
Abstract
Isolated growth hormone deficiency (GHD) is defined by growth failure in combination with retarded bone age, low serum insulin-like growth factor-1, and insufficient GH peaks in two independent GH stimulation tests. Congenital GHD can present at any age and can be associated with significant malformations of the pituitary-hypothalamic region or the midline of the brain. In rare instances, genetic analysis reveals germline mutations of transcription factors involved in embryogenesis of the pituitary gland and the hypothalamus. Acquired GHD is caused by radiation, inflammation, or tumor growth. In contrast to organic GHD, idiopathic forms are more frequent and remain unexplained.
There is a risk of progression from isolated GHD to combined pituitary hormone deficiency (> 5% for the total group), which is clearly increased in children with organic GHD, especially with significant malformation of the pituitary gland. Therefore, it is prudent to exclude additional pituitary hormone deficiencies in the follow-up of children with isolated GHD by clinical and radiological observations and endocrine baseline tests. In contrast to primary disorders of endocrine glands, secondary deficiency is frequently milder in its clinical manifestation. The pituitary hormone deficiencies can develop over time from mild insufficiency to severe deficiency. This review summarizes the current knowledge on diagnostics and therapy of additional pituitary hormone deficits occurring during rhGH treatment in children initially diagnosed with isolated GHD. Although risk factors are known, there are no absolute criteria enabling exclusion of children without any risk of progress to combined pituitary hormone deficiency. Lifelong monitoring of the endocrine function of the pituitary gland is recommended in humans with organic GHD. This paper is the essence of a workshop of pediatric endocrinologists who screened the literature for evidence with respect to evolving pituitary deficits in initially isolated GHD, their diagnosis and treatment.
Similar content being viewed by others

Background
Childhood-onset growth hormone deficiency (GHD) is frequently occurring as an isolated hormone deficiency [1], but in a subgroup of these children, additional pituitary hormone deficiencies evolve over time. This development implies either a genetically determined vulnerability or masked insufficiency of pituitary or hypothalamic cells or an acquired damage of the hypothalamic-pituitary axis with progressive dysfunction. Treatment with recombinant human GH (rhGH) may unmask TSH deficiency in some patients with GHD [2]. Additional pituitary deficits also arise over time in monogenic hypopituitarism caused by mutations in PROP1, POU1F1, HESX1, and other developmental genes [3]. Acquired GHD due to suprasellar tumor, cranial irradiation, or other causes of damage is accompanied or followed by deficits of other pituitary hormones, which frequently evolve in a characteristic pattern [4]. Therefore, the diagnosis of GHD may implicate the potential rise of additional pituitary hormone deficits.
There is no consensus on how to monitor and identify those patients with GHD in need for treatment with additional hormones and which diagnostic and prognostic markers are informative. The reported prevalence of evolving pituitary deficits in GHD clearly depends on the study inclusion criteria (idiopathic versus organic GHD), the duration of follow-up, and the clinical criteria used for the indication of dynamic testing. It seems rational to assume a higher likelihood of upcoming additional pituitary hormone deficiencies in children with monogenic disorders, with developmental disorders of the pituitary gland and the midline, after cranial irradiation, and with an inflammatory process or tumor in the hypothalamic-pituitary region. This paper is the essence of a workshop of pediatric endocrinologists who screened the literature for evidence with respect to evolving pituitary deficits in initially isolated GHD, their diagnosis and treatment.
Prognostic markers
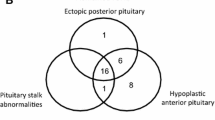
Large long-term follow-up studies evaluating major risk factors that predict the occurrence of additional pituitary hormone deficiencies in isolated GHD are missing. The largest study with this target evaluating data from the Genesis study (n = 5805) had a relatively short median follow-up of 4 years in a subgroup with 1757 patients with an incidence of combined pituitary hormone deficiency (CPHD) of 5.5% [5]. An analysis of the literature on prognostic risk factors for the progression from isolated GHD to CPHD was recently reported by Cerbone and Dattani [4]. Accordingly, children with organic GHD etiology are more likely to develop CPHD than those with idiopathic GHD. Especially, children with structural abnormalities of the pituitary gland and the brain midline like absent pituitary stalk, ectopic posterior pituitary, abnormal corpus callosum, empty sella, and septo-optic dysplasia spectrum are prone to develop CPHD. Similarly, the relatively rare genetic defects in transcription factors such as HESX1, PROP1, POU1F1, LHX3, LHX4, SOX2, and SOX3 are associated with variable phenotypes (some only presenting with isolated GHD) and frequently associated with the development of CPHD. This is even true for children with the dominantly inherited isolated GHD type 2 with splice site mutations of GH1, who may develop additional pituitary hormone deficiencies in rare instances [6]. Similarly, acquired damage of the hypothalamic-pituitary axis is very likely to proceed from isolated GHD to multiple pituitary hormone deficiency [7], and in specific damages like the post-radiation condition, prediction is possible based on the dose of radiation [8]. The severity of isolated GHD may also be a hint to additional hormone deficiencies evolving over time [5]. In contrast, children with partial GHD or mild idiopathic GHD are rarely affected by additional pituitary hormone deficiencies.
