
Abstract
Background
RUNX1 is a transcription factor and a master regulator for the specification of the hematopoietic lineage during embryogenesis and postnatal megakaryopoiesis. Mutations and rearrangements on RUNX1 are key drivers of hematological malignancies. In humans, this gene is localized to the ‘Down syndrome critical region’ of chromosome 21, triplication of which is necessary and sufficient for most phenotypes that characterize Trisomy 21.
Main body
Individuals with Down syndrome show a higher predisposition to leukemias. Hence, RUNX1 overexpression was initially proposed as a critical player on Down syndrome-associated leukemogenesis. Less is known about the functions of RUNX1 in other tissues and organs, although growing reports show important implications in development or homeostasis of neural tissues, muscle, heart, bone, ovary, or the endothelium, among others. Even less is understood about the consequences on these tissues of RUNX1 gene dosage alterations in the context of Down syndrome. In this review, we summarize the current knowledge on RUNX1 activities outside blood/leukemia, while suggesting for the first time their potential relation to specific Trisomy 21 co-occurring conditions.
Conclusion
Our concise review on the emerging RUNX1 roles in different tissues outside the hematopoietic context provides a number of well-funded hypotheses that will open new research avenues toward a better understanding of RUNX1-mediated transcription in health and disease, contributing to novel potential diagnostic and therapeutic strategies for Down syndrome-associated conditions.
Similar content being viewed by others
Introduction
RUNX genetics and functions
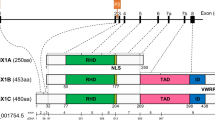
The RUNX family of transcription factors (TFs) play critical functions during development and adult organ homeostasis and are responsible for several human diseases. Runt-related genes (RUNX-1, -2, -3 and CBFβ) are members of the core-binding factor (CBF) family, and participate in a wide array of processes in several cell types and tissues. The three RUNX genes exhibit a highly conserved genomic organization across metazoans (Fig. 1A; reviewed in [1]). In vertebrates, RUNX genes are characterized by the presence of two alternative promoters, P1 (distal) and P2 (proximal) [2, 3] (Fig. 1B). RUNX TFs also feature an evolutionarily conserved Runt homology domain (RHD) in their N-terminal half [3] (Fig. 1A). The RHD is responsible for interaction with the RUNX-cognate DNA motif-PyGPyGGTPy-[4, 5], but also mediates protein–protein interactions [6, 7] and harbors a nuclear localization signal [8, 9]. A transactivation domain (TAD) and an inhibitory domain (ID) are found in the C-terminal portion, together with a terminal VWRPY motif, which binds the Groucho/TLE (Transducin-Like Enhancer of split) family of transcriptional corepressors [10,11,12]. Heterodimerization of RUNX with CBFβ is required to allosterically enhance the complex stability and affinity for DNA, leading to induction or repression of target genes [13,14,15,16] (Fig. 1C).

A. Domain organization of the RUNX family of transcription factors. General domain architecture of the mammalian RUNX family members. B. RUNX1 exon organization and major alternative splicing isoforms. Major RUNX1 variants produced by alternative promoter/splicing mechanisms (see main text and BOX for details). C. RUNX-CBFβ form heterodimers to interact with DNA and regulate transcription. Heterodimerization of RUNX TFs with CBFβ is required for enhanced DNA binding, stability and interaction with other transcriptional regulators, leading to induction or repression of target genes
Recent years have seen significant progress in our understanding of the functions performed by RUNX1 in several developing and postnatal mammalian tissues, a topic that has been recently covered elsewhere [17]. RUNX1, also known as Acute Myeloid Leukemia 1 protein (AML1), is widely considered the master regulator of developmental hematopoiesis because it has a critical function in the specification of the hematopoietic lineage during embryogenesis, while also playing essential roles in postnatal tissue homeostasis and in hematological malignancies.
Noteworthy, in humans, the RUNX1 gene is localized to band q22.12 of chromosome 21, which is triplicated in individuals with Trisomy 21 (T21). Hence, an extra copy of RUNX1 has been proposed to play relevant roles in some of the many phenotypic alterations associated with Down syndrome (DS). In addition to alternative promoter usage, in humans, RUNX1 mRNA splicing gives rise to multiple isoforms, of which RUNX1c (from P1), RUNX1a and RUNX1b (from P2) are the most abundant and best characterized [18, 19] (Fig. 1B). While RUNX1b and RUNX1c are functionally identical, RUNX1a is a shorter variant lacking the transactivation and inhibitory domains (Fig. 1B). For that reason, RUNX1a has been shown to have an antagonic, pro-oncogenic function (see BOX). More importantly, as reported by Komeno et al., the alternative splicing of exon 7a that gives rise to RUNX1a is primate-specific and thus, mice do not naturally express the RUNX1a isoform [19]. In summary, because mouse models of Down syndrome lack the RUNX1a isoform, which may be critical to T21-associated leukemogenesis, they may not be the best system to study RUNX1 function in Down syndrome (Fig. 1B and BOX [19,20,21,22,23,24,25,26,27,28,29,30,31,32,33,34,35,36]).

RUNX1 activities in normal megakaryopoiesis and hematopoietic stem cell maintenance, and their relations to hematological diseases, have been extensively studied for many years and, therefore, will not be covered in the present review. On that regard, for additional information refer to the very complete literature published in recent years (e.g., [37, 38]. Nevertheless, DS individuals show a disproportionately higher predisposition to specific hematological malignancies, and RUNX1 overexpression has been proposed to contribute to such dysregulation. Therefore, we will briefly touch on the DS-specific hematological implications of RUNX1. On the other side, much less is known about the functions of this transcription factor in other developmental and homeostatic processes, and how these could contribute to DS comorbidities, which will be the major topics of the following sections.
RUNX1 in leukemia
Mutations of RUNX1 are the underlying cause of familial platelet disorders that precede several myeloid malignancies. Somatic mutations and chromosomal rearrangements involving RUNX1 loss-of-function or changes of its transcriptional activity are frequently observed in myelodysplastic syndrome and leukemias of myeloid and lymphoid lineages, such as acute myeloid leukemia (AML), acute lymphoblastic leukemia (ALL), and chronic myelomonocytic leukemia (CMML). This topic is very well understood and therefore, for additional information refer to the excellent available reviews on this [39, 40]. On the other hand, recent studies suggest that wild-type RUNX1 is also required for growth and survival of certain types of AML cell lines [41]. Similarly, Gialesaki et al. observed a strong RUNX1-dependency in DS-associated myeloid leukemia (ML-DS) cell lines [20], which is also seen for many myeloid and lymphoid cancer cell lines from the Cancer Dependency Map portal (https://depmap.org) (Fig. 2).

RUNX1 dependency across cancer cell lines from the Cancer Dependency Map portal (depma.org). The Chronos dependency score is based on data from a cell depletion assay. A lower score indicates a higher probability that the gene (RUNX1) is essential for a given cell line. A score of 0 indicates a gene is not essential, − 1 is comparable to the median of all pan-essential genes. Note that a significant proportion of Lymphoid and particularly Myeloid cancer cell lines—but not other cancer types- require RUNX1 expression for survival, as suggested by a Chronos dependency score below − 0.5. Importantly, RUNX1 dependency across Lymphoid and Myeloid cancer cell lines is significantly correlated to the level of RUNX1 mRNA expression in each cell line (not shown)
Down syndrome and hematopoietic disorders
Newborns with Down syndrome are predisposed to a variety of hematopoietic abnormalities, ranging from relatively benign, such as neutrophilia and macrocytosis, to more severe transient myeloproliferative disorders (TMD). In most cases, these abnormalities resolve in the first few months/years of life. Nevertheless, about 10% of DS-associated TMD patients progress into acute megakaryoblastic leukemia (AMKL). AMKL in individuals with Down syndrome is heavily associated with acquired GATA1 mutations that result in the dominant expression of a short isoform, collectively referred to as GATA1s (short) mutations [42]. GATA1s is a natural alternative splice variant lacking the transactivation domain [42]. Contrary to the full-length variant, GATA1s fails to repress pro-oncogenic MYC expression and the pro-proliferative E2F transcription network, constituting a major mechanism of leukemic transformation [43,44,45,46]. Thus, DS children have a significantly higher risk of developing leukemia [47], while showing a dramatically reduced frequency of solid tumors [48]. Children with T21 have ~ 500-fold higher chances of developing AMKL, which is relatively rare in the general population, while the risk of pediatric acute lymphoblastic leukemia (ALL) is 20 to 30-fold greater in DS kids [47]. Several genes lying within the so-called Down syndrome critical region (DSCR) at 21q22—including RUNX1 and DYRK1A—are thought to contribute to such enhanced risk to leukemias [49].
RUNX1 in DS-associated leukemia
While RUNX1 is an etiologic factor in AMKL [50], there is conflicting evidence regarding RUNX1 in ML-DS. In the first place, most of the characterized RUNX1 mutations/translocations result in loss of function, suggesting that wild-type RUNX1 plays a tumor-suppressive role. In ML-DS, RUNX1 is not usually mutated. Therefore, overexpression of wild-type RUNX1 in DS cells (due to an extra copy of the gene) is predicted to decrease, rather than augment, the risk of leukemia. Results from several in vivo DS mouse models seem to indicate that RUNX1 is not the cause for the observed abnormal hematopoiesis, myeloproliferative disease or leukemia [51,52,53]. On one hand, the Ts65Dn murine model of Down syndrome develops persistent macrocytosis and myeloproliferative disease (characterized by thrombocytosis, megakaryocyte hyperplasia with dysplastic morphology, and myelofibrosis). Restoring RUNX1 gene dosage to disomic levels (Ts65Dn/Runx1+/±) did not rescue this phenotype [51]. Carmichael et al. [52] observed that the Ts1Cje DS mouse model—also trisomic for RUNX1—has mild defects in mature blood cells, including macrocytosis and anemia, but do not develop TMD or leukemia, even in the context of a Gata1s mutation. Finally, the Tc1 mouse model of Down syndrome also exhibits macrocytic anemia and increased extramedullary hematopoiesis [53, 54]. Moreover, enhanced megakaryopoiesis was seen by concomitant GATA1s mutation in these mice, but not resulting in leukemia or TMD [53]. Noteworthy, the Tc1 model is trisomic for approximately 80% of the genes encoded on Hsa21, but not for RUNX1, ruling out its involvement in the observed Tc1 hematopoietic alterations, but allowing for the speculation of whether its triplication could still lead to a more aggressive myeloproliferative disease in this paradigm. These data are consistent with a role for RUNX1 in the potentiation of megakaryopoiesis [51].
A cooperative functional interaction between wild type GATA1 and RUNX1 is known to drive normal megakaryocytic differentiation [55, 56], and this interaction seems to be conserved in the GATA1s pre-leukemic mutant version in DS-AMKL cells [56]. Using transchromosomic mouse embryonic stem cells (ESCs) bearing an additional copy of human chromosome 21 (HSA21), De Vita, et al. [57] found that an extra copy of RUNX1 caused an increase in Tie-2/c-Kit levels, partially contributing to the disturbance of early hematopoiesis in DS. More recently, Vukadin et al. [58] proposed a role for another chromosome 21 gene, SON, as a transcriptional repressor of RUNX1 expression and subsequent megakaryocytic differentiation. They showed that SON is aberrantly overexpressed in AMKL, while depletion of SON markedly increased megakaryocytic gene expression and differentiation. In line with this, Bourquin et al. [59] previously reported SON as the second most upregulated chromosome 21-encoded transcription factor in DS-AMKL samples (vs. typical AMKL). Interestingly, they also indicated that RUNX1 expression in DS-AMKL patients is decreased, despite its increased gene dosage. Thus, since RUNX1 is essential for normal megakaryopoiesis, reduction of RUNX1 expression by SON in DS-AMKL could contribute to impaired megakaryocytic differentiation. Work from two other groups recently showed that the RUNX1 gene is significantly hypermethylated in neonatal blood samples from DS newborns [60, 61]. Altogether, several pieces of evidence, including data from 3 different murine models of DS, appear to suggest that RUNX1 triplication might not play a crucial role in DS-associated leukemia, but perhaps in milder phenotypes related to megakaryopoiesis and erythropoiesis.
Despite of this, a recent report has provided important evidence challenging the idea that RUNX1 overexpression may not play a role in leukemia. Gialesaki et al. [20], uncovered a strong and specific RUNX1-dependency in ML-DS and non-DS AMKL cells. They further demonstrated that expression of the short RUNX1a splicing isoform (Fig. 1B and BOX), which lacks the transactivation domain, is elevated in patients with ML-DS. Mechanistic studies using murine ML-DS models and patient-derived xenografts revealed that during leukemogenesis, elevation of RUNX1a displaced the full-length RUNX1c variant from its endogenous binding sites, inducing oncogenic programs through direct interaction with MYC/MAX, and thus, synergizing with the pathognomonic Gata1s mutation. Hence, overexpression of the shorter alternative splicing variant RUNX1a, with opposed functions to the full-length protein, could indeed contribute to the leukemic phenotype in specific DS cell types. Since RUNX1a is not expressed in rodents [19], interpretation of experiments on RUNX1 functions from mouse models of Down syndrome and their extrapolation to humans should be done with great caution.
