
Abstract
Background
Germline variants of ten keratin genes (K1, K2, K5, K6A, K6B, K9, K10, K14, K16, and K17) have been reported for causing different types of genodermatoses with an autosomal dominant mode of inheritance. Among all the variants of these ten keratin genes, most of them are missense variants. Unlike pathogenic and likely pathogenic variants, understanding the clinical importance of novel missense variants or variants of uncertain significance (VUS) is the biggest challenge for clinicians or medical geneticists. Functional characterization is the only way to understand the clinical association of novel missense variants or VUS but it is time consuming, costly, and depends on the availability of patient’s samples. Existing databases report the pathogenic variants of the keratin genes, but never emphasize the systematic effects of these variants on keratin protein structure and genotype-phenotype correlation.
Results
To address this need, we developed a comprehensive database KVarPredDB, which contains information of all ten keratin genes associated with genodermatoses. We integrated and curated 400 reported pathogenic missense variants as well as 4629 missense VUS. KVarPredDB predicts the pathogenicity of novel missense variants as well as to understand the severity of disease phenotype, based on four criteria; firstly, the difference in physico-chemical properties between the wild type and substituted amino acids; secondly, the loss of inter/intra-chain interactions; thirdly, evolutionary conservation of the wild type amino acids and lastly, the effect of the substituted amino acids in the heptad repeat. Molecular docking simulations based on resolved crystal structures were adopted to predict stability changes and get the binding energy to compare the wild type protein with the mutated one. We use this basic information to determine the structural and functional impact of novel missense variants on the keratin coiled-coil heterodimer. KVarPredDB was built under the integrative web application development framework SSM (SpringBoot, Spring MVC, MyBatis) and implemented in Java, Bootstrap, React-mutation-mapper, MySQL, Tomcat. The website can be accessed through http://bioinfo.zju.edu.cn/KVarPredDB. The genomic variants and analysis results are freely available under the Creative Commons license.
Conclusions
KVarPredDB provides an intuitive and user-friendly interface with computational analytical investigation for each missense variant of the keratin genes associated with genodermatoses.
Similar content being viewed by others


Background
Human epidermis is the outermost layer of the skin, which plays a protective role in human body. The epidermis is majorly composed of keratinocytes (> 90%) [1]. To achieve the environment-barrier function, it should be intact and undisturbed with an integrated cytoskeletal network [2]. The cytoskeleton majorly consists of three groups of filaments: intermediate filaments (IFs), microfilaments (MFs), and microtubules (MTs) [3]. Based on the requirements for different activities, usage, and developmental stages, the structure of cytoskeleton varies from rigid to flexible. Recent research showed that the mechanical properties of keratinocytes are largely affected by the capability of the IFs [4], especially keratins [1], which are the most abundant structural proteins in keratinocytes [5].
Keratin (K), which is one of the largest gene families with 54 genes, is divided between type I (K9-K40/acidic) and type II (K1-K8, K71-K86/neutral or basic) keratins [6]. Type I and type II keratin usually bind together to form a coiled-coil heterodimer, the unit of the keratin cytoskeleton. Germline variants of ten keratin genes (K1, K2, K5, K6A, K6B, K9, K10, K14, K16, and K17) exert dominant-negative effect in the formation of keratin coiled-coil heterodimer, which finally leads to a large group of clinically and genetically heterogeneous genodermatoses [1, 7, 8]. Keratin genes associated genodermatoses usually manifested with keratinocyte fragility, blistering, and thickening of the palmoplantar epidermis [9]. Location of the missense mutation is also a significant factor for understanding the clinical severity of keratin-related genodermatoses. Keratin missense variants located in the helix initiation motif of 1A domain, termination motif of 2B domain and the L12 linker domain have been reported for causing more severe clinical manifestations. Hundreds of mutations in these ten keratin genes have been reported for causing many types of genodermatoses [10].
Clinical significance of previously reported pathogenic variants as well as likely pathogenic (majorly loss-of-function) variants are very easy to understand because loss-of-function mutations always cause loss of protein product. Meanwhile, understanding the clinical importance of novel missense variants or variants of uncertain significance (VUS) is the biggest challenge due to the difficulty of interpreting whether it is associated with disease or not without doing structural or functional analysis [11]. Note that as recommended in Standards and Guidelines for the Interpretation of Sequence Variants: A Joint Consensus Recommendation of the ACMG [12], the terms “mutation” and “polymorphism” are recommended to be replaced by the term “variant” with the following modifiers: (1) pathogenic, (2) likely pathogenic, (3) uncertain significance, (4) likely benign, or (5) benign. In this paper, we thus practice these updated standards and guidelines for classification of sequence variants.
