
Chang LY, McCurnin D, Yoder B, Shaul PW, Clyman RI (2008) Ductus arteriosus ligation and alveolar growth in preterm baboons with a patent ductus arteriosus. Pediatr Res 63:299–302
Waleh N, McCurnin DC, Yoder BA, Shaul PW, Clyman RI (2011) Patent ductus arteriosus ligation alters pulmonary gene expression in preterm baboons. Pediatr Res 69:212–216
Liebowitz M, Katheria A, Sauberan J et al (2019) Lack of equipoise in the PDA-TOLERATE trial: a comparison of eligible infants enrolled in the trial and those treated outside the trial. J Pediatr 213:222-226.e2
Clyman RI, Liebowitz M, Kaempf J et al (2019) PDA-TOLERATE trial: an exploratory randomized controlled trial of treatment of moderate-to-large patent ductus arteriosus at 1 week of age. J Pediatr 205:41-48.e6
Benitz WE, Committee on Fetus and Newborn, Watterberg KL et al (2016) Patent ductus arteriosus in preterm infants. Pediatrics 137:e20153730
Bose CL, Laughon M (2006) Treatment to prevent patency of the ductus arteriosus: beneficial or harmful? J Pediatr 148:713–714
Lakshminrusimha S, Steinhorn RH (1999) Pulmonary vascular biology during neonatal transition. Clin Perinatol 26:601–619
Lakshminrusimha S (2012) The pulmonary circulation in neonatal respiratory failure. Clin Perinatol 39:655–683
Lakshminrusimha S (2021) Neonatal and postneonatal pulmonary hypertension. Children 8:131
Bhat R, Salas AA, Foster C, Carlo WA, Ambalavanan N (2012) Prospective analysis of pulmonary hypertension in extremely low birth weight infants. Pediatrics 129:e682-689
Kim D-H, Kim H-S, Choi CW, Kim E-K, Kim BI, Choi J-H (2012) Risk factors for pulmonary artery hypertension in preterm infants with moderate or severe bronchopulmonary dysplasia. Neonatology 101:40–46
Cerro MJ, Abman S, Diaz G et al (2011) A consensus approach to the classification of pediatric pulmonary hypertensive vascular disease: report from the PVRI Pediatric Taskforce, Panama 2011. Pulm circ 1:286–298
Hansmann G, Sallmon H, Roehr CC, Kourembanas S, Austin ED, Koestenberger M, for the European Pediatric Pulmonary Vascular Disease Network (EPPVDN) (2021) Pulmonary hypertension in bronchopulmonary dysplasia. Pediatr Res 89:446–455
Mourani PM, Sontag MK, Younoszai A, Miller JI, Kinsella JP, Baker CD, Poindexter BB, Ingram DA, Abman SH (2015) Early pulmonary vascular disease in preterm infants at risk for bronchopulmonary dysplasia. Am J Respir Crit Care Med 191:87–95
Weismann CG, Asnes JD, Bazzy-Asaad A, Tolomeo C, Ehrenkranz RA, Bizzarro MJ (2017) Pulmonary hypertension in preterm infants: results of a prospective screening program. J Perinatol 37:572–577
Khemani E, McElhinney DB, Rhein L, Andrade O, Lacro RV, Thomas KC, Mullen MP (2007) Pulmonary artery hypertension in formerly premature infants with bronchopulmonary dysplasia: clinical features and outcomes in the surfactant era. Pediatrics 120:1260–1269
Pammi M, Lal CV, Wagner BD et al (2019) Airway microbiome and development of bronchopulmonary dysplasia in preterm infants: a systematic review. J Pediatr 204:126-133.e2
Wagner BD, Sontag MK, Harris JK, Miller JI, Morrow L, Robertson CE, Stephens M, Poindexter BB, Abman SH, Mourani PM (2017) Airway microbial community turnover differs by BPD severity in ventilated preterm infants. PLoS ONE 12:e0170120
Lal CV, Travers C, Aghai ZH et al (2016) The airway microbiome at birth. Sci Rep 6:31023
Payne MS, Goss KCW, Connett GJ, Kollamparambil T, Legg JP, Thwaites R, Ashton M, Puddy V, Peacock JL, Bruce KD (2010) Molecular microbiological characterization of preterm neonates at risk of bronchopulmonary dysplasia. Pediatr Res 67:412–418
Lohmann P, Luna RA, Hollister EB, Devaraj S, Mistretta T-A, Welty SE, Versalovic J (2014) The airway microbiome of intubated premature infants: characteristics and changes that predict the development of bronchopulmonary dysplasia. Pediatr Res 76:294–301
Cantey JB, Huffman LW, Subramanian A, Marshall AS, Ballard AR, Lefevre C, Sagar M, Pruszynski JE, Mallett LH (2017) Antibiotic exposure and risk for death or bronchopulmonary dysplasia in very low birth weight infants. J Pediatr 181:289-293.e1
Novitsky A, Tuttle D, Locke R, Saiman L, Mackley A, Paul D (2014) Prolonged early antibiotic use and bronchopulmonary dysplasia in very low birth weight infants. Amer J Perinatol 32:043–048
Gentle SJ, Lal CV (2020) Predicting BPD: Lessons Learned From the Airway Microbiome of Preterm Infants. Front Pediatr 7:564
Lal CV, Kandasamy J, Dolma K et al (2018) Early airway microbial metagenomic and metabolomic signatures are associated with development of severe bronchopulmonary dysplasia. Am J Physiol Lung Cell Mol Physiol 315:L810–L815
Balany J, Bhandari V (2015) Understanding the impact of infection, inflammation, and their persistence in the pathogenesis of bronchopulmonary dysplasia. Front Med. https://doi.org/10.3389/fmed.2015.00090
Hofer N, Kothari R, Morris N, Müller W, Resch B (2013) The fetal inflammatory response syndrome is a risk factor for morbidity in preterm neonates. Am J Obstet Gynecol 209:542.e1-542.e11
Zanardo V, Savio V, Giacomin C, Rinaldi A, Marzari F, Chiarelli S (2002) Relationship between neonatal leukemoid reaction and bronchopulmonary dysplasia in low-birth-weight infants: a cross-sectional study. Am J Perinatol 19:379–386
Menon R, Taylor RN, Fortunato SJ (2010) Chorioamnionitis – a complex pathophysiologic syndrome. Placenta 31:113–120
Smulian J (1999) Clinical chorioamnionitis and histologic placental inflammation. Obstet Gynecol 94:1000–1005
