
Abstract
Osmotic diarrhea and abdominal pain in humans are oftentimes associated with carbohydrate malabsorption in the small intestine due to loss of function of microvillar disaccharidases. Disaccharidases are crucial for the digestion and the subsequent absorption of carbohydrates. This review focuses on sucrase-isomaltase as the most abundant intestinal disaccharidase and the primary or induced pathological conditions that affect its physiological function. Congenital defects are primary factors which directly influence the transport and function of sucrase-isomaltase in a healthy epithelium. Based on the mutation type and the pattern of inheritance, a mutation in the sucrase-isomaltase gene may exert a variety of symptoms ranging from mild to severe. However, structure and function of wild type sucrase-isomaltase can be also affected by secondary factors which influence its structure and function either specifically via certain inhibitors and therapeutic agents or generally as a part of intestinal pathogenesis, for example in the inflammatory responses. Diagnosis of sucrase-isomaltase deficiency and discriminating it from other gastrointestinal intolerances can be latent in the patients because of common symptoms observed in all of these cases.
Here, we summarize the disorders that implicate the digestive function of sucrase-isomaltase as well as the diagnostic and therapeutic strategies utilized to restore normal intestinal function.
Similar content being viewed by others
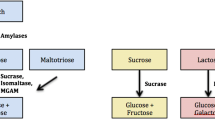

Dietary carbohydrates as human energy source
Carbohydrates are one of the vital mammalian nutrients which have been increasingly consumed by human populations since the agricultural revolution and serve as important calorie sources [1]. Dietary carbohydrates mainly include sucrose and a variety of plant starches which are composed of different α-linked sugars. Sugar transporters in the intestine are only capable of transporting monosaccharides. Therefore, accessibility of higher carbohydrates to the human body as energy sources necessitates their hydrolysis to simple monosaccharides. Starch digestion is initiated by salivary and later pancreatic α-amylases that break it down to smaller units composed of two, three, or four sugar residues. The final step of starch digestion takes place in the intestinal lumen by α-glucosidases that are localized on the brush border membrane (BBM) of the intestinal epithelium. This family of digestive enzymes includes sucrase-isomaltase (SI), maltase-glucoamylase (MGAM) and trehalase [2]. Due to its high abundance and its wide substrate specificity in hydrolyzing α-1,2, α-1,6, and α-1,4 glucosidic bonds, human SI is responsible for almost all sucrase activity and about 60 to 80 % of maltase activity in the intestinal lumen [3].
Physiological and functional requirements for carbohydrate processing
Before SI can fulfill its hydrolytic function in the intestinal lumen, it needs to be intracellularly processed and properly transported to the BBM surface of the epithelial cells. Both active subunits are then oriented into the intestinal lumen [4]. SI is a type II membrane glycoprotein. It is N-glycosylated in the endoplasmic reticulum (ER) and then transported to the Golgi apparatus where it is complex N- and O-glycosylated. Both of these modifications have a crucial role in the folding, acquisition of the functional capacity, and later appropriate sorting of the SI molecules to the BBM [5]. The latter process is mediated via association of SI molecules with cholesterol- and sphingolipid-enriched membrane microdomains, known as lipid rafts [6], in the trans-Golgi network for which proper O-glycosylation is essentially required [5].
Etiology and common symptoms of intestinal disorders by SI malfunction
The clinical presentation of intestinal disorders caused by SI malfunction is oftentimes an osmotic-fermentative diarrhea upon ingestion of carbohydrates associated with symptoms such as vomiting, flatulence and abdominal pain [2]. An increased osmotic load due to unabsorbed carbohydrates in the intestinal lumen and colon causes a flow of water and electrolytes into the lumen, thus affecting the gut motility by accelerating the small intestinal transit leading to chronic diarrhea. Other symptoms like gaseous abdominal distension, flatulence, and cramps are caused by bacterial fermentation of unabsorbed carbohydrates which additionally increase the osmotic pressure [3]. The accelerated transit further increases the degree of maldigestion of starch and monosaccharides and also affects the absorption of other nutrients, leading to a risk of malnutrition and failure to thrive in patients [7]. Additional factors contributing to the onset of disease are the quantity of ingested carbohydrates and other food components, the rate of gastric emptying, the metabolic activity of fermenting bacteria, the degree of the residual enzyme activity of SI and other intestinal disaccharidases as well as coexisting intestinal disorders or inflammation.
