
Abstract
Background
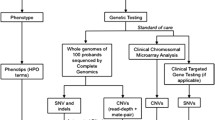
Next-generation sequencing (NGS) has revolutionized genetic research and offers enormous potential for clinical application. Sequencing the exome has the advantage of casting the net wide for all known coding regions while targeted gene panel sequencing provides enhanced sequencing depths and can be designed to avoid incidental findings in adult-onset conditions. A HaloPlex panel consisting of 180 genes within commonly altered chromosomal regions is available for use on both the Ion Personal Genome Machine® (PGMTM) and MiSeq platforms to screen for causative mutations in these genes.
Methods
We used this Haloplex ICCG panel for targeted sequencing of 15 patients with clinical presentations indicative of an abnormality in one of the 180 genes. Sequencing runs were done using the Ion 318 Chips on the Ion Torrent PGM. Variants were filtered for known polymorphisms and analysis was done to identify possible disease-causing variants before validation by Sanger sequencing. When possible, segregation of variants with phenotype in family members was performed to ascertain the pathogenicity of the variant.
Results
More than 97 % of the target bases were covered at >20×. There was an average of 9.6 novel variants per patient. Pathogenic mutations were identified in five genes for six patients, with two novel variants. There were another five likely pathogenic variants, some of which were unreported novel variants.
Conclusions
In a cohort of 15 patients, we were able to identify a likely genetic etiology in six patients (40 %). Another five patients had candidate variants for which further evaluation and segregation analysis are ongoing. Our results indicate that the HaloPlex ICCG panel is useful as a rapid, high-throughput and cost-effective screening tool for 170 of the 180 genes. There is low coverage for some regions in several genes which might have to be supplemented by Sanger sequencing. However, comparing the cost, ease of analysis, and shorter turnaround time, it is a good alternative to exome sequencing for patients whose features are suggestive of a genetic etiology involving one of the genes in the panel.
Similar content being viewed by others


Background
Congenital disorders comprise conditions present at birth or those that developed during infancy or early childhood. Presentations include structural abnormalities, neuromuscular disorders, developmental delay, and intellectual disability which collectively affect more than 10 % of children. The European Surveillance of Congenital Anomalies (EUROCAT) reported the prevalence of major congenital anomalies to be about 2.4 % of live births [1], while the Center for Disease Control and Prevention (CDC) reported 3.3 % for birth defects [2]. The prevalence of developmental disabilities is reported to be 13.9 % in the USA [3].
Less than half of these disorders have an identifiable cause such as aneuploidy, metabolic disorder, maternal infection, parental exposure to teratogenic agents, or intrapartum events. The remaining cases are thought to have a genetic etiology such as submiscroscopic chromosomal abnormalities or rare single/multiple nucleotide changes. The former can be detected by using chromosomal microarray analysis (CMA) which is now the recommended first-tier test for children with dysmorphism, multiple congenital anomalies, developmental delay/intellectual disability, and/or autism spectrum disorder [4]. Although CMA is more sensitive than conventional karyotyping, the diagnostic yield for this group of disorders is still only about 20 % in multiple studies [5–7]. Genetic causes for the rest are likely due to small deletions and insertions, balanced translocations involving gene disruptions, and point mutations which cannot be detected by commonly used CMA platforms.
With massively parallel sequencing, many regions and even the entire genome can be interrogated simultaneously to identify such mutations. Although the cost of whole genome sequencing has become progressively lower in the last few years, data analysis and interpretation remain challenging. Due to the large number of short-reads, the sequence data has to be mapped back to the reference genome and filtered through known databases to identify variants for each individual, leading to long turnaround time from clinic testing to reporting. There is also the issue of incidental findings unrelated to the indication for testing and the American College of Medical Genetics and Genomics (ACMG) have recommended the reporting of pathogenic variants for 56 genes [8]. Subsequently, the ACMG recommended that patients be given the choice of opting out of receiving such information [9]. For these reasons, many laboratories still use Sanger sequencing of single or a few genes when there are known causal genes for the suspected disorders.
Exome sequencing can partly overcome the issue of data throughput but not the possibility of incidental findings. Targeted gene panels can address both by focusing on a set of relevant candidate genes with known diagnostic yield, while providing cost-related advantage as well as easier data analysis without the need for specialized computing infrastructure and expertise. The American Society of Human Genetics (ASHG) also recommends that gene testing should be limited to single genes or targeted gene panels based on the clinical presentations of the patient [10]. Compared to Sanger sequencing of single genes, targeted gene panel sequencing has much higher throughput, but each design needs to be evaluated for coverage and sensitivity before being put to routine clinical diagnostic use.
