
Abstract
Background
Mitochondrial diseases (MDs) are a group of clinically and genetically heterogeneous disorders characterized by defects in oxidative phosphorylation. Since clinical phenotypes of MDs may be non-specific, genetic diagnosis is crucial for guiding disease management. In the current study, whole-exome sequencing (WES) was performed for our paediatric-onset MD cohort of a Southern Chinese origin, with the aim of identifying key disease-causing variants in the Chinese patients with MDs.
Methods
We recruited Chinese patients who had paediatric-onset MDs and a minimum mitochondrial disease criteria (MDC) score of 3. Patients with positive target gene or mitochondrial DNA sequencing results were excluded. WES was performed, variants with population frequency ≤ 1% were analysed for pathogenicity on the basis of the American College of Medical Genetics and Genomics guidelines.
Results
Sixty-six patients with pre-biopsy MDC scores of 3–8 were recruited. The overall diagnostic yield was 35% (23/66). Eleven patients (17%) were found to have mutations in MD-related genes, with COQ4 having the highest mutation rate owing to the Chinese-specific founder mutation (4/66, 6%). Twelve patients (12/66, 18%) had mutations in non-MD-related genes: ATP1A3 (n = 3, two were siblings), ALDH5A1, ARX, FA2H, KCNT1, LDHD, NEFL, NKX2-2, TBCK, and WAC.
Conclusions
We confirmed that the COQ4:c.370G>A, p.(Gly124Ser) variant, was a founder mutation among the Southern Chinese population. Screening for this mutation should therefore be considered while diagnosing Chinese patients suspected to have MDs. Furthermore, WES has proven to be useful in detecting variants in patients suspected to have MDs because it helps to obtain an unbiased and precise genetic diagnosis for these diseases, which are genetically heterogeneous.
Similar content being viewed by others

Background
Mitochondrial diseases (MDs) are a common group of inborn errors of metabolism (IEMs) that are caused by defects in oxidative phosphorylation and have an estimated birth prevalence of 1:5000 [1] and premature mortality rate of 14% [2]. Inherited and de novo pathogenic variants have been reported in 37 mitochondrial genes (i.e. in mtDNA) and 295 nuclear genes (i.e. in nDNA) [3]. The clinical manifestations of MDs are highly variable and indicate organ-specific or multi-systemic involvement [4]. MDs are difficult to diagnose because of their clinical and genetic heterogeneity. Involvement of novel genes, mitochondrial mimicry, and tissue specificity of mtDNA heteroplasmy further complicate the diagnosis [5].
A consensus statement on the diagnosis and management of MDs was published in 2015. As per this statement, patients suspected to have MDs should undergo clinical evaluation along with biochemical testing of the blood, urine, and cerebrospinal fluid. For investigating mtDNA mutations, deletion, duplication, and depletion, next-generation sequencing (NGS)-based methods were suggested. Furthermore, it stated that muscle or liver samples should also be assessed in cases where negative results were obtained with blood samples. The statement provides limited information on the genetic testing of nDNA. Gene panel-based NGS was listed as the preferred method for nDNA testing. If the NGS results are negative, whole-exome sequencing (WES) should be considered. Tissue biopsies and subsequent electron transport chain (ETC) enzymology should be performed only when DNA testing cannot provide a diagnosis [6].
Despite the recommendations provided in the consensus statement, multiple subsequent studies used WES or NGS for diagnosing MDs. The cohort of Wortmann et al. [7] included 109 patients up to 27 years of age who had no pathogenic variants detected by mtDNA investigations. Data were first filtered using a virtual MD gene panel comprising 238 genes. If the results were negative, WES was performed. The overall diagnostic yield was 38% (42/109), with an equal number of diagnoses being obtained for cases involving MD-related and non-MD-associated genes (19%, 21/109). Pronicka et al. [8] used WES for a cohort of 113 paediatric patients who had MDs and in whom pathogenic variants were not found. The diagnostic rate was 59% (67/113), with 17% of the pathogenic nDNA variants (19/113) being identified in non-MD-associated genes. Kohda et al. [9] and Theunissen et al. [10] both used NGS-based mtDNA sequencing followed by WES. In the cohort of Kohda et al. [9] of 142 patients with childhood-onset mitochondrial respiratory chain complex deficiencies, the WES diagnostic yield was 25% (35/142). WES helped diagnose 49% (57/117) of the patients in the study by Theunissen et al. [10], in which 86 and 31 patients were suspected to have MDs and neuromuscular diseases, respectively.
