
Abstract
In recent years, the rapid development of next-generation sequencing (NGS) technologies has led to a significant reduction in sequencing cost with improved accuracy. In the area of liquid biopsy, NGS has been applied to sequence circulating tumor DNA (ctDNA). Since ctDNA is the DNA fragments released by tumor cells, it can provide a molecular profile of cancer. Liquid biopsy can be applied to all stages of cancer diagnosis and treatment, allowing non-invasive and real-time monitoring of disease development. The most promising aspects of liquid biopsy in cancer applications are cancer screening and early diagnosis because they can lead to better survival results and less disease burden. Although many ctDNA sequencing methods have enough sensitivity to detect extremely low levels of mutation frequency at the early stage of cancer, how to effectively implement them in population screening settings remains challenging. This paper focuses on the application of liquid biopsy in the early screening and diagnosis of cancer, introduces NGS-related methods, reviews recent progress, summarizes challenges, and discusses future research directions.
Similar content being viewed by others
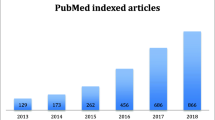


Introduction
Cancer has a significant impact on public health worldwide. One strategy to lower its burden is through cancer screening and early diagnosis. It is well known that patients have a higher cure rate and 5-year survival if diagnosed at early stages [1]. Medical expense increases dramatically with the stage [2, 3]. Tissue biopsy is the most widely-used tool for cancer detection, staging, and prognosis, but sometimes tumor tissue can be difficult to obtain, especially in metastatic diseases like late-stage lung cancer. Moreover, it is unrealistic to use tissue biopsy for cancer screening and early diagnosis when the tumors have not formed yet. Currently, there are some screening methods proved to be useful for cancer prevention. For example, the mammogram is the best way to detect breast cancer; Pap test is used for early detection of cervical cancer; regular colorectal cancer screening and low-dose computed tomography are recommended to reducing mortality from colorectal cancer and lung cancer, respectively [4]. However, all of these screening methods have limited sensitivity and specificity and are only applicable to a unique cancer type. In order to perform large-scale cancer screening among healthy individuals in the future, a more general and cost-effective approach is needed. In recent years, many scientists and companies have cast their eyes on liquid biopsy [5,6,7,8]. Blood contains many types of biological materials like circulating cells, platelets, extracellular vesicles, mRNA, miRNA, protein, and cell-free DNA (cfDNA) [9]. From the blood of cancer patients, a portion of the cfDNA is released by tumor cells through apoptosis, necrosis, or active release [10], and this DNA is called circulating tumor DNA (ctDNA). The tumor-specific mutations in ctDNA sequence can act as a new type of cancer biomarker and help to identify cancer patients from a group of healthy individuals. Compared to traditional cancer diagnosis using tissue biopsy, liquid biopsy is more feasible and less invasive and is more comprehensive than tissue biopsy to evaluate tumor heterogeneity [11] because all tumor sites will release ctDNA into the blood. Facilitated by the rapid development of next-generation sequencing (NGS) technologies, nowadays, ctDNA sequencing can achieve much higher sensitivity than tissue biopsy and can be designed for different purposes [12].
Applications
Screening and early diagnosis
Liquid biopsy is a powerful technique that can be applied to different stages of cancer screening and treatment. Among the asymptomatic population, it can be used to identify cancer patients to improve early diagnosis and better intervention. Nevertheless, using ctDNA sequencing for cancer screening and early diagnosis faces great obstacles. Firstly, the concentration of ctDNA is only about 1 to 10 ng/mL in asymptomatic individuals [12]. Therefore, in order to achieve 95% sensitivity, it was shown that around 150 to 300 ml blood sample per test is needed for breast cancer screening [13]. Secondly, apart from the tumor cells, normal healthy cells and hematopoietic cells also contribute to the cfDNA in the blood, resulting in increased false positives when applying ctDNA assays for cancer diagnosis [14]. Great efforts are being made to meet the sensitivity and specificity requirements for cancer screening and early diagnosis [15,16,17]. Currently, several ctDNA assays can achieve higher sensitivity and specificity than cancer-derived antigens like prostate-specific antigen, carcinoembryonic antigen, carbohydrate antigen (CA) 19-9, CA 15-3, and CA-125 [18]. There are several lines of evidence supporting the further application of ctDNA on screening. Some cohort studies have shown that ctDNA can be used for early lung cancer diagnosis (stage I or II) and can achieve relatively high sensitivity and specificity [16]. Mutations like KRAS and TP53 may be detected in stored sputum samples from individuals up to 1 year before cancer diagnosis [19]. In another prospective study, KRAS and TP53 mutations were detected in the cfDNA of healthy subjects up to 2 years before cancer diagnosis [20]. Apart from DNA mutation, quantification of cfDNA levels [21] and DNA methylation [22] can be combined to provide robust and consistent results. SEPT9 gene methylation detection is the first US Food and Drug Administration (FDA)-approved blood-based screening test for colorectal cancer (CRC) [23, 24]. It exhibited higher sensitivity and specificity than protein markers [25].
Treatment selection and prognosis
After cancer diagnosis, ctDNA sequencing enables tumor-specific molecular profile of the patients to guide targeted therapy for precision medicine. The half-life of cfDNA in the circulation is between 16 min and 2.5 h [26]. That is why ctDNA can be considered “real-time” snapshot to reflect the overall evolution of lesions [12]. This enables real-time and long-term monitoring of the treatment effect, allowing feasible treatment adjustment and better prognosis. Moreover, ctDNA facilitates dynamic monitoring of the clonal evolution and helps to identify the emergence of resistant subclones [12]. Up to now, the European Medicines Agency [27] and the FDA [28] have approved epidermal growth factor receptor (EGFR) mutation testing using ctDNA for therapy guidance among non-small cell lung cancer (NSCLC) patients. Moreover, some newly approved immunotherapies are known to produce different tumor response patterns from other systemic treatments. Using the regular practice of monitoring therapeutic efficacy might not be appropriate anymore. For patients treated with immune checkpoint inhibitors for NSCLC, ctDNA was shown to be an early marker of therapeutic efficacy and could better predict survival outcomes [29].
Residual disease and risk of relapse
Even when treatment is successful, relapse is still a significant threat to many cancer patients, and it is hard to detect the residual disease in time using imaging or tissue biopsy. Very few effective and reliable markers are available currently. Recent studies demonstrated that ctDNA assays were able to detect residual disease several weeks earlier than radiologic imaging [30], and ctDNA-positive patients were at higher risk of relapse and exhibited worse outcome (like shorter overall survival and disease-free survival time) compared to the ctDNA-negative group [31]. In addition, it was shown that phylogenetic ctDNA profiling could be used to track the subclonal nature of lung cancer relapse and metastasis [15]. With the collected profiles, cancer patients could be stratified into different adjuvant therapies to prevent overtreatment [12].
