
Abstract
Childhood asthma derives from complex host-environment interactions occurring in the perinatal and infant period, a critical time for lung development. Sphingolipids are bioactive molecules consistently implicated in the pathogenesis of childhood asthma. Genome wide association studies (GWAS) initially identified a link between alleles within the 17q21 asthma-susceptibility locus, childhood asthma, and overexpression of the ORMDL sphingolipid biosynthesis regulator 3 (ORMDL3), an inhibitor of de novo sphingolipid synthesis. Subsequent studies of pediatric asthma offer strong evidence that these asthma-risk alleles correlate with early-life aberrancies of sphingolipid homeostasis and asthma. Relationships between sphingolipid metabolism and asthma-related risk factors, including maternal obesity and respiratory viral infections, are currently under investigation. This review will summarize how these perinatal and early life exposures can synergize with 17q21 asthma risk alleles to exacerbate disruptions of sphingolipid homeostasis and drive asthma pathogenesis.
Similar content being viewed by others

Introduction
Asthma is a heterogenous, chronic condition clinically identified by episodic shortness of breath, wheeze, and sometimes cough [1]. Once thought of as a single disease, asthma is now recognized as a spectrum of immunopathology culminating in a final common pathway of chronic airway inflammation, reversible airway obstruction, increased mucus production, and airway hyperreactivity. Globally, over 300 million people are affected by asthma and though the disease can occur at any age, it most often develops in childhood [2, 3]. Globally, asthma is a leading cause of childhood chronic illness [4]. The epidemiologic burden, which traditionally fell to metropolitan areas in high-income countries, is now increasing in low-income countries who also shoulder a disproportionate amount of asthma-related morbidity and mortality [2]. Available interventions to both prevent and treat severe asthma require frequent and expensive interactions with the health care system which limit school participation, work productivity, and overall quality of life [5]. Children with severe asthma are more likely to have symptoms persist through adulthood. The risk of adult-onset chronic obstructive pulmonary disease (COPD) is strongly associated with childhood deficiencies in lung function, measured by spirometry [2, 6, 7]. It remains unclear if the trajectory connecting childhood asthma and long-term respiratory morbidity can be reversed [8]. However, there is mounting evidence that exposures in the peri-natal and infant period serve as priming events for abnormal lung growth and lung inflammation, reflecting a possible avenue for childhood asthma prevention.
Classification systems for pediatric asthma have evolved significantly, and with them, the spectrum of asthma investigation. Chronic airway inflammation was previously considered the foundation of the two other key disease features, airway hyperresponsiveness and airway remodeling. Previous characterizations focused on the presence or absence of atopic, T-helper type 2 (Th2) cell inflammation [9,10,11,12]. Briefly, the Th2 pathway begins with allergen stimulation of Th2 cytokines (IL-4, IL-5, IL-9, IL-13) which trigger IgE release by B cells, which together promote histamine and leukotriene release by mast cells and eosinophilic inflammation [13]. General dampening of this inflammatory response by steroids or therapies targeting specific mediators within this pathway, i.e., leukotriene inhibitors and anti-IgE, IL4, or monoclonal antibodies, have been greatly effective for some but also revealed a broad group of pediatric non-responders with seemingly non-atopic and/or non-steroid responsive disease [14, 15].
The limitations of allergy-based asthma therapies have driven efforts to develop more personalized methods for disease monitoring and treatment, beginning with the characterization of disease “endotypes.” With the advancement of high throughput technologies evolved a comprehensive approach, including genetic, metabolic, molecular, and clinical characteristics, to define more granular endotypes (Fig. 1) [8, 10, 16, 17]. This led to the increased recognition of non-Th2 inflammatory pathways including, Th1 and Th17, and the complex regulation of cells and cell mediators traditionally considered Th2. A subgroup of children with asthma display airway eosinophilia without associated Th2 cytokines [18]. Sputum transcriptomics has linked this type of airway eosinophilia to gene signatures from metabolic, ubiquitination, and mitochondrial function pathways [19]. The full range of asthma endotypes is beyond the scope of this review and is described in detail elsewhere [15, 20,21,22]. Despite the application of multi-omic technologies and related advances in asthma classification and treatment algorithms, therapy-resistant phenotypes persist, and early-life therapies have yet to change the long-term disease trajectory. Given the significant implications of childhood asthma on life-long respiratory health, there is an urgent need to address the origins of childhood disease. It is within this framework that sphingolipid metabolism has become a topic of interest for a fresh look on pathogenesis and therapies of childhood asthma.

Expanding landscape of pediatric asthma investigation. Clinical features of wheeze, breathlessness, and cough were first related to the triad of airway inflammation, bronchoconstriction, and mucus production, followed by increased understanding of T-helper (Th) cell-associated pathways linking allergen exposure and Th2 cell cytokines to airway eosinophilia and mast cell degranulation. Neutrophil dominant airway inflammation was also identified and related Th1 and Th17 pathways, mediated by interferon γ and IL-17, respectively. Applied to samples from large pediatric asthma cohorts, technologies based on high throughput sequencing and mass spectrometry have revealed surprising, but correlative, genomic, and metabolic disturbances in connection with constitutive elements of the multiple inflammatory pathways. This includes the association of 17q21 SNPs with alterations to sphingolipid gene expression and metabolism in children with non-atopic asthma.
Sphingolipids are bioactive molecules increasingly recognized in lung inflammation and airway hyperreactivity. Besides asthma, sphingolipids have been implicated in a host of chronic pulmonary disorders including bronchopulmonary dysplasia, chronic obstructive pulmonary disease (COPD), and cystic fibrosis [4]. In the context of asthma, attention turned to sphingolipids after genome-wide association studies (GWAS) reproducibly associated childhood asthma, and early-life wheeze with single-nucleotide polymorphisms (SNPs) within the region of chromosome 17q21 and increased expression of the sphingolipid synthesis regulator ORMDL3 [23,24,25]. Since then, aberrations in sphingolipid metabolism and gene expression have been seen in pediatric asthma cohorts [26, 27]. Animal models and in vitro studies have connected sphingolipid metabolism to clinical features of asthma, including airway hyperreactivity [28, 29]. This review will summarize the principal findings supporting a pathway from genetic and perinatal disruptions of sphingolipid metabolism to childhood asthma.