To summarize, six studies during 2014–2019 suggested using WES and/or NGS-based mtDNA sequencing over panel-based diagnostic methods for patients suspected to have MDs, on the basis of the high diagnostic yields (35–66%) of the former approach [5, 7,8,9,10,11]. The diagnostic yield will be higher if there is a high proportion of familial and consanguineous cases [11], if the patients have decreased activities of multiple respiratory chain complexes [8], in neonatal-onset cases [7], and in cases with severe presentation [12]. This was further supported by the findings of Pronicka et al. [8] wherein the percentage of positive WES results increased with increase in the mitochondrial disease criteria (MDC) score of patients.
In the case of MD genes, there was very little overlap across five studies during 2014–2018. Only 11 known MD nuclear genes, i.e. SCO2, MTFMT, TAZ, NDUFS7, RRMB2B, MTO1, EARS2, RARS2, C12orf65, PC, and FBXL4, were identified in three or more studies [7,8,9,10,11]. WES-positive findings were obtained in a high percentage of cases involving non-MD genes. Up to 19% of the cohort in a study by Wortmann et al. [7] was found to have mutations in non-MD genes. These findings show that gene panels will be inadequate for identifying variants in novel genes or non-MD genes.
Founder mutations have been identified in certain ethnic populations. Recurrent rare variants found in FBLX4, ACAD9, and CLPB in Polish patients with MDs suggested the ethnic specificity of these variants [8].
Therefore, in the current study, using WES, we aimed to identify the disease-causing mutations in patients who were of a Chinese descent and had paediatric-onset MDs. The findings provide improved understanding of variants and their prevalence in the Chinese population, which may have been underrepresented in previous studies.
Results
Clinical presentation
We recruited 66 patients, including 42 male and 24 female individuals (male to female ratio, 1.75:1). The age of onset ranged from the neonatal period to 9 years, with the median age of onset being 1.75 years of age. Nineteen patients presented at the neonatal stage. The patients recruited were from 63 non-consanguineous Chinese families, with a positive family history being found in five cases (siblings = 3 cases, siblings and mother = 1 case, paternal cousin = 1 case). There were 18 and 48 cases of syndromic and non-syndromic MD, respectively. The most frequently identified clinical feature was global developmental delay (GDD; 50%; 33/66), followed by epilepsy/seizure (44%; 29/66) and dystonia (40%, 26/66). Myopathy was noted in 27% of the cohort (18/66), and 23% and 18% of the patients had visual issues (15/66) and oromotor dysfunction (12/66), respectively. Cardiopulmonary, auditory, renal, and hepatic issues were less common. Neuroimaging was performed for 65 patients; of these patients, 50 had abnormal neuroimaging features. Among all the anomalies, features including signal changes in the basal ganglia (18%; 12/66), cerebral or cerebellar atrophy (15%; 10/66), and optic atrophy (2%; 1/66) were noteworthy. Furthermore, 36% of the cohort (24/66) had lactate acidosis. For clinical evaluation, Mitochondrial Disease Criteria (MDC) were used to guide and standardize the diagnosis for paediatric-onset MD. This score is based on various clinical criteria including muscular presentations, central nervous system (CNS) abnormalities, multisystemic involvement, metabolic and neuroimaging studies, and histological morphology. Each case can be classified into unlikely, possible, probable, or definite MD subgroups [13]. On the basis of the pre-biopsy MDC score, 14% (9/66) of the patients were classified into the possible, 38% (25/66) into the probable, and 48% (32/66) into the definite MD subgroups. Mitochondrial enzyme analysis was performed for 45 patients, with muscle (n = 40), skin (n = 39), and liver (n = 1) being used as tissue sources. The most common deficiency was a complex IV deficiency (13%; 6/45; five from muscle and one from muscle and skin), followed by complex II + III (11%; 5/45; all from skin), complex I + IV (7%; 3/45; all from muscle), and complex I (4%; 2/45; one from muscle and one from skin) deficiencies. Muscle biopsy was performed for 40 patients (61%); of these, seven showed an increase in MDC score after the biopsy sample analysis, but only one patient was re-classified from the probable to the definite MD subgroup.