Sequencing techniques
The concentration of ctDNA in plasma has been shown to correlate with tumor size [32] and stage [33]. Patients having stage I disease with various cancer types had fewer than 10 copies per 5 ml of tumor mutations in plasma. In contrast, the copy number increased 10 to 100 times among late-stage patients [34]. Thus, ctDNA assays used for early cancer diagnosis should be highly sensitive. However, highly sensitive assays are always expensive, making large-scale practical applications unrealistic. For late-stage cancer tumor typing, the sensitivity can be moderate because the concentration of ctDNA is much larger. At the same time, the cost of the ctDNA assay is acceptable, and there are several commercial platforms available (Table 1). There is always a tradeoff between sensitivity and cost. Various methods have been proposed to reduce cost, background noise, and errors induced in the amplification step. Those methods can be categorized in different ways. Based on the techniques, there are PCR-based sequencing and NGS-based sequencing. Based on the assay panel size, there are single-locus/multiplexed assays, targeted sequencing, and genome-wide sequencing. PCR-based sequencing can be used for single-locus/multiplexed assays and targeted panel, while NGS-based sequencing can be applied to any panel size.
PCR-based methods
PCR-based methods are most widely used and can achieve extremely high sensitivity. PCR-based methods can be divided into three major categories: real-time quantitative PCR (qPCR), digital PCR (dPCR), and the mass-spectrometry-based method. qPCR is commonly used since it is fast and relatively inexpensive [46]. However, it can only detect mutant allele fraction (MAF) that is greater than 10% [47]. Several variations have been developed to improve the sensitivity of qPCR. For example, co-amplification at lower denaturation temperature (COLD-PCR) can preferentially amplify mutant sequences by controlling the denaturation temperature. It was proved to be a robust method to detect MAF of approximately 0.1% [48, 49].
dPCR has a similar principle as qPCR except it partitions the sample into thousands of parallel PCR reactions to reduce background noise. Thus, it can detect MAF that is less than 0.1% [50]. The sensitivity can be further enhanced by using multiplexed patient-specific panels [51] or molecular barcoding [52] to reduce background sequencing error rates. Among variants of dPCR, BEAMing (on the basis of four of its principal components: beads, emulsion, amplification, and magnetics) is considered to be the most sensitive approach with the detection rate of 0.02% [53]. Nevertheless, the protocol is complicated, and it is relatively expensive for routine clinical usage. It uses primer-bound beads to combine DNA template and distribute the mix in oil detergent to create many aqueous compartments that contain no more than one template or bead. Then, the whole system undergoes conventional PCR. Since each template is distributed into a separated reaction space, the amplification of template is more specific and fewer errors are induced. In the end, fluorescent hybridization and flow cytometry are applied to distinguish and count different templates.
Apart from qPCR and dPCR, the mass-spectrometry-based method is an adaptation of the conventional PCR method with a unique advantage in multiplex detection. For example, UltraSEEK can detect mutant sequence mixtures with MAF as low as 0.1%. It first applies multiplex PCR to amplify all mixtures at the same time. Then, mutations are captured with the labeled chain terminators for sing-base extension and identified using matrix-assisted laser desorption/ionization time-of-flight mass spectrometry [54].
NGS-based methods
Although PCR-based methods are sensitive and inexpensive, they can only screen for known variants, and the input and speed are limited. NGS has high throughput and can screen unknown variants. Currently, NGS is able to detect MAF < 1% [55]. Furthermore, many methods like unique molecular identifiers [29] or unique barcodes [16] can help to increase the sensitivity and reduce the false negatives. These methods are able to detect 59% of stage I or II lung cancer patients with MAF around 0.1% [16] and have good agreement between ctDNA response and radiographic response [29]. NGS can be applied to the targeted panel for specific and highly sensitive detection of targeted ctDNA mutations. Many methods are applying NGS to target panel, namely Tagged-Amplicon deep sequencing (TAm-seq), Safe-Sequencing System (Safe-SeqS), CAncer Personalized Profiling by deep sequencing (CAPP-Seq), and Ion Torrent.
For TAm-seq, researchers first design special primers to amplify regions of interest. In order to control sampling errors and allelic loss, the primers are first used to bind to the template during a preamplification step to amplify the original signal. Next, the templates undergo individual amplification for purification. Benefiting from this two-step amplification design, TAm-seq may able to identify mutations ~ 2% MAF with sensitivity over 97% [56]. The enhanced version of TAm-Seq, named eTAm-Seq™ can detect MAF as low as 0.25% with a sensitivity of 94%. In addition, it has been revised to identify single-nucleotide variants (SNVs), short insertions/deletions (indels), and copy number variants (CNVs) [57].
For Safe-SeqS, the key idea is adding a unique identifier (UID) to each template. After amplification, if a mutation does not appear in most of the same UID-connected sequences, it is likely to be induced by other errors. In this way, Safe-SeqS reduces the sequencing errors by at least 70-fold [58] and has sensitivity as high as ~ 98% for detecting tumor mutations [59].
CAPP-Seq is a combination of the library preparation method and a specialized bioinformatics workflow. The library generates many hybrid affinity captures of recurrently mutated genomic regions from the population of interest to create a “selector.” The “selector” is applied on tumor DNA to identify individual-specific mutations as prior knowledge. Then, it is applied ctDNA for quantification [52, 60]. CAPP-Seq can detect MAF ~ 0.02% with a sensitivity of nearly 100% among stage II-IV NSCLC patients [61].
Ion Torrent is an NGS platform developed by Thermo Fisher Scientific. It allows CNVs, single-nucleotide polymorphisms (SNPs), indels, and fusion detection with as little as 1 ng DNA input [62]. One study applied this platform covering 2800 COSMIC (the Catalogue Of Somatic Mutations In Cancer) mutations from 50 cancer genes to successfully identify 71% of metastatic breast cancer patients [63]. Another study covered more than 6800 COSMIC mutations of 46 genes. About 97% of mutations identified in metastasis biopsies were detected in matched ctDNA in the study [64]. However, researchers who compared dPCR with Ion Torrent concluded that dPCR was more sensitive and can detect smaller MAF for some targeted panels [65].
Although targeted panels may be preferred for their high sensitivity and low cost, they can only detect point mutations and indels. One unique advantage of NGS is that it can be applied to the untargeted panel to find genome-wide DNA variation. Whole-genome-sequencing (WGS) is usually used to get the whole genomic profile of tumor DNA including point mutations, indels, rearrangements, and CNVs [46]. Although WGS provides us with abundant information, it is expensive and less sensitive. Whole-exome sequencing (WES) is a popular alternative of WGS. It is less expensive by only sequencing the exons. Nevertheless, both WGS and WES require high input sample volume, hindering their application in screening and early diagnosis when the concentration of ctDNA is considerably low. Many genome-wide sequencing methods have been proposed for different variation types like PARE (personalized analysis of rearranged ends) for the detection of rearrangement, digital Karyotyping for DNA content quantification, and FAST-SeqS (Fast Aneuploidy Screening Test-Sequencing System) for the detection of CNVs.