Kunzmann S, Collins JJP, Kuypers E, Kramer BW (2013) Thrown off balance: the effect of antenatal inflammation on the developing lung and immune system. Am J Obstet Gynecol 208:429–437
Galinsky R, Hooper SB, Wallace MJ, Westover AJ, Black MJ, Moss TJM, Polglase GR (2013) Intrauterine inflammation alters cardiopulmonary and cerebral haemodynamics at birth in preterm lambs: Intrauterine inflammation and neonatal haemodynamics. J Physiol 591:2127–2137
Moss TJM, Newnham JP, Willett KE, Kramer BW, Jobe AH, Ikegami M (2002) Early gestational intra-amniotic endotoxin: lung function, surfactant, and morphometry. Am J Respir Crit Care Med 165:805–811
Shrestha AK, Bettini ML, Menon RT, Gopal VYN, Huang S, Edwards DP, Pammi M, Barrios R, Shivanna B (2019) Consequences of early postnatal lipopolysaccharide exposure on developing lungs in mice. Am J Physiol Lung Cell Mol Physiol 316:L229–L244
Pan J, Zhan C, Yuan T, Wang W, Shen Y, Sun Y, Wu T, Gu W, Chen L, Yu H (2018) Effects and molecular mechanisms of intrauterine infection/inflammation on lung development. Respir Res 19:93
Yoon BH, Romero R, Jun JK, Park KH, Park JD, Ghezzi F, Kim BI (1997) Amniotic fluid cytokines (interleukin-6, tumor necrosis factor-α, interleukin-1β, and interleukin-8) and the risk for the development of bronchopulmonary dysplasia. Am J Obstet Gynecol 177:825–830
Plakkal N, Soraisham AS, Trevenen C, Freiheit EA, Sauve R (2013) Histological chorioamnionitis and bronchopulmonary dysplasia: a retrospective cohort study. J Perinatol 33:441–445
Van Marter LJ, Dammann O, Allred EN, Leviton A, Pagano M, Moore M, Martin C (2002) Chorioamnionitis, mechanical ventilation, and postnatal sepsis as modulators of chronic lung disease in preterm infants. J Pediatr 140:171–176
Hartling L, Liang Y, Lacaze-Masmonteil T (2012) Chorioamnionitis as a risk factor for bronchopulmonary dysplasia: a systematic review and meta-analysis. Arch Dis Child Fetal Neonatal Ed 97:F8–F17
Kalikkot Thekkeveedu R, Guaman MC, Shivanna B (2017) Bronchopulmonary dysplasia: A review of pathogenesis and pathophysiology. Respir Med 132:170–177
Thomas W, Speer CP (2014) Chorioamnionitis is essential in the evolution of bronchopulmonary dysplasia – The case in favour. Paediatr Respir Rev 15:49–52
Lacaze-Masmonteil T (2014) That chorioamnionitis is a risk factor for bronchopulmonary dysplasia – the case against. Paediatr Respir Rev 15:53–55
Jensen EA, Schmidt B (2014) Epidemiology of bronchopulmonary dysplasia. Birth Defects Res A Clin Mol Teratol 100:145–157
Albertine KH, Jones GP, Starcher BC, Bohnsack JF, Davis PL, Cho S-C, Carlton DP, Bland RD (1999) Chronic lung injury in preterm lambs: disordered respiratory tract development. Am J Respir Crit Care Med 159:945–958
Speer CP (2009) Chorioamnionitis, postnatal factors and proinflammatory response in the pathogenetic sequence of bronchopulmonary dysplasia. Neonatology 95:353–361
Papagianis PC, Pillow JJ, Moss TJ (2019) Bronchopulmonary dysplasia: Pathophysiology and potential anti-inflammatory therapies. Paediatr Respir Rev 30:34–41
Londhe VA, Maisonet TM, Lopez B, Jeng J-M, Xiao J, Li C, Minoo P (2011) Conditional deletion of epithelial IKKβ impairs alveolar formation through apoptosis and decreased VEGF expression during early mouse lung morphogenesis. Respir Res 12:134
Köksal N, Kayık B, Çetinkaya M, Özkan H, Budak F, Kılıç Ş, Canıtez Y, Oral B (2012) Value of serum and bronchoalveolar fluid lavage pro- and anti-inflammatory cytokine levels for predicting bronchopulmonary dysplasia in premature infants. Eur Cytokine Netw 23:29–35
Kroon AA, Wang J, Huang Z, Cao L, Kuliszewski M, Post M (2010) Inflammatory response to oxygen and endotoxin in newborn rat lung ventilated with low tidal volume. Pediatr Res 68:63–69
Coalson JJ, Winter VT, Siler-Khodr T, Yoder BA (1999) Neonatal chronic lung disease in extremely immature baboons. Am J Respir Crit Care Med 160:1333–1346
Hillman NH, Polglase GR, Jane Pillow J, Saito M, Kallapur SG, Jobe AH (2011) Inflammation and lung maturation from stretch injury in preterm fetal sheep. Am J Physiol Lung Cell Mol Physiol 300:L232–L241
Kalymbetova TV, Selvakumar B, Rodríguez-Castillo JA et al (2018) Resident alveolar macrophages are master regulators of arrested alveolarization in experimental bronchopulmonary dysplasia: Macrophages mediate aberrant lung alveolarization. J Pathol 245:153–159
Bonikos DS, Bensch KG, Ludwin SK, Northway WH (1975) Oxygen toxicity in the newborn. The effect of prolonged 100 per cent O2 exposure on the lungs of newborn mice. Lab Invest 32:619–635
Bhandari V, Elias JA (2006) Cytokines in tolerance to hyperoxia-induced injury in the developing and adult lung. Free Radical Biol Med 41:4–18
Liao J, Kapadia VS, Brown LS, Cheong N, Longoria C, Mija D, Ramgopal M, Mirpuri J, McCurnin DC, Savani RC (2015) The NLRP3 inflammasome is critically involved in the development of bronchopulmonary dysplasia. Nat Commun 6:8977
Bry K, Whitsett JA, Lappalainen U (2007) IL-1β disrupts postnatal lung morphogenesis in the mouse. Am J Respir Cell Mol Biol 36:32–42
Nadeau-Vallée M, Chin P-Y, Belarbi L et al (2017) Antenatal suppression of IL-1 protects against inflammation-induced fetal injury and improves neonatal and developmental outcomes in mice. JI. 198:2047–2062
Johnston CJ, Wright TW, Reed CK, Finkelstein JN (1997) Comparison of adult and newborn pulmonary cytokine mrna expression after hyperoxia. Exp Lung Res 23:537–552
Wagenaar GTM, ter Horst SAJ, van Gastelen MA, Leijser LM, Mauad T, van der Velden PA, de Heer E, Hiemstra PS, Poorthuis BJHM, Walther FJ (2004) Gene expression profile and histopathology of experimental bronchopulmonary dysplasia induced by prolonged oxidative stress. Free Radical Biol Med 36:782–801