RUNX1 in other DS-related phenotypes
As previously discussed in section "RUNX1 in DS-associated leukemia.", the implications of RUNX1 in Trisomy 21 have been mostly studied in the context of hematological malignancies. Nevertheless, several groups have started to shed light into potential functions of this pleiotropic transcription factor in other DS-related alterations. Mollo et al. [62] re-analyzed mouse gene expression data obtained after over-expression of individual Hsa21 genes (GEO GSE19836 [63]) and saw that the transcription factor Runx1 can induce upregulation of extracellular matrix (ECM) genes. Moreover, the ECM was consistently among the most affected GO categories by gene set enrichment analysis (GSEA) from public data (including experiments with either RUNX1 upregulation or downregulation). Interestingly, they quantified that ~ 80% of ECM genes overexpressed in T21 hearts had RUNX1 consensus sequences. Furthermore, they saw that in T21 human fetal fibroblasts there is increased expression of both RUNX1 and several ECM genes, which decreased upon RUNX1 silencing. These results suggest potential implications for RUNX1 in ECM alterations as an underlying cause for DS-related congenital heart defects [64], Hirschsprung’s disease [65] and/or pulmonary hypertension [66], among other ECM-mediated pathologic conditions.
Halevy et al. [67] characterized five independent DS-derived embryonic stem cell lines to show that differentiation of neural progenitor cells (NPCs) displayed increased apoptosis, with upregulation of mitochondrial apoptotic-related genes and downregulation of forebrain developmental genes. Analysis of differentially expressed genes (DEGs) suggested that RUNX1 overexpression may disrupt different molecular pathways during neural development in DS-NPCs. RUNX1 disruption by genome editing resulted in reduced apoptosis and neuron migration. In line with these findings, Liu et al. [68] reported that RUNX1 expression levels in DS-iPSCs were significantly higher than in controls and correlated with impaired mitochondrial functions and increased apoptosis, while inhibition of RUNX1 expression improved the mitochondrial function in DS-iPSCs. Altogether, the roles of this important transcription factor in DS-associated alterations are just starting to emerge, warranting future efforts to clearly elucidate the implications of RUNX1 and its alternative splicing regulation on the different pathophysiological features of Trisomy 21.
RUNX1 beyond blood
Neurodevelopment
Although very little is known about the roles of RUNX1 triplication in DS-related neurological alterations [67], several groups have studied the functions of this transcription factor in normal neural development and homeostasis. Theriault et al. [69] showed Runx1 expression in specific neurons of the embryonic central and peripheral nervous systems of the mouse, including cholinergic branchial and visceral motor neurons of the hindbrain, certain spinal cord somatic motor neurons, and in nociceptive and mechanoreceptor neurons from the trigeminal and vestibulocochlear ganglia. Runx1 knock-out resulted in defects on specific sensory neurons in the trigeminal and vestibulocochlear ganglia, as well as loss of cholinergic neurons in the hindbrain mantle layer. In a follow-up study [70], these authors also showed Runx1 expression in proliferating cells of the olfactory epithelium, and that in vivo Runx1 impairment resulted in premature and ectopic olfactory receptor neuronal differentiation. Ectopic Runx1 expression increased cell proliferation of olfactory and cortical neural progenitors. These observations are further supported by the fact that RUNX1 is induced in a subpopulation of adult neural stem or progenitor cells NS/PCs after brain injury in mice [71]. Furthermore, Runx1 downregulation in neurosphere cultures of adult mouse subventricular zone NS/PC inhibited proliferation without affecting differentiation. Noteworthy, Runx1 overexpression induced adult NS/PC differentiation predominantly toward a neuronal lineage, without reducing proliferation. On the other hand, Fukui et al. [72] showed that overexpression of Runx1 in cultured adult hippocampal precursor cells reduced proliferation, increased survival (reduced apoptosis), and enhanced neuronal differentiation, while slightly reducing dendritic morphology and complexity. Finally, Shrestha et al. [73] demonstrated that Runx1 controls the diversity of auditory sensory neurons—spiral ganglion neurons (SGNs)—in mice. The authors found that Runx1 is enriched in Ib/Ic SGN precursors by late embryogenesis, while embryonic loss of Runx1 resulted in greater numbers of Ia identity at the expense of Ib/Ic fates. Similarly, during mouse embryonic development, Runx1 is expressed in most nociceptor neurons, but becomes restricted to Ret+ nociceptors in adult mice [74]. In this line, Huang et al. [75] also reported that the Runx1/CBFβ transcriptional complex is essential for NGF/TrkA-dependent differentiation of nonpeptidergic nociceptors of the dorsal root ganglion (DRG). Of note, Kramer et al. [76] had previously characterized a role for Runx1 in diversification of the DRG sensory neurons. As a summary, while there are cell context-specific differences and interpretations, all these studies confirm that RUNX1 promotes survival, proliferation and/or differentiation of several neuronal populations. Importantly, except for the work from Halevy et al. [67], all of these reports were performed in mouse models that do not express the Runx1a isoform. For that reason, it is worth remembering that ectopic expression of Runx1a in cultured mouse Neural Crest Stem Cells undergoing neural differentiation predominantly resulted in glial differentiation with concomitant reductions of neuronal fates [29], while Runx1a-overepressing pups exhibited dystrophic DRGs and megacolon, presumably due to impaired survival or migration of the enteric nervous system precursors along the gastrointestinal tract (Hirschsprung’s disease). These results would suggest that contrary to murine Runx1, Runx1a inhibits neuronal survival, proliferation and/or differentiation, which is in agreement with the idea that RUNX1a acts as an endogenous antagonist of RUNX1b/c transcriptional functions [20, 21]. Therefore, the potential species-specific differences in RUNX1 alternative splicing and function during the development and differentiation of neuronal populations remains to be elucidated, a topic of profound relevance, as many of the above-mentioned RUNX1-dependent neurodevelopmental processes are frequently altered in Down syndrome individuals and experimental models (reviewed in [77]), such as the olfactory neuroepithelium [78, 79], subventricular zone neurogenesis [80, 81], the NGF-dependent cholinergic system [82], motor-neurons [83] or the cochlear sensory neurons [84, 85], among many others.
Bone and cartilage
RUNX1 functions in bone and cartilage development and homeostasis are very well established and have recently been reviewed elsewhere [86]. Therefore, we will only briefly summarize the most relevant literature and focus on potential links to Down syndrome-associated conditions. Initial reports in the 2000’s indicated that Runx1 may contribute to the early stages of murine craniofacial bone development and skeletogenesis, while continuing to function in the progenitor cells of tissues that support bone formation in the adult [87, 88]. Since then, a wealth of literature has confirmed and explored in more detail the mechanisms by which RUNX1 modulates both chondrogenesis and osteogenesis. In this line, Soung et al. [89] examined the potential role of Runx1 in osteoclast formation and function. To this end, the authors deleted Runx1 expression in myeloid osteoclast precursors (Runx1f/f; CD11b-Cre) and observed significant loss of femoral trabecular and cortical bone mass compared with control littermates. These and other experiments indicated that Runx1 expression in preosteoclasts negatively regulates osteoclast formation and activity, and contributes to overall bone mass. In parallel, mice with chondrocyte-specific deletion of Runx1 (Runx1f/f; Col2α1-Cre) manifested alterations in cartilage formation, reduced bone density, and an osteoporotic phenotype, suggesting that Runx1 is required for both chondrogenetic and osteogenetic gene expression programs [90]. Moreover, Runx1 ablation in osteoblast precursors and differentiating chondrocytes (Runx1f/f; Osx-Cre mice) resulted in an osteoporotic phenotype and decreased bone density in the long bones and skull compared to controls [91]. The authors also found that Runx1 upregulates the expression of multiple bone-specific genes and plays an indispensable role in bone formation and homeostasis for both trabecular and cortical bones. Furthermore, Tang et al. [92] generated a doubly transgenic mesenchymal progenitor-specific (Runx1f/f; Twist2-Cre) and osteoblast-specific (Runx1f/f; Col1α1-Cre) double conditional knockout (Runx1 dCKO) mouse model. Runx1 dCKO resulted in decreased osteogenesis and increased adipogenesis, while molecular analysis demonstrated that Runx1 maintains adult bone homeostasis though up-regulating Bmp7/ATF4 and WNT/β-Catenin signaling pathways. Similarly, osteoclast-specific conditional knock-out of Runx1 (Runx1f/f; LysM‐Cre mice) compromised murine fracture healing due to progressive woven bone loss and hindered remodeling of the cartilage [93]. Finally, in an experimental model of osteoarthritis using Runx1f/f; Col2α1-Cre mice, Zhou et al. [94] observed that chondrocyte-specific Runx1 knockout aggravated cartilage destruction, decreased chondrocyte proliferative capacity and enhanced loss of bone matrix.
People with Down syndrome are predisposed to skeletal deficits, including lower body height, reduced mineral density in the bone, and increased frequency of early-onset osteoporosis, with significant alterations in skeletal and craniofacial development, bone morphology and homeostasis, and age-related bone loss and fragility (reviewed in [95]). However, the genetic origins of DS-skeletal phenotypes remain unclear, although several pieces of evidence suggest a contributing role for the chromosome 21-encoded gene DYRK1A [95,96,97]. Moreover, alterations in chondrogenesis [98] and a predisposition to Down syndrome-associated arthritis [99] have been reported. Surprisingly, the potential implications of RUNX1 triplication or changes in its alternative splicing on bone/cartilage homeostasis in the context of Down syndrome have not been reported. Noteworthy, and as previously pointed, newborn mice overexpressing Runx1a were smaller than the control littermates [29], again suggesting a potential negative regulatory function over the longer isoforms Runx1b/c during embryonic development of the murine skeleton. This observation, together with the very well-known roles played by RUNX1 in bone and cartilage biology, and the important skeletal alterations seen in Down syndrome individuals warrant for future efforts to elucidate the interconnections linking RUNX1 gene triplication to bone homeostasis and disease in Trisomy 21.
Heart
During embryonic development, RUNX1 is expressed in the mesenchymal tissue of the heart and vascular tissues [100]. Runx1 KO mice have an underdeveloped coronary plexus and smaller ventricular free wall vessels, accompanied by changes in heart structure, such as ventricular septal defects and thinner myocardium [101]. Neonatal cardiomyocytes exhibited significantly increased Runx1 mRNA and other markers of cardiac differentiation (Myh7, Nkx2.5, Dab2, Destrin) than adult counterparts [102]. Górnikiewicz et al. [103] performed global DNA methylation profiles in whole murine hearts and found that promoter regions with increased DNA methylation at post-natal day 7 (compared to day 1) were significantly enriched for Runx1 binding motifs and included genes critical for heart maturation and muscle development. In the adult heart, RUNX1 is silenced, but becomes upregulated under cardiac pathologic conditions (reviewed by [104]), such as ischemic cardiomyopathy [105], chronic dilated cardiomyopathy [106, 107] and in animal models of diabetic cardiomyopathy and pressure overload [108, 109]. Recent work from Loughrey’s group shows that Runx1 ablation [107] or pharmacologic inhibition [110] has a protective effect on heart function after myocardial infarction. More recently, Swift et al. [111] reported that Runx1 plays a critical role in cardiomyocyte ploidy dynamics and cell division, in both developmental and injury contexts. Overall, RUNX1 is required for the proper development of cardiovascular tissues and its expression correlates with disease in the adult heart; therefore, it is somewhat unexpected that RUNX1 gene triplication has not been widely associated to Down syndrome-related congenital heart disease. In this context, changes in composition of the extracellular matrix (ECM) are suggested to contribute to heart defects in Trisomy 21 [112]. Moreover, Conti et al. [113] analyzed the transcriptional profile of human fetal hearts from DS fetuses and found that genes coding for ECM proteins are the most over-represented among the upregulated ones. Finally, Mollo et al. [62] found that about 80% of the ECM genes upregulated in DS fetal hearts had RUNX1 consensus sites in their promoter regions, while experimentally confirmed the role for RUNX1 as a main controller of ECM gene expression in T21 fetal fibroblasts.
About 50% of newborns with DS have some kind of congenital heart disease (CHD), including ~ 43% atrioventricular septal defects (AVSD), ~ 32% ventricular septal defects (VSD), ~ 19% isolated secundum atrial septal defects (ASD), ~ 7% isolated persistent patent ductus arteriosus (PDA) and ~ 6% tetralogy of Fallot (TOF), among others [114, 115]. Historically, this has been one of the major causes of reduced life expectancy in individuals with Trisomy 21, whereas advances in diagnosis, treatment, and surgery for CDH in DS individuals has led to a dramatic improvement in survival over the last 60 years [116]. Yet, the genetic and molecular mechanisms underlying DS-CDH are only starting to emerge (reviewed in [117, 118]). In this sense, several chromosome 21-encoded genes have been associated with cardiovascular defects, such as DYRK1A, COL6A1-2, KCNJ6 and RCAN1 (reviewed in [118]). Altogether, more research is needed to clearly define the implications of RUNX1 overexpression in DS heart development and homeostasis, which could allow more precise interventions to correct the genetic programs underlying DS-CHD.