Molecular findings
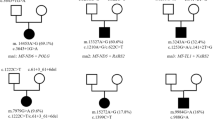
WES was performed for 55 patients as singletons, four as duos (three pairs of affected siblings and an affected mother-child pair), and seven as trios. Segregation analysis was performed after WES for 16 singletons and two siblings.
Disease-causing variants were detected in 10 patients by using singleton WES analysis. Segregation analysis helped classify the variants in 12 cases as pathogenic or likely pathogenic. The reanalysis performed in 2019 helped diagnose one more case in our cohort, leading to an overall diagnostic yield of 35% (23/66). Muscle biopsy was not required in any of the cases to confirm the pathogenicity of the variant. In one case, the pathogenic variant in the OPA1 gene did not fully explain the phenotype because the patient did not have an optic atrophy by that time and he had more severe manifestations beyond optic problems, with generalized dystonia and bilateral basal ganglion lesions being noted in neuroimaging studies (patient 11). The WES findings for two patients (patient 10 and patient 6) showed disease-causing variants in OPA1 and SURF1, respectively, and these findings were correlated with their corresponding syndromic presentations of autosomal dominant optic atrophy (ADOA) and Leigh syndrome. The highest diagnostic yield was obtained in the possible MD subgroup (44%; 4/9), followed by the definite (34%; 11/32) and probable MD (32%; 8/25) subgroups. Patients with either normal muscle biopsy or normal neuroimaging results had a higher diagnostic yield (33% [11/33] and 40% [6/15], respectively) than patients with abnormal findings (Fig. 1). Pathogenic variants were identified in 37% (7/19) of the neonatal cases.

Diagnostic yield according to the MDC subgroup, muscle biopsy findings, and neuroimaging findings
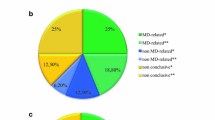
In our first-tier analysis using MitoCarta, 11 patients were found to have mutations in genes included in the panel. In our second-tier open-exome analysis, 12 more patients were diagnosed. Eleven patients (17%) were found to carry disease-causing mutations in six MD-related genes, i.e. COQ4, COQ7, NDUFA9, OPA1, SURF1, and TAZ. Twelve patients (18%) had mutations in 10 non-MD-associated genes, i.e. ATP1A3, ALDH5A1, ARX, FA2H, KCNT1, LDHD, NEFL, NKX2-2, TBCK, and WAC. From all the genes identified (MD and non-MD), five showed autosomal dominant inheritance, nine showed autosomal recessive inheritance, and two showed X-linked inheritance. The recurrent disease-causing genes were COQ4 (n = 4), OPA1 (n = 2), and ATP1A3 (n = 3, two from siblings). The findings are summarized in Tables 1 and 2.
The gene showing the highest number of recurrent mutations was COQ4 (n = 4), with the c.370G>A, p.(Gly124Ser) variant, being found in three patients. Patients with the COQ4 mutation can present with symptoms at either neonatal or infantile stages. Clinical presentations including GDD, seizures, generalized dystonia, bilateral cortical visual impairment, oromotor dysfunction, hearing problems, and lactate acidosis were noted in our study. Basal ganglion changes and cerebral atrophy were observed on analysing the magnetic resonance imaging (MRI) findings of patients with COQ4 mutations.
Discussion
In this study, WES was performed for 66 Chinese patients with paediatric-onset MDs. Despite the lack of a local diagnostic facility for a respiratory chain enzyme analysis, an overall diagnostic yield of 35% (23/66) was achieved, which was comparable to that of similar studies in recent years [7,8,9,10,11]. Among the cases identified by WES, 48% (11/23) had mutations in MD genes, while the remaining (12/23) had mutations in non-MD genes.