Wu S, Capasso L, Lessa A, Peng J, Kasisomayajula K, Rodriguez M, Suguihara C, Bancalari E (2008) High tidal volume ventilation activates Smad2 and upregulates expression of connective tissue growth factor in newborn rat lung. Pediatr Res 63:245–250
Mižíková I, Alejandre Alcazar MA, Thébaud B (2021) Pathogenesis of bronchopulmonary dysplasia. In: Sinha IP, Bhatt JM, Cleator A, Wallace H (eds) Respiratory diseases of the newborn infant. European Respiratory Society, Sheffield, United Kingdom, pp 50–67
Choi CW, Kim BI, Hong J-S, Kim E-K, Kim H-S, Choi J-H (2009) Bronchopulmonary dysplasia in a rat model induced by intra-amniotic inflammation and postnatal hyperoxia: morphometric aspects. Pediatr Res 65:323–327
Nold MF, Mangan NE, Rudloff I et al (2013) Interleukin-1 receptor antagonist prevents murine bronchopulmonary dysplasia induced by perinatal inflammation and hyperoxia. Proc Natl Acad Sci USA 110:14384–14389
Tang J-R, Seedorf GJ, Muehlethaler V, Walker DL, Markham NE, Balasubramaniam V, Abman SH (2010) Moderate postnatal hyperoxia accelerates lung growth and attenuates pulmonary hypertension in infant rats after exposure to intra-amniotic endotoxin. Am J Physiol Lung Cell Mol Physiol 299:L735–L748
Hurskainen M, Mižíková I, Cook DP et al (2021) Single cell transcriptomic analysis of murine lung development on hyperoxia-induced damage. Nat Commun 12:1565
Shrestha AK, Menon RT, El-Saie A, Barrios R, Reynolds C, Shivanna B (2020) Interactive and independent effects of early lipopolysaccharide and hyperoxia exposure on developing murine lungs. Am J Physiol Lung Cell Mol Physiol 319:L981–L996
El Saie A, Fu C, Grimm SL et al (2022) Metabolome and microbiome multi-omics integration from a murine lung inflammation model of bronchopulmonary dysplasia. Pediatr Res 92:1580–1589
Bland RD, Ertsey R, Mokres LM, Xu L, Jacobson BE, Jiang S, Alvira CM, Rabinovitch M, Shinwell ES, Dixit A (2008) Mechanical ventilation uncouples synthesis and assembly of elastin and increases apoptosis in lungs of newborn mice.: Prelude to defective alveolar septation during lung development? Am J Physiol Lung Cell Mol Physiol 294:L3–L14
Nardiello C, Mižíková I, Silva DM, Ruiz-Camp J, Mayer K, Vadász I, Herold S, Seeger W, Morty RE (2017) Standardisation of oxygen exposure in the development of mouse models for bronchopulmonary dysplasia? Dis Models Mech 10(2):185-196
Kroon AA, Wang J, Kavanagh B, Huang Z, Kuliszewski M, van Goudoever JB, Post M (2011) Prolonged mechanical ventilation induces cell cycle arrest in newborn rat lung. PLoS ONE 6:e16910
O’Reilly M, Möbius MA, Vadivel A, Ionescu L, Fung M, Eaton F, Greer JJ, Thébaud B (2020) Late rescue therapy with cord-derived mesenchymal stromal cells for established lung injury in experimental bronchopulmonary dysplasia. Stem Cells Dev 29:364–371
May M, Strobel P, Preisshofen T, Seidenspinner S, Marx A, Speer CP (2004) Apoptosis and proliferation in lungs of ventilated and oxygen-treated preterm infants. Eur Respir J 23:113–121
Hargitai B, Szabó V, Hajdú J, Harmath Á, Pataki M, Farid P, Papp Z, Szende B (2001) Apoptosis in various organs of preterm infants: histopathologic study of lung, kidney, liver, and brain of ventilated infants. Pediatr Res 50:110–114
Chambers HM, van Velzen D (1989) Ventilator-related pathology in the extremely immature lung. Pathology 21:79–83
Hillman NH, Kallapur SG, Pillow JJ, Moss TJM, Polglase GR, Nitsos I, Jobe AH (2010) Airway injury from initiating ventilation in preterm sheep. Pediatr Res 67:60–65
Polglase GR, Hillman NH, Pillow JJ, Cheah F-C, Nitsos I, Moss TJM, Kramer BW, Ikegami M, Kallapur SG, Jobe AH (2008) Positive end-expiratory pressure and tidal volume during initial ventilation of preterm lambs. Pediatr Res 64:517–522
Coalson JJ, Kuehl TJ, Escobedo MB, Leonard Hilliard J, Smith F, Meredith K, Null DM, Walsh W, Johnson D, Robotham JL (1982) A baboon model of bronchopulmonary dysplasia. Exp Mol Pathol 37:335–350
Maniscalco WM, Watkins RH, O’Reilly MA, Shea CP (2002) Increased epithelial cell proliferation in very premature baboons with chronic lung disease. Am J Physiol Lung Cell Mol Physiol 283:L991–L1001
Yee M, Buczynski BW, O’Reilly MA (2014) Neonatal hyperoxia stimulates the expansion of alveolar epithelial type II cells. Am J Respir Cell Mol Biol 50:757–766
Yee M, Domm W, Gelein R, de Bentley KL, M, Kottmann RM, Sime PJ, Lawrence BP, O’Reilly MA, (2017) Alternative progenitor lineages regenerate the adult lung depleted of alveolar epithelial type 2 cells. Am J Respir Cell Mol Biol 56:453–464
Yee M, Vitiello PF, Roper JM, Staversky RJ, Wright TW, McGrath-Morrow SA, Maniscalco WM, Finkelstein JN, O’Reilly MA (2006) Type II epithelial cells are critical target for hyperoxia-mediated impairment of postnatal lung development. Am J Physiol Lung Cell Mol Physiol 291:L1101–L1111
Xu S, Xue X, You K, Fu J (2016) Caveolin-1 regulates the expression of tight junction proteins during hyperoxia-induced pulmonary epithelial barrier breakdown. Respir Res 17:50
Davidovich N, DiPaolo BC, Lawrence GG, Chhour P, Yehya N, Margulies SS (2013) Cyclic stretch–induced oxidative stress increases pulmonary alveolar epithelial permeability. Am J Respir Cell Mol Biol 49:156–164
De Paepe ME, Mao Q, Powell J, Rubin SE, DeKoninck P, Appel N, Dixon M, Gundogan F (2006) Growth of pulmonary microvasculature in ventilated preterm infants. Am J Respir Crit Care Med 173:204–211