SI deficiencies can be categorized as (a) primary or congenital SI deficiencies which are caused by genetic alterations in the SI gene and are already present at birth and (b) induced or secondary SI deficiencies which occur later in life and are mediated by environmental factors or arise as a consequence of other genetic deficiencies which negatively influence the intestinal physiology in general and SI function in particular.
Congenital sucrase-isomaltase deficiency
Congenital sucrase-isomaltase deficiency (CSID) is a genetically determined primary defect of SI which is associated with carbohydrate malabsorption [3]. CSID is elicited by single-nucleotide polymorphism in the SI gene leading to single amino acid exchanges in the protein. These mutations can affect each of the SI domains but do not per se lead to complete lack of SI or its activities in the BBM essentially. Requirements for proper functioning of SI, including intact glycosylation and folding, correct intracellular trafficking, association with lipid rafts and apical delivery to the cell surface are directly or indirectly correlated to each other and are initially regulated at the amino acid sequence level [5]. Malfolding of SI mutants may affect the trafficking or even lead to degradation via the endoplasmic reticulum-associated protein degradation (ERAD) pathway [8]. Other mutants of SI proteins with impaired or delayed trafficking though with normal hydrolytic properties cannot fulfill their physiological functions due to mislocalization. Mutations can affect one or more of these features depending on their position within SI. Biochemical, cellular and functional analyses of a number of SI mutations constituted the basis for a classification of these mutants into seven phenotypes depending on the intracellular localization and function of SI [9] (Table 1). Phenotypes I and II include mutations which are blocked in the ER (I) or ER-Golgi intermediate compartment (ERGIC) and cis-Golgi compartments (II). A normal trafficking but absent enzymatic function characterizes mutations of phenotype III. SI mutants grouped in phenotype IV exhibit sorting defects to the microvillar membranes and that of phenotypes V and VI are intracellularly cleaved. Phenotype VII is characterized by altered folding, increased turnover rate, and partial missorting [9]. The most common mutations with an estimated frequency of 83 % in European descendants with CSID are Gly1073Asp, Val577Gly, Phe1745Cys and Arg1124Stop [10]. These mutations are intracellularly blocked in the ER and belong to phenotype I. A decisive factor in occurrence and severity of CSID is whether only one or both alleles of the gene are affected by mutations. Initially reported cases of CSID that were characterized by genetic sequencing mostly illustrated homozygote or compound heterozygote patterns of inheritance [11]. In the recent years, feasibility of gene sequencing has led to identification of many novel cases of CSID, among which also simple heterozygote subjects with only one affected allele exist [10].
Based on the mutation type, the combinatorial effects of two mutations or of one mutation with the wild-type protein can set the course of the disease and generate a mild to severe gradient of symptoms. The CSID symptoms are similar to those of other intestinal diseases and oftentimes lead to misdiagnosis or late diagnosis after childhood [3]. The high phenotypic diversity, genetic heterogeneity and common symptoms with other intestinal diseases lead to the assumption that CSID is more common than what has been initially estimated.
Secondary deficiencies of SI activity
The major types of secondary or induced SI deficiencies can be categorized into three main groups: (a) those that are induced by physical injuries to the intestine and disrupt the intestinal epithelium, (b) those that are caused via inhibitory function of some dietary components or therapeutic agents on the function of SI, and (c) those that are connected to infections or autoimmune disorders. In what follows, we will discuss the role of therapeutic agents and autoimmune disorders on SI deficiency in more details.
-
i)
Therapeutic agents that inhibit the SI function
There exist a large number of reports on the natural or synthetic compounds for deliberate reduction of disaccharidase activities in the intestine in order to control obesity or diabetic conditions. Apart from such compounds, our focus here is on medications that induce SI inhibition as an adverse effect. For example, N-butyldeoxynojirimycin (miglustat) that is used for treatment of lysosomal storage diseases can heavily inhibit SI and thus result in gastrointestinal symptoms in the majority of the patients [12]. Codeine as a pain medication and ranitidine with antihistamine effect can also inhibit sucrase activity in the intestine [13, 14]. Furthermore, several herbal folk remedies, especially those with polyphenolic components, also exert an inhibitory effect on SI [15].