Genetic dysregulation of sphingolipid metabolism in childhood asthma
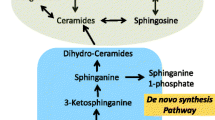
ORMDLs regulate de novo sphingolipid synthesis, which begins with the condensation of serine and palmitoyl CoA by serine palmitoyltransferase (SPT) in the endoplasmic reticulum (Fig. 2). In humans, ORMDL3 engages SPT, blocking its substrate pathway and suppressing its activity [30, 31]. GWAS studies showed ORMDL3 expression is increased with asthma risk alleles [23, 32], suggesting SPT inhibition is relevant to asthma pathogenesis. Though the genetic regulation of sphingolipid homeostasis is complex, and the mechanisms linking ORMDL3 to asthma are incompletely understood, there is mounting evidence that shifts in sphingolipid hemostasis have an important role in childhood asthma and early-life wheeze.

Pathways of Sphingolipid Metabolism. De novo synthesis begins with condensation of serine and palmitoyl CoA by serine palmitoyl-CoA transferase (SPT), the rate-limiting step in the production of 3-ketosphinganine, which is immediately reduced to sphinganine by ketodihydrosphingosine reductase (KDHR). Sphinganine can be phosphorylated by sphingosine kinases (SPHK) to sphinganine-1-phosphate or acylated by ceramide synthases (CERS) to form dihydroceramides. Dihydroceramides are converted to ceramides by dihydroceramide desaturase (DEGS), which are subsequently recycled as sphingosine, also by CERS, or converted to sphingomyelins by sphingomyelin synthase (SMS). Additional abbreviations SPP-1 (S1P phosphatase), and SMase (sphingomyelinase).
Sphingolipids are a ubiquitous and diverse class of amphipathic lipids comprised of a hydrophobic sphingoid base attached to a hydrophilic group which can consist of hydroxyl groups or, for more complex species, phosphates, and sugar residues [33]. Sphingolipids gain their complexity from the variable arrangement of these component parts. In mammalian cells, there are five known sphingoid bases with over twenty known arrangements of fatty acids, alkyl chain lengths, degrees of saturation, and hydroxylation. Sphinganine, the reduced product of the SPT catalyzed reaction, can be phosphorylated or deacylated to generate sphinganine-1-phosphate (Sa1P) or dihydroceramides, respectively. During the final step of the de novo synthesis, dihydroceramides are converted to ceramides. Ceramides, the nexus of sphingolipid metabolism, are the precursor to sphingomyelins, sphingosine, and sphingosine-1-phosphate (S1P) (Fig. 2) and more complex glycosphingolipids (not shown).
Studies measuring sphingolipid levels show consistent differences between children with asthma and children without asthma, though the relative direction of this difference varies by tissue compartment. Higher levels of ceramides and dihydroceramides were detected in exhaled breath condensates [34], serum [35], and plasma [27] of children with asthma. In a cohort of 5–17-year-old children, those with asthma displayed lower levels of sphingolipids in their blood cells. This finding was especially pronounced in children with non-allergic asthma. In this cohort, reduced blood sphingolipids are also associated with 17q21 asthma-associated risk alleles, specifically SNPs rs7216389 and rs8076131. Using heavy-isotope serine, metabolic labeling of the peripheral blood mononuclear cells from these children showed asthma and genotype-dependent decrease in de novo sphingolipid synthesis [27].
A study of two longitudinal mother-baby cohorts, the Copenhagen Prospective Study of Asthma in Childhood (COPSAC2010) and the Vitamin D Antenatal Asthma Reduction Trial (VDAART), also associated sphingolipid levels with early-life asthma. The study included plasma metabolomics at 6 months, 1 and 6 years, and transcriptomic analysis of nasal brushings at age 6. Interestingly, at age 6, asthma risk alleles were associated with reduced expression of the SPT subunits Sptlc1 and ssSPTa. The human SPT complex is composed of two large subunits, Sptlc1 and Sptlc2, and one small regulatory subunit, ssSPTa. This small subunit stabilizes the catalytic subunit Sptlc2 by altering its substrate specificity and greatly increases enzyme activity [31]. The association of the 17q21 risk alleles with lower expression of SPT subunits could point to an additional sphingolipid-regulatory mechanism associated with these genotypes. Relatedly, at age 6, there was also an inverse relationship between plasma Sa1P and airway resistance; and lower plasma Sa1P levels positively associated with 17q21 asthma risk alleles. Most interesting however is that the authors found a correlation between lower plasma sphingolipids (ceramides and sphingomyelins) at 6 months and the likelihood of asthma onset by age 3. The study clearly associates childhood asthma, 17q21 asthma genotypes with de novo pathway, but additionally suggests that disruptions in sphingolipid synthesis are present in infancy and predictive of later disease [26].
Sphingolipids are an integral part of plasma membranes, where they form discreet domains responsible for cellular processes including protein trafficking, signal transduction, and virus budding [36]. Outside of the plasma membrane, sphingolipids also serve as signaling molecules for a host of pathways including apoptosis [37], cytoskeletal reorganization, and cellular inflammation [36, 38]. While clinical studies correlate childhood asthma with alterations in sphingolipid production, animal and in vitro studies offer more granular insight into specific sphingolipid-lung interactions.
Animal models have connected ORMDL3 and sphingolipids to clinical features of asthma. ORMDL3 overexpressing mice display increased airway reactivity and airway remodeling, including increased airway smooth muscle, subepithelial fibrosis, and mucus [34, 39, 40]. Similar effects are seen with targeted inhibition of de novo sphingolipid synthesis. Both SPT haploinsufficient mice and wild-type mice treated with the SPT inhibitor myriocin display increased airway reactivity in the absence of allergic sensitization and airway inflammation [9, 28], suggesting a negative effect of lower sphingolipid synthesis on airway smooth muscle cells.
Increased ORMDL3 expression, both in mice and human lung epithelial cells, is associated with increased ceramide levels [34]. The sphingolipid mediator S1P is one of the most extensively studied sphingolipids in asthma [33, 41]. S1P is generated from the phosphorylation of sphingosine by one of two sphingosine kinases (SphK1 and SphK2). S1P modulates an array of biological processes and functions as both, intracellular second messenger and extracellular ligand for five known G protein-coupled receptors, S1PR1-5, [8]. SphKs and S1PRs are ubiquitously expressed, including in bronchial epithelial and airway smooth muscle cells [42]. Both S1P and SphK are associated with key pathogenic features of asthma including airway smooth muscle cell hyperresponsiveness and lung inflammation [9, 43]. In mice, administration of exogenous S1P increased airway resistance, bronchial contraction, and recruitment of inflammatory cells, namely mast cells and eosinophils [44]. Both ceramides and S1P emerge from the recycling/salvage pathway of sphingolipid synthesis. There is evidence to suggest that inhibition of the de novo sphingolipid synthesis pathway results in a compensatory upregulation of the recycling/salvage pathways [45]. In context with the observations from clinical cohorts, there is strong evidence that childhood asthma can evolve from a complex, integrated disruption of sphingolipid hemostasis.