Tomashefski JF, Oppermann HC, Vawter GF, Reid LM (1984) Bronchopulmonary dysplasia: a morphometry study with emphasis on the pulmonary vasculature. Pediatr Pathol 2:469–487
Thébaud B, Ladha F, Michelakis ED et al (2005) Vascular endothelial growth factor gene therapy increases survival, promotes lung angiogenesis, and prevents alveolar damage in hyperoxia-induced lung injury: evidence that angiogenesis participates in alveolarization. Circulation 112:2477–2486
Mokres LM, Parai K, Hilgendorff A, Ertsey R, Alvira CM, Rabinovitch M, Bland RD (2010) Prolonged mechanical ventilation with air induces apoptosis and causes failure of alveolar septation and angiogenesis in lungs of newborn mice. Am J Physiol Lung Cell Mol Physiol 298:L23–L35
Balasubramaniam V, Mervis CF, Maxey AM, Markham NE, Abman SH (2007) Hyperoxia reduces bone marrow, circulating, and lung endothelial progenitor cells in the developing lung: implications for the pathogenesis of bronchopulmonary dysplasia. Am J Physiol Lung Cell Mol Physiol 292:L1073–L1084
Maniscalco WM, Watkins RH, D’Angio CT, Ryan RM (1997) Hyperoxic injury decreases alveolar epithelial cell expression of vascular endothelial growth factor (VEGF) in neonatal rabbit lung. Am J Respir Cell Mol Biol 16:557–567
Maniscalco WM, Watkins RH, Pryhuber GS, Bhatt A, Shea C, Huyck H (2002) Angiogenic factors and alveolar vasculature: development and alterations by injury in very premature baboons. Am J Physiol Lung Cell Mol Physiol 282:L811–L823
Bhatt AJ, Pryhuber GS, Huyck H, Watkins RH, Metlay LA, Maniscalco WM (2001) Disrupted pulmonary vasculature and decreased vascular endothelial growth factor, Flt-1, and TIE-2 in human infants dying with bronchopulmonary dysplasia. Am J Respir Crit Care Med 164:1971–1980
Lassus P, Ristimäki A, Ylikorkala O, Viinikka L, Andersson S (1999) Vascular endothelial growth factor in human preterm lung. Am J Respir Crit Care Med 159:1429–1433
Lassus P, Turanlahti M, Heikkilä P, Andersson LC, Nupponen I, Sarnesto A, Andersson S (2001) Pulmonary vascular endothelial growth factor and Flt-1 in fetuses, in acute and chronic lung disease, and in persistent pulmonary hypertension of the newborn. Am J Respir Crit Care Med 164:1981–1987
MacRitchie AN, Albertine KH, Sun J et al (2001) Reduced endothelial nitric oxide synthase in lungs of chronically ventilated preterm lambs. Am J Physiol Lung Cell Mol Physiol 281:L1011–L1020
De Paepe ME, Patel C, Tsai A, Gundavarapu S, Mao Q (2008) Endoglin (CD105) up-regulation in pulmonary microvasculature of ventilated preterm infants. Am J Respir Crit Care Med 178:180–187
Bhandari V, Choo-Wing R, Lee CG et al (2006) Hyperoxia causes angiopoietin 2–mediated acute lung injury and necrotic cell death. Nat Med 12:1286–1293
Ren X, Ustiyan V, Guo M, Wang G, Bolte C, Zhang Y, Xu Y, Whitsett JA, Kalin TV, Kalinichenko VV (2019) Postnatal alveologenesis depends on FOXF1 signaling in c-KIT + endothelial progenitor cells. Am J Respir Crit Care Med 200:1164–1176
Ahlfeld SK, Conway SJ (2012) Aberrant signaling pathways of the lung mesenchyme and their contributions to the pathogenesis of bronchopulmonary dysplasia. Birth Defects Res A 94:3–15
Husain AN, Siddiqui NH, Stocker JT (1998) Pathology of arrested acinar development in postsurfactant bronchopulmonary dysplasia. Hum Pathol 29:710–717
Pierce RA, Albertine KH, Starcher BC, Bohnsack JF, Carlton DP, Bland RD (1997) Chronic lung injury in preterm lambs: disordered pulmonary elastin deposition. Am J Physiol Lung Cell Mol Physiol 272:L452–L460
Mižíková I, Ruiz-Camp J, Steenbock H, Madurga A, Vadász I, Herold S, Mayer K, Seeger W, Brinckmann J, Morty RE (2015) Collagen and elastin cross-linking is altered during aberrant late lung development associated with hyperoxia. Am J Physiol Lung Cell Mol Physiol 308:L1145–L1158
Mižíková I, Pfeffer T, Nardiello C et al (2018) Targeting transglutaminase 2 partially restores extracellular matrix structure but not alveolar architecture in experimental bronchopulmonary dysplasia. FEBS J 285:3056–3076
Hilgendorff A, Parai K, Ertsey R et al (2012) Neonatal mice genetically modified to express the elastase inhibitor elafin are protected against the adverse effects of mechanical ventilation on lung growth. Am J Physiol Lung Cell Mol Physiol 303:L215–L227
Benjamin JT, Smith RJ, Halloran BA, Day TJ, Kelly DR, Prince LS (2007) FGF-10 is decreased in bronchopulmonary dysplasia and suppressed by Toll-like receptor activation. Am J Physiol Lung Cell Mol Physiol 292:L550–L558
Ambalavanan N, Novak ZE (2003) Peptide growth factors in tracheal aspirates of mechanically ventilated preterm neonates. Pediatr Res 53:240–244
Jankov RP, Luo X, Campbell A, Belcastro R, Cabacungan J, Johnstone L, Frndova H, Lye SJ, Tanswell AK (2003) Fibroblast growth factor receptor-1 and neonatal compensatory lung growth after exposure to 95% oxygen. Am J Respir Crit Care Med 167:1554–1561
Gouveia L, Betsholtz C, Andrae J (2018) PDGF-A signaling is required for secondary alveolar septation and controls epithelial proliferation in the developing lung. Development. 145:dev161976
Li C, Lee MK, Gao F, et al (2019) The secondary crest myofibroblast PDGFRα controls elastogenesis pathway via a secondary tier of signaling networks during alveologenesis. Development 146(15):dev176354
Lindahl P, Karlsson L, Hellström M, Gebre-Medhin S, Willetts K, Heath JK, Betsholtz C (1997) Alveogenesis failure in PDGF-A-deficient mice is coupled to lack of distal spreading of alveolar smooth muscle cell progenitors during lung development. Development 124:3943–3953