Therefore, it seems reasonable to estimate the risk for developing combined pituitary hormone deficiency individually and to judge clinical and laboratory data based on this specific risk estimate. Moderately reduced serum hormone concentrations should be controlled earlier in organic GHD than in idiopathic GHD. Stimulatory tests for the detection of additional pituitary hormone deficits should earlier be performed in organic GHD than in idiopathic GHD. Such timing would enable early detection of patients in need of additional hormone therapy and also avoid false positives, which may be falsely substituted with hormones.
Nevertheless, it has to be stressed that the diagnosis of idiopathic isolated GHD does not exclude the occurrence of additional hormone deficiencies in the future implicating a thorough follow-up of the pituitary axes in all children with isolated GHD.
Diagnosis of TSH deficiency
Thyroid-stimulating hormone (TSH) deficiency is the most frequent secondary pituitary hormone deficiency in children initially diagnosed with GHD, with a reported prevalence of 6.3% during the first 2 years of treatment [2]. The clinical characteristics of TSH deficiency are milder than in primary hypothyroidism. An insufficient increase of IGF-1 and a low growth response to rhGH treatment have been suggested as possible signs of evolving TSH deficiency [9], but this has not been studied systematically. The ultrasound of the thyroid gland may be normal in TSH deficiency at diagnosis. After 1-year substitutional treatment of children and adolescents with TSH deficiency, the volume of the thyroid gland did not change [10].
The laboratory confirmation of TSH deficiency is done by consecutive measurements of basal serum fT4 which stay below the reference interval in the presence of a low-normal to moderately elevated serum TSH concentration [11]. The TRH challenge is not reliable, does not provide additional information, and should not be used [11, 12]. TSH deficiency should be distinguished from the frequently and early occurring decrease of fT4 during the first year of rhGH treatment, which is probably a reflection of an enhanced conversion of thyroxine to triiodothyronine and associated with a rise of fT3 [13, 14]. The consecutive fT4 nadir is transient and disappears after 6 months of rhGH treatment in the majority of treated children [10, 15]. However, it must be stressed that both TSH deficiency and enhanced conversion of thyroxine to triiodothyronine can cause fT4 serum levels below the reference interval after 3 months on rhGH. In the absence of a significant malformation of the pituitary gland or other historical, clinical, or genetic findings implying high likelihood of the occurrence of TSH deficiency, the experts considered it rational to wait and control fT4 3-monthly before treatment with l-thyroxine is initiated. The exception is any very low fT4 concentration that is unlikely to be transient.
Treatment of TSH deficiency
The treatment with l-thyroxine can cause a life-threatening adrenal crisis in patients with untreated ACTH deficiency [12, 16]. Therefore, hypocortisolism should be excluded before thyroid hormone replacement. In the absence of randomized trials, experts recommend a low l-thyroxine starting dose (i.e., 25 to 50 μg/day total) that should be increased weekly to reach the target of around 1.5 μg/kg day (for school children) within approx. 1 month. The main aim is a serum fT4 concentration in the upper normal range (> 50th percentile) accompanied by a normal fT3. Experts’ opinions are heterogeneous with respect to TSH serum values [12, 17]; most emphasize the invalidity of this parameter in the presence of TSH deficiency. Nevertheless, it must be stressed that suppressed TSH is a very frequent finding in well-treated children with TSH deficiency and certainly in the majority of cases not a sign of overdosing. In children with GHD and TSH deficiency on rhGH, the introduction of l-thyroxine treatment was shown to increase resting energy expenditure and shorten the myocardial isovolumic contraction time, which was correlated to total T3, but not total T4 [18]. It seems rational to reevaluate the thyroid axis by a drug holiday for 4 weeks, especially if GHD is not confirmed in adolescence.
Diagnosis of ACTH deficiency
Adrenocorticotropic hormone (ACTH) deficiency rarely occurs as an evolving pituitary deficit after the diagnosis of isolated GHD, and frequently, it is the last one becoming deficient. Reported prevalence ranges from less than 1 to 12%, with higher numbers in children with concomitant TSH deficiency or pituitary malformations [5, 19, 20]. ACTH deficiency has been described in patients with hypopituitarism caused by PROP1, POU1F1, LHX3, LHX4, and GLI2 mutations. In these instances, the diagnosis was frequently made at the initial presentation. Rarely, ACTH deficiency has been reported in children with autosomal dominant GHD due to GH1 mutations [6].
ACTH deficiency can evolve in acquired hypopituitarism after cranial irradiation > 30 Gy [21], sellar and suprasellar tumors, hemochromatosis, sarcoidosis, Langerhans cell histiocytosis and after tumor surgery. Here, indeed, hypopituitarism may start with isolated GHD and then proceed to CPHD including ACTH deficiency. Central hypocortisolism causes the same signs and symptoms as Addison’s disease, but in contrast to primary hypocortisolism, it is characterized by normal or hypopigmented skin and normal mineralocorticoid reserve. In addition, the clinical phenotype may be milder and adrenal crisis at the time of diagnosis less frequent than in primary adrenal insufficiency [22]. Routine monitoring of adrenal sufficiency by measuring morning serum cortisol may minimize this risk in children with organic GHD.
The diagnosis of ACTH deficiency should be based on testing and clinical suspicion, which may be raised by failure to thrive, loss of appetite, loss of weight, hypopigmentation, low blood pressure, dizziness, decreased vitality, and other findings. The recommended baseline test is measurement of the morning cortisol [23]. Morning serum cortisol concentrations > 275 nmol/l are considered to exclude ACTH deficiency, and concentrations < 85 nmol/l confirm ACTH deficiency [23]. DHEAS production of the adrenal gland is also ACTH dependent. Whereas the measurement of serum DHEAS is a good baseline test in postpubertal adolescents, this is not the case in younger children and adolescents with still rising DHEAS production [24].