External factors influencing sphingolipid homeostasis
17q21 asthma risk alleles alter sphingolipid synthesis gene expression, leaving these pathways vulnerable to further disruption. Extrinsic factors, separately related to asthma, can influence sphingolipid synthesis. This includes perturbations of the host microbiome, maternal diet and obesity, and respiratory viruses. In the presence of 17q21 risk alleles, these elements may synergize to become the “second hit” necessary to shift sphingolipid metabolism toward asthma pathogenesis.
Maternal obesity during pregnancy
The in utero period is a critical time in lung development with long-term consequences for respiratory disorders [46,47,48,49,50,51]. There is strong evidence to support a link between childhood asthma and maternal obesity during pregnancy [50,51,52,53,54,55,56,57,58,59,60,61,62,63]. Large cohorts of mother-child dyads have shown an association between maternal obesity and early life bronchodilator use [58], but not atopic eczema or hay fever [55], suggesting that maternal obesity confers a non-atopic asthma phenotype. Interestingly, the VDAART study also found a relationship between childhood asthma and maternal sphingolipids in the third trimester. The risk of asthma correlated positively with maternal blood sphingomyelins levels and was inverse with maternal blood Sa1P [64]. The relationship between maternal obesity and sphingolipids requires further investigation, as both are strongly linked to non-atopic childhood asthma and early life wheeze.
Host microbiome
Since the proposal of the “hygiene hypothesis” by David Strachan in 1989, there have been extensive efforts to determine the contribution of the host-microbiome to asthma pathogenesis and immune dysregulation. Strachan postulated that improved standards of living and hygiene followed the reduction in household infections and also, increased risk of allergy [65]. Lack of infection resulted in poorly developed mechanisms of immune regulation including an unchecked Th2 dominant response. Interestingly, the Protection against Allergy Study in Rural Environment (PASTURE) found that the 17q21 genotypes that provide a risk for the development of asthma in wheezing infants also allow for environmental protection to allergen exposure [66]. It is well documented that patients with asthma display a relative dysbiosis of their lung, nasopharyngeal, and gut microbiomes [67, 68], even before the onset of the disease [69, 70]. Multiple studies have correlated the bacterial profile of infant stool, including colonization with Clostridium difficile and Escherichia coli and low levels of Bifidobacteria, with asthma development [71,72,73]. In both humans and mouse models, shifts in the gut microbiota have been associated with alterations to immune cell composition [74] and inflammatory mediators [75]. The field has progressed beyond bacterial community characterization to mapping host-microbe interaction and with that the metabolic consequences of the bacterial dysbiosis, including altered sphingolipid metabolism [76].
Bacterial sphingolipid synthesis is limited to members of the Bacteroidetes and selected Proteobacteria species. These bacteria are abundant in the mammalian gut where they can engage in a metabolic cross-talk with the host [77]. Previous studies have demonstrated the immunomodulatory activity of B. fragilis derived polysaccharides stimulation of CD4+ T cells and correction of Th1/Th2 imbalances [78]. A recent study found Bacteroides-derived sphingolipids are both sensed and incorporated into gut epithelial cell sphingolipid pathways [77]. In the gut, these bacterial sphingolipids can drive the recruitment and proliferation of invariant natural killer cells [76], a subset of T cells linked to multiple models of asthma [79]. Importantly, a recent report from the Baby Biome study found Cesarean section and intrapartum antibiotic use can significantly reduce the presence of Bacteroides species in the infant fecal microbiome [80]. These findings connect the intrapartum environment to microbiome-derived disturbances of sphingolipid homeostasis with meaningful implications for asthma development.
The infant gut microbiota is sensitive to multiple perinatal and early life exposures including maternal obesity, mode of delivery, gestational age, systemic antibiotics, breast vs formula feeding, cigarette smoke, household members, and pets. The relative contribution of each is currently under investigation. Efforts to reconstitute with gut microbiome with probiotics supplementation of Lactobacillus and Bifidobacterium with some studies showing reduction of asthma severity and others showing no effect [81].
Recently, studies have also shown that changes in the airway microbiome are associated with bronchiolitis in infants and young children [82,83,84]. Bronchiolitis shares several features of asthma, including airway inflammation and wheezing. Metabolomic analysis of nasopharyngeal samples from a cohort of infants hospitalized with bronchiolitis found a correlation between severe disease and upregulation of sphingolipid metabolism. Streptococcus, a dominant genus in the airway of infants with bronchiolitis, is positively associated with ceramide (18:2/16:0) and sphingomyelin (16:1/16:0) [85]. In the case of bronchiolitis and asthma, it remains to be determined if changes to sphingolipid metabolism precede or follow changes to the microbiome. It is clear however that the airway microbiome and metabolome are altered in the setting of early-life lower respiratory disease.
Respiratory viruses
Viral pathogens are responsible for most acute asthma attacks. There is substantial evidence that common respiratory viruses are not only a source of asthma-related morbidity, but also critical to disease inception. Infection with respiratory syncytial viral (RSV) [86] or human rhinovirus (HRV) [87] in the first 3 years of life significantly increases the risk of asthma later in childhood [88]. Epidemiologic studies have unmasked temporal relationships between early life viral infection and later allergen sensitization. Animal models have further revealed enhanced allergen sensitization and allergic airway inflammation following infection with influenza [89], RSV and HRV [90].
Both RSV and HRV interact with the sphingolipids during infection. RSV utilizes ganglioside GM1 in the assembly and release of viral particles [91]. RSV can also stimulate neutral ceramidase and SphK1 in lung epithelial cells prolonging their survival and in term, viral infection [92]. GWAS studies found early-life RV illness significantly strengthened the relationship between 17q21 asthma risk alleles and childhood asthma [93], suggesting RV is an important catalyst in asthma development. In vitro studies have exposed interactions between RV and sphingolipid synthesis. RV infection increases ceramide sphingolipids in epithelial cells [94]. Silencing of ORMDL3 in airway epithelial cells increases de novo sphingolipid synthesis and decreases expression of ICAM-1, the receptor for the majority of RV strains [95]. Inhibiting SPT also increases epithelial cell ICAM-1 expression [95] and RV replication [96]. These studies suggest genetic dampening of SPT activity may augment cellular responses to RV, allowing viral infection to further disrupt sphingolipid synthesis.