Muscle
Initial reports suggested that Runx1 is not detected during embryonic muscle development [100, 119] or in the healthy adult muscle [120]. Nevertheless, Runx1 becomes highly upregulated in muscles exposed to myopathic damage. Hence, similarly to what is observed in the heart (see previous section), RUNX1 expression is significantly increased in samples of muscle dystrophies, including mouse models of Duchenne muscular dystrophy (DMD) [121] and amyotrophic lateral sclerosis [122], myopathy patients—including DMD, Emery-Dreifuss Muscular Dystrophy and Acute Quadriplegic Myopathy—[123], as well as in cardiotoxin-treated muscles [124] and during ischemia reperfusion-induced muscle injury [125]. Nevertheless, studies on the role of Runx1 in myoblasts have reached conflicting conclusions [126,127,128]. To address this, Umansky et al. [129, 130] generated mice specifically lacking Runx1 in the muscle (Runx1f/f; Myf5-cre) and found that Runx1 expression is switched on in response to muscle damage and cooperates with the MyoD and AP-1/c-Jun transcription factors during muscle regeneration by preventing premature myoblasts differentiation. Thus, Runx1-deficient primary myoblasts differentiated prematurely, reducing the number and size of regenerating myofibers and impairing muscle regeneration. Overexpression of Runx1 in C2C12 cells inhibited myogenic differentiation, while promoting myoblast proliferation [125]. Consistent with this, they found that Runx1 expression correlated with that of Pax7 in undifferentiated satellite cells, suggesting that Runx1 regulates muscle regeneration by promoting proliferation of satellite cells. These results are consistent with other reports in the context of cardiomyocyte injury and regeneration (i.e., see [111] and section "Heart"). More recently, Stefanowicz et al. [131] confirmed that Runx1 is required for myoblast proliferation and subsequent differentiation. Finally, in muscular fibroadipogenic progenitors, Runx1 expression is restricted by miR-206. Runx1 upregulation in these cells induces a transcriptional program that leads to their differentiation into adipocytes, resulting in intramuscular fatty deposits, which are characteristic of muscular dystrophies and aging [132].
Among many other features, Down syndrome individuals are characterized by muscle hypotonia and associated motor and postural deficits [133, 134]. Morphometric analysis demonstrated a larger size of myofibers in the quadriceps muscles of Ts65Dn mouse model of Down syndrome. However, myofibrils were thinner and contained higher amounts of mitochondria and intramuscular fatty deposits (lipid droplets), typical alterations of early aging [135]. Interestingly, Pawlikowski et al. [136] reported a failure in satellite (muscle stem) cell expansion in Ts65Dn mice that resulted in impaired muscle regeneration. Ts65Dn satellite cells appeared to accumulate DNA damage, prompting the idea that stem cell dysfunction is a common contributor to multiple Down syndrome phenotypes. Since RUNX1 expression is induced in and required for proliferation of myoblasts in the injured muscle, it is tempting to speculate that in the context of trisomy 21, overexpression of the antagonic RUNX1a isoform could lead to a reduced proliferation and premature differentiation of muscle progenitors. Alternatively, elevated basal/constitutive expression of full-length RUNX1 in satellite cells could potentially result in exhaustion of this population, leading to the observed phenotype. In conclusion, triplication of RUNX1 could be associated with pathological and regenerative states of the muscle, a tissue largely affected in the Down syndrome population. Therefore, future work should address the specific roles of RUNX1 in DS muscle health and disease, with a particular focus on the potential implications of the alternatively spliced variants, not present in previously used murine models.
Eye/endothelium
Stewart et al. [137] reported Runx1 expression in post-mitotic cells of the mouse inner retina at embryonic day 13.5, overlapping with the early amacrine and ganglion cell marker Islet1. More recently, single-cell RNA-seq analyses have confirmed RUNX1 expression in specific retinal ganglion cell subpopulations [138]. Lam et al. [139] identified RUNX1 as a gene upregulated in vascular endothelial cells from human Proliferative Diabetic Retinopathy (PDR) fibrovascular membranes (FVMs). In vitro studies using human retinal microvascular endothelial cells (hRMVECs) showed increased RUNX1 RNA and protein in response to high glucose, while RUNX1 inhibition reduced cell migration, proliferation, and tube formation. Immuno-staining assays showed that RUNX1 upregulation is a hallmark of aberrant retinal angiogenesis, later confirmed by others [140, 141]. Finally, the RUNX1 small-molecule inhibitor Ro5-3335 induced a significant reduction of neovascular tufts in oxygen-induced retinopathy models. In a follow-up study, RUNX1 upregulation was proposed to drive epithelial-to-mesenchymal transition (EMT) in primary cultures from human Proliferative Vitreoretinopathy (PVR) membranes. Importantly, topical administration of a Ro5‐3335 nano-emulsion curbed the progression of disease in a novel rabbit model of mild PVR [142]. Furthermore, Whitmore et al. [143] suggested that RUNX1 upregulation in PDR fibrovascular membranes is mediated through a TNFα/JNK signaling module. These authors further characterized RUNX1 expression in critical cell types involved in a laser-induced murine model of Choroidal Neovascularization (CNV) and provided data supporting RUNX1 inhibition as a new potential therapy for neovascular age-related macular degeneration [144]. In parallel, Xing et al. [145] confirmed RUNX1’s role as a mediator of high-glucose-induced proliferation and migration of hRMVECs. On the other hand, RUNX1 and SMAD3 are required for maintenance of corneal epithelial identity and homeostasis, as their depletion inhibits PAX6 and induces limbal stem/progenitor cells to differentiate into epidermal-like epithelial cells [146]. Finally, Voronov et al. [147] reported a role for Runx1/3 during lacrimal gland (LG) morphogenesis, where Runx1 was restricted to the epithelium, with highest levels of expression in ductal and centroacinar cells. Downregulation of Runx1-3 expression abolished LG growth and branching. Noteworthy, RUNX1 is known to play important roles in angiogenesis outside the retina. In this context, Runx1-deficient embryos show imperfect angiogenesis in head, pericardium, and liver [148]. Runx1 loss of function in zebrafish embryos leads to hematopoietic and vasculogenic defects [149]. Runx1 is suggested to directly bind to the VEGFA promoter and repress its transcription [150]. At last, Runx1 can induce endothelial cell differentiation and maturation, as well as vascular network formation by repression of IGFBP3 expression (151). To the best of our knowledge, RUNX1 expression and activity on retinal and lacrimal gland development and function have never been studied in the context of Trisomy 21, a topic that should be taken in consideration toward designing better diagnostic and therapeutic innovations for ophthalmologic alterations of Down syndrome individuals.
People with trisomy 21 manifest a wide range of ophthalmologic features, including strabismus, amblyopia, accommodation defects, refractive error, eyelid abnormalities, nasolacrimal duct obstruction, nystagmus, keratoconus, cataracts, retinal abnormalities, optic nerve defects, and glaucoma. In this regard, for additional information refer to recent reviews that cover this topic in depth [152, 153], while we will only focus on those features that more closely relate to RUNX1 functions in the eye, such us anomalies of the retinal vasculature, optic nerves, or the lacrimal gland (see below). Children with Down syndrome show an increased incidence in retinal abnormalities, with reports ranging from 1.7 to 40% [154, 155]. Williams et al. [156] first described a significantly increased density of retinal vessels crossing the margin of the optic nerve head in T21 cases. Sherk and Williams [157] noted an increased number of large vessels crossing the optic disk in DS patients, while Parsa and Almer [158] suggested that the abnormal optic disk vessels in DS eyes may be due to an extra copy of the chromosome 21-encoded gene endostatin, a potent inhibitor of angiogenesis, endothelial cell proliferation, and migration. Optic nerve abnormalities are also frequently associated with T21, with one study reporting a 14% incidence in a cohort of 806 children with Down syndrome [159]. Several other clinical studies have confirmed and extended these observations, characterizing a spectrum of retinal abnormalities in Down syndrome individuals [160,161,162,163]. Finally, nasolacrimal duct obstruction (NLDO) is another common congenital finding in DS children, and is caused by different anatomic etiologies, such as membranous obstruction at the valve of Hasner or general stenosis of the duct [164,165,166].
Ovary
Ovulation is originated with a peak of luteinizing hormone (LH) from the pituitary, resulting in a transcriptional reprogramming of ovarian granulosa cells leading to structural remodeling of the ovarian follicle and oocyte meiotic maturation. Up-regulation of RUNX1 mRNA and protein was initially detected in preovulatory follicles after human chorionic gonadotropin (hCG) injection in immature rats as well as after the luteinizing hormone surge in cycling animals. Moreover, in primary cultures of rat preovulatory granulosa cells, hCG also induced Runx1 mRNA expression, while knockdown of Runx1 decreased progesterone secretion and reduced expression of preovulatory signature genes [167], such as Ptgs2 [168] and Hapln1 [169]. Altogether, these pieces of evidence suggest that RUNX1 could mediate the LH-dependent shift from estrogen to progesterone production, an essential step in ovulation. More recently, Runx1 was also characterized as a critical female determinant gene, contributing to early sex determination by directing the developmental program for ovarian commitment [170]. In this regard, Runx1 expression is found in early mouse ovaries, but not testes. As stated above, the role of RUNX1 in the ovary goes beyond embryonic development, i.e., in granulosa cells of pre-ovulatory rodent follicles [167, 171]. Similar results were observed in human ovaries [172]. RUNX1 expression is also detected in cumulus cells, a non-luteinizing type of granulosa cells that surround the oocyte, forming the cumulus oocyte complex (COC), released at ovulation [173]. Nicol et al. [174] reported that RUNX1 expression fetal ovaries of rainbow trout, turtle, mouse, goat, and human. In mice, pre-granulosa cells express RUNX1 as the gonads differentiate. The authors proposed a RUNX1-FOXL2 cooperative interaction for the identity maintenance of fetal granulosa cells, disruption of which led to masculinization of fetal ovaries. Interestingly, Bridges et al. [175] demonstrated that conditional loss of Runx1 in ovarian somatic cells led to increased prevalence of ovarian tumors in aged mice. At younger ages, abnormal follicle-like lesions where abundant, with quiescent granulosa cells and differentiation defects, supporting the idea that Runx1 is essential for granulosa cell differentiation and inhibition of ovarian tumorigenesis. More recently, Dinh et al. [176] reported an ovary-specific Progesterone Receptor (PGR)-RUNX1 interaction in 70% of PGR-bound regions, key for the induction of essential ovulatory genes. Finally, the RUNX-binding partner CBFβ was also shown to play a prominent role in ovarian granulosa cells and female fertility [177, 178]. Noteworthy, conditional knock-out of Runx1 in decidual stromal cells—a transient uterine tissue that supports the fetus during pregnancy—leads to defects in uterine angiogenesis, trophoblast differentiation, and vascular remodeling, resulting in fetal lethality during placentation [179].
Initial studies on follicular development in ovaries from Down syndrome girls suggested absence or retardation of follicle growth compared to aged-matched controls. Reductions in the number and size of follicles occurs earlier in life, potentially due to a hormonal imbalance [180]. According to Hsiang et al. [181], primary gonadal deficiency is common in DS, progressing from birth to adolescence, and clearly manifest in adult patients (both male and female). Cento et al. [182] reported a significantly higher incidence of anovulation and luteal defects in a group of regularly menstruating DS women, while showing reduced estradiol plasma concentrations along the cycle and lower progesterone levels in the mid-luteal phase. In a follow-up study, the authors administered follicle-stimulating hormone (FSH) to a group of normo-ovulatory DS vs. control individuals and found a significant impairment in the ovarian sensitivity to FSH, as measured by estradiol production [183], and potentially contributing to gonadal disfunction in T21 women [184]. Alternatively, Angelopoulou et al. [185] suggested that primary gonadal dysfunction in Down syndrome might possibly result from an adrenal gland disorder. Finally, as reviewed by Parizot et al. [186], puberty appears to be normal in women with DS and starts at the same average age as in the general population, while women with DS are fertile; however, data have highlighted early menopause in women with DS [187] and a significant reduction of anti-Müllerian hormone [188].
The gene networks underlying developmental and functional alterations in Trisomy 21 ovaries remain virtually unexplored. In summary, the potential functions of RUNX1 during ovarian development and reproduction in the context of Down syndrome remain to be fully elucidated, a field that demands future attention due to the important implications on female endocrine development and fertility of girls and women with Trisomy 21.
Other tissues
Recent work from several groups has started to address the contribution of RUNX1 in development and/or homeostasis of several other organs, tissues, and cellular functions, which are also altered in Down syndrome individuals and models, again suggesting potential implications for RUNX1 triplication as an underlying cause of such deregulated functions.
Nonalcoholic fatty liver disease (NAFLD)
Kaur et al. [189] explored the role of RUNX1 in the pathogenesis of non-alcoholic steatohepatitis (NASH). RUNX1 expression was significantly correlated with inflammation, fibrosis, and NASH activity score in patients, while in vitro analyses suggested that steatosis-induced RUNX1 levels lead to upregulation of adhesion and angiogenic molecules in endothelial cells and may be involved in enhancing inflammation and disease severity in NASH. Alternatively, Marcher et al. [190] showed that the transcriptional regulators ETS1 and RUNX1 act as drivers of NASH-associated hepatic stellate cell activation and transcriptional plasticity, key aspects of NASH pathophysiology. More recently, Bertran et al. [191] analyzed RUNX1 expression in a well-characterized cohort of women with morbid obesity and NAFLD and reported that hepatic RUNX1 expression increases in the first steps of NAFLD but decreases in advanced stages of the disease. Subsequently, these authors applied a systems biology mathematical model that simulates NAFLD pathophysiology concluding that RUNX1 has a high relationship with hepatic injury-liver fibrosis, and a medium relationship with lipotoxicity and insulin resistance [192]. Earlier in vitro studies by Bertrand-Philippe et al. [193] using hepatic stellate cells, featured RUNX1 as a critical regulator of tissue inhibitor of metalloproteinase 1 (TIMP1), a key mediator of liver fibrosis [194, 195]. Importantly, the authors observed that overexpression of full-length RUNX1b isoform repressed TIMP1 promoter activity, whereas the truncated RUNX1a isoform and RUNX2 exerted the opposite effect, raising the possibility that a deregulation in RUNX1 splicing, rather than its overexpression, could contribute to a higher NAFLD prevalence in children with Trisomy 21. Finally, Guo et al. [196] suggest that RUNX1 induces liver fibrosis progression by directly regulating TGF-β/SMAD signaling.