Next-generation sequencing technologies were performed to identify the candidate gene with their disease-causing variants in patients with genodermatoses. During data analysis and variant interpretation, we usually identified many novel missense variants with low minor allele frequency (MAF), but we were unable to understand their pathogenicity and possible potential for causing genodermatoses. Functional characterization is the only way to understand the clinical association of those novel missense variants or VUS but it is time consuming, costly, and depends on the availability of patient’s samples. At present, none of the database or web servers assessed and predicted the potential pathogenicity of keratin missense variants, i.e., their effect on keratin protein structure and genotype-phenotype correlation.
In addition, target-based therapeutic or precision medicine to cure these genodermatoses is remaining a great challenge [13]. Hence, due to incurability of keratin-related genodermatoses, prenatal diagnosis is the best way to prevent the occurrence of such inherited diseases. A comprehensive database focusing on the human genodermatoses as well as predicting pathogenicity for novel missense variants of uncertain or unknown significance allows geneticists to evaluate the effect of the variants. It finally enables the clinicians for timely and accurate diagnosis, management, and follow-up.
So far, the most comprehensive database for keratins is the Human Intermediate filament Database (Interfil) [14]. It provides general information of intermediate filament encoded genes including mRNA sequence, protein sequence, domain information, and germline mutations associated with disease in a tabular collection format. Unfortunately, no updates have appeared since March 2017. There are also several commonly used databases providing the information of all the reported missense variants of keratin genes, such as dbSNP [15], The Human Gene Mutation Database (HGMD) [16, 17], and LOVD [18]. None of these databases provides any information regarding the effect of missense variants on keratin structure and genotype-phenotype correlation, which is highly important for investigating the genetic effect and the epigenetic interference behind the phenotype. In order to aid users to quest a missense variant of keratin gene to understand the molecular basis of genodermatoses and its genotype-phenotype correlation, we present a more comprehensive and detailed online database for human genodermatoses related keratin genes with an intuitive and user-friendly interface, namely, KVarPredDB.
We visualized manually curated missense sequence variants of all ten keratin genes, diseases, and the analyses of changes in physico-chemical characteristics between wild types and substituted amino acids as well as protein structural effect on the keratin coiled-coil heterodimer complex for each missense variant. The resulting predicted tendencies could be validated by in vivo/vitro experiments and provides guidance to genetic counselors and clinicians for accurate prenatal diagnosis.
Results
Data coverage and statistics
KVarPredDB contains missense variant information of ten keratin genes (K1, K2, K5, K6A, K6B, K9, K10, K14, K9, K16, and K17) reported to be associated with genodermatoses, including 400 reported pathogenic missense variants and 4629 missense VUS. Table 1 showed the number of missense sequence variants in ten keratin genes, respectively.
Except for sorting out 27 diseases and ethnic information of the patient harboring pathogenic missense variants, we also analyze the changes in the physico-chemical characteristics, inter/intra-chain interatomic interaction, evolutionary conservation, and heptad repeat location on the stability and assembly competence of the mutated keratin coiled-coil heterodimer based on our previous studies [19, 20]. We complete the molecular docking simulation of three protein structures, i.e., K1/K10-2B (4ZRY) [5], K5/K14-2B (3TNU) [21], and K1/K10-1B (6EC0) [22] and 332 missense sequence variants on them.
Querying the database
To facilitate the data interpretation, graphical representations with interactive features were developed. Querying the database is primarily based on either “cDNA Variant” or “Disease” under the “Search” menu. From the “cDNA Variant” page, users can quickly access the database by choosing “Gene Symbol,” “Pathogenic Missense Variants,” or “Uncertain Significance Missense Variants” which are implemented by a drop-down list box. Here, the term “Uncertain Significance Missense Variants” refers to missense sequence variants of the VUS, while “Pathogenic Missense Variants” indicates reported missense sequence variants reported to cause genodermatoses. For each missense variant entry, a lollipop plot, gene symbol, protein structural analysis (including changes in physico-chemical properties, inter/intra-chain interaction, evolutionary conservation, location in heptad repeats analyses), patient’s ethnicity/population information, related disease, molecular-docking result, and related references are displayed by default. On the “Disease” page under the “Search” menu, users are asked to select one of 27 types of genodermatoses curated in our database, which leads to the result page showing disease descriptions and missense variations in different keratin genes that cause the corresponding disease by lollipop plot together with related information on the variation in a tabular collection format. If different keratin genes cause the disease, these genes can be switched on the lollipop plot.