PARE first uses next-generation mate-paired sequence analysis to identify individualized rearrangements from tumor tissue. Then, it applies PCR for quantitatively monitoring the detected rearrangements. It is highly sensitive for detecting ctDNA lower than 0.001% in patient plasma samples [66]. Some studies suggested that ctDNA at levels > 0.75% could be detected in cancer patients with sensitivity over 90% and specificity over 99%. Even a single copy of rearrangement from ctDNA can be detected without false positives [67].
Digital karyotyping is a quantitative approach to detecting genome-wide abnormalities at high resolution, including unknown chromosomal changes, altered regions, and DNA sequences [68]. It uses two enzymes to cut the DNA into short fragments around 10 kb and ligates each fragment with a tag. The tags help to align the DNA fragments back to the genome and detect abnormalities in DNA sequence through their density. Orthodenticle homologue 2 (OTX2) amplification was identified in medulloblastomas using digital karyotyping. The overexpression of OTX2 was later confirmed to be causal for certain medulloblastomas type [69].
FAST-SeqS (Fast Aneuploidy Screening Test-Sequencing System) can discriminate as low as 4% of trisomy 21 DNA from euploid samples. The key is simplifying the library preparation steps by only using one designed single primer pair to amplify the repeat regions of interest, so that the cost can be controlled while increases the throughput [70]. There is an updated version of FAST-SeqS called modified FAST-SeqS (mFAST-SeqS). Unlike methods like PARE for quantification of target mutations predetermined by sequencing tumor tissue, mFAST-SeqS is an untargeted method to monitor residual disease or treatment response. Compared with the targeted approaches that can detect MAF as low as 0.01% to 0.5%, untargeted approaches can only detect MAF > 10%. Nevertheless, untargeted approaches require no prior knowledge and can develop genome-wide copy number pattern or assess mutation spectra [71, 72].
Methylation sequencing
Cancer screening not only requires knowing whether the person has cancer or not, but also needs to find the cancerous site for follow-up diagnosis and treatment. Somatic mutation alone may not provide adequate information about the tumor site. Epigenetic information like methylation [73] or protein biomarkers combined with ctDNA [74] has been proved to help determine the tumor origin at an early stage. It is especially useful when the primary site of cancer is unknown. Researchers found that the tumor- and-tissue-specific pattern from methylome data can help with disease classification [75, 76]. It has been shown that methylation profiles of hepatocellular carcinoma tumor DNA and matched plasma ctDNA were highly correlated [75] and could be used to differentiate breast, colon, liver, and lung cancer in diagnosis and prognosis [77].
Methylation sequencing techniques usually have a preprocessing step before sequencing. In addition to DNA conversion, the intention of the preprocessing step is enriching and selecting sequencing targets to reduce the cost. For example, some protocols use immunoprecipitation against 5-methylcytosine to allow much lower levels of input DNA while maintaining high sensitivity [73, 78]. In some other cases, methylation-sensitive restriction enzymes are used to analyze DNA methylation changes [79, 80].
Similar to DNA variants detection, the limited concentration of methylation variants poses great challenges for the balance between coverage, cost, and sensitivity while controlling the technical errors introduced during sequencing. Various methods have been proposed to address the trade-off. For example, the locus-specific techniques like methylation-specific PCR [81] and MethyLight [82] can achieve high sensitivity. However, they can only provide semi-quantitative information for a particular pattern of DNA methylation. PCR-based target selection can achieve high accuracy with a low level of input [81, 83]. However, it cannot be easily applied to the whole-genome level. On the other hand, bisulfite sequencing facilitated by NGS [61, 84, 85] can achieve genome-wide coverage. Adoptions of bisulfite sequencing like Padlock probes can enrich arbitrary target set [86], and DREAMing can detect ultra-rare heterogeneously methylated epialleles variants [87].
Challenges
Biological challenges
ctDNA is highly fragmented, ranging from 100 to 10,000 bp. It is challenging to isolate ctDNA from the blood for quantitation since the small fragments are easy to lose or degrade [88]. Although the concentration of ctDNA will increase with the stage and tumor size, the total percentage of ctDNA in the blood is extremely low, putting many requirements on the sample processing procedure. Also, it has been shown that both concentration and stability of ctDNA could be influenced by the form, release, degradation, and clearance of cfDNA [89]. Up to now, very few studies have discussed the clearance rate and biological mechanism of ctDNA. Another significant obstacle at present is the lack of biological knowledge and experimental evidence to support the quantitative relationship between ctDNA and early cancer development. The pathological evidence is hard to find. Since by the time of using the ctDNA assay for cancer screening or early detection, no knowledge of tissue samples or symptoms of cancer is available. Much remains for us to understand the fundamental biology of ctDNA before we can further push forward the clinical applications of liquid biopsy.
Panel design
It is challenging to find the optimal panel of biomarkers (in most cases, this refers to genetic mutations) according to different objectives, which may demand different tests and impose different requirements [22]. For example, screening requires high sensitivity and high coverage, while monitoring will focus more on the specificity of given mutations. Traditionally, the candidate gene mutation panel is decided on limited biological or clinical knowledge. Nowadays, bioinformatics and biostatistics tools are broadly used to guide the panel design. Information from databases like COSMIC [90] or The Cancer Genome Atlas (TCGA) [91] can be integrated to find differential expression genes or cancer-related mutants among cancer patients and healthy controls. Nevertheless, published studies often applied different methods to select the mutation panels, and there are no systematic criteria on how to choose the optimal combination.
Recently, some researches combined ctDNA mutations with other biomarkers like protein or methylation to improve the overall sensitivity. It was shown that the combination of ctDNA and protein biomarkers could dramatically increase the sensitivity [17]. However, it could be difficult to find the optimal combination of other biomarkers that can maximize the overall detection performance. Biostatistical approaches allow us to effectively identify the relationship between biomarkers like the correlation pattern to guide the panel selection. For example, a study has shown that using KRAS mutations with four protein biomarkers can increase the sensitivity from 30 to 64% and TP53 provided little improvement to the panel since it was highly correlated with KRAS [74].
Sample processing
In a recent review [92], it was proposed that the pre-analytical sample processing including collection, handling, transport, processing, and storage of a specimen is crucial to the final result of ctDNA assay since they would increase degradation of cell-free DNA or increase contamination. The recovery of smaller DNA fragments is particularly important in ctDNA analyses. Many approaches have been explored to improve sample processing quality. For example, plasma has been proven to be the superior source of ctDNA [93]. Standard lavender top tubes with anticoagulant EDTA are most suitable for sample collection [94, 95]. To conclude, a standard operating procedure for ctDNA pre-analytical sample processing is essential to allow more robust and comparable results. However, many published studies were retrospective studies and used archived serum or plasma with distinct pre-analytical procedures [96,97,98]. Little is known so far on how those variables would influence the accuracy of the test.
Data analysis
ctDNA sequencing, especially using NGS, will produce large amounts of data. In addition, in the context of disease monitoring, repeated measurements of clinical variables and outcomes and sequencing data will be collected. The large data size and complex date challenge to statistical analysis. First, researchers need to decide on the lower limits before conducting tests. However, the optimal lower limits of detection may vary depending on the intended use of the ctDNA assay, and there are no standard criteria for choosing the lower limits [99]. Some articles found that ctDNA was highly concordant with tumor DNA while others did not [100,101,102]. It was suggested that the discordant results might depend on the genetic tests applied [100] apart from variation in biosource.