As reviewed by De Matteo and Vajro [197], the liver is frequently affected in persons with Down syndrome, as evidenced at birth by transient myeloproliferative disorder (TMD)/abnormal myelopoiesis (TAM) [198] and neonatal cholestasis [199], while at later stages by celiac disease-/autoimmunity-related liver diseases [200], and gallstone formation due to gallbladder hypomotility [201]. Noteworthy, hepatic inflammation was also recently described in the Down syndrome mouse model Dp16, which was exacerbated by chronic inflammation with poly(I:C) and reduced by JAK1 inhibition [202]. Valentini et al. [203] reported a higher incidence of non-alcoholic fatty liver disease (NAFLD) in 84 children with Trisomy 21 independently of overweight/obese phenotype, in agreement with older reports suggesting an elevated frequency of hepatic steatosis in DS children [204, 205]. In a follow-up study, these authors found a prevalence of 64.3% of liver steatosis in T21 children, which was correlated to the PNPLA3 rs738409 variant and IL6 levels [206]. Altogether, these pieces of evidence point to a plausible role for RUNX1 triplication in the predisposition of individuals with Down syndrome to develop liver diseases, a topic that requires future elucidation.
Hair follicle stem cells
Initial studies by Raveh et al. [207] found prominent Runx1 expression in epithelial cells of the hair follicle, including the keratin forming layers of the hair shaft and the bulge, where Runx1 is co-expressed with the hair follicle stem cell (HFSC) marker Keratin15. During early hair morphogenesis, Runx1 is also expressed in a discrete dermal mesenchymal layer, while at later stages it is present in a hair cycle-dependent pattern in the dermal papilla. Mice with epidermal conditional knockout of Runx1 displayed structural alterations of the zigzag hair type. In parallel, Soma et al. [208] observed intense Runx1 immunoreactivity in the keratinizing area of human anagen hair follicles, and suggested a potential role in transcriptional regulation of the keratin-associated protein hKAP5.1. Subsequent work from Osorio et al. [209] showed that keratinocyte-specific conditional Runx1 knock-out impairs HFSC activation, but not their maintenance, proliferation, or differentiation. Adult mutant mice exhibited impaired de novo production of hair shafts and all temporary hair cell lineages, owing to a prolonged quiescent phase of the first hair cycle. In a follow-up report they found that in adulthood, Runx1 plays a direct, non-essential function in promoting anagen onset and HFSC proliferation by blocking the expression of the cyclin-dependent kinase (CDK) inhibitor p21, providing evidence for a role in mouse skin tumorigenesis [210]. Further work from this group showed that Runx1 is required for correct adult HFSC differentiation and skin integrity [211]. Lee et al. [212] analyzed in more detail the links between RUNX1 and p21, to find that Runx1 represses transcription of p21 (and other CDK inhibitor genes) in HFSC in vivo, while p21 appears important for the timely onset of quiescence in HFSC. Subsequent studies reported that forced Runx1 expression induces hair degeneration (catagen) and simultaneously promotes a reversible differentiation of bulge SCs toward ‘early progenitor hair germ’ (EP-HG) cell fate, further proposing that changes in endogenous Runx1 levels ensure a proper balance between SC and EP populations during niche restructuring in normal tissue homeostasis [213]. Finally, Li et al. [214] showed that Runx1 ablation in the skin epithelium not only delayed stem cell activation and hair cycle progression, as previously seen, but also increased the density of vasculature in the horizontal plexus under the hair germ. The authors propose a model whereby increased vasculature near the HFSC activation zone is inhibitory to stem cell activation and prolongs quiescence by delaying progression from telogen into anagen.
Despite dramatic increases in life span, people with Down syndrome manifest accelerated aging signs, including hair graying, hair loss, and skin wrinkling. Hair loss in DS is typically associated to Alopecia areata [215, 216], an autoimmune condition in which hair follicles are attacked by the immune cells, likely secondary to complex immune dysregulation caused by trisomy of chromosome 21 genes, such as Autoimmune Regulator—AIRE—[217] and 4 interferon receptor genes [218, 219]. Given the well-established implications of RUNX1 in hair follicle stem cell biology, we hypothesize a potential contribution for this transcription factor in the altered hair follicle homeostasis of people with Trisomy 21. In this context, Baars et al. [220] showed that RUNX1 acts as a transcriptional regulator of RASGRP1, expression of which is inversely correlated to autoimmunity. Moreover, in patients with autoimmunity, CD4+ T cells exhibited reduced RUNX1 and RASGRP1 expression, which was correlated to active inflammation. Similarly, RUNX1 is induced by Influenza A virus infection, acting as a negative modulator of the IFN signaling pathway [221]. Lastly, Zezulin et al. [222] observed that RUNX1 plays a role in limiting toll-like receptor 4 (TLR4)-dependent inflammatory response in neutrophils, including type I IFN signaling. In summary, several pieces of evidence indicate a role for RUNX1 as a negative regulator of inflammation and autoimmunity. Thus, we hypothesize that overexpression of the functionally antagonistic RUNX1a isoform in DS immune cells could promote an increased autoimmune response that contributes to Alopecia areata, and other autoimmune reactions associated with Trisomy 21.
Pulmonary hypertension (PH)
Immunohistochemistry staining by Levanon et al. [100] showed Runx1 expression in several organs of the developing mouse embryo (E14.5-E16.5), including the lung bronchi. Haley et al. [223] found that RUNX1 is highly expressed in human fetal lung, predominantly in cartilage and epithelium. Tang et al. [224] observed RUNX1 expression in mouse respiratory epithelial and non-epithelial cells at E18.5, postnatal day 1 and 8 weeks. RUNX1 protein was 4.6-fold higher in the adult lung than at E18.5. Furthermore, the authors showed that conditional Runx1 deletion in respiratory epithelial cells led to a mild delay in lung maturation, with no impact in survival and postnatal lung maturation, but exhibiting increased respiratory distress, inflammation, and pro-inflammatory cytokines upon intra-tracheal LPS administration. Liang et al. [225] reported that during PH development in mice, endothelial precursor cells (EPCs) mobilize from the bone marrow into the circulation, and contribute to the remodeled pulmonary vasculature. Inhibition of endothelial-to-hematopoietic transition (EHT) by blocking Runx1 -a master regulator of EHT-, prevented disease progression in two experimental mouse models of PH. Lastly, they found high levels of Runx1 expression in circulating CD34+ CD133+ EPCs isolated from blood of PH patients, supporting the clinical relevance of this mechanism. In a follow-up study, they extended their observations to a more established rat model of PH, where they confirmed that Runx1 inhibition prevented and reverted PH. Finally, they showed that tissue-specific conditional deletion of Runx1 gene in adult endothelium or in pulmonary macrophages also protected the mice from developing experimental PH [226].
As extensively reviewed by Bush and Ivy [227], Down syndrome individuals have an enhanced frequency of pulmonary hypertension (PH), largely associated with congenital heart disease and persistent pulmonary hypertension of the newborn (PPHN); however, there likely are multiple other genetic contributions to such increased prevalence. While the molecular mechanisms underlying PH predisposition in Trisomy 21 remain to be fully elucidated, we speculate that RUNX1 overexpression and/or splicing imbalance could be a plausible contributing factor. Given that small-molecule inhibitors against RUNX1 have proven effective in experimental models of PH [226] and retinopathy (see section "Eye/endothelium"), future work should address their efficacy in the context of DS-associated PH.
Other cellular functions
Recent work from several groups has suggested critical roles for RUNX1 in Epithelial-Mesenchymal Transition-EMT-(recently reviewed by [228]), in DNA damage response (reviewed by [229]), in browning of white adipose tissue [230] and in diverse types of cancer (reviewed in [231]). The potential implications that DS-associated RUNX1 overexpression or alternative splicing deregulation could play in these paradigms are yet to be elucidated.
Concluding remarks and future perspectives
While initially characterized as a master regulator of hematopoiesis and blood cell differentiation, RUNX1 is emerging as a critical transcription factor participating in a wide spectrum of developmental and homeostatic functions, most of which manifest frequent alterations in people with Down syndrome (summarized in Fig. 3). Although little is known about most of these ‘non-canonical’ RUNX1 roles across different tissues and organs in Trisomy 21, our review should set the stage for the research community to validate the many hypotheses proposed here. More importantly, the primate-specific alternative splicing that gives rise to RUNX1a should be taken into consideration when interpreting data from murine models. Finally, the fact that RUNX1 can be pharmacologically inhibited is an added motivation to test RUNX1 as a potential druggable target in specific pathological conditions associated with Down syndrome that currently have no treatment and represent important limitations in health and life quality for individuals with Trisomy 21 and their families.

Summary of tissues, organs, and phenotypes where RUNX1 plays important developmental or homeostatic functions, and frequently altered or observed in individuals with Down syndrome and/or T21 model animals. Made with BioRender
Availability of data and materials
Not applicable.
References
Mevel R, Draper JE, Lie-A-Ling M, Kouskoff V, Lacaud G. RUNX transcription factors: Orchestrators of development. Development. 2019. https://doi.org/10.1242/dev.148296.
Levanon D, Groner Y. Structure and regulated expression of mammalian RUNX genes. Oncogene. 2004. https://doi.org/10.1038/sj.onc.1207670.
Rennert J, Coffman JA, Mushegian AR, Robertson AJ. The evolution of Runx genes I. A comparative study of sequences from phylogenetically diverse model organisms. BMC Evolut Biol. 2003;3(1):4. https://doi.org/10.1186/1471-2148-3-4.
Kamachi Y, Ogawa E, Asano M, Ishida S, Murakami Y, Satake M, Ito Y, Shigesada K. Purification of a mouse nuclear factor that binds to both the A and B cores of the polyomavirus enhancer. J Virol. 1990. https://doi.org/10.1128/jvi.64.10.4808-4819.1990.
Wang SW, Speck NA. Purification of core-binding factor, a protein that binds the conserved core site in murine leukemia virus enhancers. Mol Cell Biol. 1992. https://doi.org/10.1128/MCB.12.1.89.
Lilly AJ, Costa G, Largeot A, Fadlullah MZH, Lie-A-Ling M, Lacaud G, et al. Interplay between SOX7 and RUNX1 regulates hemogenic endothelial fate in the yolk sac. Development. 2016. https://doi.org/10.1242/dev.140970.
Nagata T, Gupta V, Sorce D, Kim WY, Sali A, Chait BT, et al. Immunoglobulin motif DNA recognition and heterodimerization of the PEBP2/CBF Runt domain. Nat Struct Biol. 1999;6(7):615–9.
Michaud J, et al. In vitro analyses of known and novel RUNX1/AML1 mutations in dominant familial platelet disorder with predisposition to acute myelogenous leukemia: implications for mechanisms of pathogenesis. Blood. 2002. https://doi.org/10.1182/blood.V99.4.1364.
Telfer JC, Hedblom EE, Anderson MK, Laurent MN, Rothenberg EV. Localization of the domains in Runx transcription factors required for the repression of CD4 in thymocytes. J Immunol. 2004. https://doi.org/10.4049/jimmunol.172.7.4359.
Levanon D, Goldstein RE, Bernstein Y, Tang H, Goldenberg D, Stifani S, et al. Transcriptional repression by AML1 and LEF-1 is mediated by the TLEGroucho corepressors [Internet]. Vol. 95, Biochemistry Communicated by Leo Sachs. 1998. Available from: www.pnas.org.
Seo W, Tanaka H, Miyamoto C, Levanon D, Groner Y, Taniuchi I. Roles of VWRPY motif-mediated gene repression by Runx proteins during T-cell development. Immunol Cell Biol. 2012. https://doi.org/10.1038/icb.2012.6.
Yarmus M, Woolf E, Bernstein Y, Fainaru O, Negreanu V, Levanon D, et al. Groucho/transducin-like enhancer-of-split (TLE)-dependent and -independent transcriptional regulation by Runx3. Proc Natl Acad Sci U S A. 2006. https://doi.org/10.1073/pnas.0602470103.
Bravo J, Li Z, Speck NA, Warren AJ. The leukemia-associated AML1 (Runx1)-CBFβ complex functions as a DNA-induced molecular clamp [Internet]. 2001. Available from: http://structbio.nature.com.
Huang G, Shigesada K, Ito K, Wee HJ, Yokomizo T, Ito Y. Dimerization with PEBP2β protects RUNX1/AML1 from ubiquitin-proteasome-mediated degradation. EMBO J. 2001. https://doi.org/10.1093/emboj/20.4.723.
Tang YY, Crute BE, Iii JJK, Huang X, Yan J, Shi J, et al. Biophysical characterization of interactions between the core binding factor K and L subunits and DNA. FEBS Lett. 2000. https://doi.org/10.1016/S0014-5793.
Yan J, Liu Y, Lukasik SM, Speck NA, Bushweller JH. A R T I C L E S CBFβ allosterically regulates the Runx1 Runt domain via a dynamic conformational equilibrium Core binding factors (CBFs) are heterodimeric transcription factors consisting of a DNA-binding CBF subunit and non-DNA-binding CBF subunit. The CBF subunit increases the affinity of the DNA-binding Runt domain of CBF for DNA while making no direct contacts to the DNA. We present evidence for conformational exchange in the. Nat Struct Mol Biol [Internet]. 2004;11(9). Available from: http://www.nature.com/natstructmolbiol.