In the case of borderline low morning cortisol values (< 275, but > 85 nmol/l) and clinical suspicion, a short Synacthen test is recommended. There is still discussion about the best way to this test. Some experts recommended the low-dose Synacthen test (1 μg i.v. absolute) because of increased accuracy [25]. Others prefer the standard Synacthen test (125 μg or 250 μg i.v. absolute) claiming that the exact administration of 1 μg Synacthen by diluting it from an ampoule containing 250 μg may be technically challenging, even as Synacthen sticks to plastic and glass surfaces [26]. A systematic review and meta-analysis did not detect an advantage of the low-dose versus the high-dose Synacthen dose [27]. Recommended cutoffs vary. Kazlauskaite and Maghnie recommended for both tests a stimulated serum cortisol concentration > 490 nmol/l for exclusion and < 360 nmol/l for confirmation of ACTH deficiency [23]. In dubious cases with values in between, an additional insulin-hypoglycemia test (ITT) or glucagon test is recommended. The ITT cutoff is 490 or 550 nmol/l [23, 28]. The ITT is potentially dangerous, and its use should be restricted to older children.
Sometimes, hormone substitution may precipitate ACTH deficiency: treatment with rhGH attenuates 11β-HSD-type-1 isoenzyme activity, thereby leading to reduced cortisone-to-cortisol conversion [29]. Treatment with l-thyroxine can unmask ACTH deficiency and provoke adrenal crisis [16].
Treatment of ACTH deficiency
Oral hydrocortisone is the first choice of treatment of secondary adrenal insufficiency in childhood. The recommended dose is 8–10 mg/m2 BSA TID daily [30]. Traditionally, used hydrocortisone doses are lower than in primary adrenal insufficiency [31]. Residual ACTH secretion, concomitant therapies, and diseases may influence the replacement dose; patients with hypopituitarism on treatment with rhGH and estrogens may need higher glucocorticoid dose as compared to patients in whom GHD and hypogonadism are not replaced [32].
The strategy of personalized dosing as well as the teaching of the patient and caregivers regarding stress-dose and emergency glucocorticoid administration does not differ from primary adrenal insufficiency. However, published guidelines for children do not exist [33]. In female adolescents with central adrenal insufficiency that suffer from depression or lack of libido, the additional hormone replacement with 25 mg DHEA per day is beneficial according to a randomized controlled trial [34].
The incidence of adrenal crisis in individuals with adrenal insufficiency when treated is approximately 5–10 events per 100 patient years in adults [35]. The risk of adrenal crisis seems to be lower in secondary than in primary adrenal insufficiency, but pediatric data are lacking [36]. Death in children monitored during rhGH treatment could be traced back to adrenal crisis related to ACTH deficiency in 12–25% of the cases [37].
There are no good biochemical markers for monitoring glucocorticoid replacement in patients with ACTH deficiency. Glucocorticoid replacement should be based on the general dose recommendations, well-being and physical vitality of the patient, physiological increase in body weight, normal fat distribution, normal height velocity, and normal blood pressure as suggested for patients with primary adrenal insufficiency [38]. The hydrocortisone dose should be kept as low as possible.
Diagnosis of gonadotropin deficiency
Gonadotropin deficiency is the second most frequent pituitary hormone deficiency evolving in children with isolated GHD; the estimated prevalence ranges from 4.5 to 38% [20, 39]. However, as gonadotropin deficiency is clinically silent in childhood (at least in girls), estimates based on cohorts of children of any age are likely underestimations. Some hypothalamic syndromes like Prader-Willi syndrome (PWS) are characterized by frequent combination of both GH and gonadotropin deficiency [40]. Mutations of the developmental gene SOX2 can cause anophthalmia, esophageal atresia, and genital tract abnormalities associated with GH and gonadotropin deficiency [41]. The most frequent monogenic defect of pituitary development with recessive inheritance is caused by PROP1 mutations, which is characterized by the combination of GH, TSH, ACTH, and gonadotropin deficiency [42].
Micropenis and bilateral cryptorchidism are important clinical clues in male infants. The earliest window of opportunity to diagnose gonadotropin deficiency by hormone measurements is minipuberty during the first 4 to 6 months of life [43] when sex hormones and gonadotropins reach midpubertal serum levels in both genders during this time period [44]. There are examples for dissociation between minipuberty and puberty in certain disorders (Prader-Willi syndrome, hypogonadotropic hypogonadism due to KISS1R loss of function) that must be kept in mind [45].
At pubertal age, the manifestation of gonadotropin deficiency comprises the broad spectrum from no entry into puberty to an uncompleted or prolonged puberty. In adolescents with IGHD, the differential diagnosis of delayed puberty between constitutional delay in growth and puberty (CDGP) or hypogonadotropic hypogonadism can be difficult. In the presence of idiopathic IGHD and a positive family history for CDGP, the likelihood of gonadotropin deficiency is low. The likelihood increases in organic isolated GHD, in the absence of a positive family history for CDGP, and in boys with a history of bilateral orchidopexy or micropenis. The distinction on biochemical parameters includes the baseline measurement of inhibin B, LH, FSH, and sex hormones [46]. Reference intervals and cutoffs, however, heavily rely on the assays used: therefore, one should prefer immunoassays or liquid chromatography-tandem MS assays with adequate reference data for the pediatric population. The most accurate dynamic test is the GnRH agonist challenge with an LH cutoff at 4 h being approx. 5 IU/l in adolescent boys [47]. For girls, data are sparse. According to a recent small study, stimulated FSH after GnRH challenge is more discriminatory than LH in adolescent girls; the cutoff was 11 IU/l [48]. In adolescents with isolated GHD, it is crucial to diagnose gonadotropin deficiency as early as possible, as treatment should be offered when the peer group goes into puberty.