COQ4 mutation was the most important cause of MDs caused by nDNA mutations in the Southern Chinese patients
Eleven patients (17%) were found to have mutations in MD-related genes, with COQ4 having the highest mutation rate (4/66, 6%). The COQ4 missense variants were classified as VUSs at the initial stage of this study. The pathogenicity was supported by the segregation study and functional analysis of the skin fibroblasts, which showed complex II + III deficiency and low CoQ concentration. COQ4 is associated with primary coenzyme Q10 deficiency type 7 [14]. Subsequent to the first Chinese study published in 2019 by Lu et al. [15], which described a pair of siblings with Leigh syndrome caused by homozygous COQ4 c.370G>A, p.(Gly124Ser), our team published the largest COQ4 cases series with 11 Chinese patients from nine families (including patients 1 to 4 in this study) in collaboration with a Taiwanese group [16]. Our collaborative study proved that CoQ10 deficiency could occur in neonates, infants, or children with variable phenotypes, which altered the original notion that CoQ10 deficiency shows only neonatal onset [14, 17]. Furthermore, we found that 10 of the 11 patients carried the c.370G>A, p.(Gly124Ser) mutation. Using a single nucleotide polymorphism (SNP) array, we have previously shown that this mutation is a Chinese-specific founder mutation. In the same year of 2019, Ling et al. [18] also described three unrelated Chinese patients with this founder mutation. Currently, 16 Chinese COQ4 cases have been reported, with 15 patients in 12 non-consanguineous Chinese families carrying the c.370G>A Chinese founder mutation.
Performing multiple studies with patients from the same ethnic background can aid in effectively identifying recurrent founder mutations. In a study by Piekutowska-Abramczuk et al. [19], the c.845_846delCT variant was found in 77.6% of the SURF1 alleles in the cohort of Polish patients; however, it was found in a much lower percentage (9%) in the non-Polish population. In 2013, Pronicka et al. [20] studied a cohort with a similar ethnic background; a homozygous c.1541G>A variant in SCO2 (a known MD gene) was identified in an extremely high proportion of the cohort (35/36). In 2016, the detection of recurrent rare pathogenic variants of FBXL4, ACAD9, and CLPB further extended the scope of the suspected Polish-specific MD mutations. The ethnic specificity (Polish) observed in the MD-causing genes [8] parallels the situation of this study in which the COQ4 variant is strongly enriched in cohorts of Chinese patients. A recent study on IEMs have also identified other founder mutations, e.g. GCDH and SLC25A20, in the Southern Chinese population [21]. It is postulated that, when more cases are identified in the Chinese population, the frequency of the c.370G>A, p.(Gly124Ser) variant, or other MD-causing mutations will also subsequently increase. Therefore, we suggest that more studies should be performed in Chinese populations to identify any ethnic-specific variants in MD genes.
Benefits of using WES for first-tier genetic testing
According to the 2015 consensus statement [6], patients should undergo WES only if positive results are not obtained with targeted gene sequencing, gene panel analysis, and mtDNA sequencing. However, in our study, as well as in several previous studies [7,8,9,10,11], WES was chosen as the diagnostic method instead of gene panel-based NGS because of the sensitivity of WES. On comparing genes identified in our first-tier results with genes on MitoCarta, we found that 56% (9/16; one MD and eight non-MD genes) of the genes identified in our study were not included in the MitoCarta gene panel. Furthermore, 44% (7/16) of the genes would have been missed if the mitochondrial disorders panel of Genomics England PanelApp (gene panel curated by UK Genetic Testing Network–Association for Clinical Genome Science) [22] would have been used (seven non-MD genes). If a panel-based NGS method had been adopted instead of WES, 52% (12/23) of the patients found to be positive by WES would not have been identified in our study. This statistic is backed by four studies that were conducted after 2015 and used WES [7,8,9,10], wherein 24–49% of the genes identified would have been missed if gene panel-based NGS based on MitoCarta had been used. The false-negative rates of MitoCarta panel-based NGS range from 16 to 45%.