Wang CQ, Mok MMH, Yokomizo T, Tergaonkar V, Osato M. RUNX Proteins in Development and Cancer. Vol. 962, Advances in Experimental Medicine and Biology. 2017.
Miyoshi H, Ohira M, Shimizu K, Mitani K, Hirai H, Imai T, et al. Alternative splicing and genomic structure of the AML1 gene involved in acute myeloid leukemia. Nucleic Acids Res. 1995;23(14):2762–9.
Komeno Y, Yan M, Matsuura S, Lam K, Lo MC, Huang YJ, et al. Runx1 exon 6-related alternative splicing isoforms differentially regulate hematopoiesis in mice. Blood. 2014;123(24):3760–9.
Gialesaki S, Bräuer-Hartmann D, Issa H, Bhayadia R, Alejo-Valle O, Verboon L, et al. RUNX1 isoform disequilibrium promotes the development of trisomy 21–associated myeloid leukemia. Blood. 2023;141(10).
Tanaka T, Tanaka K, Ogawa S, Kurokawa M, Mitani K, Nishida J, et al. An acute myeloid leukemia gene, AML 1, regulates hemopoietic myeloid cell differentiation and transcriptional activation antagonistically by two alternative spliced forms. EMBO J. 1995;14(2):341–50.
Tsuzuki S, Hong D, Gupta R, Matsuo K, Seto M, Enver T. Isoform-specific potentiation of stem and progenitor cell engraftment by AML1/RUNX1. PLoS Med. 2007;4(5):0880–96.
Tsuzuki S, Seto M. Expansion of functionally defined mouse hematopoietic stem and progenitor cells by a short isoform of RUNX1/AML1. Blood. 2012;119(3):727–35.
Liu X, Zhang Q, Zhang DE, Zhou C, Xing H, Tian Z, et al. Overexpression of an isoform of AML1 in acute leukemia and its potential role in leukemogenesis. Leukemia. 2009;23(4):739–45.
Guo H, Ma O, Speck NA, Friedman AD. Runx1 deletion or dominant inhibition reduces Cebpa transcription via conserved promoter and distal enhancer sites to favor monopoiesis over granulopoiesis. Blood. 2012;119(19):4408–18.
Ran D, Shia WJ, Lo MC, Fan JB, Knorr DA, Ferrell PI, et al. RUNX1a enhances hematopoietic lineage commitment from human embryonic stem cells and inducible pluripotent stem cells. 2013; Available from: http://ashpublications.org/blood/article-pdf/121/15/2882/1365983/2882.pdf.
Sakurai H, Harada Y, Matsui H, Nakajima H, Kitamura T, Komatsu N, et al. Overexpression of RUNX1 short isoform plays a pivotal role in the development of myelodysplastic syndromes/myeloproliferative neoplasms. Exp Hematol. 2015;43(9).
Sakurai H, Harada Y, Ogata Y, Kagiyama Y, Shingai N, Doki N, et al. Overexpression of RUNX1 short isoform has an important role in the development of myelodysplastic/myeloproliferative neoplasms. Blood Adv. 2017;1(18):1382–6.
Kanaykina N, Abelson K, King D, Liakhovitskaia A, Schreiner S, Wegner M, et al. In vitro and in vivo effects on neural crest stem cell differentiation by conditional activation of Runx1 short isoform and its effect on neuropathic pain behavior. Ups J Med Sci. 2010;115(1):56–64.
BECKER LE, MITO T, TAKASHIMA S, ONODERA K, FRIEND WC. Association of phenotypic abnormalities of Down syndrome with an imbalance of genes on chromosome 21. APMIS. 1993;101(40 S).
Moore SW. Advances in understanding the association between Down syndrome and Hirschsprung disease (DS-HSCR). Pediatr Surg Int. 2018;34:1127–37. https://doi.org/10.1007/s00383-018-4344-z.
Roper RJ, VanHorn JF, Cain CC, Reeves RH. A neural crest deficit in Down syndrome mice is associated with deficient mitotic response to Sonic hedgehog. Mech Develop. 2009;126(3–4):212–9. https://doi.org/10.1016/j.mod.2008.11.002.
Wu H, Zheng J, Deng J, Zhang L, Li N, Li W, Li F, Lu J, Zhou Y. LincRNA-uc002yug.2 involves in alternative splicing of RUNX1 and serves as a predictor for esophageal cancer and prognosis. Oncogene. 2014. https://doi.org/10.1038/onc.2014.400.
Davis AG, Einstein JM, Zheng D, Jayne ND, Fu XD, Tian B, et al. A CRISPR RNA-binding protein screen reveals regulators of RUNX1 isoform generation. Blood Adv. 2021;5(5):1310–23.
Atas-Ozcan H, Brault V, Duchon A, Herault Y. Dyrk1a from Gene function in development and physiology to dosage correction across life span in Down syndrome. Genes. 2021;2021(12):1833.
Nguyen TL, Duchon A, Manousopoulou A, Loaëc N, Villiers B, Pani G, et al. Correction of cognitive deficits in mouse models of Down syndrome by a pharmacological inhibitor of DYRK1A. DMM Dis Mod Mech. 2018. https://doi.org/10.1242/dmm.035634.
Bonifer C, Levantini E, Kouskoff V, Lacaud G. Runx1 structure and function in blood cell development. In: Advances in Experimental Medicine and Biology. 2017.
Gao L, Tober J, Gao P, Chen C, Tan K, Speck NA. RUNX1 and the endothelial origin of blood. Exper Hemat. 2018;68:2–9. https://doi.org/10.1016/j.exphem.2018.10.009.
Sood R, Kamikubo Y, Liu P. Review Series TRANSCRIPTION FACTORS IN HEMATOPOIESIS AND HEMATOLOGIC DISEASE Role of RUNX1 in hematological malignancies. Available from: http://ashpublications.org/blood/article-pdf/129/15/2070/1400307/blood687830.pdf.
Yokota A, Huo L, Lan F, Wu J, Huang G. The clinical, molecular, and mechanistic basis of RUNX1 mutations identified in hematological malignancies. Mol cells. 2020.
Goyama S, Schibler J, Cunningham L, Zhang Y, Rao Y, Nishimoto N, et al. Erratum: transcription factor RUNX1 promotes survival of acute myeloid leukemia cells. J Clin Invest. 2013. https://doi.org/10.1172/JCI73313.
Bhatnagar N, Nizery L, Tunstall O, Vyas P, Roberts I. Transient Abnormal Myelopoiesis and AML in Down Syndrome: an Update. 2016.
Rylski M, Welch JJ, Chen YY, Letting DL, Diehl JA, Chodosh LA, et al. GATA-1-Mediated Proliferation Arrest during Erythroid Maturation. Mol Cell Biol. 2003;23(14).
Klusmann JH, Godinho F, Pushpanathan T, Reinhardt D, Orkin SH, Li Z. GATA1s Mutant Protein Contributes to “Down” Syndrome Megakaryoblastic Leukemia by Derepression of E2F Targets. Blood. 2008;112(11).
Klusmann JH, Godinho FJ, Heitmann K, Maroz A, Koch ML, Reinhardt D, et al. Developmental stage-specific interplay of GATA1 and IGF signaling in fetal megakaryopoiesis and leukemogenesis. Genes Dev. 2010;24(15).
Maroz A, Stachorski L, Emmrich S, Reinhardt K, Xu J, Shao Z, et al. GATA1s induces hyperproliferation of eosinophil precursors in Down syndrome transient leukemia. Leukemia. 2014. https://doi.org/10.1038/leu.2013.373.
Hitzler JK, Zipursky A. Origins of leukaemia in children with down syndrome. Nat Rev Cancer. 2005. https://doi.org/10.1038/nrc1525.
Hasle H, Friedman JM, Olsen JH, Rasmussen SA. Low risk of solid tumors in persons with Down syndrome. Genet Med. 2016. https://doi.org/10.1038/gim.2016.23.
Gupte A, Al-Antary ET, Edwards H, Ravindranath Y, Ge Y, Taub JW. The paradox of Myeloid Leukemia associated with Down syndrome. Biochem Pharmacol. 2022. https://doi.org/10.1016/j.bcp.2022.115046.
Roy A, Roberts I, Norton A, Vyas P. Acute megakaryoblastic leukaemia (AMKL) and transient myeloproliferative disorder (TMD) in Down syndrome: a multi-step model of myeloid leukaemogenesis. British J Haematol. 2009. https://doi.org/10.1111/j.1365-2141.2009.07789.x.
Kirsammer G, Jilani S, Liu H, Davis E, Gurbuxani S, Le Beau MM, et al. Highly penetrant myeloproliferative disease in the Ts65Dn mouse model of Down syndrome. 2008; Available from: http://ashpublications.org/blood/article-pdf/111/2/767/1220455/zh800208000767.pdf.
Carmichael CL, Majewski IJ, Alexander WS, Metcalf D, Hilton DJ, Hewitt CA, et al. Hematopoietic defects in the Ts1Cje mouse model of Down syndrome. Blood. 2009;113:1929–37.
Alford KA, Slender A, Vanes L, Li Z, Fisher EMC, Nizetic D, et al. Perturbed hematopoiesis in the Tc1 mouse model of Down syndrome. Blood. 2010;115(14):2928–37.
Alford KA, Vanes L, Li Z, Orkin SH, Fisher EMC, Tybulewicz VLJ. A myeloproliferative disorder in the Tc1 mouse model of Down syndrome. Blood. 2008. https://doi.org/10.1182/blood.V112.11.2790.2790.
Elagib KE, Racke FK, Mogass M, Khetawat R, Delehanty LL, Goldfarb AN. RUNX1 and GATA-1 coexpression and cooperation in megakaryocytic differentiation. Blood. 2003. https://doi.org/10.1182/blood-2002-09-2708.
Xu G, Kanezaki R, Toki T, Watanabe S, Takahashi Y, Terui K, et al. Physical association of the patient-specific GATA1 mutants with RUNX1 in acute megakaryoblastic leukemia accompanying Down syndrome. Leukemia. 2006;20(6):1002–8.
De Vita S, Canzonetta C, Mulligan C, Delom F, Groet J, Baldo C, et al. Trisomic dose of several chromosome 21 genes perturbs haematopoietic stem and progenitor cell differentiation in Down’s syndrome. Oncogene. 2010;29(46):6102–14.
Vukadin L, Kim JH, Park EY, Stone JK, Ungerleider N, Baddoo MC, et al. SON inhibits megakaryocytic differentiation via repressing RUNX1 and the megakaryocytic gene expression program in acute megakaryoblastic leukemia. Cancer Gene Ther. 2021;28(9):1000–15.
Bourquin JP, Subramanian A, Langebrake C, Reinhardt D, Bernard O, Ballerini P, et al. Identification of distinct molecular phenotypes in acute megakaryoblastic leukemia by gene expression profiling. 2006. Available from: www.ncbi.nlm.nih.govgeo
Laufer BI, Hwang H, Jianu JM, Mordaunt CE, Korf IF, Hertz-Picciotto I, et al. Low-pass whole genome bisulfite sequencing of neonatal dried blood spots identifies a role for RUNX1 in down syndrome DNA methylation profiles. Hum Mol Genet. 2020;29(21):3465–76.
Muskens IS, Li S, Jackson T, Elliot N, Hansen HM, Myint SS, et al. The genome-wide impact of trisomy 21 on DNA methylation and its implications for hematopoiesis. Nat Commun. 2021. https://doi.org/10.1038/s41467-021-21064-z.
Mollo N, Aurilia M, Scognamiglio R, Zerillo L, Cicatiello R, Bonfiglio F, et al. Overexpression of the Hsa21 transcription factor RUNX1 modulates the extracellular matrix in trisomy 21 cells. Front Genet. 2022;10:13.
De Cegli R, Romito A, Iacobacci S, Mao L, Lauria M, Fedele AO, et al. A mouse embryonic stem cell bank for inducible overexpression of human chromosome 21 genes. Genome Biol. 2010. https://doi.org/10.1186/gb-2010-11-6-r64.
Stoll C, Dott B, Alembik Y, Roth MP. Associated congenital anomalies among cases with Down syndrome. Eur J Med Genet. 2015. https://doi.org/10.1016/j.ejmg.2015.11.003.
Heuckeroth RO. Hirschsprung’s disease, Down syndrome, and missing heritability: too much collagen slows migration. J Clin Invest. 2015. https://doi.org/10.1172/JCI85003.
Thenappan T, Chan SY, Kenneth Weir E. Role of extracellular matrix in the pathogenesis of pulmonary arterial hypertension. Rev Extracell Matrix Cardiovasc Pathophysiol Am J Physiol Heart Circ Physiol. 2018;315:1322–31.
Halevy T, Biancotti JC, Yanuka O, Golan-Lev T, Benvenisty N. Molecular characterization of Down syndrome embryonic stem cells reveals a role for RUNX1 in neural differentiation. Stem Cell Rep. 2016;7(4):777–86.
Liu Y, Zhang Y, Ren Z, Zeng F, Yan J. Molecules and Cells RUNX1 Upregulation Causes Mitochondrial Dysfunction via Regulating the PI3K-Akt Pathway in iPSC from Patients with Down Syndrome. Available from: https://doi.org/10.14348/molcells.2022.0095www.molcells.org.