Therapeutic manipulation of the sphingolipid pathway
Collectively, these studies suggest infancy and the perinatal period represent a vulnerable time for children with 17q21 asthma risk alleles. The maternal metabolome, with its many influences, along with common intrapartum and early-life exposures can irrecoverably offset their suboptimal sphingolipid homeostasis. The question that emerges is if the sphingolipid synthesis pathway can serve as a novel therapeutic target for both, prevention, and treatment of childhood asthma. Pharmacologic modification of sphingolipid metabolism in mice can attenuate asthma symptoms. Intranasal administration of FTY720, a structural analog of sphingosine, which can be phosphorylated by SphK and then act as an antagonist for S1PRs, reduces airway inflammation and hyperreactivity [34]. Similar effects can be elicited by inhibition of SphK1 [97]. To overcome the effects of decreased sphingolipid de novo synthesis a recent study trialed fenretinide, a dihydroceramide desaturase inhibitor that indirectly stimulates the de novo pathway, and GlyH-101, a chloride channel blocker that increases levels of multiple sphingolipids by an unknown mechanism [29, 98]. Both agents increased de novo sphingolipid metabolites in lung epithelial cells and reduced agonist-induced contraction in proximal and peripheral airways [29]. These studies suggest pharmacologic both induction of the de novo pathway and antagonizing some effects S1P are viable options for mitigating airway hyperreactivity.
Conclusion
Asthma is a major cause of morbidity for children around the world. Multi-omic analyses of large pediatric cohorts have exposed several connections between sphingolipids and asthma/early life wheeze. These suggest asthma evolves from dynamic shifts in sphingolipid homeostasis, beginning with 17q21 asthma risk alleles and advancing with critical perinatal exposures that exacerbate genetic disruptions of sphingolipid metabolism. Maternal factors including weight, diet, mode of delivery, and intrapartum antibiotic use can directly and indirectly, via the gut microbiome, alter sphingolipid production. Post-partum, respiratory viral infections, and alterations of the airway microbiome can worsen these aberrations (Fig. 3). Together, these factors appear to tip the homeostatic balance toward lower de novo sphingolipid synthesis and increasing S1P.

Schematic model for the genetic and extrinsic exposures affecting sphingolipid-driven asthma pathogenesis in children. 17q21 asthma risk alleles induce suboptimal de novo sphingolipid synthesis, which is exacerbated by perinatal and early life exposures including maternal obesity, mode of delivery, and intrapartum care, alterations to the host microbiome and respiratory viral infections. Together, these factors appear to tip the homeostatic balance away from the protective effects of a normal sphingolipid synthesis and toward the pathogenic effects of decreased de novo synthesis and increased S1P. Pregnant woman illustration author Sudowoodo, title "Bathroom and medical people icons stock illustration, USA, icon, people, women, men." Child with inhaler illustration author Irina_Strelnikova title "Asthma stock illustration: Asthmatic, Child, Inhaling, Illness, Asthma Inhaler", both provided by istockphoto.com
Since the initial identification of 17q21 as an asthma risk region for childhood asthma, much attention has focused on how factors regulated and expressed in this region relate to the pathogenesis of childhood asthma. As a basic mechanism, genetically altered sphingolipid metabolism in children who are carriers of 17q21 asthma risk genotypes is thought to lead to functional effects on airway resistance and may act as a predisposing factor for the development of asthma. Study results in recent years suggest a strong association of 17q SNPs with the phenotype of persistent and intermediate wheezing in childhood, but not to allergic disease. It is possible that a specific form of childhood asthma exists that is characterized by decreased sphingolipid concentrations associated with 17q21 gene variants. Animal models suggest direct pharmacologic manipulation of the sphingolipid pathway can reset this balance. More work is needed to understand the role of sphingolipids in childhood asthma, as means of both preventing and treating this common disease.
Availability of data and materials
Not applicable
Abbreviations
- COPD:
-
Chronic obstructive pulmonary disease
- GWAS:
-
Genome-wide association studies
- SNP:
-
Single-nucleotide polymorphism
- ORMDL3:
-
ORMDL sphingolipid biosynthesis regulator 3
- SPT:
-
Serine palmitoyl transferase
- Sa1P:
-
Sphinganine-1-phosphate
- S1P:
-
Sphingosine-1-phosphate
- COPSAC2010 :
-
Copenhagen Prospective Study of Asthma in Childhood
- VDAART:
-
Vitamin D Antenatal Asthma Reduction Trial
- SphK:
-
Sphingosine kinases
- RV:
-
Human rhinoviruses
References
Pavord ID, Beasley R, Agusti A, Anderson GP, Bel E, Brusselle G, Cullinan P, Custovic A, Ducharme FM, Fahy JV, Frey U, Gibson P, Heaney LG, Holt PG, Humbert M, Lloyd CM, Marks G, Martinez FD, Sly PD, von Mutius E, Wenzel S, Zar HJ, Bush A (2018) After asthma: redefining airways diseases. Lancet 391:350–35Ѐ
Ferrante G, La Grutta S (2018) The Burden of Pediatric Asthma. Front Pediatr 6:186
Centers for disease control and prevention. data query from the child and adolescent health measurement initiative. Center for Child and Adolescent Health website [serial online] 2020. Available from: https://www.cdc.gov/asthma/most_recent_national_asthma_data.htm