Another statistical challenge is building the classification model. Since the sample size is usually small compared to the number of biomarkers, selecting a subset of most important biomarkers helps to avoid overfitting. Different methods have been used for biomarker selection and model training in published studies [17, 75]. However, some of the procedures were not appropriate. For example, one of the most commonly seen mistakes is using all data for model training and testing, which might induce bias and appear to have high accuracy. Although many model selection methods are available, without appropriate training, testing, model comparison, and diagnosis procedure, the results could be biased and invalid.
The third problem is how to integrate data from different resources. This is especially challenging for cancer screening where we can collect longitudinal data regarding ctDNA sequencing, other biomarkers like protein and methylation, demographic data, medical record, living habits, and so on. Combining available information can help distinguish different populations and improve diagnostic accuracy. A model like CancerSEEK uses both mutation data and protein data to achieve high classification accuracy [17]. One drawback of CancerSEEK is that it transforms all ctDNA mutation data into a single omega score and puts it into the model with other protein biomarker data instead of directly using all the information contained in ctDNA mutations. There are few methods available to build such a model that can integrate different data types, track the change over time with suitably selected predictors, and maximize the usage of all available information.
Clinical applications
Two paradigms are proposed for demonstrating clinical validity and utility using ctDNA [92]. First, prospective clinical trials can be used to test ctDNA as an independent test. Alternatively, the information provided by ctDNA and tissue samples can be assessed to compare their similarity. Both paradigms face many challenges especially in the context of disease screening and early diagnosis. For the first one, the validation of the assay quantitation of tumor burden is technically challenging due to the sample processing issues discussed above. In addition, absolute quantitation is hard to obtain. Most of the methods only obtain relative measures, and few studies conducted cross-platform comparisons. Even when the accurate measure can be obtained, clinical validation requires large-scale prospective trials including both healthy people and cancer patients for treatment guidance and outcome evaluation.
For the second paradigm, the concordance between tumor tissue and ctDNA are not consistent across different studies. A significant number of studies showed that the correlation between plasma mutation status and response rates to therapy was almost the same as that of tumor tissue [103,104,105,106,107,108]. Nevertheless, other studies showed covariates such as disease stage, tumor type, and tumor heterogeneity and whether the variant was clonal or subclonal could influence the concordance between tissue and plasma mutation status [21, 92, 109, 110]. These observations suggest that although it is necessary to develop the concordance between tissue and ctDNA, directly relating ctDNA mutation profiles to clinical measurements of cancer may be another strategy. Last but not least, there is the concern of false positives and overdiagnosis brought by cancer screening. Some patients will not become symptomatic, or their tumors can be benign even they are tested positive. Whether the benefits overwhelm the additional cost and the medical pressure brought by the practice of liquid biopsy remains to be carefully examined [111].
Up to now, there are many liquid biopsy-based assays designed for disease detection, diagnosis, profiling, and treatment selection. Some of them have already been used commercially on cancer patients (Table 1). However, most studies about liquid biopsy were observational, and some of them lacked healthy controls. To date, no studies have shown any improvement in patients outcomes or medical cost using liquid biopsy compared with standard-of-care monitoring methods [92]. Moreover, few studies have evaluated the treatment outcome only based on ctDNA assay-guided targeted therapy. Few of previous studies were intended for cancer screening and early diagnosis. Nevertheless, many large-scale prospective studies are undergoing to demonstrate the clinical validity and utility of ctDNA assays rigorously. For example, powered by Illumina, a company named GRAIL planned to start the SUMMIT study enrolling approximately 50,000 participants without cancer from a high-risk population. They aimed to develop an affordable blood test to detect multiple types of cancer at the same time (Table 1).
Conclusion
Up to now, ctDNA has shown many promising results for cancer classification, monitoring, prognosis, and treatment selection. However, using ctDNA for cancer screening and early detection remained to be solved. The biggest challenge is the low concentration of ctDNA in the blood. Although some NGS-based protocols improve the sensitivity of ctDNA assays in many different ways, the trade-off between sensitivity and cost is still the greatest concern in practice. In the future, other sources of information apart from ctDNA should be combined to increase sensitivity and specificity. Moreover, applying ctDNA sequencing to cancer screening provides us with a good opportunity to collect longitudinal data to create a better disease classification model. As the price for sequencing continues to decrease, using liquid biopsy for cancer prevention and treatment hold promise in the future.
Availability of data and materials
Not applicable
Abbreviations
- CA:
-
Carbohydrate antigen
- CAPP-Seq:
-
CAncer Personalized Profiling by deep sequencing
- cfDNA:
-
Cell-free DNA
- CNV:
-
Copy number variant
- COLD-PCR:
-
CO-amplification at Lower Denaturation temperature
- COSMIC:
-
The Catalogue Of Somatic Mutations In Cancer
- CRC:
-
Colorectal cancer
- ctDNA:
-
Circulating tumor DNA
- FAST-SeqS:
-
Fast Aneuploidy Screening Test-Sequencing System
- FDA:
-
The US Food and Drug Administration
- indel:
-
Insertion or deletion
- MAF:
-
Mutant allele fraction
- mFAST-SeqS:
-
Modified FAST-SeqS
- NGS:
-
Next-generation sequencing
- NSCLC:
-
Non-small cell lung cancer
- OTX2:
-
Orthodenticle homologue 2
- PARE:
-
Personalized analysis of rearranged ends
- PCR dPCR:
-
Digital PCR
- PCR TAm-seq:
-
Tagged-Amplicon deep sequencing
- qPCR:
-
Real-time quantitative
- Safe-SeqS:
-
Safe-Sequencing System
- SNP:
-
Single-nucleotide polymorphisms
- SNV:
-
Single-nucleotide variant
- TCGA:
-
The Cancer Genome Atlas
- UID:
-
Unique identifier
- WES:
-
Whole-exome-sequencing
- WGS:
-
Whole-genome-sequencing
References
World Health Organization. WHO. Cancer. 2019. https://www.who.int/news-room/fact-sheets/detail/cancer, [Accessed 12 Sept 2018].
Blumen H, Fitch K, Polkus V. Comparison of treatment costs for breast cancer, by tumor stage and type of service. Am Health Drug Benefits. 2016;9(1):23.
Cipriano LE, Romanus D, Earle CC, Neville BA, Halpern EF, Gazelle GS, McMahon PM. Lung cancer treatment costs, including patient responsibility, by disease stage and treatment modality, 1992 to 2003. Value Health. 2011;14(1):41–52.
Center for Disease Control CDC and Prevention. How to prevent cancer or find it early: screening tests. 2019. https://www.cdc.gov/cancer/dcpc/prevention/screening.htm, [Accessed 02 May 2018].
Kwapisz D. The first liquid biopsy test approved. is it a new era of mutation testing for non-small cell lung cancer? Ann Transl Med. 2017;5(3):46.