Under the “Browse” menu, the user can navigate “Keratin Genes/Proteins,” “Disease List,” and “Protein Structure & Molecular Docking.” “Keratin Genes/Proteins” provides a comprehensive overview of selected keratin genes/proteins in terms of protein description, coding sequence for downloading, missense variations distributed on the linearized keratin protein and its domains by lollipop plot, and lists of pathogenic missense variants and VUS. “Disease List” leads to collection of 27 types of genodermatoses, with respective OMIM ID, which is clickable and will take users to the corresponding disease page. A three-dimensional presentation of the determined crystal structure (1B and 2B domains of K1/K10, and 2B domain of K5/K14 heterodimers) is displayed on the “Protein structure & Molecular Docking” page, powered by NGL Viewer for 3D structure viewing. Lower panel on the same page, the “Molecular Docking Results” section shows all keratin gene missense sequence variants’ binding energy of wild type and the lowest binding energy score of mutant models.
The “API” menu contains all the application-programming interfaces. The application API provides a way to programmatically access our data including molecular docking information for each variant, related missense variants for each disease, and so on. Users, especially researchers, can provide novel keratin missense sequence variants to us by the “Submit” menu. We will verify and update the missense sequence variant information.
Instructions on how to query KVarPredDB are provided on the “Tutorials” page linked from the home page. At the same time, the “About” menu also contains KVarPredDB introduction information. The KVarPredDB runs in most common web browsers. Every element like number, image, or plot in the database is interactive with clickable buttons, and is thus far more user friendly and provides more easily accessible information.
Discussion
KVarPredDB mainly works on integration and the prediction of pathogenicity of germline missense sequence variants of ten keratin genes associated with genodermatoses. As we noted above, keratin is one of the largest multi-gene families encoding structural proteins, with extremely high tissue-specific expression. Keratin-related genodermatoses are rare, incurable, and with an autosomal dominant mode of inheritance. Existing databases have reported the pathogenic missense variants of these genes, but have never emphasize on the systematic effect of these missense mutations on keratin protein structure and genotype-phenotype correlation. This hampers the investigation of the genetic effect and the epigenetic interference behind the phenotype.
Within this database, we visualized manually curated keratin gene missense sequence variants, diseases, patients’ ethnicity, and the analyses of changes in physico-chemical characteristics between the wild and mutant amino acids as well as protein structural effects on the keratin coiled-coil heterodimer for each missense variant. KVarPredDB also provides computational analysis results for users to assess the potential pathogenicity of missense VUS, based on a more comprehensive systematic analyses than common pathogenicity prediction tools such as SIFT [23], Polyphen-2 [24], DUET [25], and MutationAssessor [26, 27]. The resulting pathogenic tendencies could be further validated by in vivo/vitro experiments and provide guidance to genetic counselors and clinicians for easy and accurate prenatal diagnosis. These in silico tools aid in the interpretation of missense sequence variants. The algorithms used by each tool may differ from each other but could be divided into two main categories: one predicts whether the variant is damaging to the resultant protein function or structure, and the other predicts the effect on splicing at the nucleotide level. None of these tools is sufficient to reveal the possible mechanism behind the phenotype from both physic-chemical and structural points of view. Searching in our database, every user can easily get the information related to pathogenicity of missense sequence variants of keratin genes. It will give keratin-related-disease families a chance for getting a healthy baby, and provide new ideas for genetic counseling. It will be a valuable way for minimizing the risk of occurrence of keratin-related genodermatoses.
At present, only three partial crystal structures of keratin proteins have been published. We will continue to finish the work with K1/K10-1B domain (6EC0) [22]. With the aids of crystallization and cyto-electron microscopy (Cryo-EM) techniques, a high resolution of the class of intermediate filament proteins will allow us to perform more accurate predictions. For the protein domains without crystal structure, analysis will be conducted via homology modeling owing to their high structural similarities. Next, we will continue to update the missense sequence variants and related information to ensure the timeliness of the database.
Conclusions
KVarPredDB provides the computational analytical investigation for each missense variant of the keratin genes, which could be further validated by in vivo/vitro experiments or provide guidance to genetic counselors and clinicians for easy and accurate prenatal diagnosis. Meanwhile, we will investigate the protein structural and assembly characteristics of more types of coding variations to further improve our analysis strategies for the pathogenicity prediction.