Among several pre-designed catalog panels for pediatric congenital disorders, there is one comprising 180 genes located within chromosomal regions with a high frequency of cytogenetic abnormalities in constitutional disorders [11] according to publicly available data from the International Collaboration for Clinical Genomics (ICCG—previously known as International Standards for Cytogenomic Arrays or ISCA) [12, 13]. To assess the coverage and sensitivity of this ICCG gene panel for high-throughput next-generation sequencing in congenital disorders, we used the Ion Torrent PGM platform to perform mutation screening of 15 pediatric patients with suspected genetic disorders.
Materials and methods
Ethics statement
The patients were previously recruited under two separate projects (CIRB Ref: 2007/831/F and 2010/238/F). Approval to conduct this sequencing study was provided by the SingHealth Central Institutional Review Board (CIRB Ref: 2013/798/F). All the subjects were minors, and written informed consent had been obtained from the parents.
Study samples
The 15 patients were previously recruited from the hospital’s Genetics Clinics for testing of chromosomal imbalance using human 400 K CGH arrays (Agilent Technologies Inc., Santa Clara, USA). No significant pathogenic copy number changes were identified in all 15. Inclusion criteria include developmental delay/intellectual disability and multiple congenital anomalies. Each patient had been followed up and examined by a clinical geneticist. All of them have clinical features suggestive of a disorder associated with one of the 180 genes, although the features may not have been typical or completely fulfilled the clinical criteria of a specific syndrome at the time of recruitment.
DNA extraction
Genomic DNA was manually extracted from peripheral blood collected in EDTA tubes using the Gentra Puregene Blood Kit (Qiagen Inc., Valencia, USA) according to the manufacturer’s instructions. DNA quality and quantity were measured on a Nanodrop Spectrophotometer (Thermo Scientific, Wilmington, USA).
Library construction, sequencing, and data analysis
Genomic DNA (225 ng gDNA) was digested with 16 different restriction enzymes at 37 °C for 30 min to create a library of gDNA restriction fragments. Both ends of the targeted fragments were selectively hybridized to biotinylated probes from the HaloPlex ICCG panel (Agilent Technologies Inc., Santa Clara, CA, USA), which resulted in direct fragment circularization. During the 16-h hybridization process, HaloPlex ION Barcodes and Ion Torrent sequencing motifs were incorporated into the targeted fragments. Circularized target DNA-HaloPlex probe hybrids containing biotin were then captured by HaloPlex Magnetic Beads on the Agencourt SPRIPlate Super magnet magnetic plate. DNA ligase was added to close the nicks in the hybrids, and freshly-prepared NaOH was used to elute the captured target libraries. The target libraries were then amplified with 18 PCR cycles and purified using AMPure XP beads. Amplicons ranging from 150 to 550 bp were then quantified using an Agilent BioAnalyzer High Sensitivity DNA Assay kit on the 2100 Bioanalyzer to validate the enrichment of the libraries. Library preparation took approximately 1½ days.
Equimolar amounts of four multiplexed bar-coded libraries were pooled and clonally amplified by emulsion PCR, using the Ion PGM Template OT2 200 Kit 9 (Life Technologies, Carlsbad, CA, USA). The template-positive Ion Sphere Particles (ISPs) were then enriched with the Ion OneTouchTM ES and loaded on an Ion 318TM Chip v1. Four separate runs were performed for the 15 samples, with one sample sequenced twice on two different chips. Sequencing was carried out in the Ion PGMTM System using the Ion PGMTM Sequencing 200 Kit v2 according to the manufacturer’s instructions with 500 flow runs.
The data from the sequencing runs were analyzed using the Torrent Suite v4.0.2 analysis pipeline, which includes raw sequencing data processing (DAT processing), splitting of the reads according to the barcode for the individual sample output sequence, classification, signal processing, base calling, read filtering, adapter trimming, and alignment QC. Single-nucleotide polymorphisms (SNP), multi-nucleotide polymorphisms (MNPs), insertions, and deletions were identified across the targeted subset of the reference using a plug-in Torrent Variant Caller (v4.0-r76860), with the parameter settings optimized for germ-line high frequency variants and minimal false positive calls. The output variant call format (VCF) file was then annotated through the web-based user-interfaced GeneTalk (GeneTalk GmbH, Berlin, Germany) and Ensembl Variant Effect Predictor [14].