Crowley E, Di Nicolantonio F, Loupakis F, Bardelli A. Liquid biopsy: monitoring cancer- genetics in the blood. Nat Rev Clin Oncol. 2013;10(8):472.
Yoshioka Y, Kosaka N, Konishi Y, Ohta H, Okamoto H, Sonoda H, Nonaka R, Yamamoto H, Ishii H, Mori M, et al. Ultra-sensitive liquid biopsy of circulating extracellular vesicles using exoscreen. Nat Commun. 2014;5:3591.
Pantel K, Alix-Panabières C. Real-time liquid biopsy in cancer patients: fact or fiction? Cancer Res. 2013;73(21):6384–8.
Labib M, Mohamadi RM, Poudineh M, Ahmed SU, Ivanov I, Huang C-L, Moosavi M, Sargent EH, Kelley SO. Single-cell mrna cytometry via sequence-specific nanoparticle clustering and trapping. Nat Chem. 2018;10(5):1.
Stroun M, Lyautey J, Lederrey C, Olson-Sand A, Anker P. About the possible origin and mechanism of circulating DNA: apoptosis and active DNA release. Clin Chim Acta. 2001;313(1–2):139–42.
Siena S, Sartore-Bianchi A, Garcia-Carbonero R, Karthaus M, Smith D, Tabernero J, Van Cutsem E, Guan X, Boedigheimer M, Ang A, et al. Dynamic molecular analysis and clinical correlates of tumor evolution within a phase ii trial of panitumumab-based therapy in metastatic colorectal cancer. Ann Oncol. 2017;29(1):119–26.
Wan JCM, Massie C, Garcia-Corbacho J, Mouliere F, Brenton JD, Caldas C, Pacey S, Baird R, Rosenfeld N. Liquid biopsies come of age: towards implementation of circulating tumour DNA. Nat Rev Cancer. 2017;17(4):223.
Tangvarasittichai O, Jaiwang W, Tangvarasittichai S. The plasma DNA concentration as a potential breast cancer screening marker. Indian J Clin Biochem. 2015;30(1):55–8.
Lehmann-Werman R, Neiman D, Zemmour H, Moss J, Magenheim J, Vaknin-Dembinsky A, Rubertsson S, Nellgård B, Blennow K, Zetterberg H, et al. Identification of tissue-specific cell death using methylation patterns of circulating DNA. Proc Natl Acad Sci. 2016;113(13):E1826–34.
Abbosh C, Birkbak NJ, Wilson GA, Jamal-Hanjani M, Constantin T, Salari R, Le Quesne J, Moore DA, Veeriah S, Rosenthal R, et al. Phylogenetic ctDNA analysis depicts early-stage lung cancer evolution. Nature. 2017;545(7655):446.
Phallen J, Sausen M, Adleff V, Leal A, Hruban C, White J, Anagnostou V, Fiksel J, Cristiano S, Papp E, et al. Direct detection of early-stage cancers using circulating tumor DNA. Sci Transl Med. 2017;9(403):eaan2415.
Cohen JD, Li L, Wang Y, Thoburn C, Afsari B, Danilova L, Douville C, Javed AA, Wong F, Mattox A, et al. Detection and localization of surgically resectable cancers with a multi-analyte blood test. Science. 2018;359(6378):eaar3247.
Donaldson J, Park BH. Circulating tumor DNA: measurement and clinical utility. Ann Rev Med. 2018;69:223–34.
Mao L, Hruban RH, Boyle JO, Tockman M, Sidransky D. Detection of oncogene mutations in sputum precedes diagnosis of lung cancer. Cancer Res. 1994;54(7):1634–7.
Gormally E, Vineis P, Matullo G, Veglia F, Caboux E, Le Roux E, Peluso M, Garte S, Guarrera S, Munnia A, et al. Tp53 and kras2 mutations in plasma DNA of healthy subjects and subsequent cancer occurrence: a prospective study. Cancer Res. 2006;66(13):6871–6.
Gorgannezhad L, Umer M, Islam MN, Nguyen N-T, Shiddiky MJA. Circulating tumor DNA and liquid biopsy: opportunities, challenges, and recent advances in detection technologies. Lab Chip. 2018;18(8):1174–96.
Ossandon MR, Agrawal L, Bernhard EJ, Conley BA, Dey SM, Divi RL, Guan P, Lively TG, McKee TC, Sorg BS, et al. Circulating tumor DNA assays in clinical cancer research. JNCI: J Natl Cancer Inst. 2018;110(9):929–34.
Church TR, Wandell M, Lofton-Day C, Mongin SJ, Burger M, Payne SR, Castaños-Vélez E, Blumenstein BA, Rösch T, Osborn N, et al. Prospective evaluation of methylated sept9 in plasma for detection of asymptomatic colorectal cancer. Gut. 2013;63(2):317–25.
Song L, Li Y. Sept9: a specific circulating biomarker for colorectal cancer. Adv Clin Chem. 2015;72:171–204 Elsevier.
Song L, Jia J, Peng X, Xiao W, Li Y. The performance of the sept9 gene methylation assay and a comparison with other crc screening tests: a meta-analysis. Sci Rep. 2017;7(1):3032.
Diehl F, Schmidt K, Choti MA, Romans K, Goodman S, Li M, Thornton K, Agrawal N, Sokoll L, Szabo SA, et al. Circulating mutant DNA to assess tumor dynamics. Nat Med. 2008;14(9):985.
European Medicines Agency. EMA. Tarceva. 2018. https://www.ema.europa.eu/en/medicines/human/EPAR/tarceva, [Accessed 27 July 2018].
US Food FDA and Drug Administration. Premarket approval P150044 Cobas EGFR MUTATION TEST V2. 2018. https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfpma/pma.cfm?id=P150044, [Accessed 27 July 2018].
Goldberg SB, Narayan A, Kole AJ, Decker RH, Teysir J, Carriero NJ, Lee A, Nemati R, Nath SK, Mane SM, et al. Early assessment of lung cancer immunotherapy response via circulating tumor DNA. Clin Cancer Res. 2018;24(8):1341.
Reinert T, Schøler LV, Thomsen R, Tobiasen H, Vang S, Nordentoft I, Lamy P, Kannerup A-S, Mortensen FV, Stribolt K, Hamilton-Dutoit S, Nielsen HJ, Laurberg S, Pallisgaard N, Pedersen JS, Ørntoft TF, Andersen CL. Analysis of circulating tumour DNA to monitor disease burden following colorectal cancer surgery. Gut. 2016;65(4):625–34.
Lecomte T, Berger A, Zinzindohoué F, Micard S, Landi B, Blons H, Beaune P, Cugnenc P-H, Laurent-Puig P. Detection of free-circulating tumor-associated DNA in plasma of colorectal cancer patients and its association with prognosis. Int J Cancer. 2002;100(5):542–8.
Thierry AR, Mouliere F, Gongora C, Ollier J, Robert B, Ychou M, Del Rio M, Molina F. Origin and quantification of circulating DNA in mice with human colorectal cancer xenografts. Nucleic Acids Res. 2010;38(18):6159–75.