Theriault FM, Roy P, Stifani S. AML1/Runx1 is important for the development of hindbrain cholinergic branchiovisceral motor neurons and selected cranial sensory neurons. Proc Natl Acad Sci U S A. 2004;101(28):10343–8.
Theriault FM, Nuthall HN, Dong Z, Lo R, Barnabe-Heider F, Miller FD, et al. Role for Runx1 in the proliferation and neuronal differentiation of selected progenitor cells in the mammalian nervous system. J Neurosci. 2005;25(8):2050–61.
Logan TT, Rusnak M, Symes AJ. Runx1 promotes proliferation and neuronal differentiation in adult mouse neurosphere cultures. Stem Cell Res. 2015;15(3):554–64.
Fukui H, Rünker A, Fabel K, Buchholz F, Kempermann G. Transcription factor Runx1 is pro-neurogenic in adult hippocampal precursor cells. PLoS One. 2018. https://doi.org/10.1371/journal.pone.0190789.
Shrestha BR, Wu L, Goodrich LV. Runx1 controls auditory sensory neuron diversity in mice. Dev Cell. 2023;58(4):306–19.
Chen CL, Broom DC, Liu Y, De Nooij JC, Li Z, Cen C, et al. Runx1 determines nociceptive sensory neuron phenotype and is required for thermal and neuropathic pain. Neuron. 2006;49(3):365–77.
Huang S, O’Donovan KJ, Turner EE, Zhong J, Ginty DD. Extrinsic and intrinsic signals converge on the Runx1/CBFβ transcription factor for nonpeptidergic nociceptor maturation. Elife. 2015. https://doi.org/10.7554/eLife.10874.
Kramer I, Sigrist M, De Nooij JC, Taniuchi I, Jessell TM, Arber S. A role for Runx transcription factor signaling in dorsal root ganglion sensory neuron diversification. Neuron. 2006;49(3):379–93.
Citation Klein JA, Haydar TF. Open access edited by Neurodevelopment in Down syndrome: Concordance in humans and models. 2022.
Brozzetti L, Scambi I, Bertoldi L, Zanini A, Malacrida G, Sacchetto L, et al. RNAseq analysis of olfactory neuroepithelium cytological samples in individuals with Down syndrome compared to euploid controls: a pilot study. Neurol Sci. 2023. https://doi.org/10.1007/s10072-022-06500-2.
Guidi S, Bianchi P, Stagni F, Giacomini A, Emili M, Trazzi S, et al. Lithium restores age-related olfactory impairment in the Ts65Dn mouse model of Down syndrome. CNS Neurol Disord Drug Targets. 2016. https://doi.org/10.2174/1871527315666160801143108.
Bianchi P, Ciani E, Guidi S, Trazzi S, Felice D, Grossi G, et al. Early pharmacotherapy restores neurogenesis and cognitive performance in the Ts65Dn mouse model for down syndrome. J Neurosci. 2010. https://doi.org/10.1523/JNEUROSCI.0534-10.2010.
Guidi S, Stagni F, Bianchi P, Ciani E, Giacomini A, De Franceschi M, et al. Prenatal pharmacotherapy rescues brain development in a Down’s syndrome mouse model. Brain. 2014. https://doi.org/10.1093/brain/awt340.
Do Carmo S, Kannel B, Cuello AC. Nerve growth factor compromise in Down syndrome. Front Aging Neurosci. 2021. https://doi.org/10.3389/fnagi.2021.719507.
Watson-Scales S, Kalmar B, Lana-Elola E, Gibbins D, La Russa F, Wiseman F, et al. Analysis of motor dysfunction in Down syndrome reveals motor neuron degeneration. PLoS Genet. 2018. https://doi.org/10.1371/journal.pgen.1007383.
Inagaki T, Morita N, Cureoglu S, Schachern PA, Nomiya S, Nomiya R, et al. Peripheral vestibular system in Down syndrome: quantitative assessment of vestibular histopathology. Otolaryngol Head Neck Surg. 2011;144(2):280–3.
Di CG, Li L, McCall A, Ding D, Xing Z, Yu YE, et al. Hearing impairment in murine model of Down syndrome. Front Genet. 2022;4:13.
Liu Y, Huang C, Bai M, Pi C, Zhang D, Xie J. The roles of Runx1 in skeletal development and osteoarthritis: a concise review. vol. 8, Heliyon. Elsevier Ltd; 2022.
Yamashiro T, Åberg T, Levanon D, Groner Y, Thesleff I. Expression of Runx1, -2 and -3 during tooth, palate and craniofacial bone development. Mech Dev. 2002. https://doi.org/10.1016/S0925-4773(03)00101-1.
Lian JB, Balint E, Javed A, Drissi H, Vitti R, Quinlan EJ, et al. Runx1/AML1 hematopoietic transcription factor contributes to skeletal development in vivo. J Cell Physiol. 2003;196(2):301–11.
Soung DY, Kalinowski J, Baniwal SK, Jacome-Galarza CE, Frenkel B, Lorenzo J, et al. Runx1-mediated regulation of osteoclast differentiation and function. Mol Endocrinol. 2014;28(4):546–53.
Tang CY, Chen W, Luo Y, Wu J, Zhang Y, McVicar A, et al. Runx1 up-regulates chondrocyte to osteoblast lineage commitment and promotes bone formation by enhancing both chondrogenesis and osteogenesis. Biochem J. 2020;477(13):2421–38.
Tang J, Xie J, Chen W, Tang C, Wu J, Wang Y, et al. Runt-related transcription factor 1 is required for murine osteoblast differentiation and bone formation. J Biol Chem. 2020;295(33):11669–81.
Tang CY, Wu M, Zhao D, Edwards D, McVicar A, Luo Y, et al. Runx1 is a central regulator of osteogenesis for bone homeostasis by orchestrating BMP and WNT signaling pathways. PLoS Genet. 2021. https://doi.org/10.1371/journal.pgen.1009233.
Paglia DN, Diaz-Hernandez ME, Roberts JL, Kalinowski J, Lorenzo J, Drissi H. Deletion of Runx1 in osteoclasts impairs murine fracture healing through progressive woven bone loss and delayed cartilage remodeling. J Orthop Res. 2020;38(5):1007–15.
Zhou C, Cui Y, Yang Y, Guo D, Zhang D, Fan Y, et al. Runx1 protects against the pathological progression of osteoarthritis. Bone Res. 2021. https://doi.org/10.1038/s41413-021-00173-x.
Thomas JR, Roper RJ. Current analysis of skeletal phenotypes in Down Syndrome. Curr Osteoporosis Rep. 2021. https://doi.org/10.1007/s11914-021-00674-y.
Blazek JD, Abeysekera I, Li J, Roper RJ. Rescue of the abnormal skeletal phenotype in Ts65Dn Down syndrome mice using genetic and therapeutic modulation of trisomic Dyrk1a. Hum Mol Genet. 2015;24(20):5687–96.
Sloan K, Thomas J, Blackwell M, Voisard D, Lana-Elola E, Watson-Scales S, et al. Genetic dissection of triplicated Hsa21 orthologs produces differential skeletal phenotypes in Down syndrome mouse models. Dis Models Mech. 2023. https://doi.org/10.1242/dmm.049927.
Garcia-Ramírez M, Toran N, Carrascosa A, Audi L. Down’s syndrome: altered chondrogenesis in fetal rib. Pediatr Res. 1998. https://doi.org/10.1203/00006450-199807000-00015.
Jones JT, Kitchen J, Talib N. Down syndrome-associated arthritis (DA): diagnostic and management challenges. Pediatric Health Med Ther. 2022;13:53–62.
Levanon D, Brenner O, Negreanu V, Bettoun D, Woolf E, Eilam R, et al. Spatial and temporal expression pattern of Runx3 (Aml2) and Runx1 (Aml1) indicates non-redundant functions during mouse embryogenesis [Internet]. Available from: www.elsevier.com/locate/modo.
Lluri G, Huang V, Touma M, Liu X, Harmon AW, Nakano A. Hematopoietic progenitors are required for proper development of coronary vasculature. J Mol Cell Cardiol. 2015;1(86):199–207.
Eulalio A, Mano M, Ferro MD, Zentilin L, Sinagra G, Zacchigna S, et al. Functional screening identifies miRNAs inducing cardiac regeneration. Nature. 2012;492(7429):376–81.
Górnikiewicz B, Ronowicz A, Krzemiński M, Sachadyn P. Changes in gene methylation patterns in neonatal murine hearts: Implications for the regenerative potential. BMC Genom. 2016. https://doi.org/10.1186/s12864-016-2545-1.
Riddell A, McBride M, Braun T, Nicklin SA, Cameron E, Loughrey CM, et al. RUNX1: an emerging therapeutic target for cardiovascular disease. Cardiovasc Res. 2020. https://doi.org/10.1093/cvr/cvaa034.
Gattenlöhner S, Waller C, Ertl G, Bültmann BD, Müller-Hermelink HK, Marx A. NCAM(CD56) and RUNX1(AML1) are up-regulated in human ischemic cardiomyopathy and a rat model of chronic cardiac ischemia. Am J Pathol. 2003. https://doi.org/10.1016/S0002-9440(10)63467-0.
Kubin T, Pöling J, Kostin S, Gajawada P, Hein S, Rees W, et al. Oncostatin M is a major mediator of cardiomyocyte dedifferentiation and remodeling. Cell Stem Cell. 2011. https://doi.org/10.1016/j.stem.2011.08.013.
McCarroll CS, He W, Foote K, Bradley A, McGlynn K, Vidler F, et al. Runx1 deficiency protects against adverse cardiac remodeling after myocardial infarction. Circulation. 2018;137(1):57–70.
Ikeda S, Mukai R, Mizushima W, Zhai P, Ichi OS, Nakamura M, et al. Yes-associated protein (YAP) facilitates pressure overload-induced dysfunction in the diabetic heart. JACC Basic Transl Sci. 2019. https://doi.org/10.1016/j.jacbts.2019.05.006.
Zhang X, Ma S, Zhang R, Li S, Zhu D, Han D, et al. Oncostatin M-induced cardiomyocyte dedifferentiation regulates the progression of diabetic cardiomyopathy through B-Raf/Mek/Erk signaling pathway. Acta Biochim Biophys Sin. 2015. https://doi.org/10.1093/abbs/gmv137.
Martin TP, Macdonald EA, Bradley A, Watson H, Saxena P, Rog-Zielinska EA, et al. Inhibiting Runx1 protects heart function after myocardial infarction. 2022. https://doi.org/10.1101/2022.02.17.480749.
Swift SK, Purdy AL, Kolell ME, Andresen KG, Lahue C, Buddell T, et al. Cardiomyocyte ploidy is dynamic during postnatal development and varies across genetic backgrounds. 2023.
Gittenberger-De Groot AC, Bartram U, Oosthoek PW, Bartelings MM, Hogers B, Poelmann RE, et al. Collagen type VI expression during cardiac development and in human fetuses with trisomy 21. Anat Record A Discover Mol Cell Evolut Biol. 2003. https://doi.org/10.1002/ar.a.10126.
Conti A, Fabbrini F, D’Agostino P, Negri R, Greco D, Genesio R, et al. Altered expression of mitochondrial and extracellular matrix genes in the heart of human fetuses with chromosome 21 trisomy. BMC Genom. 2007. https://doi.org/10.1186/1471-2164-8-268.
Freeman SB, Taft LF, Dooley KJ, Allran K, Sherman SL, Hassold TJ, et al. Population-based study of congenital heart defects in Down syndrome. Am J Med Genet. 1998;80(3).
Roizen NJ, Patterson D. Down’ s syndrome. Seminar. 2003. https://doi.org/10.1016/S0140-6736(03)12987-X.
Bittles AH, Bower C, Hussain R, Glasson EJ. The four ages of Down syndrome. Eur J Public Health. 2007. https://doi.org/10.1093/eurpub/ckl103.
Zhang H, Liu L, Tian J. Molecular mechanisms of congenital heart disease in down syndrome. Genes Dis. 2019. https://doi.org/10.1016/j.gendis.2019.06.007.
Venegas-Zamora L, Bravo-Acuña F, Sigcho F, Gomez W, Bustamante-Salazar J, Pedrozo Z, et al. New molecular and organelle alterations linked to Down syndrome heart disease. Front Genet. 2022. https://doi.org/10.3389/fgene.2021.792231.
Simeone A, Daga A, Calabi F. Expression of runt in the mouse embryo. Develop Dyn. 1995. https://doi.org/10.1002/aja.1002030107.
Zhu X, Yeadon JE, Burden SJ. AML1 is expressed in skeletal muscle and is regulated by innervation. Mol Cell Biol. 1994. https://doi.org/10.1128/MCB.14.12.8051.
Porter JD, Merriam AP, Leahy P, Gong B, Khanna S. Dissection of temporal gene expression signatures of affected and spared muscle groups in dystrophin-deficient (mdx) mice. Human Mol Genet. 2003. https://doi.org/10.1093/hmg/ddg197.
Gonzalez De Aguilar JL, Niederhauser-Wiederkehr C, Halter B, De Tapia M, Di Scala F, Demougin P, et al. Gene profiling of skeletal muscle in an amyotrophic lateral sclerosis mouse model. Phys Gen. 2008. https://doi.org/10.1152/physiolgenomics.00017.2007
Bakay M, Wang Z, Melcon G, Schiltz L, Xuan J, Zhao P, et al. Nuclear envelope dystrophies show a transcriptional fingerprint suggesting disruption of Rb-MyoD pathways in muscle regeneration. Brain. 2006. https://doi.org/10.1093/brain/awl023.