Treatment of gonadotropin deficiency
The choice of drug in girls with gonadotropin deficiency is estradiol. In a recent international consensus statement, transdermal estradiol substitution was preferably recommended started at low doses (0.06 μg/kg nocturnally) and increased slowly over 24 months to ensure normal breast development. Estradiol dosing is thereby aiming to mimic estradiol levels during gonadarche. A typical target dose in adulthood would be 50 μg estradiol daily by patch, or one to two pumps of 0.06% estradiol gel [49]. Subsequently, cyclic progestin should be added for 14 days. The pleaded advantages in comparison to oral treatment are minimization of the first path effect, higher IGF-1 serum levels [50], better feminization [51], and potentially a better cardiovascular risk profile in adulthood [52, 53]. Major disadvantages are the lack of suitable estradiol patches, the lack of long-term trials comparing transdermal and oral treatment, the lack of knowledge with respect to adherence, and the absence of approval for the transdermal indication. In a recent paper, Donaldson et al. offered detailed instructions how the recommended low estradiol doses can be reached by cutting the available estradiol patches into small pieces and proposed a protocol for estradiol dosing during a 3-year pubertal induction [54]. Fertility treatment is successful in the majority of women with hypogonadotropic hypogonadism [55].
In male hypogonadotropic hypogonadism, the induction of puberty is classically achieved with intramuscular depot testosterone (testosterone enanthate). The alternative is the combined subcutaneous administration of HCG and rhFSH enabling almost normal testicular growth and timely spermatogenesis [56]. Both treatments can be offered also during minipuberty in boys with micropenis due to hypogonadotropic hypogonadism [57]. Puberty induction by testosterone enanthate is based on long clinical practice and associated with high adherence, but inhibits testicular growth and spermatogenesis. Induction of puberty by gonadotropins promotes spermatogenesis and testicular volume, but is dependent on good adherence and lacks long clinical experience. It remains to be proven whether a higher rate and an earlier onset of fertility in adulthood can be achieved by puberty induction using gonadotropins, but evidence is missing yet. Predictors of reduced fertility in male hypogonadotropic hypogonadism are inborn cryptorchidism as well as low testicular volume and low inhibin B serum levels at the start of puberty induction [58].
Diagnosis of ADH deficiency
Anti-diuretic hormone (ADH) deficiency very rarely evolves after the diagnosis of isolated GHD. The highest prevalence reported in a Brazilian group of children with severe GHD was 5% [20]; the lowest 0.2% in a big cohort with strictly defined isolated GHD from the Genesis observational trial [5]. Probably, the more frequent pattern of evolution is ADH deficiency followed by GH deficiency [59]. In the presence of GHD, ADH deficiency may be associated with septo-optic dysplasia and other severe midline defects such as holoprosencephaly. More frequently, pituitary and hypothalamic surgery, infundibulitis, craniopharyngioma, germinoma, histiocytosis, severe brain trauma, or acute brain insults are causative [60]. Therefore, the occurrence of diabetes insipidus in children with isolated GHD should always arouse suspicion of the presence of histiocytosis, germinoma, or infundibulitis, which all can cause GHD. In such children, a lesion of the pituitary stalk and the pituitary gland should be excluded by cranial magnetic resonance imaging (cMRI) with gadolinium contrast [61].
The main clinical symptoms are polyuria and polydipsia, which should systematically be monitored after exclusion of diabetes mellitus and hypercalciuria. The definition of polydipsia varies; a fluid intake > 2 l/m2 per day is the most common one [62]. Adrenal insufficiency and hypothyroidism can mask ADH deficiency. Traditionally, complete ADH deficiency is confirmed by insufficient increase in urinary molarity at the end of a water deprivation test (< 300 mosm/kg) and a significant increase of urinary molarity after subsequent administration of desmopressin (> 750 mosm/kg). A marker of ADH secretion is copeptin, the C-terminal portion of the precursor peptide of arginine vasopressin, which is secreted in equimolar amounts from the posterior pituitary and more stable in serum. First experiences in a pediatric cohort, however, attest a relatively low specificity at 67% for the distinction from primary polydipsia [63].
A triphasic pattern in the disturbance of ADH secretion, which is characterized by diabetes insipidus and syndrome of inappropriate ADH secretion (SIADH), may occur after neurosurgery in the hypothalamic-pituitary region. A transient postoperative polyuria is followed by oliguria and then often permanent polyuria [64].