Sequence variants were compared with data in dbSNP, 1000 Genomes and Human Genome Mutation Database. Variants not previously reported in healthy controls or previously classified as pathogenic were evaluated for coverage depth and also visually inspected using the Integrative Genomics Viewer before validation by dideoxy sequencing using standard protocol for BigDye® Terminator v3.1 Cycle Sequencing Kit (Life Technologies, Carlsbad, CA, USA). Segregation analysis was performed when DNA from family members was available. Sequencing was carried out on the Applied Biosystems® 3130 Genetic Analyzer (Life Technologies, Carlsbad, CA, USA). In addition, SIFT (sift.bii.a-star.edu.sg) and Polyphen2 (genetics.bwh.harvard.edu/pph2) were used to check the likely functional significance of missense variants for clinical interpretation.
Results
An average of 790 Mb was generated per chip (range 748–828 Mb). Loading densities of the targeted sequencing of four libraries (four samples were multiplexed in each library) ranged from 75 to 81 %. The total number of reads (usable sequence) ranged from 5.8 to 6.4 M, and average read length ranged from 124 to 131 bp. After filtering out polyclonal, low quality, and primer dimers, the percentage of usable reads ranged from 69 to 73 %. On average, each sample yielded 196 M bases from 1.5 M reads (Table 1 and Fig. 1) from 58,670 amplicons with a mean read length of 128 bp. One sample was sequenced twice, with near identical output obtained for both runs. The numbers of reads were 1,552,042 and 1,556,202 for total reads and 1,522,728 and 1,524,576 for mapped reads, and total numbers of bases sequenced were 199,024,281 and 200,813,003.

Percentage of bases at the different read depths
Approximately 97.4 % of the reads were aligned to the reference genome (hg19) and 91 % mapped to the target regions, with average base coverage ranging from 203× to 256× for individual samples. 97.7 % of the targets had minimum read depth of 20×, 95.6 % at >50× and 88.2 % at >100×. Full coverage was achieved for more than 95 % of targets in all 15 samples, and most (approximately 89.9 %) target bases did not show any bias toward forward or reverse strand read alignment. The average total coverage of all targeted bases was 95.7 % at 20× and 82.38 % at 100×. Coverage was also uniform across all samples. More than 88 % of called bases had a quality score of ≥Q20 (Table 1).
At the gene level, 137 of the 180 genes had mean coverage of at least 20×, of which 99 had a mean of >50× and 40 had a mean of >100× (Table 2). Despite the high target region coverage, amplification failed for at least 26 exons across the 180 genes. Thirteen genes (CFC1, CHRNA7, CYP21A2, EHMT1, F8, HBA1, HBA2, IKBKG, NOTCH2, PKD1, SGCE, SRY, TSC2) had at least one region that was not amplified and therefore not sequenced (lowest number of reads “0” in Table 2). The sequencing coverage of CFC1, IKBKG, HBA1, and HBA2 was low with >50 % of these genes sequenced at >20× (Table 3). The gene with the highest mean coverage was SALL1 (358×). The poorest coverage was for CFC1. Mean read depth for individual exons for three different genes were shown in Figs. 2, 3, and 4.

Average target base read depth for exons 2–38 of CHD7

Average target base read depth for exons 1–4 of MECP2

Average target base read depth for exons 2–11 of SATB2
Overall, 2326 single-nucleotide variants (SNVs) and 25 indels were identified in the 15 patients. These variants identified from the Ion Reporter had an average coverage of 595× and an average Qscore of 38. Variant annotation indicated that 2203 were common variants present in dbSNP and 1000 Genome Project databases. The number of variants ranged from 154 to 175 per patient, with an average of 9.6 novel variants each. Synonymous variants were the most common.
Variants were prioritized for Sanger confirmation based on the individual’s clinical presentations. Pathogenic variants were confirmed in six patients. The identified CHD7 (two patients), SHH, TCF4, TSC2, and MECP2 variants and the clinical features of these six patients are listed in Table 4. Another five patients had candidate variants for which further evaluation and segregation analysis are ongoing.
Discussion
The HaloPlex ICCG panel is a pre-designed made-to-order panel targeting 180 genes. It follows the ICCG recommendations for design and resolution and is available through SureDesign from Agilent Technologies. The targeted panel includes genes in the most commonly altered chromosomal regions according to the ISCA/ICCG database. The 180 genes are covered by 2509 target regions which range in size from 2 to 6575 nucleotides. Depending on its size, a region is covered by between 1 and 547 amplicons.