Asher I, Pearce N (2014) Global burden of asthma among children. Int J Tuberc Lung Dis 18:1269–1278
Nurmagambetov T, Kuwahara R, Garbe P (2018) The economic burden of asthma in the United States, 2008-2013. Ann Am Thorac Soc 15:348–356
Berry CE, Billheimer D, Jenkins IC, Lu ZJ, Stern DA, Gerald LB, Carr TF, Guerra S, Morgan WJ, Wright AL, Martinez FD (2016) A distinct low lung function trajectory from childhood to the fourth decade of life. Am J Respir Crit Care Med 194:607–612
Bui DS, Lodge CJ, Burgess JA, Lowe AJ, Perret J, Bui MQ, Bowatte G, Gurrin L, Johns DP, Thompson BR, Hamilton GS, Frith PA, James AL, Thomas PS, Jarvis D, Svanes C, Russell M, Morrison SC, Feather I, Allen KJ, Wood-Baker R, Hopper J, Giles GG, Abramson MJ, Walters EH, Matheson MC, Dharmage SC (2018) Childhood predictors of lung function trajectories and future COPD risk: a prospective cohort study from the first to the sixth decade of life. Lancet Respir Med 6:535–544
Bush A (2019) Pathophysiological Mechanisms of Asthma. Front Pediatr 7:68
Ono JG, Worgall TS, Worgall S (2015) Airway reactivity and sphingolipids-implications for childhood asthma. Mole Cell Pediatr 2:13
Sturgill JL (2018) Sphingolipids and their enigmatic role in asthma. Adv Biol Regul 70:74–81
McGrath KW, Icitovic N, Boushey HA, Lazarus SC, Sutherland ER, Chinchilli VM, Fahy JV (2012) A large subgroup of mild-to-moderate asthma is persistently noneosinophilic. Am J Respir Crit Care Med 185:612–619
Peters MC, Mekonnen ZK, Yuan S, Bhakta NR, Woodruff PG, Fahy JV (2014) Measures of gene expression in sputum cells can identify TH2-high and TH2-low subtypes of asthma. J Allergy Clin Immunol 133:388–394
Gans MD, Gavrilova T (2020) Understanding the immunology of asthma: pathophysiology, biomarkers, and treatments for asthma endotypes. Paediatr Respir Rev 36:118–127
O'Byrne PM (2011) Therapeutic strategies to reduce asthma exacerbations. J Allergy Clin Immunol 128:257–263 quiz 264-255
Conrad LA, Cabana MD, Rastogi D (2021) Defining pediatric asthma: phenotypes to endotypes and beyond. Pediatr Res 90:45–51
Villaseñor A, Eguiluz-Gracia I, Moreira A, Wheelock CE, Escribese MM (2021) Metabolomics in the Identification of Biomarkers of Asthma. Metabolites 11
Papamichael MM, Katsardis C, Sarandi E, Georgaki S, Frima ES, Varvarigou A, Tsoukalas D (2021) Application of Metabolomics in Pediatric Asthma: Prediction, Diagnosis and Personalized Treatment. Metabolites 11
Bossley CJ, Fleming L, Gupta A, Regamey N, Frith J, Oates T, Tsartsali L, Lloyd CM, Bush A, Saglani S (2012) Pediatric severe asthma is characterized by eosinophilia and remodeling without T(H)2 cytokines. J Allergy Clin Immunol 129:974–982.e913
Kuo CS, Pavlidis S, Loza M, Baribaud F, Rowe A, Pandis I, Sousa A, Corfield J, Djukanovic R, Lutter R, Sterk PJ, Auffray C, Guo Y, Adcock IM, Chung KF (2017) T-helper cell type 2 (Th2) and non-Th2 molecular phenotypes of asthma using sputum transcriptomics in U-BIOPRED. Eur Respir J 49
Moore WC, Meyers DA, Wenzel SE, Teague WG, Li H, Li X, D'Agostino R Jr, Castro M, Curran-Everett D, Fitzpatrick AM, Gaston B, Jarjour NN, Sorkness R, Calhoun WJ, Chung KF, Comhair SA, Dweik RA, Israel E, Peters SP, Busse WW, Erzurum SC, Bleecker ER (2010) Identification of asthma phenotypes using cluster analysis in the severe asthma research program. Am J Respir Crit Care Med 181:315–323
Teague WG, Phillips BR, Fahy JV, Wenzel SE, Fitzpatrick AM, Moore WC, Hastie AT, Bleecker ER, Meyers DA, Peters SP, Castro M, Coverstone AM, Bacharier LB, Ly NP, Peters MC, Denlinger LC, Ramratnam S, Sorkness RL, Gaston BM, Erzurum SC, Comhair SAA, Myers RE, Zein J, DeBoer MD, Irani AM, Israel E, Levy B, Cardet JC, Phipatanakul W, Gaffin JM, Holguin F, Fajt ML, Aujla SJ, Mauger DT, Jarjour NN (2018) Baseline features of the severe asthma research program (SARP III) cohort: differences with age. J Allergy Clin Immunol Pract 6:545–554.e544
Zoratti EM, Krouse RZ, Babineau DC, Pongracic JA, O'Connor GT, Wood RA, Khurana Hershey GK, Kercsmar CM, Gruchalla RS, Kattan M, Teach SJ, Sigelman SM, Gergen PJ, Togias A, Visness CM, Busse WW, Liu AH (2016) Asthma phenotypes in inner-city children. J Allergy Clin Immunol 138:1016–1029
Moffatt MF, Kabesch M, Liang L, Dixon AL, Strachan D, Heath S, Depner M, von Berg A, Bufe A, Rietschel E, Heinzmann A, Simma B, Frischer T, Willis-Owen SA, Wong KC, Illig T, Vogelberg C, Weiland SK, von Mutius E, Abecasis GR, Farrall M, Gut IG, Lathrop GM, Cookson WO (2007) Genetic variants regulating ORMDL3 expression contribute to the risk of childhood asthma. Nature 448:470–473
Stein MM, Thompson EE, Schoettler N, Helling BA, Magnaye KM, Stanhope C, Igartua C, Morin A, Washington C 3rd, Nicolae D, Bonnelykke K, Ober C (2018) A decade of research on the 17q12-21 asthma locus: piecing together the puzzle. J Allergy Clin Immunol 142(749-764):e743
Halapi E, Gudbjartsson DF, Jonsdottir GM, Bjornsdottir US, Thorleifsson G, Helgadottir H, Williams C, Koppelman GH, Heinzmann A, Boezen HM, Jonasdottir A, Blondal T, Gudjonsson SA, Thorlacius T, Henry AP, Altmueller J, Krueger M, Shin HD, Uh ST, Cheong HS, Jonsdottir B, Ludviksson BR, Ludviksdottir D, Gislason D, Park CS, Deichmann K, Thompson PJ, Wjst M, Hall IP, Postma DS, Gislason T, Kong A, Jonsdottir I, Thorsteinsdottir U, Stefansson K (2010) A sequence variant on 17q21 is associated with age at onset and severity of asthma. Eur J Human Genet 18:902–908