We compared our study with five other studies that were performed during 2014–2018 and used WES for MD diagnosis [7,8,9,10,11]. A total of 92 MD genes were identified in those five studies; however, only 31 (34%) of these genes were reported more than once. Among those 31 genes, only five were also reported in our study indicating that there was minimal overlap (Table 3). More importantly, all studies identified the disease-causing mutations in non-MD genes in patients with suspected MDs. In our study, 12 patients (18%) had non-MD gene mutations. Non-MD gene involvement has also been significant in previous studies [7,8,9,10,11], ranging from 2 to 19%, but only MECP2 was reported in more than one of the five studies published during 2014–2018 (Table 4). Non-MD genes would not have been detected if only gene panel-based NGS, e.g. MitoCarta, had been used. We therefore recommend WES as a first-tier genetic test for patients with MDs because of the high genetic heterogeneity noted in such cases.
Although analysis of the respiratory chain enzyme activity may provide an effective approach for screening out potential patients with MDs, this test is currently unavailable in Hong Kong. Furthermore, the protocol is not standardized, which may lead to discrepancies because of variation in the assay conditions and control values used by different laboratories [24]. The use of WES for first-tier analysis can help avoid unnecessary invasive biopsies. In our study, 20% of such procedures could have been avoided in one MD and eight non-MD cases because molecular diagnosis was directly established through WES. Furthermore, secondary complex IV deficiency was detected in two non-MD patients with ARX and LDHD mutations. These findings indicate that WES is a more comprehensive diagnostic approach than the use of enzymology alone to diagnose MDs. On the other hand, the traditional way of initially investigating a patient suspected of a MD biochemically followed by WES might reveal enzymatic OXPHOS defects in non-MD genes previously linked to well-defined syndromes but not brought into association with mitochondrial failure before. Understanding such pathophysiology with functional studies would hopefully close this knowledge gap. The lack of clear phenotype–genotype relationships and the fact that ongoing studies constantly identify novel mutations make it inefficient to determine what additional gene panel(s) should be used in conjunction with an MD gene panel. This is because non-MD-associated genes could be involved. The use of WES can help fill in existing knowledge gaps and prevent delay in achieving an accurate genetic diagnosis.
With the improvements achieved in bioinformatics analysis, mitochondrial DNA variants can be identified using the off-target read from WES with high recall rate and precision [5]. Overall, the findings indicate that WES should be used for first-tier genetic testing in our local setting. We also propose that WES results should be incorporated in the diagnostic criteria of MDs.
Limitations of our study and future directions
There are certain shortfalls of using WES. Pathogenic variants located in the non-coding region could not be detected. Single/multiple deletions, or depletion of mtDNA, and low heteroplasmy variants could have been missed [5]. With technological improvements, it is expected that whole-genome sequencing (WGS) will also be used in the future, when reductions in cost and increased accuracy of variant interpretation are achieved [3].
Conclusions
To our knowledge, this is the largest WES study on Chinese paediatric-onset MDs. Primary coenzyme Q10 deficiency type 7 was found to be the most common MD caused by nDNA mutations in our cohort, owing to the Chinese founder mutation, c.370G>A, p.(Gly124Ser). Clinicians should actively consider the possibility of COQ4 mutation and provide biochemical and genetic workups for Chinese patients suspected to have MDs. Owing to the highly variable phenotypes and genes involved in paediatric-onset MDs, we recommend the use of WES as a first-tier test to maximize the diagnostic yield. The molecular diagnoses obtained using WES will help to guide diagnosis and management and should be incorporated into future diagnostic criteria of MDs; this is especially relevant in geographical regions where enzyme testing is not available.