The recommended minimum read depth for clinical diagnostic sequencing is 20× [15, 16], which was achieved for over 90 % of the target for 170 genes. For CHD7, even the exon with the poorest coverage had a mean of 36 (Fig. 2). Of the remaining ten, four genes had 80–90 % coverage, and the other six (CFC1, CYP21A, HBA1, HBA2, IKBKG, NOTCH2, PLP1) had <80 %. More than half of the targets in these individual genes are within GC-rich regions. Less efficient PCR for these templates might have resulted in sequencing failure during library preparation, or insufficient sequence data were produced [17]. In addition, the HaloPlex protocol uses restriction enzymes which are sequence-dependent and nonrandom, this method might have contributed further to uneven coverage and also gaps in coverage [18]. For IKBKG, the presence of a pseudogene might have caused non-specific alignment and contributed to the low capture of target sequences [19]. Nijman et al. have almost no mapped reads in IKBKG in their targeted sequencing, and generally poor coverage of CFC1 and IKBKG had been reported in multiple studies [20–22]. For the gene with the poorest coverage CFC1, all six exons had no reads across all 15 samples. This gene is associated with the generation of left-right asymmetry via the TGF pathway. There were 23 mutations in HGMD, 13 of which were found in patients with congenital heart disease [23]. This panel would not be useful for patients with clinical suspicion of CFC1 gene mutations.
The first exon of 64 genes was not included in the design (indicated with “*” in Table 2). All the 64 genes have one or more non-coding exon. The entire exon 1 of these genes (and additional exons for some others) contains only untranslated regions. In general, amplification of exon 1 of some genes was problematic because of the generally higher GC content and sequence complexity [24–26]. Our results showed that MECP2 had an average target base read depth of 118×. The coverage for exon 1 is the lowest among all, but it is still two times that of the minimum of 20× recommended for clinical diagnostics (Fig. 3). SATB2 had an average target base read depth of 300×, but exon 1 was not covered in the design (Fig. 4). Nevertheless, including non-coding exons in the design might improve the yield of NGS as variants affecting splicing of non-coding exons have been reported to be disease-causing [27].
Many congenital disorders do not have unique and exclusive features, and the presentations may be non-specific. Even for syndromic disorders, there are overlapping features, and the phenotypic features in some patients may be atypical, making it challenging for the clinical geneticists to come to a diagnosis based on clinical history and examination. All the 15 patients in this study have constitutional disorders and suspicion of chromosomal disorders, but CMA did not find any pathogenic copy number abnormality. With this targeted panel, we were able to reach a molecular diagnosis for six patients after reviewing the results with their primary physicians (Table 4). Pathogenic CHD7 variants were detected in two patients with clinical features consistent with CHARGE syndrome. Both CHD7 variants identified (p.R2613X and p.Q201X) have been previously reported in other CHARGE patients [28]. A pathogenic p.R255X MECP2 variant was detected in a patient with clinical features of Rett syndrome. This variant has also been reported previously [29]. The patients with the truncating TSC2 variant and the missense SHH variant also showed clinical features consistent with the respective causative genes. These two variants are novel and the missense variant is predicted to be pathogenic according to both SIFT and Polyphen. Similarly, the clinical features of the patient with the TCF4 variant are found to be consistent with Pitt-Hopkins syndrome upon retrospective review of the patient’s progressive features by the attending physician. This p.R580Q TCF4 variant has been reported as pathogenic in patients with Pitt-Hopkins syndrome [30].
The identification of a patient’s causative mutation has the translational benefit of providing the parents with an answer for their child’s condition. In addition, it provides a guide to the attending clinician on the management and prognosis of the patient. A molecular diagnosis would also facilitate access to clinical trials and programs for special needs children. The use of appropriate gene panels obviates the need for subjective clinical decision on which gene(s) to test in each patient, and may lead to a standard testing workflow for each group of disorders. Generally for those whose diagnosis can be narrowed down to a few suspected genetic syndromes, targeted gene panels would be superior to exome sequencing which has more limitations in the diagnostic setting due to coverage deficiencies in some genes and longer turnaround time. Higher-average read depth could be attained at a lower cost, making it superior to exome sequencing in terms of cost, sensitivity, and expected diagnostic yield [31, 32].