-
ii)
Role of gut autoimmune disorders in SI deficiency
Intestinal lymphatic tissue, named gut-associated lymphoid tissue (GALT), is known to be the largest compartment of the immune system. The intestinal tract is highly and directly exposed to foreign material through the ingestion of food. On the other hand, the commensal bacteria in the colon, the gut microbiota, can be differently modulated by diet or physiological conditions of the gut and can consequently influence the gut immune system [16]. All of these factors have rendered the gut tissue highly susceptible to autoimmune disorders and the subsequent tissue pathogenesis.
-
iii)
Celiac disease
Celiac disease (CD) is one of the most common causes of chronic intestinal malabsorption and leads to occurrence of conventional gastrointestinal symptoms as a result of villus atrophy [17]. The disease is an autoimmune disorder that is triggered by hypersensitivity to ingested gliadins from wheat and other cereals [18]. The frequency of this disease can be up to 3 % in the different populations, but this ratio was detected to be as high as 11 % among patients with type 1 diabetes mellitus [19]. As a direct consequence of villus atrophy in CD, the expression and activity levels of intestinal disaccharidases including SI are significantly reduced. Therefore, SI expression has been used as an indicator to diagnose and later evaluate the physiological response to different treatments of the patients [20, 21]. Disaccharidase deficiencies can be also detected in CD patients with intact villi, which may represent latent development of CD [22].
-
iv)
Inflammatory bowel disease
Inflammatory bowel disease (IBD) which is mostly diagnosed in the forms of Crohn’s disease and ulcerative colitis is an autoimmune disease in which imbalance between anti- and pro-inflammatory cytokines results in severe organ pathology and loss of barrier function in the intestinal tissue [23]. Induced colitis in animal models revealed local colon inflammation affecting the intestinal function in the ileal and jejunal areas and resulting in loss of SI expression and activity [24]. Besides that, the existing SI molecules are not properly expressed at the apical cell surface and reveal a predominant intracellular localization [25]. In a Caco-2 cell model of the intestinal epithelium, treatment with interleukin 6 and interferon gamma led to a significant decrease in the biosynthesis of SI [26]. Therefore, it can be concluded that SI deficiency in IBD patients occurs as an indirect effect of tissue injury alongside the direct effects of the inflammatory cytokines on the expression and function of SI.
-
v)
Human immunodeficiency virus infection
Gastrointestinal intolerances are common symptoms in human immunodeficiency virus (HIV)-infected patients [27]. In contrast to healthy HIV-positive individuals, severe SI and lactase-phlorizin hydrolase deficiencies are identified in most of the jejunal specimens from patients with acquired immune deficiency syndrome (AIDS) [27]. The gastrointestinal symptoms are associated with rapid small bowel transit [28] and are not influenced by anti-retroviral therapy [29]. Infection of rhesus monkeys with simian immunodeficiency virus (SIV) is a known experimental animal model for HIV. As shown in this model, gut-associated lymphoid tissue is one of the first targets of SIV infection [30]. Negative regulation of the IL-6-STAT3 pathway seems to be responsible for the persistent inflammation in the jejunum and colon of these animals [31]. Development of disaccharidase deficiencies in later stages of HIV infection can be also associated with immunodeficiency or infection with opportunistic enteropathogens [30].
-
vi)
Giardiasis
Giardiasis can also trigger autoimmune responses that cause mucosal alterations in the intestine. In this case, the microvilli are shortened and the level of SI as well as other disaccharidases is reduced in a direct relation to the number of parasites [32]. In a study using isolated T cells from infected mice, Scott et al. could show that the CD8(+) T cells mediate the loss of brush border area and reduce disaccharidase levels in the uninfected recipient mice [33].