Methods
Patient recruitment
A total of 66 patients under clinical management at the Queen Mary Hospital and the Duchess of Kent Children’s Hospital, which are affiliated with the University of Hong Kong (HKU), were recruited during 2011–2018. The inclusion criteria were as follows: (1) suspicion of MDs by the study authors (F.C.W. and C.H.Y.B.), along with a minimum pre-biopsy MDC score of 3 (MD subgroups: possible, score of 3–4; probable, score of 5–7; or definite, score of 8–12) (13); (2) absence of large-scale mtDNA deletions and recurrent point mutations in the mitochondrial genome; and (3) normal chromosome microarray results.
The cohort is representative of the heterogeneous patient population noted during genetic and neurometabolic consultations provided by the authors as part of the Hong Kong health care system, which ranged from neonatal intensive care unit (NICU) cases to out-patient cases.
Clinical evaluation
Clinical evaluation was performed as per the recommendations of our paediatric neurologist (F.C.W.) and clinical geneticist (C.H.Y.B.). Most of the clinical, metabolic, histological, electrophysiological, and radiological analyses were performed locally. The only exception was the analysis of oxidative phosphorylation enzyme activities (OXPHOS) in freshly obtained muscle and fibroblast samples from our cohort, which was performed at the Radboud Center for Mitochondrial Medicine, Nijmegen [25], as such technology is unavailable in Hong Kong. During the same period of 2011–2018, three other patients were diagnosed with MDs by using a molecular diagnostic approach in the routine clinical setting and were therefore excluded from the study. These patients included two with the MT-ND5 m.13513G>A mutation and one with the MT-ND5 m.13052G>A mutation, presenting as mitochondrial encephalopathy, lactic acidosis, and stroke-like episodes (MELAS; n = 2) and non-syndromic MD, respectively.
Whole-exome sequencing
Genomic DNA was extracted from peripheral blood samples by using the Qiagen Blood Mini Kit (Qiagen). For the first 41 patients, WES was performed at Genome Diagnostics, Radboud University, Nijmegen, using previously described methodology [7]. For the subsequent 25 cases, WES was performed at the Department of Paediatric and Adolescent Medicine of HKU [26]. The exome library was prepared using the TruSeq Rapid Exome Library Prep Kit (Illumina) and sequenced using the Illumina NextSeq500 system. The in-house bioinformatics pipeline was used for data analysis. Raw reads were aligned to the reference human genome [hg19] with the Burrows–Wheeler Aligner (BWA) 0.7.10 [27] and processed following the Genome Analysis Toolkit (GATK) best practices [28]. Rare variants were analysed for pathogenicity according to the American College of Medical Genetics and Genomics (ACMG) guidelines [29]. Data analysis consisted of an initial filtering step involving a virtual MD gene panel based on MitoCarta 2.0; this panel consisted of 1158 genes which encode proteins that have been strongly correlated with mitochondrial localization [30]. If the results were negative, the entire exome was analysed. This approach would include analysis on secondary MD-associated genes and non-MD-associated genes, and the latter have also been reported to be involved in MD in previous studies [7,8,9,10]. For variants of uncertain significance (VUSs), the phenotype was reassessed in the same way in order to curate possible novel disease-causing variants. The data for the initially negative WES results were reanalysed in July 2019 after considering the findings of other recent studies on MDs. The subsequent variant confirmation and segregation analysis were performed at our department in HKU.
Availability of data and materials
The datasets used and/or analysed during the current study are available from the corresponding author on reasonable request.
References
Skladal D, Halliday J, Thorburn DR. Minimum birth prevalence of mitochondrial respiratory chain disorders in children. Brain. 2003;126(Pt 8):1905–12.
Eom S, Lee HN, Lee S, Kang H-C, Lee JS, Kim HD, et al. Cause of death in children with mitochondrial diseases. Pediatr Neurol. 2017;66:82–8.
Thompson K, Collier JJ, Glasgow RIC, Robertson FM, Pyle A, Blakely EL, et al. Recent advances in understanding the molecular genetic basis of mitochondrial disease. J Inherit Metab Dis. 2020;43(1):36–50.
Chi C-S, Lee H-F, Tsai C-R, Lee H-J, Chen L-H. Clinical manifestations in children with mitochondrial diseases. Pediatr Neurol. 2010;43(3):183–9.