Ruiz‐Camp J, Quantius J, Lignelli E, et al (2019) Targeting miR‐34a/ Pdgfra interactions partially corrects alveologenesis in experimental bronchopulmonary dysplasia. EMBO Mol Med. https://doi.org/10.15252/emmm.201809448
Fulton CT, Cui TX, Goldsmith AM, Bermick J, Popova AP (2018) Gene Expression Signatures Point to a Male Sex-Specific Lung Mesenchymal Cell PDGF Receptor Signaling Defect in Infants Developing Bronchopulmonary Dysplasia. Sci Rep 8:17070
Alejandre-Alcázar MA, Michiels-Corsten M, Vicencio AG et al (2008) TGF-β signaling is dynamically regulated during the alveolarization of rodent and human lungs. Dev Dyn 237:259–269
Chen F, Desai TJ, Qian J, Niederreither K, Lü J, Cardoso WV (2007) Inhibition of Tgfβ signaling by endogenous retinoic acid is essential for primary lung bud induction. Development 134:2969–2979
Kotecha S, Wangoo A, Silverman M, Shaw RJ (1996) Increase in the concentration of transforming growth factor beta-1 in bronchoalveolar lavage fluid before development of chronic lung disease of prematurity. J Pediatr 128:464–469
Alejandre-Alcázar MA, Kwapiszewska G, Reiss I et al (2007) Hyperoxia modulates TGF-β/BMP signaling in a mouse model of bronchopulmonary dysplasia. Am J Physiol Lung Cell Mol Physiol 292:L537–L549
Nakanishi H, Sugiura T, Streisand JB, Lonning SM, Roberts JD (2007) TGF-β-neutralizing antibodies improve pulmonary alveologenesis and vasculogenesis in the injured newborn lung. Am J Physiol Lung Cell Mol Physiol 293:L151–L161
Bruce MC, Schuyler M, Martin RJ, Starcher BC, Tomashefski JF, Wedig KE (1992) Risk factors for the degradation of lung elastic fibers in the ventilated neonate: implications for impaired lung development in bronchopulmonary dysplasia. Am Rev Respir Dis 146:204–212
Thibeault DW, Mabry SM, Ekekezie II, Truog WE (2000) Lung elastic tissue maturation and perturbations during the evolution of chronic lung disease. Pediatrics 106:1452–1459
Kumarasamy A, Schmitt I, Nave AH et al (2009) Lysyl oxidase activity is dysregulated during impaired alveolarization of mouse and human lungs. Am J Respir Crit Care Med 180:1239–1252
Bruce MC, Honaker C, Karathanasis P (1996) Postnatal age at onset of hyperoxic exposure influences developmentally regulated tropoelastin gene expression in the neonatal rat lung. Am J Respir Cell Mol Biol 14:177–185
Bhandari V (2013) The potential of non-invasive ventilation to decrease BPD. Semin Perinatol 37:108–114
Fischer HS, Schmölzer GM, Cheung P-Y, Bührer C (2018) Sustained inflations and avoiding mechanical ventilation to prevent death or bronchopulmonary dysplasia: a meta-analysis. Eur Respir Rev 27:180083
Stevens TP, Blennow M, Myers EH, Soll R (2007) Early surfactant administration with brief ventilation vs. selective surfactant and continued mechanical ventilation for preterm infants with or at risk for respiratory distress syndrome. Cochrane Database Syst Rev. https://doi.org/10.1002/14651858.CD003063.pub3
Aldana-Aguirre JC, Pinto M, Featherstone RM, Kumar M (2017) Less invasive surfactant administration versus intubation for surfactant delivery in preterm infants with respiratory distress syndrome: a systematic review and meta-analysis. Arch Dis Child Fetal Neonatal Ed 102:F17–F23
Isayama T, Chai-Adisaksopha C, McDonald SD (2015) Noninvasive ventilation with vs without early surfactant to prevent chronic lung disease in preterm infants: a systematic review and meta-analysis. JAMA Pediatr 169:731
Rigo V, Lefebvre C, Broux I (2016) Surfactant instillation in spontaneously breathing preterm infants: a systematic review and meta-analysis. Eur J Pediatr 175:1933–1942
Pierro M, Van Mechelen K, van Westering-Kroon E, Villamor-Martínez E, Villamor E (2022) Endotypes of Prematurity and Phenotypes of Bronchopulmonary Dysplasia: Toward Personalized Neonatology. JPM 12:687
Logan JW, Lynch SK, Curtiss J, Shepherd EG (2019) Clinical phenotypes and management concepts for severe, established bronchopulmonary dysplasia. Paediatr Respir Rev 31:58–63
Shepherd EG, Clouse BJ, Hasenstab KA, Sitaram S, Malleske DT, Nelin LD, Jadcherla SR (2018) Infant pulmonary function testing and phenotypes in severe bronchopulmonary dysplasia. Pediatrics 141:e20173350
Collaco JM, McGrath-Morrow SA (2018) Respiratory phenotypes for preterm infants, children, and adults: bronchopulmonary dysplasia and more. Annals ATS 15:530–538
Wu KY, Jensen EA, White AM, Wang Y, Biko DM, Nilan K, Fraga MV, Mercer-Rosa L, Zhang H, Kirpalani H (2020) Characterization of disease phenotype in very preterm infants with severe bronchopulmonary dysplasia. Am J Respir Crit Care Med 201:1398–1406
McGrath-Morrow SA, Collaco JM (2019) Bronchopulmonary dysplasia: what are its links to COPD? Ther Adv Respir Dis 13:175346661989249
Baraldi E, Bonetto G, Zacchello F, Filippone M (2005) Low exhaled nitric oxide in school-age children with bronchopulmonary dysplasia and airflow limitation. Am J Respir Crit Care Med 171:68–72
McElrath TF, Hecht JL, Dammann O et al (2008) Pregnancy disorders that lead to delivery before the 28th week of gestation: an epidemiologic approach to classification. Am J Epidemiol 168:980–989
Villamor-Martinez E, Álvarez-Fuente M, Ghazi AMT, Degraeuwe P, Zimmermann LJI, Kramer BW, Villamor E (2019) Association of chorioamnionitis with bronchopulmonary dysplasia among preterm infants: a systematic review, meta-analysis, and metaregression. JAMA Netw Open 2:e1914611
Cohen M, Giladi A, Gorki A-D et al (2018) Lung single-cell signaling interaction map reveals basophil role in macrophage imprinting. Cell 175:1031-1044.e18