-
vii)
Other secondary factors influencing the SI function
Additionally reported factors influencing the function of SI and thereby triggering carbohydrate malabsorption symptoms are summarized in this part. Rotavirus infections or acute Yersinia enterocolitica infections affect the microvillar cytoskeleton structure of the intestinal enterocytes and thereby cause a decreased disaccharidase expression and function at the BBM [34, 35]. Also, the Shiga toxin (Stx) is reported to affect SI function by inhibiting the synthesis of BBM proteins as well as damaging the intestinal epithelium directly [36]. If the intestinal tissue is subjected to ischemia with blocked or decreased blood flow, a decrease in disaccharidase activities can be detected [37]. Chronic psychological stresses as well as deficiencies of iron and vitamin A are other factors that are associated with loss of the sucrase activity in the intestine [38–41].
Diagnosis
The gold standard for the diagnosis of intestinal disorders associated with carbohydrate malabsorption due to primary or secondary defects in SI are endoscopic small bowel biopsies and assessment of disaccharidase activities exvivo. The additional histological examination of the intestinal biopsy specimen allows distinguishing between primary and secondary disaccharidase disorders [42]. In primary disorders, normal intestinal morphology concomitant with reduced enzymatic activities are expected while secondary disorders oftentimes go along with an impaired intestinal morphology. Confocal laser endomicroscopy (CLE) is a novel endoscopic technique to visualize the intestinal morphology in vivo [43]. A non-invasive but not always reliable diagnostic approach is the hydrogen breath test that measures the exhaled H2 levels produced by bacterial fermentation after ingestion of a test carbohydrate. An increased hydrogen of more than 20 part per million is considered to indicate carbohydrate malabsorption [44]. Mutations in the SI gene causing CSID can be identified by genetic screenings using saliva or blood of patients. Prior to the mentioned diagnostic methods, a dietary assessment by food exclusion and observation of symptom improvement can give first hints to carbohydrate malabsorption. Blood and fecal tests can additionally provide information on possible intestinal inflammation [45].
Therapy
There are different treatment options of disaccharide malabsorption due to SI malfunction. One possibility is dietary management by a life-long sucrose- and starch-restricted diet adapted to the requirements of the patient. A good alternative to the avoidance of malabsorbed sugars, which may not be easy especially for children, is the enzyme replacement therapy to compensate the malfunction of SI. Sacrosidase or invertase (Sucraid) which is a liquid preparation from Saccharomyces cerevisiae is frequently used as therapy for intestinal disorders associated with carbohydrate malabsorption [3]. Prevention and treatment of secondary intestinal disorders with indirect influence on the SI function can be achieved by the ingestion of probiotics which are beneficial microorganisms to the human health [46]. The normal intestinal flora or microbiota provides an effective barrier against pathogenic microorganism. Disruption of this healthy microflora can increase the susceptibility for pathogenic infections which may in turn affect the BBM and thereby negatively influence the SI function. Intake of prebiotics (poorly digestible carbohydrates) and probiotics supports the maintenance or reestablishment of a healthy gut microflora [46, 47].
Conclusions
SI is a unique enzyme of the intestinal epithelium due to its high prevalence and its wide substrate specificity for digestion of different dietary carbohydrates. Therefore, deficiencies in the SI function can substantially disrupt the intestinal physiology, a fact associated with gastrointestinal symptoms, weight loss, and immunological disorders mediated by altered gut microbiota. On the other hand, organ pathologies that generally influence the intestinal tissue, such as IBD or specific inhibition of SI by therapeutic or dietary substances can substantially reduce the capacity of the intestinal lumen in SI activity (Fig. 1). If not treated, the outcome of this condition can result in progressive and life-threatening gastrointestinal symptoms. Therefore, it is legitimate to consider a major role for the proper function of SI in the maintenance of the intestinal physiology.