Treatment of ADH deficiency
If ADH deficiency cannot be compensated by moderately increased fluid intake or causes severe sleep disturbance, the synthetic ADH analog desmopressin is the treatment of choice. Administration routes are multiple; the preferred one is orally, as water intoxication and hyponatremia occur less likely than after intranasal administration [65, 66]. The bioavailability of the intranasal desmopressin administration is ten times less than the intravenous administration, and ten times more than the oral administration. The new desmopressin lyophilisate for oral absorption is approximately 1.7 times more potent than the conventional tablet. Overdosing of desmopressin and/or excessive fluid intake independent of thirst can cause water intoxication and potentially life-threatening brain edema [65]. Therefore, teaching of the patient and caregivers regarding individual fluid intake and situational adaptation of the desmopressin dose is paramount. Interactions of other drugs with endogenous ADH secretion and tubular action need to be considered [65].
Conclusions
Treatment of children with isolated GHD demands regular monitoring of the other pituitary hormone axes, because over time isolated GHD can progress to CPHD. This dynamic can be slow, and its detection is mainly based on the measurement of pituitary and peripheral hormones (Table 1). Relevant risk factors for this development are organic GHD, especially significant malformations of the pituitary-hypothalamic region, and acquired GHD. The manifestation of TSH deficiency is most frequent, and ADH deficiency is rare. Treatment is based on the experience made with peripheral gland disorders. However, treatment monitoring should abstain from the conventional judgment of the pituitary hormone level, because this one is deficient. Lifelong monitoring of the pituitary gland function is recommended in humans with organic GHD.
Availability of data and materials
Not applicable.
Abbreviations
- GHD:
-
Growth hormone deficiency
- CPHD:
-
Combined pituitary hormone deficiency
- rhGH:
-
Recombinant human GH
- TSH:
-
Thyroid-stimulating hormone
- ACTH:
-
Adrenocorticotropic hormone
- ITT:
-
Insulin-hypoglycemia test
- CDGP:
-
Constitutional delay in growth and puberty
- ADH:
-
Anti-diuretic hormone
- cMRI:
-
Cranial magnetic resonance imaging
- SIADH:
-
Syndrome of inappropriate ADH secretion
References
Growth Hormone Research Society (2000) Consensus guidelines for the diagnosis and treatment of growth hormone (GH) deficiency in childhood and adolescence: summary statement of the GH Research Society. GH Research Society. J Clin Endocrinol Metab 85(11):3990–3993
van Iersel L, van Santen HM, Zandwijken GRJ, Zwaveling-Soonawala N, Hokken-Koelega ACS, van Trotsenburg ASP (2018) Low FT4 concentrations around the start of recombinant human growth hormone treatment: predictor of congenital structural hypothalamic-pituitary abnormalities? Horm Res Paediatr 89(2):98–107
Schoenmakers N, Alatzoglou KS, Chatterjee VK, Dattani MT (2015) Recent advances in central congenital hypothyroidism. J Endocrinol 227(3):R51–R71
Cerbone M, Dattani MT (2017) Progression from isolated growth hormone deficiency to combined pituitary hormone deficiency. Growth Horm IGF Res 37:19–25
Blum WF, Deal C, Zimmermann AG, Shavrikova EP, Child CJ, Quigley CA, Drop SL, Cutler GB Jr, Rosenfeld RG (2013) Development of additional pituitary hormone deficiencies in pediatric patients originally diagnosed with idiopathic isolated GH deficiency. Eur J Endocrinol 170(1):13–21
Mullis PE, Robinson ICAF, Salemi S, Eblé A, Besson A, Vuissoz J-M, Deladoey J, Simon D, Czernichow P, Binder G (2005) Isolated autosomal dominant growth hormone deficiency: an evolving pituitary deficit? A multicenter follow-up study. J Clin Endocrinol Metab 90(4):2089–2096
van Iersel L, Li Z, Srivastava DK, Brinkman TM, Bjornard KL, Wilson CL, Green DM, Merchant TE, Pui CH, Howell RM, Smith SA, Armstrong GT, Hudson MM, Robison LL, Ness KK, Gajjar A, Krull KR, Sklar CA, van Santen HM, Chemaitilly W (2019) Hypothalamic-pituitary disorders in childhood cancer survivors: prevalence, risk factors and long-term health outcomes. J Clin Endocrinol Metab 104(12):6101–6115
Gebauer J, Higham C, Langer T, Denzer C, Brabant G (2019) Long-term endocrine and metabolic consequences of cancer treatment: a systematic review. Endocr Rev 40(3):711–767
Laurberg P, Jakobsen PE, Hoeck HC, Vestergaard P (1994) Growth hormone and thyroid function: is secondary throid failure and underdiagnosed in growth hormone deficient patients? Thyroidol Clin Exp 6:73–79
Keskin M, Bayramoglu E, Aycan Z (2017) Effects of 1-year growth hormone replacement therapy on thyroid volume and function of the children and adolescents with idiopathic growth hormone deficiency. J Pediatr Endocrinol Metab 30(11):1187–1190
Mehta A, Hindmarsh PC, Stanhope RG, Brain CE, Preece MA, Dattani MT (2003) Is the thyrotropin-releasing hormone test necessary in the diagnosis of central hypothyroidism in children? J Clin Endocrinol Metab 88(12):5696–5703
Beck-Peccoz P, Rodar G, Giavoli C, Lania A (2017) Central hypothyroidism-a neglected thyroid disorder. Nat Rev Endocrinol 13(10):588–598
Grunfeld C, Sherman BM, Cavalieri RR (1988) The acute effects of human growth hormone administration on thyroid function in normal men. J Clin Endocrinol Metab 67(5):1111–1114
Wyatt DT, Gesundheit N, B Sherman BM (1998) Changes in thyroid hormone levels during growth hormone therapy in initially euthyroid patients: lack of need for thyroxine supplementation. J Clin Endocrinol Metab 83(10):3493-3497.