Zhao P, Iezzi S, Carver E, Dressman D, Gridley T, Sartorelli V, et al. Slug is a novel downstream target of MyoD Temporal profiling in muscle regeneration. J Biol Chem. 2002. https://doi.org/10.1074/jbc.M202668200.
Bao M, Liu S, Yu XY, Wu C, Chen Q, Ding H, et al. Runx1 promotes satellite cell proliferation during ischemia-Induced muscle regeneration. Biochem Biophys Res Commun. 2018;503(4):2993–7.
Blum R, Dynlacht BD. The role of MyoD1 and histone modifications in the activation of muscle enhancers. Epigenetics. 2013. https://doi.org/10.4161/epi.25441.
Philipot O, Joliot V, Ait-Mohamed O, Pellentz C, Robin P, Fritsch L, et al. The core binding factor CBF negatively regulates skeletal muscle terminal differentiation. PLoS One. 2010. https://doi.org/10.1371/journal.pone.0009425.
MacQuarrie KL, Yao Z, Young JM, Cao Y, Tapscott SJ. MiR-206 integrates multiple components of differentiation pathways to control the transition from growth to differentiation in rhabdomyosarcoma cells. Skelet Muscle. 2012. https://doi.org/10.1186/2044-5040-2-7.
Umansky KB, Feldmesser E, Groner Y. Genomic-wide transcriptional profiling in primary myoblasts reveals Runx1-regulated genes in muscle regeneration. Genom Data. 2015;1(6):120–2.
Umansky KB, Gruenbaum-Cohen Y, Tsoory M, Feldmesser E, Goldenberg D, Brenner O, et al. Runx1 Transcription Factor Is Required for Myoblasts Proliferation during Muscle Regeneration. PLoS Genet. 2015. https://doi.org/10.1371/journal.pgen.1005457.
Stefanowicz M, Nikołajuk A, Matulewicz N, Strączkowski M, Karczewska-Kupczewska M. Skeletal muscle RUNX1 is related to insulin sensitivity through its effect on myogenic potential. Eur J Endocrinol. 2022;187(1):143–57.
Wosczyna MN, Perez Carbajal EE, Wagner MW, Paredes S, Konishi CT, Liu L, et al. Targeting microRNA-mediated gene repression limits adipogenic conversion of skeletal muscle mesenchymal stromal cells. Cell Stem Cell. 2021;28(7):1323-1334.e8.
Foley C, Killeen OG. Musculoskeletal anomalies in children with Down syndrome: an observational study. Arch Dis Child. 2019;104(5):482–7.
Jain PD, Nayak A, Karnad SD, Doctor KN. Gross motor dysfunction and balance impairments in children and adolescents with Down syndrome: a systematic review. Clin Exp Pediatr. 2022;65(3):142–9.
Cisterna B, Sobolev AP, Costanzo M, Malatesta M, Zancanaro C. Combined microscopic and metabolomic approach to characterize the skeletal muscle fiber of the Ts65Dn mouse, a model of Down syndrome. Microsc Microanal. 2020;26(5):1014–23.
Pawlikowski B, Betta ND, Elston T, Williams DA, Olwin BB. Muscle stem cell dysfunction impairs muscle regeneration in a mouse model of Down syndrome. Sci Rep. 2018. https://doi.org/10.1038/s41598-018-22342-5.
Stewart L, Potok MA, Camper SA, Stifani S. Runx1 expression defines a subpopulation of displaced amacrine cells in the developing mouse retina. J Neurochem. 2005;94(6):1739–45.
Rheaume BA, Jereen A, Bolisetty M, Sajid MS, Yang Y, Renna K, et al. Single cell transcriptome profiling of retinal ganglion cells identifies cellular subtypes. Nat Commun. 2018. https://doi.org/10.1038/s41467-018-05134-3.
Lam JD, Oh DJ, Wong LL, Amarnani D, Park-Windhol C, Sanchez AV, et al. Identification of RUNX1 as a mediator of aberrant retinal angiogenesis. Diabetes. 2017;66(7):1950–6.
Zou W, Zhang Z, Luo S, Cheng L, Huang X, Ding N, et al. P38 promoted retinal micro-angiogenesis through up-regulated RUNX1 expression in diabetic retinopathy. 2020. Biosci Rep. https://doi.org/10.1042/BSR20193256.
Zhang W, Zhang D, Cheng Y, Liang X, Wang J. Runx1 regulates Tff1 expression to expedite viability of retinal microvascular endothelial cells in mice with diabetic retinopathy. Exp Eye Res. 2022;1:217.
Delgado-Tirado S, Amarnani D, Zhao G, Rossin EJ, Eliott D, Miller JB, et al. Topical delivery of a small molecule RUNX1 transcription factor inhibitor for the treatment of proliferative vitreoretinopathy. Sci Rep. 2020.
Whitmore HAB, Amarnani D, O’Hare M, Delgado-Tirado S, Gonzalez-Buendia L, An M, et al. TNF-α signaling regulates RUNX1 function in endothelial cells. FASEB J. 2021.
Gonzalez-Buendia L, Delgado-Tirado S, An M, O’Hare M, Amarnani D, Whitmore HAB, et al. Treatment of experimental choroidal neovascularization via RUNX1 inhibition. Am J Pathol. 2021;191(3):418–24.
Xing X, Wang H, Niu T, Jiang Y, Shi X, Liu K. RUNX1 can mediate the glucose and O-GlcNAc-driven proliferation and migration of human retinal microvascular endothelial cells. BMJ Open Diabetes Res Care. 2021;9(1).
Li M, Huang H, Li L, He C, Zhu L, Guo H, et al. Core transcription regulatory circuitry orchestrates corneal epithelial homeostasis. Nat Commun. 2021;12(1):420.
Voronov D, Gromova A, Liu D, Zoukhri D, Medvinsky A, Meech R, et al. Transcription factors Runx1 to 3 are expressed in the lacrimal gland epithelium and are involved in regulation of gland morphogenesis and regeneration. Invest Ophthalmol Vis Sci. 2013. Available from: www.iovs.org.
Suda T, Takakura N. RoLe of hematopoietic stem cells in angiogenesis. Int J Hematol. 2001;74(3).
Kalev-Zylinska ML, Horsfield JA, Flores MVC, Postlethwait JH, Vitas MR, Baas AM, et al. Runx1 is required for zebrafish blood and vessel development and expression of a human RUNX-1-CBF2T1 transgene advances a model for studies of leukemogenesis. Development. 2002.
Liu C, Xu D, Xue B, Liu B, Li J, Huang J. Upregulation of RUNX1 Suppresses proliferation and migration through repressing VEGFA expression in hepatocellular carcinoma. Pathol Oncol Res. 2020;26(2).
Iwatsuki K, Tanaka K, Kaneko T, Kazama R, Okamoto S, Nakayama Y, et al. Runx1 promotes angiogenesis by downregulation of insulin-like growth factor-binding protein-3. Oncogene. 2005;24(7).
Sun E, Kraus CL. The ophthalmic manifestations of Down syndrome. Children. MDPI; 2023.
Haseeb A, Huynh E, ElSheikh RH, ElHawary AS, Scelfo C, Ledoux DM, et al. Down syndrome: a review of ocular manifestations. Ther Adv Ophthalmol. 2022;14:251584142211017.
Liza-Sharmini AT, Azlan ZN, Zilfalil BA. Ocular findings in Malaysian children with Down syndrome. Singapore Med J. 2006;47(1).
Adio AO, Wajuihian SO. Ophthalmic manifestations of children with Down syndrome in Port Harcourt, Nigeria. Clinical Ophthalmol. 2012;6(1).
Williams EJ, McCormick AQ, Tischler B. Retinal vessels in Down’s syndrome. Arch Ophthalmol. 1973. https://doi.org/10.1001/archopht.1973.01000040271001.
Sherk MC, Williams TD. Disc vascularity in down’s syndrome. Optomet Vis Sci. 1979. https://doi.org/10.1097/00006324-197908000-00005.
Parsa CF, Almer Z. Supranumerary optic disc vessels may indicate reduced systemic angiogenesis in Down syndrome. British J Ophthalmol. 2008. https://doi.org/10.1136/bjo.2007.124248.
Schneier AJ, Heidary G, Ledoux DM. Optic nerve appearance in patients with down syndrome. J Pediatric Ophthalmol Strabismus. 2013. https://doi.org/10.3928/01913913-20121214-01.
Postolache L. Abnormalities of the optic nerve in down syndrome and associations with visual acuity. Front Neurol. 2019. https://doi.org/10.3389/fneur.2019.00633.
Al-Hemidan AI, Al-Hazzaa SAF, Chavis P, Al-Hussein H. Optic disc elevation in Down syndrome. Ophthalmic Genet. 1999. https://doi.org/10.1076/opge.20.1.45.2297.
Catalano RA, Simon JW. Optic disk elevation in Down’s syndrome. Am J Ophthalmol. 1990. https://doi.org/10.1016/S0002-9394(14)76933-8.
Ugurlu A, Altinkurt E. Ophthalmologic manifestations and retinal findings in children with Down syndrome. J Ophthalmol. 2020. https://doi.org/10.1155/2020/9726261.
Landau Prat D, Tadros S, Revere KE, Katowitz JA, Katowitz WR. Management of congenital nasolacrimal duct obstruction in down syndrome. Eye. 2022. https://doi.org/10.1038/s41433-022-02047-w.
Li Y, Wei M, Liu X, Zhang L, Song X, Xiao C. Dacryoendoscopy-assisted incision of Hasner’s valve under nasoendoscopy for membranous congenital nasolacrimal duct obstruction after probing failure: a retrospective study. BMC Ophthalmol. 2021. https://doi.org/10.1186/s12886-021-01948-w.
Lim CS, Martin F, Beckenham T, Cumming RG. Nasolacrimal duct obstruction in children: outcome of intubation. J AAPOS. 2004. https://doi.org/10.1016/j.jaapos.2004.06.013.
Jo M, Curry TE. Luteinizing hormone-induced RUNX1 regulates the expression of genes in granulosa cells of rat periovulatory follicles. Mol Endocrinol. 2006;20(9):2156–72.
Liu J, Park ES, Jo M. Runt-related transcription factor 1 regulates luteinized hormone-induced prostaglandin-endoperoxide synthase 2 expression in rat periovulatory granulosa cells. Endocrinology. 2009;150(7):3291–300.
Liu J, Park ES, Curry TE, Jo M. Periovulatory expression of hyaluronan and proteoglycan link protein 1 (Hapln1) in the rat ovary: hormonal regulation and potential function. Mol Endocrinol. 2010;24(6):1203–17.
Naillat F, Yan W, Karjalainen R, Liakhovitskaia A, Samoylenko A, Xu Q, et al. Identification of the genes regulated by Wnt-4, a critical signal for commitment of the ovary. Exp Cell Res. 2015. https://doi.org/10.1016/j.yexcr.2015.01.010.
Jo M, Gieske MC, Payne CE, Wheeler-Price SE, Gieske JB, Ignatius I V., et al. Development and application of a rat ovarian gene expression database. Endocrinology. 2004;145(11).
Park ES, Lind AK, Dahm-Kähler P, Brännström M, Carletti MZ, Christenson LK, et al. RUNX2 transcription factor regulates gene expression in luteinizing granulosa cells of rat ovaries. Mol Endocrinol. 2010;24(4):846–58.
Hernandez-Gonzalez I, Gonzalez-Robayna I, Shimada M, Wayne CM, Ochsner SA, White L, et al. Gene expression profiles of cumulus cell oocyte complexes during ovulation reveal cumulus cells express neuronal and immune-related genes: does this expand their role in the ovulation process? Mol Endocrinol. 2006;20(6):1300–21.
Nicol B, Grimm SA, Chalmel F, Lecluze E, Pannetier M, Pailhoux E, et al. RUNX1 maintains the identity of the fetal ovary through an interplay with FOXL2. Nat Commun. 2019;10(1):5116.
Bridges K, Yao HHC, Nicol B. Loss of Runx1 induces granulosa cell defects and development of ovarian tumors in the mouse. Int J Mol Sci. 2022;23(22):14442.
Dinh D, Breen J, Nicol B, Smith K, Nicholls M, Emery A, et al. Progesterone receptor-A isoform interaction with RUNX transcription factors controls chromatin remodelling at promoters during ovulation. bioRxiv. 2021.
Wilson K, Park J, Curry TE, Mishra B, Gossen J, Taniuchi I, et al. Core binding factor-β knockdown alters ovarian gene expression and function in the mouse. Mol Endocrinol. 2016;30(7):733–47.
Lee-Thacker S, Choi Y, Taniuchi I, Takarada T, Yoneda Y, Ko CM, et al. Core binding factor b expression in ovarian granulosa cells is essential for female fertility. Endocrinology. 2018;159(5):2094–109.
Kannan A, Beal JR, Neff AM, Bagchi MK, Bagchi IC. Runx1 regulates critical factors that control uterine angiogenesis and trophoblast differentiation during placental development. Available from: https://doi.org/10.1101/2023.03.21.532831.
Højager B, Peters H, Byskov AG, Faber M. Follicular development in ovaries of children with Down’s syndrome. Acta Paediatr. 1978;67(5):637–43.
Hsiang YHH, Berkovitz GD, Bland GL, Migeon CJ, Warren AC. Gonadal function in patients with Down syndrome. Am J Med Genet. 1987. https://doi.org/10.1002/ajmg.1320270223.