Multifactorial causes of primary or secondary sucrase-isomaltase deficiencies. The SI function in cleaving carbohydrates depends on the correct transport of the enzymatically competent protein to the BBM. This process occurs via association of SI with lipid rafts in the trans-Golgi network vesicles that are transported to the BBM along the actin cytoskeleton. The digestive capacity of SI can be affected by primary genetic mutations that target the SI gene. On the other hand, environmental factors can also influence expression and function of SI. The induced or secondary effects on the SI function can be elicited by altered epithelial cell integrity or the cytoskeletal organization of the enterocytes as well as changes in the healthy protective microflora. In some cases, the structure and function of SI can be directly influenced by certain therapeutics or dietary components. Together, alterations in the intestinal physiology triggered for example by inflammation can exert adverse effects on the trafficking and function of SI. The maldigested carbohydrates will in turn moderate the gut microbiota differently from healthy conditions and may render subsequent negative influences on the intestinal physiology
Abbreviations
- AIDS:
-
acquired immune deficiency syndrome
- BBM:
-
brush border membrane
- CD:
-
celiac disease
- CLE:
-
confocal laser endomicroscopy
- CSID:
-
congenital sucrase-isomaltase deficiency
- ER:
-
endoplasmic reticulum
- ERAD:
-
endoplasmic reticulum-associated protein degradation
- ERGIC:
-
ER-Golgi intermediate compartment
- GALT:
-
gut-associated lymphoid tissue
- HIV:
-
human immunodeficiency virus
- IBD:
-
inflammatory bowel disease
- MGAM:
-
maltase-glucoamylase
- SI:
-
sucrase-isomaltase
- SIV:
-
simian immunodeficiency virus
- Stx:
-
Shiga toxin
References
Mann J, Cummings JH, Englyst HN, Key T, Liu S, Riccardi G, Summerbell C, Uauy R, van Dam RM, Venn B, Vorster HH, Wiseman M (2007) FAO/WHO scientific update on carbohydrates in human nutrition: conclusions. Eur J Clin Nutr 61(Suppl 1):S132–S137
Naim HY, Zimmer K-P (2008) Genetically determined disaccharidase deficiency. In: Kleinman R, Goulet O-J, Mieli-Vergani G, Sanderson I, Sherman P, Shneider B (eds) Walker’s Pediatric Gastrointestinal Disease, 5th edn, BC Decker Inc, Hamilton
Treem WR (2012) Clinical aspects and treatment of congenital sucrase-isomaltase deficiency. J Pediatr Gastroenterol Nutr 55:S7–S13
Naim HY, Sterchi EE, Lentze MJ (1988) Biosynthesis of the human sucrase-isomaltase complex. Differential O-glycosylation of the sucrase subunit correlates with its position within the enzyme complex. J Biol Chem 263(15):7242–7253
Alfalah M, Jacob R, Preuss U, Zimmer KP, Naim H, Naim HY (1999) O-linked glycans mediate apical sorting of human intestinal sucrase-isomaltase through association with lipid rafts. Curr Biol 9(11):593–596
Lindner R, Naim HY (2009) Domains in biological membranes. Exp Cell Res 315(17):2871–2878
Belmont JW, Reid B, Taylor W, Baker SS, Moore WH, Morriss MC, Podrebarac SM, Glass N, Schwartz ID (2002) Congenital sucrase-isomaltase deficiency presenting with failure to thrive, hypercalcemia, and nephrocalcinosis. BMC Pediatr 2:4
Bernasconi R, Molinari M (2011) ERAD and ERAD tuning: disposal of cargo and of ERAD regulators from the mammalian ER. Curr Opin Cell Biol 23(2):176–183
Naim HY, Heine M, Zimmer KP (2012) Congenital sucrase-isomaltase deficiency: heterogeneity of inheritance, trafficking, and function of an intestinal enzyme complex. J Pediatr Gastroenterol Nutr 55 Suppl 2:S13–S20
Uhrich S, Wu Z, Huang J-Y, Scott CR (2012) Four mutations in the SI gene are responsible for the majority of clinical symptoms of CSID. J Pediatr Gastroenterol Nutr 55:S34–S35