Kalina-Faska B, Kalina M, Koehler B (2004) Effects of recombinant growth hormone therapy on thyroid hormone concentrations. Int J Clin Pharmacol Ther 42(1):30–34
Shaikh MG, Lewis P, Kirk JM (2004) Thyroxine unmasks Addison’s disease. Acta Paediatr 93(12):1663–1665
Carrozza V , Csako G, Yanovski JA, Skarulis MC, Nieman L, Wesley R, Pucino F (1999) Levothyroxine replacement therapy in central hypothyroidism: a practice report. pharmacotherapy 19(3):349-55.
Martins MRA, Doin FC, Komatsu WR, Barros-Neto TL, Moises VA, Abucham J (2007) Growth hormone replacement improves thyroxine biological effects: implications for management of central hypothyroidism. J Clin Endocrinol Metab 92(11):4144–4153
Walvoord EC, Rosenman MB, Eugster EA (2004) Prevalence of adrenocorticotropin deficiency in children with idiopathic growth hormone deficiency. J Clin Endocrinol Metab 89(10):5030–5034
Otto AP, França MM, Correa FA, Costalonga EF, Leite CC, Mendonca BB, Arnhold IJ, Carvalho LR, Jorge AA (2015) Frequent development of combined pituitary hormone deficiency in patients initially diagnosed as isolated growth hormone deficiency: a long term follow-up of patients from a single center. Pituitary 18(4):561–567
Sklar CA, Antal Z, Chemaitilly W, Cohen LE, Follin C, Meacham LR, Murad MH (2018) Hypothalamic-pituitary and growth disorders in survivors of childhood cancer: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab 103(8):2761–2784
UçarA BF, Saka N (2016) Diagnosis and management of pediatric adrenal insufficiency. World J Pediatr 12(3):261–274
Kazlauskaite R, Maghnie M (2010) Pitfalls in the diagnosis of central adrenal insufficiency in children. Endocr Dev 17:96–107
Guran T, Firat I, Yildiz F, Kaplan Bulut I, Dogru M, Bereket A (2015) Reference values for serum dehydroepiandrosterone-sulphate in healthy children and adolescents with emphasis on the age of adrenarche and pubarche. Clin Endocrinol (Oxf) 82(5):712–718
Agwu JC, Spoudeas H, Hindmarsh PC, Pringle PJ, Brook CG (1999) Tests of adrenal insufficiency. Arch Dis Child 80(4):330–333
Stouffer JE, Lipscomb HS (1963) Reversible binding of ACTH to a glass surface. Endocrinology 72(1):91–94
Ospina NS, Nofal AA, Bancos I, Javed A, Benkhadra K, Kapoor E, Lteif AN, Natt N, Murad MH (2016) ACTH stimulation tests for the diagnosis of adrenal insufficiency: systematic review and meta-analysis. J Clin Endocrinol Metab 101(2):427–434
Böttner A, Kratzsch J, Liebermann S, Keller A, Pfaffle RW, Kiess W, Keller E (2005) Comparison of adrenal function tests in children--the glucagon stimulation test allows the simultaneous assessment of adrenal function and growth hormone response in children. J Pediatr Endocrinol Metab 18(5):433–442
Stewart PM, Toogood AA, Tomlinson JW (2001) Growth hormone, insulin-like growth factor-I and the cortisol-cortisone shuttle. Horm Res 56(Suppl 1):1–6
Reznik Y, Barat P, Bertherat J, Bouvattier C, Castinetti F, Chabre O, Chanson P, Cortet C, Delemer B, Goichot B, Gruson D, Guignat L, Proust-Lemoine E, Sanson MR, Reynaud R, Boustani DS, Simon D, Tabarin A, Zenaty D (2018) SFE/SFEDP adrenal insufficiency French consensus: introduction and handbook. Ann Endocrinol (Paris) 79(1):1–22
Ventura M, Serra-Caetano J, Cardoso R, Dinis I, Melo M, Carrilho F, Mirante A (2019) The spectrum of pediatric adrenal insufficiency: insights from 34 years of experience. J Pediatr Endocrinol Metab 32(7):721–726
Mazziotti G, Formenti AM, Frara S, Roca E, Mortini P, Berruti A, Giustina A (2017) Management of endocrine disease: risk of overtreatment in patients with adrenal insufficiency: current and emerging aspects. Eur J Endocrinol 177(5):R231–r248
Fleseriu M, Hashim IA, Karavitaki N, Melmed S, Murad MH, Salvatori R, Samuels MH (2016) Hormonal replacement in hypopituitarism in adults: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab 101(11):3888–3921
Binder G, Weber S, Ehrismann M, Zaiser N, Meisner C, Ranke MB, Maier L, Wudy SA, Hartmann MF, Heinrich U, Bettendorf M, Doerr HG, Pfaeffle RW, Keller E (2009) Effects of dehydroepiandrosterone therapy on pubic hair growth and psychological well-being in adolescent girls and young women with central adrenal insufficiency: a double-blind, randomized, placebo-controlled phase III trial. J Clin Endocrinol Metab 94(4):1182–1190
Arlt W, Allolio B (2003) Adrenal insufficiency. Lancet 361(9372):1881–1893
Rushworth RL, Torpy DJ, Stratakis CA, Falhammar H (2018) Adrenal crises in children: perspectives and research directions. Horm Res Paediatr 89(5):341–351
Shulman DI, Palmert MR, Kemp SF (2007) Adrenal insufficiency: still a cause of morbidity and death in childhood. Pediatrics 119(2):e484–e494
Bornstein SR, Allolio B, Arlt W, Barthel A, Don-Wauchope A, Hammer GD, Husebye ES, Merke DP, Murad MH, Stratakis CA, Torpy DJ (2016) Diagnosis and treatment of primary adrenal insufficiency: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab 101(2):364–389
August GP, Lippe BM, Blethen SL, Rosenfeld RG, Seelig SA, Johanson AJ, Compton PG, Frane JW, McClellan BH, Sherman BM (1990) Growth hormone treatment in the United States: demographic and diagnostic features of 2331 children. J Pediatr 116(6):899–903
Radicioni AF, Di Giorgio G, Grugni G, Cuttini M, Losacco V, Anzuini A, Spera S, Marzano C, Lenzi A, Cappa M, Crinò A (2012) Multiple forms of hypogonadism of central, peripheral or combined origin in males with Prader-Willi syndrome. Clin Endocrinol (Oxf).76(1):72-7.