Bettegowda C, Sausen M, Leary RJ, Kinde I, Wang Y, Agrawal N, Bartlett BR, Wang H, Luber B, Alani RM, et al. Detection of circulating tumor DNA in early-and late-stage human malignancies. Sci Transl Med. 2014;6(224):224ra24.
Haque IS, Elemento O. Challenges in using ctDNA to achieve early detection of cancer. bioRxiv. 2017;237578.
Inc. Grail. ESUMMIT Study. 2018. https://grail.com/clinical-studies/summit-study/, [Accessed 21 Feb 2019].
Inc. Guardant Health. Early detection LUNAR-2. 2018. https://guardanthealth.com/solutions/#lunar-2, [Accessed 21 Feb 2019].
Freenome. Clinical studies. 2019. https://www.freenome.com/clinicalstudies [Accessed 21 Feb 2019].
Inc. Biocept. Target Selector™ ctDNA EGFR Kit*. 2019. https://biocept.com/egfr-kit/, [Accessed 21 Feb 2019].
Inivata. Our products. 2019. https://www.inivata.com/our-products/, [Accessed 21 Feb 2019].
Inc. Cynvenio Biosystems. The LiquidBiopsy® Platform. 2019. https://www.cynvenio.com/instrumentation, [Accessed 21 Feb 2019].
CellMax Life. FirstSightCRC. 2019. https://cellmaxlife.com/test-firstsight-crc/, [Accessed 21 Feb 2019].
Exosome Diagnostics. Our diagnostics: for patients. 2019. http://www.exosomedx.com/patients, [Accessed 21 Feb 2019].
Inc. Biodesix. Genomic blood test. 2019. https://www.biodesix.com/products/biodesix-lung-reflex/genestrat-2/, [Accessed 21 Feb 2019].
Inc. Personal Genome Diagnostics. Liquid biopsy. 2019. https://www.personalgenome.com/cap-clia, [Accessed 21 Feb 2019].
Cui S, Cheng Z, Qin W, Jiang L. Exosomes as a liquid biopsy for lung cancer. Lung Cancer. 2018;116:46–54.
Köhn L, Johansson M, Grankvist K, Nilsson J. Liquid biopsies in lung cancer? time to implement research technologies in routine care? Ann Transl Med. 2017;5(13):278.
Sorber L, Zwaenepoel K, Deschoolmeester V, Van Schil PEY, Van Meerbeeck J, Lardon F, Rolfo C, Pauwels P. Circulating cell-free nucleic acids and platelets as a liquid biopsy in the provision of personalized therapy for lung cancer patients. Lung Cancer. 2017;107:100–7.
Santis G, Angell R, Nickless G, Quinn A, Herbert A, Cane P, Spicer J, Breen R, McLean E, Tobal K. Screening for egfr and kras mutations in endobronchial ultrasound derived transbronchial needle aspirates in non-small cell lung cancer using COLD-PCR. PLoS One. 2011;6(9):e25191.
Castellanos-Rizaldos E, Liu P, Milbury CA, Guha M, Brisci A, Cremonesi L, Ferrari M, Mamon H, Makrigiorgos GM. Temperature-tolerant cold-pcr eliminates temperature stringency and enables robust mutation enrichment. Clin Chem. 2012;58(7):1130–8.
Pérez-Callejo D, Romero A, Provencio M, Torrente M. Liquid biopsy based biomarkers in non-small cell lung cancer for diagnosis and treatment monitoring. Transl Lung Cancer Res. 2016;5(5):455.
Narayan A, Carriero NJ, Gettinger SN, Kluytenaar J, Kozak KR, Yock TI, Muscato NE, Ugarelli P, Decker RH, Patel AA. Ultrasensitive measurement of hotspot mutations in tumor DNA in blood using error-suppressed multiplexed deep sequencing. Cancer Res. 2012;72(14):3492–8.
Newman AM, Lovejoy AF, Klass DM, Kurtz DM, Chabon JJ, Scherer F, Stehr H, Liu CL, Bratman SV, Say C, et al. Integrated digital error suppression for improved detection of circulating tumor DNA. Nat Biotechnol. 2016;34(5):547.
Dressman D, Yan H, Traverso G, Kinzler KW, Vogelstein B. Transforming single DNA molecules into fluorescent magnetic particles for detection and enumeration of genetic variations. Proc Natl Acad Sci. 2003;100(15):8817–22.
Mosko MJ, Nakorchevsky AA, Flores E, Metzler H, Ehrich M, van den Boom DJ, Sherwood JL, Nygren AOH. Ultrasensitive detection of multiplexed somatic mutations using maldi-tof mass spectrometry. J Mol Diagn. 2016;18(1):23–31.
Baer C, Kern W, Koch S, Nadarajah N, Schindela S, Meggendorfer M, Haferlach C, Haferlach T. Ultra-deep sequencing leads to earlier and more sensitive detection of the tyrosine kinase inhibitor resistance mutation t315i in chronic myeloid leukemia. Haematologica. 2016;101(7):830–8.
Forshew T, Murtaza M, Parkinson C, Gale D, Tsui DWY, Kaper F, Dawson S-J, Piskorz AM, Jimenez-Linan M, Bentley D, et al. Noninvasive identification and monitoring of cancer mutations by targeted deep sequencing of plasma DNA. Sci Transl Med. 2012;4(136):136ra68.
Gale D, Lawson ARJ, Howarth K, Madi M, Durham B, Smalley S, Calaway J, Blais S, Jones G, Clark J, et al. Development of a highly sensitive liquid biopsy platform to detect clinically-relevant cancer mutations at low allele fractions in cell-free DNA. PloS one. 2018;13(3):e0194630.
Kinde I, Wu J, Papadopoulos N, Kinzler KW, Vogelstein B. Detection and quantification of rare mutations with massively parallel sequencing. ProcNatl Acad Sci. 2011;108(23):9530–5.
Jeanne T, Kinde I, Wang Y, Wong HL, Roebert J, Christie M, Tacey M, Wong R, Singh M, Karapetis CS, et al. Circulating tumor DNA as an early marker of therapeutic response in patients with metastatic colorectal cancer. Ann Oncol. 2015;26(8):1715–22.
Chabon JJ, Simmons AD, Lovejoy AF, Esfahani MS, Newman AM, Haringsma HJ, Kurtz DM, Stehr H, Scherer F, Karlovich CA, et al. Circulating tumour DNA profiling reveals heterogeneity of egfr inhibitor resistance mechanisms in lung cancer patients. Nat Commun. 2016;7:11815.
Newman AM, Bratman SV, Jacqueline To, Wynne JF, Eclov NCW, Modlin LA, Liu CL, Neal JW, Wakelee HA, Merritt RE, et al. An ultrasensitive method for quantitating circulating tumor DNA with broad patient coverage. Nat Med. 2014;20(5):548.
Thermo Fisher Scientific. Ion AmpliSeq Panels. 2019. https://www.thermofisher.com/us/en/home/life-science/sequencing/next-generation-sequencing/ion-torrent-next-generation-sequencing-workflow/ion-torrent-next-generation-sequencing-select-targets/ampliseq-target-selection.html?SID=fr-ampliseqod-main, [Accessed 07 Feb 2019].