Wagner M, Berutti R, Lorenz-Depiereux B, Graf E, Eckstein G, Mayr JA, et al. Mitochondrial DNA mutation analysis from exome sequencing-A more holistic approach in diagnostics of suspected mitochondrial disease. J Inherit Metab Dis. 2019;42(5):909–17.
Parikh S, Goldstein A, Koenig MK, Scaglia F, Enns GM, Saneto R, et al. Diagnosis and management of mitochondrial disease: a consensus statement from the Mitochondrial Medicine Society. Genet Med. 2015;17(9):689–701.
Wortmann SB, Koolen DA, Smeitink JA, van den Heuvel L, Rodenburg RJ. Whole exome sequencing of suspected mitochondrial patients in clinical practice. J Inherit Metab Dis. 2015;38(3):437–43.
Pronicka E, Piekutowska-Abramczuk D, Ciara E, Trubicka J, Rokicki D, Karkucinska-Wieckowska A, et al. New perspective in diagnostics of mitochondrial disorders: two years' experience with whole-exome sequencing at a national paediatric centre. J Transl Med. 2016;14(1):174.
Kohda M, Tokuzawa Y, Kishita Y, Nyuzuki H, Moriyama Y, Mizuno Y, et al. A comprehensive genomic analysis reveals the genetic landscape of mitochondrial respiratory chain complex deficiencies. PLoS Genet. 2016;12(1):e1005679.
Theunissen TEJ, Nguyen M, Kamps R, Hendrickx AT, Sallevelt SCEH, Gottschalk RWH, et al. Whole exome sequencing is the preferred strategy to identify the genetic defect in patients with a probable or possible mitochondrial cause. Front Genet. 2018;9(400).
Taylor RW, Pyle A, Griffin H, Blakely EL, Duff J, He L, et al. Use of whole-exome sequencing to determine the genetic basis of multiple mitochondrial respiratory chain complex deficiencies. JAMA. 2014;312(1):68–77.
Lieber DS, Calvo SE, Shanahan K, Slate NG, Liu S, Hershman SG, et al. Targeted exome sequencing of suspected mitochondrial disorders. Neurology. 2013;80(19):1762–70.
Wolf NI, Smeitink JA. Mitochondrial disorders: a proposal for consensus diagnostic criteria in infants and children. Neurology. 2002;59(9):1402–5.
Brea-Calvo G, Haack Tobias B, Karall D, Ohtake A, Invernizzi F, Carrozzo R, et al. COQ4 Mutations cause a broad spectrum of mitochondrial disorders associated with CoQ10 deficiency. Am J Hum Genet. 2015;96(2):309–17.
Lu M, Zhou Y, Wang Z, Xia Z, Ren J, Guo Q. Clinical phenotype, in silico and biomedical analyses, and intervention for an East Asian population-specific c.370G>A (p.G124S) COQ4 mutation in a Chinese family with CoQ10 deficiency-associated Leigh syndrome. J Hum Genet. 2019;64(4):297–304.
Yu MH-C, Tsang MH-Y, Lai S, Ho MS-P, Tse DML, Willis B, et al. Primary coenzyme Q10 deficiency-7: expanded phenotypic spectrum and a founder mutation in southern Chinese. npj Genomic Med. 2019;4(1):18.
Chung WK, Martin K, Jalas C, Braddock SR, Juusola J, Monaghan KG, et al. Mutations in <em>COQ4</em>, an essential component of coenzyme Q biosynthesis, cause lethal neonatal mitochondrial encephalomyopathy. J Med Genet. 2015;52(9):627–35.
T-k L, C-y L, C-h K, N-c F, K-c W, Lee K-l, et al. A common <em>COQ4</em> mutation in undiagnosed mitochondrial disease: a local case series. Pathology. 2019;51:S112–S3.
Piekutowska-Abramczuk D, Popowska E, Pronicki M, Karczmarewicz E, Tylek-Lemanska D, Sykut-Cegielska J, et al. High prevalence of SURF1 c.845_846delCT mutation in Polish Leigh patients. Eur J Paediatr Neurol. 2009;13(2):146–53.