Kelberman D, Rizzoti K, Avilion A, Bitner-Glindzicz M, Cianfarani S, Collins J, Chong WK, Kirk JM, Achermann JC, Ross R, Carmignac D, Lovell-Badge R, Robinson IC, Dattani MT (2006) Mutations within Sox2/SOX2 are associated with abnormalities in the hypothalamo-pituitary-gonadal axis in mice and humans. Journal of Clinical Investigation 116(9):2442–2455
Blum WF, Klammt J, Amselem S, Pfäffle HM, Legendre M, Sobrier ML, Luton MP, Child CJ, Jones C, Zimmermann AG, Quigley CA, Cutler GB Jr, Deal CL, Lebl J, Rosenfeld RG, Parks JS, Pfäffle RW (2018) Screening a large pediatric cohort with GH deficiency for mutations in genes regulating pituitary development and GH secretion: frequencies, phenotypes and growth outcomes. EBioMedicine 36:390–400
Grumbach MM (2005) A window of opportunity: the diagnosis of gonadotropin deficiency in the male infant. J Clin Endocrinol Metab 90(5):3122–3127
Kuiri-Hanninen T, Sankilampi U, Dunkel L (2014) Activation of the hypothalamic-pituitary-gonadal axis in infancy: minipuberty. Hormone Research in Paediatrics 82(2):73–80
Bizzarri C, Cappa M (2020) Ontogeny of hypothalamus-pituitary gonadal axis and minipuberty: an ongoing debate? Front Endocrinol (Lausanne) 11:187
Harrington J, Palmert MR (2012) Clinical review: Distinguishing constitutional delay of growth and puberty from isolated hypogonadotropic hypogonadism: critical appraisal of available diagnostic tests. J Clin Endocrinol Metab 97(9):3056–3067
Binder G, Schweizer R, Blumenstock G, Braun R (2015) Inhibin B plus LH vs GnRH agonist test for distinguishing constitutional delay of growth and puberty from isolated hypogonadotropic hypogonadism in boys. Clin Endocrinol (Oxf) 82(1):100–105
Binder G, Schweizer R, Haber P, Blumenstock G, Braun R (2015) Accuracy of endocrine tests for detecting hypogonadotropic hypogonadism in girls. J Pediatr 167(3):674–678
Boehm U, Bouloux PM, Dattani MT, de Roux N, Dodé C, Dunkel L, Dwyer AA, Giacobini P, Hardelin JP, Juul A, Maghnie M, Pitteloud N, Prevot V, Raivio T, Tena-Sempere M, Quinton R, Young J (2015) Expert consensus document: European Consensus Statement on congenital hypogonadotropic hypogonadism--pathogenesis, diagnosis and treatment. Nat Rev Endocrinol 11(9):547–564
Ho KKY, Gibney J, Johannsson G, Wolthers T (2006) Regulating of growth hormone sensitivity by sex steroids: implications for therapy. Front Horm Res 35:115–128
Shah S, Forghani N, Durham E, Neely EK (2014) A randomized trial of transdermal and oral estrogen therapy in adolescent girls with hypogonadism. Int J Pediatr Endocrinol 2014(1):12
Menon DV, Vongpatanasin W (2006) Effects of transdermal estrogen replacement therapy on cardiovascular risk factors. Treat Endocrinol 5(1):37–51
Johnson JV, Lowell J, Badger GJ, Rosing J, Tchaikovski S, Cushman M (2008) Effects of oral and transdermal hormonal contraception on vascular risk markers: a randomized controlled trial. Obstetr Gynecol 111(2 Pt 1):278–284
Donaldson M, Kriström B, Ankarberg-Lindgren C, Verlinde S, van Alfen-van der Velden J, Gawlik A, van Gelder MMHJ, Sas T, on behalf of the European Society for Paediatric Endocrinology Turner Syndrome Working Group (2019) Optimal pubertal induction in girls with Turner syndrome using either oral or transdermal estradiol: a proposed modern strategy. Horm Res Paediatr 91(3):153–163
Kaufmann R, Dunn R, Vaughn T, Hughes G, O'Brien F, Hemsey G, Thomson B, O'Dea LS (2007) Recombinant human luteinizing hormone, lutropin alfa, for the induction of follicular development and pregnancy in profoundly gonadotrophin-deficient women. Clin Endocrinol (Oxf) 67(4):563–569
Rohayem J, Hauffa BP, Zacharin M, Kliesch S, Zitzmann M; “German Adolescent Hypogonadotropic Hypogonadism Study Group” (2017) Testicular growth and spermatogenesis: new goals for pubertal hormone replacement in boys with hypogonadotropic hypogonadism? - a multicentre prospective study of hCG/rFSH treatment outcomes during adolescence. Clin Endocrinol (Oxf) 86(1):75–87
Kohva E, Huopio H, Hietamäki J, Hero M, Miettinen PJ, Raivio T (2019) Treatment of gonadotropin deficiency during the first year of life: long-term observation and outcome in five boys. Hum Reprod 34(5):863–871