Domingo-Gonzalez R, Zanini F, Che X, Liu M, Jones RC, Swift MA, Quake SR, Cornfield DN, Alvira CM (2020) Diverse homeostatic and immunomodulatory roles of immune cells in the developing mouse lung at single cell resolution. eLife 9:e56890
Guo M, Du Y, Gokey JJ et al (2019) Single cell RNA analysis identifies cellular heterogeneity and adaptive responses of the lung at birth. Nat Commun 10:37
Hagan AS, Zhang B, Ornitz DM (2019) Identification of an FGF18-expressing alveolar myofibroblast that is developmentally cleared during alveologenesis. Development 147(2):dev181032
Li R, Bernau K, Sandbo N, Gu J, Preissl S, Sun X (2018) Pdgfra marks a cellular lineage with distinct contributions to myofibroblasts in lung maturation and injury response. eLife 7:e36865
Vila Ellis L, Cain MP, Hutchison V, Flodby P, Crandall ED, Borok Z, Zhou B, Ostrin EJ, Wythe JD, Chen J (2020) Epithelial vegfa specifies a distinct endothelial population in the mouse lung. Dev Cell 52:617-630.e6
Danopoulos S, Bhattacharya S, Mariani TJ, Al Alam D (2020) Transcriptional characterisation of human lung cells identifies novel mesenchymal lineage markers. Eur Respir J 55:1900746
Cao J, O’Day DR, Pliner HA et al (2020) A human cell atlas of fetal gene expression. Science 370:eaba7721
Miller AJ, Yu Q, Czerwinski M et al (2020) In vitro and in vivo development of the human airway at single-cell resolution. Dev Cell 53:117-128.e6
He P, Lim K, Sun D et al (2022) A human fetal lung cell atlas uncovers proximal-distal gradients of differentiation and key regulators of epithelial fates. Cell 185:4841-4860.e25
Lu J, Zhu X, Shui JE et al (2021) Rho/SMAD/mTOR triple inhibition enables long-term expansion of human neonatal tracheal aspirate-derived airway basal cell-like cells. Pediatr Res 89:502–509
Gillich A, Zhang F, Farmer CG, Travaglini KJ, Tan SY, Gu M, Zhou B, Feinstein JA, Krasnow MA, Metzger RJ (2020) Capillary cell-type specialization in the alveolus. Nature 586:785–789
Alphonse RS, Vadivel A, Fung M et al (2014) Existence, functional impairment, and lung repair potential of endothelial colony-forming cells in oxygen-induced arrested alveolar growth. Circulation 129:2144–2157
Niethamer TK, Stabler CT, Leach JP, Zepp JA, Morley MP, Babu A, Zhou S, Morrisey EE (2020) Defining the role of pulmonary endothelial cell heterogeneity in the response to acute lung injury. eLife 9:e53072
Scaffa A, Yao H, Oulhen N, Wallace J, Peterson AL, Rizal S, Ragavendran A, Wessel G, De Paepe ME, Dennery PA (2021) Single-cell transcriptomics reveals lasting changes in the lung cellular landscape into adulthood after neonatal hyperoxic exposure. Redox Biol 48:102091
Polin RA, Abman SH, Rowitch D, Benitz WE (2021) Fetal and Neonatal Physiology, 6th ed. Elsevier, Inc, Philadelphia. ISBN number: 978-0-323-71284-2
Rawlins EL, Hogan BLM (2008) Ciliated epithelial cell lifespan in the mouse trachea and lung. Am J Physiol Lung Cell Mol Physiol 295:L231–L234
Ciechanowicz A (2019) Stem cells in lungs. In: Ratajczak MZ (ed) Stem Cells. Springer International Publishing, Cham, pp 261–274
Logan CY, Desai TJ (2015) Keeping it together: pulmonary alveoli are maintained by a hierarchy of cellular programs. BioEssays 37:1028–1037
Möbius MA, Thébaud B (2017) Bronchopulmonary dysplasia: where have all the stem cells gone? Chest 152:1043–1052
Morrisey EE (2018) Basal cells in lung development and repair. Dev Cell 44:653–654
Rawlins EL (2008) Lung epithelial progenitor cells: lessons from development. Proc Am Thorac Soc 5:675–681
Kim CFB, Jackson EL, Woolfenden AE, Lawrence S, Babar I, Vogel S, Crowley D, Bronson RT, Jacks T (2005) Identification of bronchioalveolar stem cells in normal lung and lung cancer. Cell 121:823–835
Watson JK, Rulands S, Wilkinson AC, Wuidart A, Ousset M, Van Keymeulen A, Göttgens B, Blanpain C, Simons BD, Rawlins EL (2015) Clonal dynamics reveal two distinct populations of basal cells in slow-turnover airway epithelium. Cell Rep 12:90–101
Wang G, Lou HH, Salit J et al (2019) Characterization of an immortalized human small airway basal stem/progenitor cell line with airway region-specific differentiation capacity. Respir Res 20:196
Siebel C, Lendahl U (2017) Notch signaling in development, tissue homeostasis, and disease. Physiol Rev 97:1235–1294
Morimoto M, Liu Z, Cheng H-T, Winters N, Bader D, Kopan R (2010) Canonical Notch signaling in the developing lung is required for determination of arterial smooth muscle cells and selection of Clara versus ciliated cell fate. J Cell Sci 123:213–224
Avasthi P, Marshall WF (2012) Stages of ciliogenesis and regulation of ciliary length. Differentiation 83:S30–S42
Rawlins EL, Okubo T, Xue Y, Brass DM, Auten RL, Hasegawa H, Wang F, Hogan BLM (2009) The role of Scgb1a1+ clara cells in the long-term maintenance and repair of lung airway, but not alveolar, epithelium. Cell Stem Cell 4:525–534
Frank DB, Penkala IJ, Zepp JA et al (2019) Early lineage specification defines alveolar epithelial ontogeny in the murine lung. Proc Natl Acad Sci USA 116:4362–4371
Mižíková I, Thébaud B (2021) Looking at the developing lung in single-cell resolution. Am J Physiol Lung Cell Mol Physiol 320:L680–L687
Fujino N, Kubo H, Suzuki T et al (2011) Isolation of alveolar epithelial type II progenitor cells from adult human lungs. Lab Invest 91:363–378
Evans MJ, Cabral LJ, Stephens RJ, Freeman G (1973) Renewal of alveolar epithelium in the rat following exposure to NO2. Am J Pathol 70:175–198
Barkauskas CE, Cronce MJ, Rackley CR, Bowie EJ, Keene DR, Stripp BR, Randell SH, Noble PW, Hogan BLM (2013) Type 2 alveolar cells are stem cells in adult lung. J Clin Invest 123:3025–3036
