
Abstract
Intrauterine growth restriction (IUGR) is a fetal pathology which leads to increased risk for certain neonatal complications. Furthermore, clinical and experimental studies revealed that IUGR is associated with a significantly higher incidence of metabolic, renal and cardiovascular diseases (CVD) later in life. One hypothesis for the higher risk of CVD after IUGR postulates that IUGR induces metabolic alterations that then lead to CVD.
This minireview focuses on recent studies which demonstrate that IUGR is followed by early primary cardiovascular alterations which may directly progress to CVD later in life.
Similar content being viewed by others


Background
Intrauterine growth restriction (IUGR) affects 3–7% of all newborns. As a consequence of maternal, placental or fetal pathology the fetus cannot fully exploit its growth potential [1]. While the term “small for gestational age” (SGA) describes a newborn with a birth weight less than the 10th percentile, IUGR requires a pathological retardation of intrauterine growth velocity clearly highlighted by a characteristic kink in the curve of intrauterine growth [2].
Newborns with IUGR exhibit significantly increased morbidity immediately after birth (e.g. hypoglycaemia, hypothermia, infant respiratory distress syndrome). Later in life former IUGR-patients were found to have a significantly higher incidence of renal, cardiovascular and metabolic diseases, the same ailments that are also the most frequent causes of morbidity and mortality in the western world [3,4,5].
The underlying mechanisms leading from fetal undersupply to the development of diseases in adulthood are not fully understood. In this context Barker et al. hypothesize that intrauterine undernutrition compromises growth and differentiation of organs during the vulnerable phase of fetal development that results in persistent alterations of the organism and leads to the development of secondary diseases later in life [6,7,8].
The shortage of nutrients during fetal development observed in IUGR is commonly replaced by an adequate nutrient supply after birth. The hypothesis of the “thrifty phenotype” considers the mismatch between intrauterine and postnatal supply with nutrients in IUGR individuals to be the underlying cause of secondary pathologies. In a fetus adapted to shortage of nutrients during intrauterine development postnatal hyperalimentation leads initially to excessive catch up growth and later to metabolic, cardiovascular and renal diseases [9].
As IUGR is associated with a higher incidence of metabolic syndrome, it appears conceivable that cardiovascular dysfunction observed after IUGR is secondary to diabetes, dyslipidemia or hypertension [10]. However, growing evidence suggests that IUGR directly causes cardiovascular alterations independent of pre-existing metabolic disease. Recent clinical and animal studies identified candidate mechanisms that may mediate the development of cardiovascular alterations in the setting of IUGR consistent with the hypothesis of perinatal programming [11, 12].
Myocardial function and structure after IUGR
In the heart, a number of studies revealed myocardial alterations induced by IUGR, that can be detected long before the onset of metabolic disease and arterial hypertension. IUGR affected cardiac development and significantly reduced the number of cardiomyocytes at the time of birth [13, 14]. Studies in humans revealed early and persistent alterations of myocardial structure and decreased cardiac function detected by echocardiography in fetal, neonatal and juvenile patients with IUGR [15,16,17]. These results were supported by findings in an animal model of IUGR exhibiting echocardiographic signs of cardiac dysfunction accompanied by a more distensible myocardium in the absence of arterial hypertension [18]. As a possible underlying mechanism, changes in the expression pattern of Titin (Ttn) after IUGR was observed: The passive elasticity of cardiomyocytes is modulated by alternative splicing of titin, a structural protein of the myocardial sarcomere, resulting in the two isoforms N2BA and N2B. Corresponding to the echocardiographic signs of a more distensible myocardium, relative overexpression of the less rigid isoform N2BA was observed in IUGR animals [18].
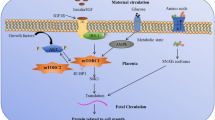
Moreover, assessment of inflammatory and profibrotic markers revealed an early induction of the expression of transforming growth factor beta (TGF-ß), connective tissue growth factor (CTGF) and microfibrillar matrix molecules in the myocardium of juvenile IUGR animals without evidence of metabolic syndrome [19]. This supports the notion of early and direct molecular changes in the cardiovascular system (Fig. 1).

Direct and indirect pathways leading to cardiovascular disease in IUGR
Atherosclerosis, vascular remodeling and fibrosis after IUGR
IUGR-induced metabolic disease (including dyslipidemia and insulin resistance) may likewise indirectly lead to atherosclerosis [10]. In addition, several studies provided evidence that IUGR is accompanied by early structural alterations in blood vessels [20,21,22,23]. Vascular remodeling favors the development of atherosclerosis [24]. In an animal model of vascular remodeling induced by unilateral ligation of the A. carotis communis significantly increased neointima formation and media thickness was observed in juvenile IUGR rats in the absence of metabolic disease [25]. Moreover, dedifferentiation of vascular smooth muscle cells (VSMC) was more prominent and collagen deposition in the media was increased after IUGR [25].