Young J, Xu C, Papadakis GE, Acierno JS, Maione L, Hietamäki J, Raivio T, Pitteloud N (2019) Clinical management of congenital hypogonadotropic hypogonadism. Endocr Rev 40(2):669–710
Di Iorgi N, Allegri AEM, Napoli F, Calcagno A, Calandra E, Fratangeli N, Vannati M, Rossi A, Bagnasco F, Haupt R, Maghnie M (2014) Central diabetes insipidus in children and young adults: etiological diagnosis and long-term outcome of idiopathic cases. J Clin Endocrinol Metab 99(4):1264–1272
Yang YH, Lin JJ, Hsia SH, Wu CT, Wang HS, Hung PC, Chou ML, Hsieh MY, Lin KL (2011) Central diabetes insipidus in children with acute brain insult. Pediatr Neurol 45(6):377–380
Liu W, Wang L, Liu M, Li G (2016) Pituitary morphology and function in 43 children with central diabetes insipidus. Int J Endocrinology 2016:6365830. https://doi.org/10.1155/2016/6365830
Maghnie M (2003) Diabetes insipidus. Horm Res 59(Suppl 1):42–54
Tuli G, Tessaris D, Einaudi S, Matarazzo P, De Sanctis L (2018) Copeptin role in polyuria-polydipsia syndrome differential diagnosis and reference range in paediatric age. Clin Endocrinol (Oxf). 88(6):873–879
Edate S, Albanese A (2015) Management of electrolyte and fluid disorders after brain surgery for pituitary/suprasellar tumours. Horm Res Paediatr 83(5):293–301
Di Iorgi N, Morana G, Napoli F, Allegri AE, Rossi A, Maghnie M (2015) Management of diabetes insipidus and adipsia in the child. Best Pract Res Clin Endocrinol Metab 29(3):415–436
Kataoka Y, Nishida S, Hirakawa A, Oiso Y, Arima H (2015) Comparison of incidence of hyponatremia between intranasal and oral desmopressin in patients with central diabetes insipidus. Endocr J 62(2):195–200
Acknowledgements
Not applicable.
Funding
The company Novo Nordisk Germany supported the meeting in Frankfurt in March 2020 financially. The sponsor did not take part in the analysis of the published data or the preparation of the manuscript. Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Contributions
GB organized the panel meeting in Frankfurt, wrote a questionnaire for each expert providing a concept for the meeting, and recorded the first draft of this paper based on the PowerPoint presentations of the co-authors. The other authors participated in the panel meeting in Frankfurt, each providing a review on diagnostics or treatment of a specific pituitary hormone deficiency, and reviewed and corrected the first draft. The author(s) read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
GB has received honoraria for lectures from Ferring, Ipsen, Lilly, Merck Serono, Novo Nordisk, Pfizer, and Sandoz and for membership in advisory boards from Ferring, Ipsen, Novo Nordisk, and Pfizer.
DS has received honoraria for lectures from Ipsen, Merck Serono, Novo Nordisk, Pfizer, and Sandoz and for membership in advisory boards from Novo Nordisk, Merck Serono, and Sandoz.
TR has received honoraria for lectures from Ipsen, Merck Serono, Novo Nordisk, Ferring, Lilly, Pfizer, and Sandoz and for membership in advisory boards from Novo Nordisk, Ferring, Merck Serono, and Sandoz.
RP has received honoraria for lectures from Sandoz, Ipsen, Novo Nordisk, Merck Serono, and Ferring and for membership in advisory boards from Sandoz, Novo Nordisk, and Lilly.
HGD has received honoraria for lectures from Ferring, Ipsen, Novo Nordisk, and Pfizer.
MB has received honoraria for lectures from Novo Nordisk, Pfizer, Ipsen, and Hexal and advisory fees from Takeda Pharmaceutical Company Limited.
BH has received honoraria for lectures from Boehringer, Ferring, Ipsen, Merck Serono, Novo Nordisk, Pfizer, and Sandoz and for membership in advisory boards from Merck Serono and Sandoz.
JW has received honoraria for lectures/presentations from Ipsen, Merck Serono, Novo Nordisk, and Pfizer and for membership in advisory boards from Ipsen and Novo Nordisk.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Binder, G., Schnabel, D., Reinehr, T. et al. Evolving pituitary hormone deficits in primarily isolated GHD: a review and experts’ consensus. Mol Cell Pediatr 7, 16 (2020). https://doi.org/10.1186/s40348-020-00108-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40348-020-00108-2