Rothé F, Laes J-F, Lambrechts D, Smeets D, Vincent D, Maetens M, Fumagalli D, Michiels S, Drisis S, Moerman C, et al. Plasma circulating tumor DNA as an alternative to metastatic biopsies for mutational analysis in breast cancer. Ann Oncol. 2014;25(10):1959–65.
Lebofsky R, Decraene C, Bernard V, Kamal M, Blin A, Leroy Q, Frio TR, Pierron G, Callens C, Bieche I, et al. Circulating tumor DNA as a non-invasive substitute to metastasis biopsy for tumor genotyping and personalized medicine in a prospective trial across all tumor types. Mol Oncol. 2015;9(4):783–90.
Guttery DS, Page K, Hills A, Woodley L, Marchese SD, Rghebi B, Hastings RK, Luo J, Pringle JH, Stebbing J, et al. Noninvasive detection of activating estrogen receptor 1 (esr1) mutations in estrogen receptor–positive metastatic breast cancer. Clin Chem. 2015;61(7):974–82.
Leary RJ, Kinde I, Diehl F, Schmidt K, Clouser C, Duncan C, Antipova A, Lee C, McKernan K, Francisco M, et al. Development of personalized tumor biomarkers using massively parallel sequencing. Sci Transl Med. 2010;2(20):20ra14.
Leary RJ, Sausen M, Kinde I, Papadopoulos N, Carpten JD, Craig D, O’shaughnessy J, Kinzler KW, Parmigiani G, Vogelstein B, et al. Detection of chromosomal alterations in the circulation of cancer patients with whole-genome sequencing. Sci Transl Med. 2012;4(162):162ra154.
Wang T-L, Maierhofer C, Speicher MR, Lengauer C, Vogelstein B, Kinzler KW, Velculescu VE. Digital karyotyping. Proc Natl Acad Sci. 2002;99(25):16156–61.
Boon K, Eberhart CG, Riggins GJ. Genomic amplification of orthodenticle homologue 2 in medulloblastomas. Cancer Res. 2005;65(3):703–7.
Kinde I, Papadopoulos N, Kinzler KW, Vogelstein B. Fast-seqs: a simple and efficient method for the detection of aneuploidy by massively parallel sequencing. PloS one. 2012;7(7):e41162.
Belic J, Koch M, Ulz P, Auer M, Gerhalter T, Mohan S, Fischereder K, Petru E, Bauernhofer T, Geigl JB, et al. Rapid identification of plasma DNA samples with increased ctDNA levels by a modified fast-seqs approach. Clin Chem. 2015;61(6):838–49.
Belic J, Koch M, Ulz P, Auer M, Gerhalter T, Mohan S, Fischereder K, Petru E, Bauernhofer T, Geigl JB, et al. mfast-seqs as a monitoring and pre-screening tool for tumor-specific aneuploidy in plasma DNA. Circ Nucleic Acids in Serum and Plasma–CNAPS IX. 2016;924:147–55 Springer.
Shen SY, Singhania R, Fehringer G, Chakravarthy A, Roehrl MHA, Chadwick D, Zuzarte PC, Borgida A, Wang TT, Li T, et al. Sensitive tumour detection and classification using plasma cell-free DNA methylomes. Nature. 2018;563(7732):579.
Cohen JD, Javed AA, Thoburn C, Wong F, Tie J, Gibbs P, Schmidt CM, Yip-Schneider MT, Allen PJ, Schattner M, et al. Combined circulating tumor DNA and protein biomarker-based liquid biopsy for the earlier detection of pancreatic cancers. Proc Natl Acad Sci. 2017;114:201704961.
Xu R-h, Wei W, Krawczyk M, Wang W, Luo H, Flagg K, Yi S, Shi W, Quan Q, Li K, et al. Circulating tumour DNA methylation markers for diagnosis and prognosis of hepatocellular carcinoma. Nat Mater. 2017;16(11):1155.
Guo S, Diep D, Plongthongkum N, Fung H-L, Zhang K, Zhang K, et al. Nat Genet. 2017;49(4):635.
Hao X, Luo H, Krawczyk M, Wei W, Wang W, Wang J, Flagg K, Hou J, Zhang H, Yi S, et al. Dna methylation markers for diagnosis and prognosis of common cancers. Proc Natl Acad Sci. 2017;114(28):7414–9.
Taiwo O, Wilson GA, Morris T, Seisenberger S, Reik W, Pearce D, Beck S, Butcher LM. Methylome analysis using medip-seq with low DNA concentrations. Nat Protoc. 2012;7(4):617.
Beikircher G, Pulverer W, Hofner M, Noehammer C, Weinhaeusel A. Multiplexed and sensitive DNA methylation testing using methylation-sensitive restriction enzymes “msre-qpcr”. In: DNA methylation protocols: Springer; 2018. p. 407–24.
Kristiansen S, Nielsen D, Sölétormos G. Detection and monitoring of hypermethylated rassf1a in serum from patients with metastatic breast cancer. Clinical Epigenetics. 2016;8(1):35.
Herman JG, Graff JR, Myöhänen SBDN, Nelkin BD, Baylin SB. Methylation-specific pcr: a novel pcr assay for methylation status of cpg islands. Proc Natl Acad Sci. 1996;93(18):9821–6.
Eads CA, Danenberg KD, Kawakami K, Saltz LB, Blake C, Shibata D, Danenberg PV, Laird PW. Methylight: a high-throughput assay to measure DNA methylation. Nucleic Acids Res. 2000;28(8):e32–00.
Bibikova M, Lin Z, Zhou L, Chudin E, Garcia EW, Wu B, Doucet D, Thomas NJ, Wang Y, Vollmer E, et al. High-throughput DNA methylation profiling using universal bead arrays. Genome Res. 2006;16(3):383–93.
Taylor KH, Kramer RS, Davis JW, Guo J, Duff DJ, Xu D, Caldwell CW, Shi H. Ultradeep bisulfite sequencing analysis of DNA methylation patterns in multiple gene promoters by 454 sequencing. Cancer Res. 2007;67(18):8511–8.
Meissner A, Gnirke A, Bell GW, Ramsahoye B, Lander ES, Jaenisch R. Reduced representation bisulfite sequencing for comparative high-resolution DNA methylation analysis. Nucleic acids Res. 2005;33(18):5868–77.
Deng J, Shoemaker R, Xie B, Gore A, LeProust EM, Antosiewicz-Bourget J, Egli D, Maherali N, Park I-H, Yu J, et al. Targeted bisulfite sequencing reveals changes in DNA methylation associated with nuclear reprogramming. Nat Biotechnol. 2009;27(4):353.
Pisanic TR, Athamanolap P, Poh W, Chen C, Hulbert A, Brock MV, Herman JG, Wang T-H. Dreaming: a simple and ultrasensitive method for assessing intratumor epigenetic heterogeneity directly from liquid biopsies. Nucleic Acids Res. 2015;43(22):e154.