Sander P, Alfalah M, Keiser M, Korponay-Szabo I, Kovacs JB, Leeb T, Naim HY (2006) Novel mutations in the human sucrase-isomaltase gene (SI) that cause congenital carbohydrate malabsorption. Hum Mutat 27(1):119
Amiri M, Naim HY (2012) Miglustat-induced intestinal carbohydrate malabsorption is due to the inhibition of alpha-glucosidases, but not beta-galactosidases. J Inherit Metab Dis 35(6):949–954
Minai-Tehrani D, Ghaffari M, Sobhani-Damavandifar Z, Minoui S, Alavi S, Osmani R, Ahmadi S (2012) Ranitidine induces inhibition and structural changes in sucrase. J Enzyme Inhib Med Chem 27(4):553–557
Minai-Tehrani D, Minoui S, Sepehre M, Sharif-Khodai Z, Aavani T (2009) Inhibitory effect of codeine on sucrase activity. Drug Metab Lett 3(1):58–60
Choi HJ, Kim NJ, Kim DH (2000) Inhibitory effects of crude drugs on alpha-glucosidase. Arch Pharm Res 23(3):261–266
Azzali G (2003) Structure, lymphatic vascularization and lymphocyte migration in mucosa-associated lymphoid tissue. Immunol Rev 195:178–189
Cornell HJ, Auricchio RS, Deritis G, Devincenzi M, Maiuri L, Raia V, Silano V (1988) Intestinal-mucosa of celiacs in remission is unable to abolish toxicity of gliadin peptides on in vitro developing fetal-rat intestine and cultured atrophic celiac mucosa. Pediatr Res 24(2):233–23
Vriezinga SL, Schweizer JJ, Koning F, Mearin ML (2015) Coeliac disease and gluten-related disorders in childhood. Nat Rev Gastroenterol Hepatol 12(9):527–536
Armstrong MJ, Hegade VS, Robins G (2012) Advances in coeliac disease. Curr Opin Gastroenterol 28(2):104–112
Prasad KK, Thapa BR, Nain CK, Sharma AK, Singh K (2008) Brush border enzyme activities in relation to histological lesion in pediatric celiac disease. J Gastroenterol Hepatol 23(8):E348–E352
Nieminen U, Kahri A, Savilahti E, Farkkila MA (2001) Duodenal disaccharidase activities in the follow-up of villous atrophy in coeliac disease. Scand J Gastroenterol 36(5):507–510
Murray IA, Smith JA, Coupland K, Ansell ID, Long RG (2001) Intestinal disaccharidase deficiency without villous atrophy may represent early celiac disease. Scand J Gastroenterol 36(2):163–168
Neurath MF (2014) Cytokines in inflammatory bowel disease. Nat Rev Immunol 14(5):329–342
Amit-Romach E, Reifen R, Uni Z (2006) Mucosal function in rat jejunum and ileum is altered by induction of colitis. Int J Mol Med 18(4):721–727
Andrews CW, Ohara CJ, Goldman H, Mercurio AM, Silverman ML, Steele GD (1992) Sucrase-isomaltase expression in chronic ulcerative-colitis and dysplasia. Hum Pathol 23(7):774–779
Ziambaras T, Rubin DC, Perlmutter DH (1996) Regulation of sucrase-isomaltase gene expression in human intestinal epithelial cells by inflammatory cytokines. J Biol Chem 271(2):1237–1242
Taylor C, Hodgson K, Sharpstone D, Sigthorsson G, Coutts M, Sherwood R, Menzies I, Gazzard B, Bjarnason I (2000) The prevalence and severity of intestinal disaccharidase deficiency in human immunodeficiency virus-infected subjects. Scand J Gastroenterol 35(6):599–606
Sharpstone D, Neild P, Crane R, Taylor C, Hodgson C, Sherwood R, Gazzard B, Bjarnason I (1999) Small intestinal transit, absorption, and permeability in patients with AIDS with and without diarrhoea. Gut 45(1):70–76
Lim SG, Menzies IS, Nukajam WS, Lee CA, Johnson MA, Pounder RE (1995) Intestinal disaccharidase activity in human-immunodeficiency-virus disease. Scand J Gastroenterol 30(3):235–241
Heise C, Miller CJ, Lackner A, Dandekar S (1994) Primary acute simian immunodeficiency virus-infection of intestinal lymphoid-tissue is associated with gastrointestinal dysfunction. J Infect Dis 169(5):1116–1120