Methods
Data acquisition
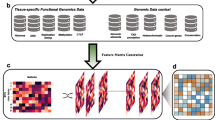
KVarPredDB contains information of ten keratin genes reported to be related to genodermatoses. All this information was integrated and manually curated from the Interfil, NCBI-dbSNP, NCBI-PubMed (before 30 October 2020) (Fig. 1). In particular, pathogenic missense variants were extracted and integrated from the Interfil. Related references in the NCBI-PubMed were searched with the keywords “mutation” and “keratin” dated from March 2017. Meanwhile, VUS were mainly extracted and curated from NCBI-dbSNP. All this variant information is presented in two ways that are easily accessible (i.e., text and lollipop diagram to the users [28]).

Schematic presentation of data integration and curation of KVarPredDB
We also integrated and enlisted all types of keratin gene associated diseases with a hyperlink to the Online Mendelian Inheritance in Man (OMIM) and classified these pathogenic missense variants according to different disease types. KVarPredDB displayed the complete keratin information in the database, including gene and protein information, such as keratin-coding sequence obtained from Ensembl and Uniprot. Also, KVarPredDB had all the ethnic or population information of the patient identified with the reported pathogenic missense variants.
Due to the particularly long fibrous structure of keratin, only part of the crystal structures was available for protein structure analyses, i.e., K1/K10-2B domain (4ZRY) [5], K5/K14-2B domain (3TNU) [21], and K1/K10-1B domain (6EC0) [22], which was retrieved from RCSB Protein Data Bank.
Pathogenicity prediction
Analyses of pathogenicity for each missense variant were performed according to two parts, i.e., our previous computational studies [19, 20] and molecular docking methods based on resolved crystal structures.
KVarPredDB is providing the detailed information including changes in physico-chemical characteristics, inter/intra-chain interaction, evolutionary conservation, and heptad repeat location to understand the stability and assembly competence of the keratin coiled-coil heterodimer upon missense variants (Fig. 2). We can use this basic information to determine the structural and functional impact of variants on the keratin coiled-coil heterodimer.

Example outputs of KVarPredDB. a A lollipop diagram is displayed to show the distribution of all the missense sequence variations on a linearized K10 protein and its domains. Y-axis represents K10 sequence variant frequency. b Structural analyses in terms of physico-chemical properties, inter/intra-chain interactions, evolutionary conservation, and location in heptad repeats. c Visualization of ddg score generated by molecular docking simulation (box plot) and structural alignment (GIF format) between the wild type and mutant keratin coiled-coil heterodimer. d Screenshot of K5/K14 heterodimer coiled-coil complex structure (3TNU) in the “NGL Viewer” GUI
Besides, molecular docking simulations were adopted to predict stability changes and get the binding energy to compare the wild type protein with the mutated one. The structure was first relaxed using the Rosetta relax application [29]. We used the relax application in order to find the most energetically favorable conformation of the protein. The Monte Carlo (MC) algorithm generates conformational structure changes; the energy of the new conformation is calculated and compared with the energy before change. If the energy is better, the change is accepted. We run 50 repeats of the relax application and choose the best one as input. We took the relaxed rada recombinase as the wild type and then used Rosetta backrub application to generate a structural model of the mutant. Rosetta Backrub application attempts to capture the tiny conformational changes with the protein. The protein backrub is first divided into multiple fragments; each of the fragments rotates around the connecting axis while immobilizing the rest of the protein. Rotational movements have six internal backbone degrees of freedom: φ, ψ, and N-Cα-C bond angle at each pivot. The side chain repackaging and energy minimization follow along with all torsion angles. We repeated this process for 1000 trials and selected the lowest energy structure. The resulting conformations were scored with the Rosetta scoring function and accepted (or rejected) according to the Metropolis criterion using a kT of 0.6. For each missense variant, at least 50 models were generated. To predict the change in stability of rada recombinase mutant protein induces by a missense variant, models are further screened with the ddG method in Rosetta Script, chain number is 2. DdG refers to binding energy and gives the differences in Rosetta energy between the wild and mutant protein structure. Rosetta energy function Talaris2014 was used. Finally, we visualize the difference between wild and mutant protein structure with box plot chart and structural alignment.