Conclusions
The Haloplex ICCG panel had good coverage except for ten of the target genes. Consideration would have to be made for the low coverage for some regions in several genes which might have to be supplemented by Sanger sequencing. However, comparing the cost, ease of analysis, and shorter turnaround time, it is a good alternative to exome sequencing for patients whose features are suggestive of a genetic etiology involving one of the genes in the panel.
References
Dolk H, Loane M, Garne E. The prevalence of congenital anomalies in Europe. Adv Exp Med Biol. 2010;686:349–64.
Birth defects [http://www.cdc.gov/ncbddd/birthdefects/index.html]. Accessed 4 Aug 2015.
Boyle CA, Boulet S, Schieve LA, Cohen RA, Blumberg SJ, Yeargin-Allsopp M, et al. Trends in the prevalence of developmental disabilities in US children, 1997–2008. Pediatrics. 2011;127:1034–42.
Miller DT, Adam MP, Aradhya S, Biesecker LG, Brothman AR, Carter NP, et al. Consensus statement: chromosomal microarray is a first-tier clinical diagnostic test for individuals with developmental disabilities or congenital anomalies. Am J Hum Genet. 2010;86:749–64.
Bartnik M, Wisniowiecka-Kowalnik B, Nowakowska B, Smyk M, Kedzior M, Sobecka K, et al. The usefulness of array comparative genomic hybridization in clinical diagnostics of intellectual disability in children. Dev Period Med. 2014;18:307–17.
Battaglia A, Doccini V, Bernardini L, Novelli A, Loddo S, Capalbo A, et al. Confirmation of chromosomal microarray as a first-tier clinical diagnostic test for individuals with developmental delay, intellectual disability, autism spectrum disorders and dysmorphic features. Eur J Paediatr Neurol. 2013;17:589–99.
Chong WW, Lo IF, Lam ST, Wang CC, Luk HM, Leung TY, et al. Performance of chromosomal microarray for patients with intellectual disabilities/developmental delay, autism, and multiple congenital anomalies in a Chinese cohort. Mol Cytogenet. 2014;7:34.
Green RC, Berg JS, Grody WW, Kalia SS, Korf BR, Martin CL, et al. ACMG recommendations for reporting of incidental findings in clinical exome and genome sequencing. Genet Med. 2013;15:565–74.
ACMG Board of Directors. ACMG policy statement: updated recommendations regarding analysis and reporting of secondary findings in clinical genome-scale sequencing. Genet Med. 2015;17:68–9.
Botkin JR, Belmont JW, Berg JS, Berkman BE, Bombard Y, Holm IA, et al. Points to consider: ethical, legal, and psychosocial implications of genetic testing in children and adolescents. Am J Hum Genet. 2015;97:6–21.
Haloplex disease research panels [http://www.chem.agilent.com/Library/datasheets/Public/HaloPlexDiseaseResearchPanels5991-2526ENa.pdf]. Accessed 4 Aug 2015.
Kaminsky EB, Kaul V, Paschall J, Church DM, Bunke B, Kunig D, et al. An evidence-based approach to establish the functional and clinical significance of copy number variants in intellectual and developmental disabilities. Genet Med. 2011;13:777–84.
Moreno-De-Luca D, Sanders SJ, Willsey AJ, Mulle JG, Lowe JK, Geschwind DH, et al. Using large clinical data sets to infer pathogenicity for rare copy number variants in autism cohorts. Mol Psychiatry. 2013;18:1090–5.
Yourshaw M, Taylor SP, Rao AR, Martin MG, Nelson SF. Rich annotation of DNA sequencing variants by leveraging the Ensembl Variant Effect Predictor with plugins. Brief Bioinform. 2014;16:255–64.
Rehm HL, Bale SJ, Bayrak-Toydemir P, Berg JS, Brown KK, Deignan JL, et al. ACMG clinical laboratory standards for next-generation sequencing. Genet Med. 2013;15:733–47.
Yohe S, Hauge A, Bunjer K, Kemmer T, Bower M, Schomaker M, et al. Clinical validation of targeted next-generation sequencing for inherited disorders. Arch Pathol Lab Med. 2015;139:204–10.
Bragg LM, Stone G, Butler MK, Hugenholtz P, Tyson GW. Shining a light on dark sequencing: characterising errors in Ion Torrent PGM data. PLoS Comput Biol. 2013;9:e1003031.
Coonrod EM, Durtschi JD, VanSant WC, Voelkerding KV, Kumanovics A. Next-generation sequencing of custom amplicons to improve coverage of HaloPlex multigene panels. Biotechniques. 2014;57:204–7.