Pronicka E, Piekutowska-Abramczuk D, Szymanska-Debinska T, Bielecka L, Kowalski P, Luczak S, et al. The natural history of SCO2 deficiency in 36 Polish children confirmed the genotype-phenotype correlation. Mitochondrion. 2013;13(6):810–6.
Hui J, Tang NL, Li CK, Law LK, To KF, Yau P, et al. Inherited metabolic diseases in the Southern Chinese population: spectrum of diseases and estimated incidence from recurrent mutations. Pathology. 2014;46(5):375–82.
Martin AR, Williams E, Foulger RE, Leigh S, Daugherty LC, Niblock O, et al. PanelApp crowdsources expert knowledge to establish consensus diagnostic gene panels. Nat Genet. 2019;51(11):1560–5.
Ben-Ari Fuchs S, Lieder I, Stelzer G, Mazor Y, Buzhor E, Kaplan S, et al. GeneAnalytics: an integrative gene set analysis tool for next generation sequencing, RNAseq and microarray data. OMICS. 2016;20(3):139–51.
Rodenburg RJ, Schoonderwoerd GC, Tiranti V, Taylor RW, Rötig A, Valente L, et al. A multi-center comparison of diagnostic methods for the biochemical evaluation of suspected mitochondrial disorders. Mitochondrion. 2013;13(1):36–43.
Rodenburg RJ. Biochemical diagnosis of mitochondrial disorders. J Inherit Metab Dis. 2011;34(2):283–92.
Tsang MH-Y, Leung GK-C, Ho AC-C, Yeung K-S, Mak CC-Y, Pei SL-C, et al. Exome sequencing identifies molecular diagnosis in children with drug-resistant epilepsy. Epilepsia Open. 2019;4(1):63–72.
Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics (Oxford, England). 2009;25(14):1754–60.
Poplin R, Ruano-Rubio V, DePristo MA, Fennell TJ, Carneiro MO, Van der Auwera GA, et al. Scaling accurate genetic variant discovery to tens of thousands of samples. bioRxiv. 2018;201178.
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405–24.
Calvo SE, Clauser KR, Mootha VK. MitoCarta2.0: an updated inventory of mammalian mitochondrial proteins. Nucleic Acids Res. 2016;44(D1):D1251–7.
Acknowledgements
We thank all the referring clinicians and all the participating patients and families.
We would like to thank The Society for the Relief of Disabled Children, the Joshua Hellmann Foundation, and The Edward and Yolanda Wong Fund for their support.
Funding
The Society for the Relief of Disabled Children, the Joshua Hellmann Foundation, and The Edward and Yolanda Wong Fund
Author information
Authors and Affiliations
Contributions
MHYT and AKYK should be considered joint first author. BHYC and CWF should be considered joint corresponding authors. BHYC and CWF designed and supervised the study. MHYT, AKYK, JLFF, MHCY, CCYM, KSY, RJTR, and JAMS performed the data analysis and interpretation. MHYT, AKYK, KLSC, and CCYM drafted the manuscript. JAMS, BHYC, and CWF evaluated the manuscript. RC, TT, JH, SSNW, SMT, VC, CKM, STHF, SPW, WKC, BHYC, and CWF recruited and managed the patients. All co-authors approved the submitted manuscript. The author(s) read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
The study was approved by the institutional review board of the University of Hong Kong/Hospital Authority Hong Kong West Cluster (IRB Ref. no: UW11-190 and UW12-211). Written informed consent was obtained from the patients or their guardians before performing WES.
Consent for publication
Written informed consent for publication was obtained from the parents of participants.
Competing interests
J.S. is the CEO of Khondrion, a pharmaceutical company developing compounds to potentially treat mitochondrial disease. Other authors declare no conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Tsang, M.H., Kwong, A.K., Chan, K.L. et al. Delineation of molecular findings by whole-exome sequencing for suspected cases of paediatric-onset mitochondrial diseases in the Southern Chinese population. Hum Genomics 14, 28 (2020). https://doi.org/10.1186/s40246-020-00278-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40246-020-00278-0