Database construction
A user-friendly web interface was developed with Java 1.8. All data was implemented with MySQL (version 5.6). The tables included diseases, pathogenic/uncertain significance missense variants, references, proteins, amino acid physico-chemical properties, and molecular mocking results. The back end used a three-tier model: customer display layer, business logic layer, and data layer, which has good flexibility, scalability, and shareability. It was built under SpringBoot (version 2.1.6), Spring MVC, and Mybatis (version 2.0.1) framework. The front end was implemented using Bootstrap (version 3.3.7) and jQuery. The pages used Ajax, which refers to a web development technology for creating interactive, fast and dynamic web applications. It can update web pages without reloading the entire web page.
A lollipop plot (also known as stick or needle plots) generated by the MutationMapper visualization tool [28, 30] displays the distribution of all missense variants of a linearized keratin protein and its domains (genome build GRCh37/hg19). An embedded module from RCSC supported by NGL viewer (ngl.js) was employed to display molecular graphics [31]. Embed Tomcat was used for the server. The website design was responsive. The database is routinely updated and, as such, the quantity of analysis and accuracy will continue to increase as more case reports and determined crystal structures are added.
Accession numbers
Atomic coordinates and structure factors for the reported crystal structures are retrieved from the Protein Data bank under accession number 3TNU, 4ZRY, and 6EC0.
Searching for keratin gene variants with NCBI-dbSNP under accession number 3848, 3849, 3852, 3853, 3854, 3857, 3858, 3861, 3868, and 3872.
Availability of data and materials
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
Lulevich V, Yang HY, Isseroff RR, Liu GY. Single cell mechanics of keratinocyte cells. Ultramicroscopy. 2010;110(12):1435–42.
Simpson CL, Patel DM, Green KJ. Deconstructing the skin: cytoarchitectural determinants of epidermal morphogenesis. Nat Rev Mol Cell Biol. 2011;12(9):565–80.
Omary MB. “IF-pathies”: a broad spectrum of intermediate filament-associated diseases. J Clin Invest. 2009;119(7):1756–62.
Robert A, Hookway C, Gelfand VI. Intermediate filament dynamics: what we can see now and why it matters. Bioessays. 2016;38(3):232–43.
Bunick CG, Milstone LM. The X-ray crystal structure of the keratin 1-keratin 10 helix 2B heterodimer reveals molecular surface properties and biochemical insights into human skin disease. J Invest Dermatol. 2017;137(1):142–50.
Toivola DM, Boor P, Alam C, Strnad P. Keratins in health and disease. Curr Opin Cell Biol. 2015;32:73–81.
Knöbel M, O’Toole EA, Smith FJ. Keratins and skin disease. Cell Tissue Res. 2015;360(3):583–9.
Elango T, Sun J, Zhu C, Zhou F, Zhang Y, Sun L, Yang S, Zhang X. Mutational analysis of epidermal and hyperproliferative type I keratins in mild and moderate psoriasis vulgaris patients: a possible role in the pathogenesis of psoriasis along with disease severity. Hum Genomics. 2018;12(1):27.
Itin PH, Fistarol SK. Palmoplantar keratodermas. Clin Dermatol. 2005;23(1):15–22.
Bowden PE. Mutations in a keratin 6 isomer (K6c) cause a type of focal palmoplantar keratoderma. J Invest Dermatol. 2010;130(2):336–8.
Atkinson SD, McGilligan VE, Liao HH, Szeverenyi I, Smith FJD, Moore CBT, McLean WHI. Development of allele-specific therapeutic siRNA for keratin 5 mutations in epidermolysis bullosa simplex. J Invest Dermatol. 2011;131(10):2079–86.
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405–24.
Chiu FP, Doolan BJ, McGrath JA, Onoufriadis A. A decade of next-generation sequencing in genodermatoses: the impact on gene discovery and clinical diagnostics. Br J Dermatol. 2020. https://doi.org/10.1111/bjd.19384.
Szeverenyi I, Cassidy AJ, Chung CW, Lee BT, Common JEA, Ogg SC, Chen H, Sim SY, Goh WLR, Ng KW, et al. The human intermediate filament database: comprehensive information on a gene family involved in many human diseases. Human Mutat. 2008;29(3):351–60.
Sherry ST, Ward MH, Kholodov M, Baker J, Phan L, Smigielski EM, Sirotkin K. dbSNP: the NCBI database of genetic variation. Nucleic Acids Res. 2001;29(1):308–11.
Stenson PD, Mort M, Ball EV, Evans K, Hayden M, Heywood S, Hussain M, Phillips AD, Cooper DN. The Human Gene Mutation Database: towards a comprehensive repository of inherited mutation data for medical research, genetic diagnosis and next-generation sequencing studies. Human Genet. 2017;136(6):665–77.