Desai TJ, Brownfield DG, Krasnow MA (2014) Alveolar progenitor and stem cells in lung development, renewal and cancer. Nature 507:190–194
Hou A, Fu J, Yang H, Zhu Y, Pan Y, Xu S, Xue X (2015) Hyperoxia stimulates the transdifferentiation of type II alveolar epithelial cells in newborn rats. Am J Physiol Lung Cell Mol Physiol 308:L861–L872
Abdelwahab EMM, Rapp J, Feller D, Csongei V, Pal S, Bartis D, Thickett DR, Pongracz JE (2019) Wnt signaling regulates trans-differentiation of stem cell like type 2 alveolar epithelial cells to type 1 epithelial cells. Respir Res 20:204
Gonzalez RF, Allen L, Dobbs LG (2009) Rat alveolar type I cells proliferate, express OCT-4, and exhibit phenotypic plasticity in vitro. Am J Physiol Lung Cell Mol Physiol 297:L1045–L1055
Danto SI, Shannon JM, Borok Z, Zabski SM, Crandall ED (1995) Reversible transdifferentiation of alveolar epithelial cells. Am J Respir Cell Mol Biol 12:497–502
Jain R, Barkauskas CE, Takeda N et al (2015) Plasticity of Hopx+ type I alveolar cells to regenerate type II cells in the lung. Nat Commun 6:6727
Basil MC, Cardenas-Diaz FL, Kathiriya JJ et al (2022) Human distal airways contain a multipotent secretory cell that can regenerate alveoli. Nature 604:120–126
Thébaud B, Abman SH (2007) Bronchopulmonary dysplasia: where have all the vessels gone? Roles of angiogenic growth factors in chronic lung disease. Am J Respir Crit Care Med 175:978–985
Gerber HP, Hillan KJ, Ryan AM, Kowalski J, Keller GA, Rangell L, Wright BD, Radtke F, Aguet M, Ferrara N (1999) VEGF is required for growth and survival in neonatal mice. Development 126:1149–1159
Galambos C, Ng Y-S, Ali A, Noguchi A, Lovejoy S, D’Amore PA, deMello DE (2002) Defective pulmonary development in the absence of heparin-binding vascular endothelial growth factor isoforms. Am J Respir Cell Mol Biol 27:194–203
Jakkula M, Le Cras TD, Gebb S, Hirth KP, Tuder RM, Voelkel NF, Abman SH (2000) Inhibition of angiogenesis decreases alveolarization in the developing rat lung. Am J Physiol Lung Cell Mol Physiol 279:L600–L607
Asikainen TM, Waleh NS, Schneider BK, Clyman RI, White CW (2006) Enhancement of angiogenic effectors through hypoxia-inducible factor in preterm primate lung in vivo. Am J Physiol Lung Cell Mol Physiol 291:L588–L595
Kunig AM, Balasubramaniam V, Markham NE, Morgan D, Montgomery G, Grover TR, Abman SH (2005) Recombinant human VEGF treatment enhances alveolarization after hyperoxic lung injury in neonatal rats. Am J Physiol Lung Cell Mol Physiol 289:L529–L535
Kunig AM, Balasubramaniam V, Markham NE, Seedorf G, Gien J, Abman SH (2006) Recombinant human VEGF treatment transiently increases lung edema but enhances lung structure after neonatal hyperoxia. Am J Physiol Lung Cell Mol Physiol 291:L1068–L1078
Aslam M, Baveja R, Liang OD, Fernandez-Gonzalez A, Lee C, Mitsialis SA, Kourembanas S (2009) Bone marrow stromal cells attenuate lung injury in a murine model of neonatal chronic lung disease. Am J Respir Crit Care Med 180:1122–1130
Pierro M, Ionescu L, Montemurro T et al (2013) Short-term, long-term and paracrine effect of human umbilical cord-derived stem cells in lung injury prevention and repair in experimental bronchopulmonary dysplasia. Thorax 68:475–484
Sutsko RP, Young KC, Ribeiro A, Torres E, Rodriguez M, Hehre D, Devia C, McNiece I, Suguihara C (2013) Long-term reparative effects of mesenchymal stem cell therapy following neonatal hyperoxia-induced lung injury. Pediatr Res 73:46–53
van Haaften T, Byrne R, Bonnet S et al (2009) Airway delivery of mesenchymal stem cells prevents arrested alveolar growth in neonatal lung injury in rats. Am J Respir Crit Care Med 180:1131–1142
Chang YS, Oh W, Choi SJ, Sung DK, Kim SY, Choi EY, Kang S, Jin HJ, Yang YS, Park WS (2009) Human umbilical cord blood-derived mesenchymal stem cells attenuate hyperoxia-induced lung injury in neonatal rats. Cell Transplant 18:869–886
Ahn SY, Chang YS, Kim JH, Sung SI, Park WS (2017) Two-year follow-up outcomes of premature infants enrolled in the phase I trial of mesenchymal stem cells transplantation for bronchopulmonary dysplasia. J Pediatr 185:49-54.e2
Ahn SY, Chang YS, Lee MH, Sung SI, Lee BS, Kim KS, Kim A-R, Park WS (2021) Stem cells for bronchopulmonary dysplasia in preterm infants: a randomized controlled phase II trial. Stem Cells Transl Med 10:1129–1137
Chang YS, Ahn SY, Yoo HS, Sung SI, Choi SJ, Oh WI, Park WS (2014) Mesenchymal stem cells for bronchopulmonary dysplasia: phase 1 dose-escalation clinical trial. J Pediatr 164:966-972.e6
Powell SB, Silvestri JM (2019) Safety of intratracheal administration of human umbilical cord blood derived mesenchymal stromal cells in extremely low birth weight preterm infants. J Pediatr 210:209-213.e2
Théry C, Witwer KW, Aikawa E et al (2018) Minimal information for studies of extracellular vesicles 2018 (MISEV2018): a position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. Journal of Extracellular Vesicles 7:1535750
Lesage F, Thébaud B (2022) Mesenchymal stromal cell-derived extracellular vesicles for neonatal lung disease: tiny particles, major promise, rigorous requirements for clinical translation. Cells 11:1176
Reis M, Willis GR, Fernandez-Gonzalez A et al (2021) Mesenchymal stromal cell-derived extracellular vesicles restore thymic architecture and T cell function disrupted by neonatal hyperoxia. Front Immunol 12:640595
Willis GR, Fernandez-Gonzalez A, Anastas J, Vitali SH, Liu X, Ericsson M, Kwong A, Mitsialis SA, Kourembanas S (2018) Mesenchymal stromal cell exosomes ameliorate experimental bronchopulmonary dysplasia and restore lung function through macrophage immunomodulation. Am J Respir Crit Care Med 197:104–116