Rago D, Pedersen CT, Huang M, Kelly RS, Gürdeniz G, Brustad N, Knihtilä H, Lee-Sarwar KA, Morin A, Rasmussen MA, Stokholm J, Bønnelykke K, Litonjua AA, Wheelock CE, Weiss ST, Lasky-Su J, Bisgaard H, Chawes BL (2021) Characteristics and mechanisms of a sphingolipid-associated childhood asthma endotype. Am J Respir Crit Care Med 203:853–863
Ono JG, Kim BI, Zhao Y, Christos PJ, Tesfaigzi Y, Worgall TS, Worgall S (2020) Decreased sphingolipid synthesis in children with 17q21 asthma-risk genotypes. J Clin Invest 130:921–926
Worgall TS, Veerappan A, Sung B, Kim BI, Weiner E, Bholah R, Silver RB, Jiang XC, Worgall S (2013) Impaired sphingolipid synthesis in the respiratory tract induces airway hyperreactivity. Sci Transl Med 5:186ra167
Heras AF, Veerappan A, Silver RB, Emala CW, Worgall TS, Perez-Zoghbi J, Worgall S (2020) Increasing Sphingolipid Synthesis Alleviates Airway Hyperreactivity. Am J Respir Cell Mol Biol 63:690–698
Wang Y, Niu Y, Zhang Z, Gable K, Gupta SD, Somashekarappa N, Han G, Zhao H, Myasnikov AG, Kalathur RC, Dunn TM, Lee C-H (2021) Structural insights into the regulation of human serine palmitoyltransferase complexes. Nat Struct Mole Biol 28:240–248
Li S, Xie T, Liu P, Wang L, Gong X (2021) Structural insights into the assembly and substrate selectivity of human SPT–ORMDL3 complex. Nat Struct Mole Biol 28:249–257
Acevedo N, Reinius LE, Greco D, Gref A, Orsmark-Pietras C, Persson H, Pershagen G, Hedlin G, Melén E, Scheynius A, Kere J, Söderhäll C (2015) Risk of childhood asthma is associated with CpG-site polymorphisms, regional DNA methylation and mRNA levels at the GSDMB/ORMDL3 locus. Hum Mol Genet 24:875–890
Breslow DK, Weissman JS (2010) Membranes in balance: mechanisms of sphingolipid homeostasis. Molecular Cell 40:267–279
Oyeniran C, Sturgill JL, Hait NC, Huang WC, Avni D, Maceyka M, Newton J, Allegood JC, Montpetit A, Conrad DH, Milstien S, Spiegel S (2015) Aberrant ORM (yeast)-like protein isoform 3 (ORMDL3) expression dysregulates ceramide homeostasis in cells and ceramide exacerbates allergic asthma in mice. J Allergy Clin Immunol 136(1035-1046):e1036
Perzanowski MS, Ono JG, Acosta LM, Kim BI, Divjan A, Miller R, Rundle A, Worgall S, Worgall TS (2017) Distinct serum sphingolipid profiles among school-aged children with exercise-induced wheeze and asthma persistence. Am J Respir Crit Care Med 195:1068–1070
Kraft ML (2016) Sphingolipid organization in the plasma membrane and the mechanisms that influence it. Front Cell Dev Biol 4:154
Yabu T, Shiba H, Shibasaki Y, Nakanishi T, Imamura S, Touhata K, Yamashita M (2015) Stress-induced ceramide generation and apoptosis via the phosphorylation and activation of nSMase1 by JNK signaling. Cell Death Different 22:258–273
Gandy KA, Canals D, Adada M, Wada M, Roddy P, Snider AJ, Hannun YA, Obeid LM (2013) Sphingosine 1-phosphate induces filopodia formation through S1PR2 activation of ERM proteins. Biochem J 449:661–672
Miller M, Rosenthal P, Beppu A, Mueller JL, Hoffman HM, Tam AB, Doherty TA, McGeough MD, Pena CA, Suzukawa M, Niwa M, Broide DH (2014) ORMDL3 transgenic mice have increased airway remodeling and airway responsiveness characteristic of asthma. J Immunol 192:3475–3487
Das S, Miller M, Broide DH (2017) Chromosome 17q21 Genes ORMDL3 and GSDMB in asthma and immune diseases. Adv Immunol 135:1–52
Hannun YA, Obeid LM (2008) Principles of bioactive lipid signalling: lessons from sphingolipids. Nat Rev Mole Cell Biol 9:139–150
Nishiuma T, Nishimura Y, Okada T, Kuramoto E, Kotani Y, Jahangeer S, Nakamura S (2008) Inhalation of sphingosine kinase inhibitor attenuates airway inflammation in asthmatic mouse model. Am J Physiol Lung Cell Mol Physiol 294:L1085–L1093
Clarke DL, Dakshinamurti S, Larsson AK, Ward JE, Yamasaki A (2009) Lipid metabolites as regulators of airway smooth muscle function. Pulm Pharmacol Ther 22:426–435
Kume H, Takeda N, Oguma T, Ito S, Kondo M, Ito Y, Shimokata K (2007) Sphingosine 1-phosphate causes airway hyper-reactivity by rho-mediated myosin phosphatase inactivation. J Pharmacol Exp Ther 320:766–773
Hagen-Euteneuer N, Lütjohann D, Park H, Merrill AH, van Echten-Deckert G (2012) Sphingosine 1-Phosphate (S1P) lyase deficiency increases sphingolipid formation via recycling at the expense of de novo biosynthesis in neurons*. J Biol Chem 287:9128–9136
Gilliland FD, Li YF, Peters JM (2001) Effects of maternal smoking during pregnancy and environmental tobacco smoke on asthma and wheezing in children. Am J Respir Crit Care Med 163:429–436
Azad MB, Moyce BL, Guillemette L, Pascoe CD, Wicklow B, McGavock JM, Halayko AJ, Dolinsky VW (2017) Diabetes in pregnancy and lung health in offspring: developmental origins of respiratory disease. Paediatr Respir Rev 21:19–26
Brauer M, Hoek G, Smit HA, de Jongste JC, Gerritsen J, Postma DS, Kerkhof M, Brunekreef B (2007) Air pollution and development of asthma, allergy and infections in a birth cohort. Eur Respir J 29:879–888
DiFranza JR, Aligne CA, Weitzman M (2004) Prenatal and postnatal environmental tobacco smoke exposure and children’s health. Pediatrics 113:1007–1015
MacDonald KD, Moran AR, Scherman AJ, McEvoy CT, Platteau AS (2017) Maternal high-fat diet in mice leads to innate airway hyperresponsiveness in the adult offspring. Physiol Rep 5
Mayor RS, Finch KE, Zehr J, Morselli E, Neinast MD, Frank AP, Hahner LD, Wang J, Rakheja D, Palmer BF, Rosenfeld CR, Savani RC, Clegg DJ (2015) Maternal high-fat diet is associated with impaired fetal lung development. Am J Physiol Lung Cell Mol Physiol 309:L360–L368
E-Lacerda RR, Teixeira CJ, Bordin S, Antunes E, Anhê GF (2019) Maternal Obesity in Mice Exacerbates the Allergic Inflammatory Response in the Airways of Male Offspring. Nutrients 11(12):2902. https://doi.org/10.3390/nu11122902