Sedlackova T, Repiska G, Celec P, Szemes T, Minarik G. Fragmentation of DNA affects the accuracy of the DNA quantitation by the commonly used methods. Biol Proced Online. 2013;15(1):5.
Thakur BK, Zhang H, Becker A, Matei I, Huang Y, Costa-Silva B, Zheng Y, Hoshino A, Brazier H, Xiang J, et al. Double-stranded DNA in exosomes: a novel biomarker in cancer detection. Cell Res. 2014;24(6):766.
Forbes SA, Bindal N, Bamford S, Cole C, Kok CY, Beare D, Jia M, Shepherd R, Leung K, Menzies A, et al. Cosmic: mining complete cancer genomes in the catalogue of somatic mutations in cancer. Nucleic Acids Res. 2010;39(suppl 1):D945–50.
Tomczak K, Czerwińska P, Wiznerowicz M. The Cancer Genome Atlas (TCGA): an immeasurable source of knowledge. Contemp Oncol. 2015;19(1A):A68.
Merker JD, Oxnard GR, Compton C, Diehn M, Hurley P, Lazar AJ, Lindeman N, Lockwood CM, Rai AJ, Schilsky RL, et al. Circulating tumor DNA analysis in patients with cancer: American society of clinical oncology and college of american pathologists joint review. Arch Pathol Lab Med. 2018;142(10):1242–53.
Zhao H, Chen K-Z, Hui B-G, Zhang K, Yang F, Wang J. Role of circulating tumor DNA in the management of early-stage lung cancer. Thoracic Cancer. 2018;9(5):509–15.
Kang Q, Henry NL, Paoletti C, Jiang H, Vats P, Chinnaiyan AM, Hayes DF, Merajver SD, Rae JM, Tewari M. Comparative analysis of circulating tumor DNA stability in k3edta, streck, and cellsave blood collection tubes. Clin Biochem. 2016;49(18):1354–60.
Lam NYL, Rainer TH, Chiu RWK, Lo YMD. Edta is a better anticoagulant than heparin or citrate for delayed blood processing for plasma DNA analysis. Clin Chem. 2004;50(1):256–7.
Xi L, Pham TH-T, Payabyab EC, Sherry RM, Rosenberg SA, Raffeld M. Circulating tumor DNA as an early indicator of response to T-cell transfer immunotherapy in metastatic melanoma. Clin Cancer Res. 2016;22(22):5480–6.
Wang R, Li X, Zhang H, Wang K, He J. Cell-free circulating tumor DNA analysis for breast cancer and its clinical utilization as a biomarker. Oncotarget. 2017;8(43):75742.
Vandeputte C, Kehagias P, El Housni H, Ameye L, Laes J-F, Desmedt C, Sotiriou C, Deleporte A, Puleo F, Geboes K, et al. Circulating tumor DNA in early response assessment and monitoring of advanced colorectal cancer treated with a multi-kinase inhibitor. Oncotarget. 2018;9(25):17756.
Dawson S-J, Tsui DWY, Murtaza M, Biggs H, Rueda OM, Chin S-F, Dunning MJ, Gale D, Forshew T, Mahler-Araujo B, et al. Analysis of circulating tumor DNA to monitor metastatic breast cancer. New England J Med. 2013;368(13):1199–209.
Kuderer NM, Burton KA, Blau S, Rose AL, Parker S, Lyman GH, Blau CA. Comparison of 2 commercially available next-generation sequencing platforms in oncology. JAMA Oncol. 2017;3(7):996–8.
Wyatt AW, Annala M, Aggarwal R, Beja K, Feng F, Youngren J, Foye A, Lloyd P, Nykter M, Beer TM, et al. Concordance of circulating tumor DNA and matched metastatic tissue biopsy in prostate cancer. JNCI: J Natl Cancer Inst. 2017;110(1):78–86.
Guo Q, Wang J, Xiao J, Lin W, Hu X, Yu W, Song G, Lou J, Chen JF. Heterogeneous mutation pattern in tumor tissue and circulating tumor DNA warrants parallel ngs panel testing. Mol Cancer. 2018;17(1):131.
Mok TS, Wu Y-L, Ahn M-J, Garassino MC, Kim HR, Ramalingam SS, Shepherd FA, He Y, Akamatsu H, Theelen WSME, et al. Osimertinib or platinum–pemetrexed in egfr t790m–positive lung cancer. New England J Med. 2017;376(7):629–40.
Douillard J-Y, Ostoros G, Cobo M, Ciuleanu T, Cole R, McWalter G, Walker J, Dearden S, Webster A, Milenkova T, et al. Gefitinib treatment in egfr mutated caucasian nsclc: circulating-free tumor DNA as a surrogate for determination of egfr status. J Thor Oncol. 2014;9(9):1345–53.
Jean-Yves D, Ostoros G, Cobo M, Ciuleanu T, McCormack R, Webster A, Milenkova T. First-line gefitinib in caucasian egfr mutation-positive nsclc patients: a phase-iv, open-label, single-arm study. Br J Cancer. 2014;110(1):55.
Oxnard GR, Thress KS, Alden RS, Lawrance R, Paweletz CP, Cantarini M, Yang JC-H, Barrett JC, Jänne PA. Association between plasma genotyping and outcomes of treatment with osimertinib (azd9291) in advanced non–small-cell lung cancer. J Clin Oncol. 2016;34(28):3375.
Jing C-W, Wang Z, Cao H-X, Ma R, Wu J-Z. High resolution melting analysis for epidermal growth factor receptor mutations in formalin-fixed paraffin-embedded tissue and plasma free DNA from non-small cell lung cancer patients. Asian Pac J Cancer Prev. 2013;14(11):6619–23.
Guo N, Lou F, Ma Y, Li J, Yang B, Chen W, Ye H, Zhang J-B, Zhao M-Y, Wu W-J, et al. Circulating tumor DNA detection in lung cancer patients before and after surgery. Sci Rep. 2016;6:33519.
Condello V, Macerola E, Ugolini C, De Napoli L, Romei C, Materazzi G, Elisei R, Basolo F. Analysis of circulating tumor DNA does not improve the clinical management of patients with locally advanced and metastatic papillary thyroid carcinoma. Head Neck. 2018;40(8):1752–8.
Chen K, Zhang J, Guan T, Yang F, Lou F, Chen W, Zhao M, Zhang J, Chen S, Wang J. Comparison of plasma to tissue DNA mutations in surgical patients with non–small cell lung cancer. J Thorac Cardiovasc Surg. 2017;154(3):1123–31.
The National Lung Screening Trial Research Team. Reduced lung-cancer mortality with low-dose computed tomographic screening. New England J Med. 2011;365(5):395–409.
Acknowledgements
Not applicable
Author information
Authors and Affiliations
Contributions
MC was the major contributor in writing the review. HZ substantially modified the review. All authors read and approved the final review of the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable
Consent for publication
Not applicable
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Chen, M., Zhao, H. Next-generation sequencing in liquid biopsy: cancer screening and early detection. Hum Genomics 13, 34 (2019). https://doi.org/10.1186/s40246-019-0220-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40246-019-0220-8