Cento RM, Ragusa L, Proto C, Alberti A, Romano C, Boemi G, et al. Basal body temperature curves and endocrine pattern of menstrual cycles in Down syndrome. Gynecol Endocrinol. 1996. https://doi.org/10.3109/09513599609097904.
Cento RM, Ragusa L, Proto C, Alberti A, Fiore G, Colabucci F, et al. Ovarian sensitivity to follicle stimulating hormone is blunted in normo-ovulatory women with Down’s syndrome. Human Reprod. 1997. https://doi.org/10.1093/humrep/12.8.1709.
Cento RM, Proto C, Guido M, Lanzone A. Postmenarchal Down’s syndrome: a theoretical model of growth hormone action on ovarian function. Gynecol Endocrinol. 2001. https://doi.org/10.1080/gye.15.1.81.88.
Angelopoulou N, Souftas V, Sakadamis A, Matziari C, Papameletiou V, Mandroukas K. Gonadal function in young women with Down syndrome. Int J Gynecol Obstet. 1999;67(1):15–21.
Parizot E, Dard R, Janel N, Vialard F. Down syndrome and infertility: what support should we provide? J Assisted Reprod Genet. 2019. https://doi.org/10.1007/s10815-019-01457-2.
Bartmann AK, Araújo FM, Iannetta O, Paneto JCC, Martelli L, Ramos ES. Down syndrome and precocious menopause. J Assist Reprod Genet. 2005. https://doi.org/10.1007/s10815-005-4878-6.
Schupf N, Zigman W, Kapell D, Lee JH, Kline J, Levin B. Early menopause in women with Down’s syndrome. J Intell Disabil Res. 1997. https://doi.org/10.1046/j.1365-2788.1997.03838.x.
Kaur S, Rawal P, Siddiqui H, Rohilla S, Sharma S, Tripathi DM, et al. Increased expression of RUNX1 in liver correlates with NASH activity score in patients with non-alcoholic steatohepatitis (NASH). Cells. 2019. https://doi.org/10.3390/cells8101277.
Marcher AB, Bendixen SM, Terkelsen MK, Hohmann SS, Hansen MH, Larsen BD, et al. Transcriptional regulation of Hepatic stellate cell activation in NASH. Sci Rep. 2019. https://doi.org/10.1038/s41598-019-39112-6.
Bertran L, Pastor A, Portillo-Carrasquer M, Binetti J, Aguilar C, Martínez S, et al. The potential protective role of runx1 in nonalcoholic fatty liver disease. Int J Mol Sci. 2021;22(10).
Bertran L, Eigbefoh‐addeh A, Portillo‐carrasquer M, Barrientos‐riosalido A, Binetti J, Aguilar C, et al. Identification of the potential molecular mechanisms linking RUNX1 activity with nonalcoholic fatty liver disease, by means of systems biology. Biomedicines. 2022;10(6).
Bertrand-Philippe M, Ruddell RG, Arthur MJP, Thomas J, Mungalsingh N, Mann DA. Regulation of tissue inhibitor of metalloproteinase 1 gene transcription by RUNX1 and RUNX2. J Biol Chem. 2004;279(23):24530–9.
Yoshiji H, Kuriyama S, Yoshii J, Ikenaka Y, Noguchi R, Nakatani T, et al. Tissue inhibitor of metalloproteinases-1 attenuates spontaneous liver fibrosis resolution in the transgenic mouse. Hepatology. 2002;36(4).
Yoshiji H, Kuriyama S, Miyamoto Y, Thorgeirsson UP, Gomez DE, Kawata M, et al. Tissue inhibitor of metalloproteinases-1 promotes liver fibrosis development in a transgenic mouse model. Hepatology. 2000;32(6):1248–54.
Guo Z, Liu X, Zhao S, Sun F, Ren W, Ma M, et al. Original article RUNX1 promotes liver fibrosis progression through regulating TGF-β signalling. Int J Exp Path. 2023. https://doi.org/10.1111/iep.12474.
De Matteo A, Vajro P. Down syndrome and pediatric nonalcoholic fatty liver disease: a causal or casual relationship? 2017. Available from: https://doi.org/10.1016/j.jpeds.2017.07.011.
Park MJ, Sotomatsu M, Ohki K, Arai K, Maruyama K, Kobayashi T, et al. Liver disease is frequently observed in Down syndrome patients with transient abnormal myelopoiesis. Int J Hematol. 2014. https://doi.org/10.1007/s12185-013-1487-5.
Arnell H, Fischler B. Population-based study of incidence and clinical outcome of neonatal cholestasis in patients with down syndrome. J Pediatr. 2012. https://doi.org/10.1016/j.jpeds.2012.04.037.
Vajro P, Cucchiara S, Vegnente A, Iorio R, De Silva C, Cipolletta L, et al. Primary sclerosing cholangitis preceding Crohn’s disease in a child with Down’s syndrome. Dig Dis Sci. 1998. https://doi.org/10.1023/A:1018800826608.
Taşdemir HA, Çetinkaya MC, Polat C, Belet U, Kalayci AG, Akbaş S. Gallbladder motility in children with Down syndrome. J Pediatr Gastroenterol Nutr. 2004;39(2):187–91.
Tuttle KD, Waugh KA, Araya P, Minter R, Orlicky DJ, Ludwig M, et al. JAK1 inhibition blocks lethal immune hypersensitivity in a mouse model of Down syndrome. Cell Rep. 2020. https://doi.org/10.1016/j.celrep.2020.108407.
Valentini D, Alisi A, Di Camillo C, Sartorelli MR, Crudele A, Bartuli A, et al. Nonalcoholic fatty liver disease in italian children with Down syndrome: prevalence and correlation with obesity-related features. J Pediatr. 2017. https://doi.org/10.1016/j.jpeds.2017.05.077.
Roosen-Runge EC. Fatty infiltration of the liver in patients with mongolism and in children with hydrocephaly and microcephaly. Am J Pathol. 1947;23(1):79.
Seeff LB, Levitsky J, Tillman PW, Perou ML, Zimmerman HJ. Histopathology of the liver in Down’s syndrome. Am J Dig Dis. 1967;12:1102–13.
Valentini D, Mosca A, Di Camillo C, Crudele A, Sartorelli MR, Scoppola V, et al. PNPLA3 gene polymorphism is associated with liver steatosis in children with Down syndrome. Nutr Metab Cardiovasc Dis. 2020;30(9):1564–72.
Raveh E, Cohen S, Levanon D, Negreanu V, Groner Y, Gat U. Dynamic expression of Runx1 in skin affects hair structure. Mech Dev. 2006;123(11):842–50.
Soma T, Ishimatsu-Tsuji Y, Tajima M, Kishimoto J. Runx1 transcription factor is involved in the regulation of KAP5 gene expression in human hair follicles. J Dermatol Sci. 2006;41(3):221–4.
Osorio KM, Lee SE, McDermitt DJ, Waghmare SK, Zhang YV, Woo HN, et al. Runx1 modulates developmental, but not injury-driven, hair follicle stem cell activation. Development. 2008;135(6):1059–68.
Hoi CSL, Lee SE, Lu SY, McDermitt DJ, Osorio KM, Piskun CM, et al. Runx1 directly promotes proliferation of hair follicle stem cells and epithelial tumor formation in mouse skin. Mol Cell Biol. 2010;30(10):2518–36.
Osorio KM, Lilja KC, Tumbar T. Runx1 modulates adult hair follicle stem cell emergence and maintenance from distinct embryonic skin compartments. J Cell Biol. 2011;193(1):235–50.
Lee J, Hoi CSL, Lilja KC, White BS, Lee SE, Shalloway D, et al. Runx1 and p21 synergistically limit the extent of hair follicle stem cell quiescence in vivo. Proc Natl Acad Sci U S A. 2013;110(12):4634–9.
Lee SE, Sada A, Zhang M, McDermitt DJ, Lu SY, Kemphues KJ, et al. High Runx1 levels promote a reversible, more-differentiated cell state in hair-follicle stem cells during quiescence. Cell Rep. 2014;6(3):499–513.
Li KN, Jain P, He CH, Eun FC, Kang S, Tumbar T. Skin vasculature and hair follicle cross-talking associated with stem cell activation and tissue homeostasis. Elife. 2019. https://doi.org/10.7554/eLife.45977.
Ryan C, Vellody K, Belazarian L, Rork JF. Dermatologic conditions in Down syndrome. Pediatric Dermatol. 2021. https://doi.org/10.1111/pde.14731.
Kranyak A, Rork J, Levy J, Burdick TE. Alopecia areata and thyroid screening in Down syndrome: leveraging epic cosmos dataset. J Am Acad Dermatol. 2023; Available from: http://www.ncbi.nlm.nih.gov/pubmed/36997068.
Tazi-Ahnini R, Cork MJ, Gawkrodger DJ, Birch MP, Wengraf D, McDonagh AJG, et al. Role of the autoimmune regulator (AIRE) gene in alopecia areata: Strong association of a potentially functional AIRE polymorphism with alopecia universalis. Tissue Antigens. 2002. https://doi.org/10.1034/j.1399-0039.2002.600604.x.
Lensing M, Jabbari A. An overview of JAK/STAT pathways and JAK inhibition in alopecia areata. Front Immunol. 2022. https://doi.org/10.3389/fimmu.2022.955035.
Rachubinski AL, Estrada BE, Norris D, Dunnick CA, Boldrick JC, Espinosa JM. Janus kinase inhibition in Down syndrome: 2 cases of therapeutic benefit for alopecia areata. JAAD Case Rep. 2019;5(4):365–7.
Baars MJD, Douma T, Simeonov DR, Myers DR, Kulhanek K, Banerjee S, et al. Dysregulated RASGRP1 expression through RUNX1 mediated transcription promotes autoimmunity. Eur J Immunol. 2021;51(2):471–82.
Hu Y, Pan Q, Zhou K, Ling Y, Wang H, Li Y. RUNX1 inhibits the antiviral immune response against influenza A virus through attenuating type I interferon signaling. Virol J. 2022. https://doi.org/10.1186/s12985-022-01764-8.
Zezulin AU, Ye D, Howell E, Yen D, Bresciani E, Diemer J, et al. RUNX1 is required in granulocyte-monocyte progenitors to attenuate inflammatory cytokine production by neutrophils. Available from: https://doi.org/10.1101/2023.01.27.525911.
Haley KJ, Lasky-Su J, Manoli SE, Smith LA, Shahsafaei A, Weiss ST, Tantisira K. RUNX transcription factors: association with pediatric asthma and modulated by maternal smoking. Am J Physiol-Lung Cell Mol Physiol. 2011;301(5):L693–701. https://doi.org/10.1152/ajplung.00348.2010.
Tang X, Sun L, Jin X, Chen Y, Zhu H, Liang Y, et al. Runt-related transcription factor 1 regulates LPS-induced acute lung injury via NF-kB signaling. Am J Respir Cell Mol Biol. 2017;57(2):174–83.
Liang OD, So EY, Egan PC, Goldberg LR, Aliotta JM, Wu KQ, et al. Endothelial to haematopoietic transition contributes to pulmonary arterial hypertension. Cardiovasc Res. 2017;113(13):1560–73.
Jeong EM, Pereira M, So EY, Wu KQ, Del Tatto M, Wen S, et al. Targeting RUNX1 as a novel treatment modality for pulmonary arterial hypertension. Cardiovasc Res. 2022;118(16):3211–24.
Bush DS, Dunbar Ivy D. Pulmonary hypertension in the population with Down syndrome. Cardiol Therapy. 2022;11(1):33–47. https://doi.org/10.1007/s40119-021-00251-5.
Voon DCC, Thiery JP. The emerging roles of RUNX transcription factors in epithelial-mesenchymal transition. In: Groner Y, Ito Y, Liu P, Neil JC, Speck NA, van Wijnen A, editors. RUNX Proteins in Development and Cancer. Singapore: Springer; 2017. p. 471–89. https://doi.org/10.1007/978-981-10-3233-2_28.
Samarakkody AS, Shin NY, Cantor AB. Role of RUNX family transcription factors in DNA damage response. Mol Cells. 2020;43(2):99–106.
Hou X, Zhang Y, Li W, Hu AJ, Luo C, Zhou W, Hu JK, Daniele SG, Wang J, Sheng J, Fan Y, Greenberg AS, Farmer SR, Hu MG. CDK6 inhibits white to beige fat transition by suppressing RUNX1. Nat Commun. 2018. https://doi.org/10.1038/s41467-018-03451-1.
Lin TC. RUNX1 and cancer. Biochimica et Biophysica Acta (BBA) Rev Cancer. 2022;1877(3):188715. https://doi.org/10.1016/j.bbcan.2022.188715.
Acknowledgements
Not applicable.
Funding
MAA and CDO are funded by an NIH R01 Research Project Grant 1R01HL156475-01 ‘Function of RUNX1 in diverse Down syndrome tissues’. MAA and EJR are funded by the Anna and John J. Sie Foundation/Global Down Syndrome Foundation. CDO is funded by a Crnic Institute Blumenthal Fellowship.
Author information
Authors and Affiliations
Contributions
EJR conceptualized the manuscript, preformed the literature research, and wrote the manuscript with support and supervision of MAA. CDO contributed to the revision and resubmission of the manuscript. EJR and MAA revised and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Rozen, E.J., Ozeroff, C.D. & Allen, M.A. RUN(X) out of blood: emerging RUNX1 functions beyond hematopoiesis and links to Down syndrome. Hum Genomics 17, 83 (2023). https://doi.org/10.1186/s40246-023-00531-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40246-023-00531-2