Caudri D, Savenije OE, Smit HA, Postma DS, Koppelman GH, Wijga AH, Kerkhof M, Gehring U, Hoekstra MO, Brunekreef B, de Jongste JC (2013) Perinatal risk factors for wheezing phenotypes in the first 8 years of life. Clin Exp Allergy 43:1395–1405
Haberg SE, Stigum H, London SJ, Nystad W, Nafstad P (2009) Maternal obesity in pregnancy and respiratory health in early childhood. Paediatr Perinat Epidemiol 23:352–362
Harpsoe MC, Basit S, Bager P, Wohlfahrt J, Benn CS, Nohr EA, Linneberg A, Jess T (2013) Maternal obesity, gestational weight gain, and risk of asthma and atopic disease in offspring: a study within the Danish National Birth Cohort. J Allergy Clin Immunol 131:1033–1040
Kumar R, Story RE, Pongracic JA, Hong X, Arguelles L, Wang G, Kuptsova-Clarkson N, Pearson C, Ortiz K, Bonzagni A, Apollon S, Fu L, Bauchner H, Wang X (2010) Maternal pre-pregnancy obesity and recurrent wheezing in early childhood. Pediatr Allergy Immunol Pulmonol 23:183–190
Leermakers ET, Sonnenschein-van der Voort AM, Gaillard R, Hofman A, de Jongste JC, Jaddoe VW, Duijts L (2013) Maternal weight, gestational weight gain and preschool wheezing: the Generation R Study. Eur Respir J 42:1234–1243
MacDonald KD, Vesco KK, Funk KL, Donovan J, Nguyen T, Chen Z, Lapidus JA, Stevens VJ, McEvoy CT (2016) Maternal body mass index before pregnancy is associated with increased bronchodilator dispensing in early childhood: a cross-sectional study. Pediatr Pulmonol 51:803–811
Oliveti JF, Kercsmar CM, Redline S (1996) Pre- and perinatal risk factors for asthma in inner city African-American children. Am J Epidemiol 143:570–577
Polinski KJ, Liu J, Boghossian NS, McLain AC (2017) Maternal obesity, gestational weight gain, and asthma in offspring. Prev Chronic Dis 14:E109
Rusconi F, Galassi C, Forastiere F, Bellasio M, De Sario M, Ciccone G, Brunetti L, Chellini E, Corbo G, La Grutta S, Lombardi E, Piffer S, Talassi F, Biggeri A, Pearce N (2007) Maternal complications and procedures in pregnancy and at birth and wheezing phenotypes in children. Am J Respir Crit Care Med 175:16–21
Wright RJ, Fisher K, Chiu YH, Wright RO, Fein R, Cohen S, Coull BA (2013) Disrupted prenatal maternal cortisol, maternal obesity, and childhood wheeze. Insights into prenatal programming. Am J Respir Crit Care Med 187:1186–1193
MacDonald KD, Scherman AJ, Vesco KK, Funk KL, Markwardt SE, Lapidus JA, Stevens VJ, McEvoy CT (2018) Prepregnancy extreme obesity is associated with increased steroid dispensing in early childhood. Pediatr Allergy Immunol Pulmonol 31:84–90
Huang M, Kelly RS, Chu SH, Kachroo P, Gurdeniz G, Chawes BL, Bisgaard H, Weiss ST, Lasky-Su J (2021) Maternal Metabolome in Pregnancy and Childhood Asthma or Recurrent Wheeze in the Vitamin D Antenatal Asthma Reduction Trial. Metabolites 11
Strachan DP (1989) Hay fever, hygiene, and household size. BMJ 299:1259–1260
Loss GJ, Depner M, Hose AJ, Genuneit J, Karvonen AM, Hyvärinen A, Roduit C, Kabesch M, Lauener R, Pfefferle PI, Pekkanen J, Dalphin JC, Riedler J, Braun-Fahrländer C, von Mutius E, Ege MJ (2016) The Early Development of Wheeze. Environmental determinants and genetic susceptibility at 17q21. Am J Respir Crit Care Med 193:889–897
Hilty M, Burke C, Pedro H, Cardenas P, Bush A, Bossley C, Davies J, Ervine A, Poulter L, Pachter L, Moffatt MF, Cookson WO (2010) Disordered microbial communities in asthmatic airways. PLoS One 5:e8578
Huang YJ, Nelson CE, Brodie EL, Desantis TZ, Baek MS, Liu J, Woyke T, Allgaier M, Bristow J, Wiener-Kronish JP, Sutherland ER, King TS, Icitovic N, Martin RJ, Calhoun WJ, Castro M, Denlinger LC, Dimango E, Kraft M, Peters SP, Wasserman SI, Wechsler ME, Boushey HA, Lynch SV (2011) Airway microbiota and bronchial hyperresponsiveness in patients with suboptimally controlled asthma. J Allergy Clin Immunol 127:372–381 e371-373
Bisgaard H, Hermansen MN, Buchvald F, Loland L, Halkjaer LB, Bønnelykke K, Brasholt M, Heltberg A, Vissing NH, Thorsen SV, Stage M, Pipper CB (2007) Childhood asthma after bacterial colonization of the airway in neonates. N Engl J Med 357:1487–1495
Liu AH (2015) Revisiting the hygiene hypothesis for allergy and asthma. J Allergy Clin Immunol 136:860–865
Johnson CC, Ownby DR (2017) The infant gut bacterial microbiota and risk of pediatric asthma and allergic diseases. Transl Res 179:60–70
Björkstén B, Sepp E, Julge K, Voor T, Mikelsaar M (2001) Allergy development and the intestinal microflora during the first year of life. J Allergy Clin Immunol 108:516–520
Penders J, Thijs C, van den Brandt PA, Kummeling I, Snijders B, Stelma F, Adams H, van Ree R, Stobberingh EE (2007) Gut microbiota composition and development of atopic manifestations in infancy: the KOALA Birth Cohort Study. Gut 56:661–667
Karimi K, Inman MD, Bienenstock J, Forsythe P (2009) Lactobacillus reuteri-induced regulatory T cells protect against an allergic airway response in mice. Am J Respir Crit Care Med 179:186–193
West CE, Rydén P, Lundin D, Engstrand L, Tulic MK, Prescott SL (2015) Gut microbiome and innate immune response patterns in IgE-associated eczema. Clin Exp Allergy 45:1419–1429
Lee-Sarwar KA, Lasky-Su J, Kelly RS, Litonjua AA, Weiss ST (2020) Gut Microbial-Derived Metabolomics of Asthma. Metabolites 10
Johnson EL, Heaver SL, Waters JL, Kim BI, Bretin A, Goodman AL, Gewirtz AT, Worgall TS, Ley RE (2020) Sphingolipids produced by gut bacteria enter host metabolic pathways impacting ceramide levels. Nat Commun 11:2471