Willis GR, Reis M, Gheinani AH et al (2021) Extracellular vesicles protect the neonatal lung from hyperoxic injury through the epigenetic and transcriptomic reprogramming of myeloid cells. Am J Respir Crit Care Med 204:1418–1432
Bellio MA, Young KC, Milberg J et al (2021) Amniotic fluid-derived extracellular vesicles: characterization and therapeutic efficacy in an experimental model of bronchopulmonary dysplasia. Cytotherapy 23:1097–1107
Sharma M, Bellio MA, Benny M et al (2022) Mesenchymal stem cell-derived extracellular vesicles prevent experimental bronchopulmonary dysplasia complicated by pulmonary hypertension. Stem Cells Transl Med 11:828–840
Abele AN, Taglauer ES, Almeda M, Wilson N, Abikoye A, Seedorf GJ, Mitsialis SA, Kourembanas S, Abman SH (2022) Antenatal mesenchymal stromal cell extracellular vesicle treatment preserves lung development in a model of bronchopulmonary dysplasia due to chorioamnionitis. Am J Physiol Lung Cell Mol Physiol 322:L179–L190
Lithopoulos MA, Strueby L, O’Reilly M et al (2022) Pulmonary and neurologic effects of mesenchymal stromal cell extracellular vesicles in a multifactorial lung injury model. Am J Respir Crit Care Med 205:1186–1201
Hennrick KT, Keeton AG, Nanua S et al (2007) Lung cells from neonates show a mesenchymal stem cell phenotype. Am J Respir Crit Care Med 175:1158–1164
Popova AP, Bozyk PD, Bentley JK, Linn MJ, Goldsmith AM, Schumacher RE, Weiner GM, Filbrun AG, Hershenson MB (2010) Isolation of tracheal aspirate mesenchymal stromal cells predicts bronchopulmonary dysplasia. Pediatrics 126:e1127–e1133
Reicherzer T, Häffner S, Shahzad T et al (2018) Activation of the NF-κB pathway alters the phenotype of MSCs in the tracheal aspirates of preterm infants with severe BPD. Am J Physiol Lung Cell Mol Physiol 315:L87–L101
Möbius MA, Freund D, Vadivel A, Koss S, McConaghy S, Ohls RK, Rüdiger M, Thébaud B (2019) Oxygen disrupts human fetal lung mesenchymal cells. Implications for Bronchopulmonary Dysplasia. Am J Respir Cell Mol Biol 60:592–600
Collins JJP, Lithopoulos MA, dos Santos CC, Issa N, Möbius MA, Ito C, Zhong S, Vadivel A, Thébaud B (2018) Impaired angiogenic supportive capacity and altered gene expression profile of resident CD146 + mesenchymal stromal cells isolated from hyperoxia-injured neonatal rat lungs. Stem Cells and Development 27:1109–1124
Mižíková I, Lesage F, Cyr-Depauw C et al (2022) Single-cell RNA sequencing-based characterization of resident lung mesenchymal stromal cells in bronchopulmonary dysplasia. Stem Cells 40:479–492
Dominici M, Le Blanc K, Mueller I, Slaper-Cortenbach I, Marini FC, Krause DS, Deans RJ, Keating A, Prockop DJ, Horwitz EM (2006) Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy 8:315–317
Gong X, Sun Z, Cui D, Xu X, Zhu H, Wang L, Qian W, Han X (2014) Isolation and characterization of lung resident mesenchymal stem cells capable of differentiating into alveolar epithelial type II cells: Isolation and characterization of LR-MSCs. Cell Biol Int 38:405–411
Rolandsson Enes S, Andersson Sjöland A, Skog I, Hansson L, Larsson H, Le Blanc K, Eriksson L, Bjermer L, Scheding S, Westergren-Thorsson G (2016) MSC from fetal and adult lungs possess lung-specific properties compared to bone marrow-derived MSC. Sci Rep 6:29160
McQualter JL, Brouard N, Williams B, Baird BN, Sims-Lucas S, Yuen K, Nilsson SK, Simmons PJ, Bertoncello I (2009) Endogenous fibroblastic progenitor cells in the adult mouse lung are highly enriched in the Sca-1 positive cell fraction. Stem Cells 27:623–633
Chu X, Lingampally A, Moiseenko A et al (2022) GLI1+ cells are a source of repair-supportive mesenchymal cells (RSMCs) during airway epithelial regeneration. Cell Mol Life Sci 79:581
Kramann R, Schneider RK, DiRocco DP, Machado F, Fleig S, Bondzie PA, Henderson JM, Ebert BL, Humphreys BD (2015) perivascular Gli1+ progenitors are key contributors to injury-induced organ fibrosis. Cell Stem Cell 16:51–66
Wu Y, Zhou X, Yuan W et al (2022) Gli1+ mesenchymal stem cells in bone and teeth. CSCR 17:494–502
Chen J, Li M, Liu A-Q et al (2020) Gli1+ cells couple with type H vessels and are required for type H vessel formation. Stem Cell Reports 15:110–124
Peng J, Li F, Wang J et al (2022) Identification of a rare Gli1+ progenitor cell population contributing to liver regeneration during chronic injury. Cell Discov 8:118
Cao H, Chen X, Hou J, Wang C, Xiang Z, Shen Y, Han X (2020) The Shh/Gli signaling cascade regulates myofibroblastic activation of lung-resident mesenchymal stem cells via the modulation of Wnt10a expression during pulmonary fibrogenesis. Lab Invest 100:363–377
Acknowledgements
Not applicable.
Funding
Open Access funding enabled and organized by Projekt DEAL. This study was supported by the German Research Foundation (Deutsche Forschungsgemeinschaft) and the Canadian Institutes of Health Research (CIHR).
Author information
Authors and Affiliations
Contributions
IM performed literature research and drafted the manuscript. TB drafted the manuscript. IM and TB discussed content, modified, and approved the final version of the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Mižíková, I., Thébaud, B. Perinatal origins of bronchopulmonary dysplasia—deciphering normal and impaired lung development cell by cell. Mol Cell Pediatr 10, 4 (2023). https://doi.org/10.1186/s40348-023-00158-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40348-023-00158-2