But even in the absence of experimental vascular disease, primary vascular alterations were detected in normotensive IUGR animals: A significantly increased expression of connective tissue growth factor (CTGF) was observed in aortas of neonatal animals after IUGR. Collagen I and collagen IV deposition in the aorta was more prominent in juvenile IUGR animals [19]. Recent studies examined IUGR-induced molecular mechanisms of endothelial dysfunction that favor the development of atherosclerosis. Oliveira et al. detected lower NO levels and increased eNOS phosphorylation in thoracic aortas of IUGR rats as a sign of endothelial dysfunction [26]. Taken together, these observations indicate that IUGR renders individuals more susceptible to the development of atherosclerotic lesions and vascular dysfunction and thus to cardiovascular-related diseases later in life.
Conclusions
IUGR favors the development of secondary cardiovascular diseases later in life which are among the most frequent causes of morbidity and mortality and constitute a significant proportion of healthcare-related expenditures in the western world. Cardiovascular disease appears not only to be a consequence of metabolic syndrome, but also caused by direct effects on cardiac and vasculature structure and function in individuals with IUGR. A more detailed knowledge of underlying disease mechanisms is likely to advance prevention and treatment of IUGR and its complications, and thereby improve long-term outcomes for patients with IUGR.
Abbreviations
- CTGF:
-
connective tissue growth factor
- eNOS:
-
endothelial NO-Synthase
- IUGR:
-
intrauterine growth restriction
- NO:
-
nitric oxide
- SGA:
-
small for gestational age
- TGF-ß:
-
transforming growth factor beta
- VSMC:
-
vascular smooth muscle cells
References
Romo A, Carceller R, Tobajas J (2009) Intrauterine growth retardation (IUGR): epidemiology and etiology. Pediatr Endocrinol Rev 6(Suppl 3):332–336
Sharma D, Shastri S, Sharma P (2016) Intrauterine growth restriction: antenatal and postnatal aspects. Clin Med Insights Pediatr 10:67–83
Legrand M, Elie C, Stefani J, Flores N, Culeux C, Delissen O, Ibanez C, Lestaevel P, Eriksson P, Dinocourt C (2016) Cell proliferation and cell death are disturbed during prenatal and postnatal brain development after uranium exposure. Neurotoxicology 52:34–45
Eriksson JG, Kajantie E, Thornburg K, Osmond C (2016) Prenatal and maternal characteristics and later risk for coronary heart disease among women. Eur J Prev Cardiol 23(4):385–390
Mierzynski R, Dluski D, Darmochwal-Kolarz D, Poniedzialek-Czajkowska E, Leszczynska-Gorzelak B, Kimber-Trojnar Z, Agnieszka W, Oleszczuk J (2016) Intra-uterine growth retardation as a risk factor of postnatal metabolic disorders. Curr Pharm Biotechnol 17(7):587–596
Barker DJ (1998) In utero programming of chronic disease. Clin Sci (Lond) 95(2):115–128
de Boo HA, Harding JE (2006) The developmental origins of adult disease (Barker) hypothesis. Aust N Z J Obstet Gynaecol 46(1):4–14
Dotsch J (2014) Perinatal programming - myths, fact, and future of research. Mol Cell Pediatr 1(1):2
Hales CN (1997) Fetal and infant growth and impaired glucose tolerance in adulthood: the “thrifty phenotype” hypothesis revisited. Acta Paediatr Suppl 422:73–77
Longo S, Bollani L, Decembrino L, Di Comite A, Angelini M, Stronati M (2013) Short-term and long-term sequelae in intrauterine growth retardation (IUGR). J Matern Fetal Neonatal Med 26(3):222–225
Zohdi V, Lim K, Pearson JT, Black MJ (2014) Developmental programming of cardiovascular disease following intrauterine growth restriction: findings utilising a rat model of maternal protein restriction. Nutrients 7(1):119–152
Sehgal A, Skilton MR, Crispi F (2016) Human fetal growth restriction: a cardiovascular journey through to adolescence. J Dev Orig Health Dis 7(6):626–635
Corstius HB, Zimanyi MA, Maka N, Herath T, Thomas W, van der Laarse A, Wreford NG, Black MJ (2005) Effect of intrauterine growth restriction on the number of cardiomyocytes in rat hearts. Pediatr Res 57(6):796–800