Mohan M, Aye PP, Borda JT, Alvarez X, Lackner AA (2007) Gastrointestinal disease in simian immunodeficiency virus-infected rhesus macaques is characterized by proinflammatory dysregulation of the interleukin-6-janus kinase/signal transducer and activator of transcription3 pathway. Am J Pathol 171(6):1952–1965
Daniels CW, Belosevic M (1995) Disaccharidase activity in male and female C57BL/6 mice infected with Giardia muris. Parasitol Res 81(2):143–147
Scott KGE, Yu LCH, Buret AG (2004) Role of CD8(+) and CD4(+) T lymphocytes in jejunal mucosal injury during murine Giardiasis. Infect Immun 72(6):3536–3542
Buret A, O'Loughlin EV, Curtis GH, Gall DG (1990) Effect of acute Yersinia enterocolitica infection on small intestinal ultrastructure. Gastroenterology 98(6):1401–1407
Jourdan N, Brunet JP, Sapin C, Blais A, Cotte-Laffitte J, Forestier F, Quero AM, Trugnan G, Servin AL (1998) Rotavirus infection reduces sucrase-isomaltase expression in human intestinal epithelial cells by perturbing protein targeting and organization of microvillar cytoskeleton. J Virol 72(9):7228–7236
Chopra P, Verma D, Khullar M, Sapru S, Mahmood S (2006) Shiga toxin exposure modulates intestinal brush border membrane functional proteins in rabbit ileum. Mol Cell Biochem 283(1-2):85–92
Varga J, Toth S Jr, Toth S, Tomeckova V, Gregova K, Vesela J (2012) The relationship between morphology and disaccharidase activity in ischemia-reperfusion injured intestine. Acta Biochim Pol 59(4):631–638
Boudry G, Jury J, Yang PC, Perdue MH (2007) Chronic psychological stress alters epithelial cell turn-over in rat ileum. Am J Physiol Gastrointest Liver Physiol 292(5):G1228–G1232
Lanzkowsky P, Karayalcin G, Miller F (1982) Disaccharidase levels in iron deficient rats at birth and during the nursing and postweaning periods: response to iron treatment. Pediatr Res 16(4 Pt 1):318–323
West AR, Oates PS (2005) Decreased sucrase and lactase activity in iron deficiency is accompanied by reduced gene expression and upregulation of the transcriptional repressor PDX-1. Am J Physiol Gastrointest Liver Physiol 289(6):G1108–G1114
Reifen R, Zaiger G, Uni Z (1998) Effect of vitamin A on small intestinal brush border enzymes in a rat. International journal for vitamin and nutrition research Internationale Zeitschrift fur Vitamin- und Ernahrungsforschung Journal international de vitaminologie et de nutrition 68(5):281–286
Robayo-Torres CC, Quezada-Calvillo R, Nichols BL (2006) Disaccharide digestion: clinical and molecular aspects. Clinical gastroenterology and hepatology : the official clinical practice journal of the American Gastroenterological Association 4(3):276–287
Dunbar K, Canto M (2008) Confocal endomicroscopy. Curr Opin Gastroenterol 24(5):631–637
Levitt MD, Donaldson RM (1970) Use of respiratory hydrogen (H2) excretion to detect carbohydrate malabsorption. J Lab Clin Med 75(6):937–945
Langhorst J, Elsenbruch S, Koelzer J, Rueffer A, Michalsen A, Dobos GJ (2008) Noninvasive markers in the assessment of intestinal inflammation in inflammatory bowel diseases: performance of fecal lactoferrin, calprotectin, and PMN-elastase, CRP, and clinical indices. Am J Gastroenterol 103(1):162–169
Peterson CT, Sharma V, Elmen L, Peterson SN (2015) Immune homeostasis, dysbiosis and therapeutic modulation of the gut microbiota. Clin Exp Immunol 179(3):363–377
Fedorak RN, Madsen KL (2004) Probiotics and prebiotics in gastrointestinal disorders. Curr Opin Gastroenterol 20(2):146–155
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Gericke, B., Amiri, M. & Naim, H.Y. The multiple roles of sucrase-isomaltase in the intestinal physiology. Mol Cell Pediatr 3, 2 (2016). https://doi.org/10.1186/s40348-016-0033-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40348-016-0033-y