Mazmanian SK, Liu CH, Tzianabos AO, Kasper DL (2005) An immunomodulatory molecule of symbiotic bacteria directs maturation of the host immune system. Cell 122:107–118
Matangkasombut P, Pichavant M, DeKruyff RH, Umetsu DT (2009) Natural killer T cells and the regulation of asthma. Mucosal Immunol 2:383–392
Shao Y, Forster SC, Tsaliki E, Vervier K, Strang A, Simpson N, Kumar N, Stares MD, Rodger A, Brocklehurst P, Field N, Lawley TD (2019) Stunted microbiota and opportunistic pathogen colonization in caesarean-section birth. Nature 574:117–121
Chunxi L, Haiyue L, Yanxia L, Jianbing P, Jin S (2020) The gut microbiota and respiratory diseases: new evidence. J Immunol Res 2020:2340670
Luna PN, Hasegawa K, Ajami NJ, Espinola JA, Henke DM, Petrosino JF, Piedra PA, Sullivan AF, Camargo CA Jr, Shaw CA, Mansbach JM (2018) The association between anterior nares and nasopharyngeal microbiota in infants hospitalized for bronchiolitis. Microbiome 6:2
Hasegawa K, Mansbach JM, Ajami NJ, Espinola JA, Henke DM, Petrosino JF, Piedra PA, Shaw CA, Sullivan AF, Camargo CA Jr (2016) Association of nasopharyngeal microbiota profiles with bronchiolitis severity in infants hospitalised for bronchiolitis. Eur Respir J 48:1329–1339
Teo SM, Mok D, Pham K, Kusel M, Serralha M, Troy N, Holt BJ, Hales BJ, Walker ML, Hollams E, Bochkov YA, Grindle K, Johnston SL, Gern JE, Sly PD, Holt PG, Holt KE, Inouye M (2015) The infant nasopharyngeal microbiome impacts severity of lower respiratory infection and risk of asthma development. Cell Host Microbe 17:704–715
Stewart CJ, Mansbach JM, Wong MC, Ajami NJ, Petrosino JF, Camargo CA Jr, Hasegawa K (2017) Associations of nasopharyngeal metabolome and microbiome with severity among infants with bronchiolitis. A multiomic analysis. Am J Respir Crit Care Med 196:882–891
Stein RT, Sherrill D, Morgan WJ, Holberg CJ, Halonen M, Taussig LM, Wright AL, Martinez FD (1999) Respiratory syncytial virus in early life and risk of wheeze and allergy by age 13 years. Lancet 354:541–545
Lemanske RF Jr, Jackson DJ, Gangnon RE, Evans MD, Li Z, Shult PA, Kirk CJ, Reisdorf E, Roberg KA, Anderson EL, Carlson-Dakes KT, Adler KJ, Gilbertson-White S, Pappas TE, Dasilva DF, Tisler CJ, Gern JE (2005) Rhinovirus illnesses during infancy predict subsequent childhood wheezing. J Allergy Clin Immunol 116:571–577
Piedimonte G (2013) Respiratory syncytial virus and asthma: speed-dating or long-term relationship? Curr Opin Pediatr 25:344–349
Wu Q, Jorde I, Kershaw O, Jeron A, Bruder D, Schreiber J, Stegemann-Koniszewski S (2020) Resolved influenza A virus infection has extended effects on lung homeostasis and attenuates allergic airway inflammation in a mouse model. Microorganisms 8:1878
Schwarze J, Gelfand EW (2002) Respiratory viral infections as promoters of allergic sensitization and asthma in animal models. Eur Respir J 19:341–349
Chang TH, Segovia J, Sabbah A, Mgbemena V, Bose S (2012) Cholesterol-rich lipid rafts are required for release of infectious human respiratory syncytial virus particles. Virology 422:205–213
Monick MM, Cameron K, Powers LS, Butler NS, McCoy D, Mallampalli RK, Hunninghake GW (2004) Sphingosine kinase mediates activation of extracellular signal-related kinase and Akt by respiratory syncytial virus. Am J Respir Cell Mol Biol 30:844–852
Caliskan M, Bochkov YA, Kreiner-Moller E, Bonnelykke K, Stein MM, Du G, Bisgaard H, Jackson DJ, Gern JE, Lemanske RF Jr, Nicolae DL, Ober C (2013) Rhinovirus wheezing illness and genetic risk of childhood-onset asthma. N Engl J Med 368:1398–1407
Dreschers S, Franz P, Dumitru C, Wilker B, Jahnke K, Gulbins E (2007) Infections with human rhinovirus induce the formation of distinct functional membrane domains. Cell Physiol Biochem 20:241–254
Zhang Y, Willis-Owen SAG, Spiegel S, Lloyd CM, Moffatt MF, Cookson WOCM (2019) The ORMDL3 Asthma gene regulates ICAM1 and has multiple effects on cellular inflammation. Am J Respir Crit Care Med 199:478–488
Liu Y, Bochkov YA, Eickhoff JC, Hu T, Zumwalde NA, Tan JW, Lopez C, Fichtinger PS, Reddy TR, Overmyer KA, Gumperz JE, Coon J, Mathur SK, Gern JE, Smith JA (2020) Orosomucoid-like 3 supports rhinovirus replication in human epithelial cells. Am J Respir Cell Mol Biol 62:783–792
Price MM, Oskeritzian CA, Falanga YT, Harikumar KB, Allegood JC, Alvarez SE, Conrad D, Ryan JJ, Milstien S, Spiegel S (2013) A specific sphingosine kinase 1 inhibitor attenuates airway hyperresponsiveness and inflammation in a mast cell-dependent murine model of allergic asthma. J Allergy Clin Immunol 131:501–511.e501
Lam M, Bourke JE (2020) Solving the riddle: targeting the imbalance of sphingolipids in asthma to oppose airway hyperresponsiveness. Am J Respir Cell Mol Biol 63:555–557
Acknowledgements
We acknowledge the generous support of Ronay Menschel, Christine and Pasco Alfaro, and Joanna Weiss.
Funding
SW is supported by the National Institute of Health grants R21 AI140724. E.W. was supported by the National Center for Advancing Translational Sciences of the NIH KL2 TR0002385.
Author information
Authors and Affiliations
Contributions
EW and SW wrote the manuscript and reviewed the literature. The authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable
Consent for publication
Not applicable
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wasserman, E., Worgall, S. Perinatal origins of chronic lung disease: mechanisms–prevention–therapy—sphingolipid metabolism and the genetic and perinatal origins of childhood asthma. Mol Cell Pediatr 8, 22 (2021). https://doi.org/10.1186/s40348-021-00130-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40348-021-00130-y