Wadley GD, McConell GK, Goodman CA, Siebel AL, Westcott KT, Wlodek ME (2013) Growth restriction in the rat alters expression of metabolic genes during postnatal cardiac development in a sex-specific manner. Physiol Genomics 45(3):99–105
Crispi F, Bijnens B, Figueras F, Bartrons J, Eixarch E, Le Noble F, Ahmed A, Gratacos E (2010) Fetal growth restriction results in remodeled and less efficient hearts in children. Circulation 121(22):2427–2436
Cruz-Lemini M, Crispi F, Valenzuela-Alcaraz B, Figueras F, Sitges M, Bijnens B, Gratacos E (2016) Fetal cardiovascular remodeling persists at 6 months in infants with intrauterine growth restriction. Ultrasound Obstet Gynecol 48(3):349–356
Sarvari SI, Rodriguez-Lopez M, Nunez-Garcia M, Sitges M, Sepulveda-Martinez A, Camara O, Butakoff C, Gratacos E, Bijnens B, Crispi F (2017) Persistence of cardiac remodeling in preadolescents with fetal growth restriction. Circ Cardiovasc Imaging 10(1):e005270
Menendez-Castro C, Toka O, Fahlbusch F, Cordasic N, Wachtveitl R, Hilgers KF, Rascher W, Hartner A (2014) Impaired myocardial performance in a normotensive rat model of intrauterine growth restriction. Pediatr Res 75(6):697–706
Menendez-Castro C, Fahlbusch F, Cordasic N, Amann K, Munzel K, Plank C, Wachtveitl R, Rascher W, Hilgers KF, Hartner A (2011) Early and late postnatal myocardial and vascular changes in a protein restriction rat model of intrauterine growth restriction. PLoS One 6(5):e20369
Brodszki J, Lanne T, Marsal K, Ley D (2005) Impaired vascular growth in late adolescence after intrauterine growth restriction. Circulation 111(20):2623–2628
Morsing E, Liuba P, Fellman V, Marsal K, Brodszki J (2014) Cardiovascular function in children born very preterm after intrauterine growth restriction with severely abnormal umbilical artery blood flow. Eur J Prev Cardiol 21(10):1257–1266
Skilton MR, Evans N, Griffiths KA, Harmer JA, Celermajer DS (2005) Aortic wall thickness in newborns with intrauterine growth restriction. Lancet 365(9469):1484–1486
Dodson RB, Rozance PJ, Petrash CC, Hunter KS, Ferguson VL (2014) Thoracic and abdominal aortas stiffen through unique extracellular matrix changes in intrauterine growth restricted fetal sheep. Am J Physiol Heart Circ Physiol 306(3):H429–H437
Chatzizisis YS, Coskun AU, Jonas M, Edelman ER, Feldman CL, Stone PH (2007) Role of endothelial shear stress in the natural history of coronary atherosclerosis and vascular remodeling: molecular, cellular, and vascular behavior. J Am Coll Cardiol 49(25):2379–2393
Menendez-Castro C, Cordasic N, Schmid M, Fahlbusch F, Rascher W, Amann K, Hilgers KF, Hartner A (2012) Intrauterine growth restriction promotes vascular remodelling following carotid artery ligation in rats. Clin Sci (Lond) 123(7):437–444
Oliveira V, de Souza LV, Fernandes T, Junior SDS, de Carvalho MHC, Akamine EH, Michelini LC, de Oliveira EM, Franco MDC (2017) Intrauterine growth restriction-induced deleterious adaptations in endothelial progenitor cells: possible mechanism to impair endothelial function. J Dev Orig Health Dis 8(6):665–673
Acknowledgements
We thank Karl F. Hilgers, Department of Nephrology and Hypertension, University Hospital of Erlangen-Nuremberg and Markus Bitzer, Division of Nephrology, University of Michigan, Ann Arbor, MI, USA for critical revision of the manuscript. Due to space restrictions and the focus of this minireview, we were not able to include all relevant articles to this topic.
Funding
This work was supported by an Erlanger Leistungsbezogene Anschubfinanzierung und Nachwuchsfoerderung (ELAN) grant from the University Hospital of Erlangen-Nuremberg to CMC.
Author information
Authors and Affiliations
Contributions
CMC reviewed the literature, CMC, WR and AH contributed to drafting and finalizing the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Menendez-Castro, C., Rascher, W. & Hartner, A. Intrauterine growth restriction - impact on cardiovascular diseases later in life. Mol Cell Pediatr 5, 4 (2018). https://doi.org/10.1186/s40348-018-0082-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40348-018-0082-5