Aksyonov SA, Bittner M, Bloom LB, Reha-Krantz LJ, Gould IR, Hayes MA, et al. Multiplexed DNA sequencing-by-synthesis. Anal Biochem. 2006;348:127–38.
Brett M, McPherson J, Zang ZJ, Lai A, Tan ES, Ng I, et al. Massively parallel sequencing of patients with intellectual disability, congenital anomalies and/or autism spectrum disorders with a targeted gene panel. PLoS One. 2014;9:e93409.
Nijman IJ, van Montfrans JM, Hoogstraat M, Boes ML, van de Corput L, Renner ED, et al. Targeted next-generation sequencing: a novel diagnostic tool for primary immunodeficiencies. J Allergy Clin Immunol. 2014;133:529–34.
Kammermeier J, Drury S, James CT, Dziubak R, Ocaka L, Elawad M, et al. Targeted gene panel sequencing in children with very early onset inflammatory bowel disease—evaluation and prospective analysis. J Med Genet. 2014;51:748–55.
Human gene mutation database [https://portal.biobase-international.com/hgmd/pro/gene.php?gene=CFC1]. Accessed 2 Sep 2015.
Gnirke A, Melnikov A, Maguire J, Rogov P, LeProust EM, Brockman W, et al. Solution hybrid selection with ultra-long oligonucleotides for massively parallel targeted sequencing. Nat Biotechnol. 2009;27:182–9.
Hu H, Wrogemann K, Kalscheuer V, Tzschach A, Richard H, Haas SA, et al. Mutation screening in 86 known X-linked mental retardation genes by droplet-based multiplex PCR and massive parallel sequencing. Hugo J. 2009;3:41–9.
Porreca GJ, Zhang K, Li JB, Xie B, Austin D, Vassallo SL, et al. Multiplex amplification of large sets of human exons. Nat Methods. 2007;4:931–6.
Eisenberger T, Neuhaus C, Khan AO, Decker C, Preising MN, Friedburg C, et al. Increasing the yield in targeted next-generation sequencing by implicating CNV analysis, non-coding exons and the overall variant load: the example of retinal dystrophies. PLoS One. 2013;8:e78496.
Bartels CF, Scacheri C, White L, Scacheri PC, Bale S. Mutations in the CHD7 gene: the experience of a commercial laboratory. Genet Test Mol Biomarkers. 2010;14:881–91.
Amir RE, Van den Veyver IB, Wan M, Tran CQ, Francke U, Zoghbi HY. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat Genet. 1999;23:185–8.
Amiel J, Rio M, de Pontual L, Redon R, Malan V, Boddaert N, et al. Mutations in TCF4, encoding a class I basic helix-loop-helix transcription factor, are responsible for Pitt-Hopkins syndrome, a severe epileptic encephalopathy associated with autonomic dysfunction. Am J Hum Genet. 2007;80:988–93.
Ankala A, da Silva C, Gualandi F, Ferlini A, Bean LJ, Collins C, et al. A comprehensive genomic approach for neuromuscular diseases gives a high diagnostic yield. Ann Neurol. 2015;77:206–14.
Consugar MB, Navarro-Gomez D, Place EM, Bujakowska KM, Sousa ME, Fonseca-Kelly ZD, et al. Panel-based genetic diagnostic testing for inherited eye diseases is highly accurate and reproducible, and more sensitive for variant detection, than exome sequencing. Genet Med. 2015;17:253–61.
Acknowledgements
This work is supported by NMRC/CG/006/2013 and NMRC/PPG/KKH12010-Theme3 from the National Medical Research Council, Ministry of Health, Republic of Singapore. We thank K Chang and CH Kuick for the use of the Ion Torrent PGM.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
All authors declare that they have no competing interests.
Authors’ contributions
ECT designed the study and obtained the funding. EL and SPL carried out the sequencing experiments. EL, MB, and SJ performed the analysis and interpretation of the sequencing data. MB, AL, EST, and IN carried out the selection of patients, clinical assessment, and phenotype correlation. EL and ECT prepared the manuscript. All authors read and approved the manuscript.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Lim, E.C.P., Brett, M., Lai, A.H.M. et al. Next-generation sequencing using a pre-designed gene panel for the molecular diagnosis of congenital disorders in pediatric patients. Hum Genomics 9, 33 (2015). https://doi.org/10.1186/s40246-015-0055-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40246-015-0055-x