Stenson PD, Ball EV, Howells K, Phillips AD, Mort M, Cooper DN. The Human Gene Mutation Database: providing a comprehensive central mutation database for molecular diagnostics and personalized genomics. Hum Genomics. 2009;4(2):69–72.
Fokkema IFAC, Taschner PEM, Schaafsma GCP, Celli J, Laros JFJ, den Dunnen JT. LOVD v.2.0: the next generation in gene variant databases. Human Mutat. 2011;32(5):557–63.
Banerjee S, Wu Q, Yu P, Qi M, Li C. In silico analysis of all point mutations on the 2B domain of K5/K14 causing epidermolysis bullosa simplex: a genotype-phenotype correlation. Mol Biosystems. 2014;10(10):2567–77.
Banerjee S, Wu Q, Ying YY, Li YN, Shirota M, Neculai D, Li C. In silico predicted structural and functional insights of all missense mutations on 2B domain of K1/K10 causing genodermatoses. Oncotarget. 2016;7(33):52766–80.
Lee CH, Kim MS, Chung BM, Leahy DJ, Coulombe PA. Structural basis for heteromeric assembly and perinuclear organization of keratin filaments. Nat Struct Mol Biol. 2012;19(7):707–15.
Eldirany SA, Ho M, Hinbest AJ, Lomakin IB, Bunick CG. Human keratin 1/10-1B tetramer structures reveal a knob-pocket mechanism in intermediate filament assembly. Embo j. 2019;38(11):e100741.
Ng PC, Henikoff S. SIFT: predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003;31(13):3812–4.
Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS, Sunyaev SR. A method and server for predicting damaging missense mutations. Nature Methods. 2010;7(4):248–9.
Pires DEV, Ascher DB, Blundell TL. DUET: a server for predicting effects of mutations on protein stability using an integrated computational approach. Nucleic Acids Res. 2014;42(W1):W314–9.
Reva B, Antipin Y, Sander C. Determinants of protein function revealed by combinatorial entropy optimization. Genome Biol. 2007;8(11):R232.
Reva B, Antipin Y, Sander C. Predicting the functional impact of protein mutations: application to cancer genomics. Nucleic Acids Res. 2011;39(17):e118.
Gao JJ, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Sun YC, Jacobsen A, Sinha R, Larsson E, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Science Signaling. 2013;6(269):pl1.
Conway P, Tyka MD, DiMaio F, Konerding DE, Baker D. Relaxation of backbone bond geometry improves protein energy landscape modeling. Protein Sci. 2014;23(1):47–55.
Cheng FX, Liu C, Jiang J, Lu WQ, Li WH, Liu GX, Zhou WX, Huang J, Tang Y. Prediction of drug-target interactions and drug repositioning via network-based inference. Plos Comput Biol. 2012;8(5):e1002503.
Rose AS, Bradley AR, Valasatava Y, Duarte JM, Prlic A, Rose PW. NGL viewer: web-based molecular graphics for large complexes. Bioinformatics. 2018;34(21):3755–8.
Acknowledgements
We thank Dr. Xiaokang Zhang in the Center of Cryo-Electron Microscopy (CCEM), Zhejiang University for his technical assistance on computer clustering. And we are deeply thankful for the computational resources provided by the Super Computer System, Human Genome Center, Institute of Medical Science, University of Tokyo, Japan.
Funding
This work has been supported by the grant from Zhejiang Provincial Natural Science Foundation of China (LY17C060003), Chinese National Natural Science Foundation (81601515), and Zhejiang Provincial Key Projects of Technology Research (WKJ-ZJ-2033).
Author information
Authors and Affiliations
Contributions
Chen Li and Jiale Qin designed the study experiments. Yuyi Ying, Lu Lu, and Santasree Banerjee collected the data and conducted the database construction. Lizhen Xu, Qiang Zhao, and Hao Wu performed data analysis. Chen Li, Yuyi Ying, Lu Lu, and Santasree Banerjee wrote the article. Dante Neculai, Yongmei Xi, Fan Yang, Jiale Qin, and Chen Li supervised the study. All authors revised and approved the article.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
N/A.
Competing interests
The authors declare no conflict of interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Ying, Y., Lu, L., Banerjee, S. et al. KVarPredDB: a database for predicting pathogenicity of missense sequence variants of keratin genes associated with genodermatoses. Hum Genomics 14, 45 (2020). https://doi.org/10.1186/s40246-020-00295-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40246-020-00295-z