
Abstract
Extracellular vesicles (EV) are blebs of cellular membranes, which entrap small portions of subjacent cytosol. They are released from a variety of cells, circulate in the blood for an unknown length of time and come to rest on endothelial surfaces. They contribute to an array of physiologic pathways, the complexity of which is still being investigated. They contribute to metastatic malignant cell implants and tumor-related angiogenesis, possibly abetted by the tissue factor that they carry. It is thought that the adherence of the EV to endothelium is dependent upon a combination of their P-selectin glycoprotein ligand-1 and exposed phosphatidylserine, the latter of which is normally hidden on the inner bilayer of the intact cellular membrane. This manuscript reviews what is known about EV origins, their clearance from the circulation and how they contribute to malignant cell implants upon endothelium surfaces and subsequent tumor growth.
Similar content being viewed by others


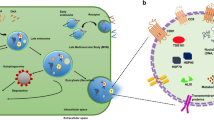
Definition of extracellular vesicles
Cells shed blebs of their phospholipid bilayer plasma membranes as byproducts of cell growth, apoptosis and in response to physiologic and pathophysiologic stimuli. These vesicles encapsulate small portions of the subjacent cytosol, creating a heterogeneous population of phospholipid-walled vesicles. These particles are referred to as extracellular vesicles (EV), but also as microparticles, microvesicles, microsomes, lipid vesicles, apoptotic blebs and exosomes [1,2,3,4,5,6].
EV’s are characterized by their size (30–100 nm for exosomes and 100–1000 nm diameter for the larger microvesicles), by their cells of origin including megakaryocytes, platelets, red blood cells, endothelial cells and others, and by their intravesicular contents [2, 3, 6, 7]. Their intravesicular contents depend upon their cells of origin and can include tissue factor, double stranded DNA, mRNA, microRNA, adhesion integrins, growth factors, protease inhibitors, P-selectin glycoprotein ligand-1 (PSGL-1) and ceramides [8,9,10,11,12,13,14,15,16,17]. They have been detected in blood plasma and other physiologic fluids [3, 4]. Gender, age of subjects and diseases influence their number in circulating blood and their size distribution [18, 19].
EVs are also released in response to pathophysiologic stimuli including thrombin, shear stress, complement activation, sepsis, hypoxia, inflammation, from malignant cells and following chemotherapy for malignancies [20,21,22]. They may also be byproducts of cell maturation with shedding of excess cell membrane [23].
The normal cell membrane is a bilayer structure with inner cytosolic layer enriched with phosphatidylserine and phosphatidyl-ethanolamine. This structure is maintained by enzymes flippase, floppase and scramblase [6]. When the EV are created, the endoplasmic reticulum releases Ca2+ which inactivates flippase and activates floppase and scramblase leading to loss of the normal asymmetry of the cell membrane and reversal of the normal order, creating an outward facing phosphatidylserine enriched layer [6, 24]. See Fig. 1. The phosphatidylserine is then available to be tethered by lactadherin (also known as Human Milk Fat globule factor 8 or MFG-E8) and Tim 4 to endothelium [25, 26]. See Fig. 2.

The normal cell membrane is an asymmetrical bilayered structure with phosphatidylserine- and phosphatidyl-ethanolamine-enriched cytosolic layers, maintained by flippase, floppase and scramblase. As extracellular vesicles are formed, the flippases is inactivated while floppase and scramblase are activated, leading to reversal of the normal asymmetry, creating an outward facing phosphatidylserine enriched layer

Extracellular vesicle attachment to endothelial cells is dependent upon their exposed PSGL-1 attaching to the P-selectin expressed from Weibel–Palade bodies and platelet alpha-granules, and upon the tethering of the exposed surface phosphatidylserine to Tim4, lactadherin/MFG-E8 and probably other cell adhesion molecules
Exosomes are unique subsets of EVs with specific biogenesis, measuring 30–100 nm [4, 17]. They are derived from endosomal multivesicular bodies, which then fuse with plasma membranes and are secreted from the cell into extracellular space, a process termed exocytosis. The primary function of these endosomes appears to be for the cellular uptake and extrusion of macromolecules from and to the extracellular fluids [1,2,3, 27]. They transport bioactive molecules including proteins, DNA, functional mRNA and microRNA from cell to cell by membranous transfer, thereby influencing other cell systems [17, 25, 28]. The currently available techniques do not allow for clean separation of these exosomes from the larger extracellular vesicles.
Apoptotic bodies may co-isolate with extracellular vesicles when isolated in the lab, but based upon origin they differ greatly from EV [29]. The EV appear to be generated continuously from normal, viable cells, while the apoptotic bodies are derived following a highly regulated programmed cell death, in which the caspase pathway plays a central role. Apoptosis involves cell shrinkage and nuclear fragmentation, with the debris separated into nuclear or cytoplasmic vesicles of various sizes, which may be as small as the EV, but also may be up to 5000 nm diameter. They do express the “eat me” cellular signal to facilitate their phagocytosis by macrophages in the vicinity of the dead cell, using some of the same signals as the other EV [29].
Methods of isolation
The methods of isolation and preservation of the EVs are improving, but have not reached the ultimate goal of allowing analysis of homogeneous EV subpopulations and precise study of their targets [3,4,5, 30]. Currently EVs are defined predominantly by the techniques used for their isolation (flow cytometry, density gradient separation, differential centrifugation, size-exclusion chromatography, immunoaffinity isolation, polymer precipitation and others) as they affect the size and density of the isolates. Also, pre-analytical manipulation of samples influence the study results [31]. As of now there is overlap of the size subclasses and cells of origin [4, 5]. For reporting purposes the EV can be further described by flow cytometry, mass spectroscopy and by their specific contents such as DNA, microRNA and mRNA [3, 32, 33].
A new technique for isolation of EV was reported using affinity-based methodology taking advantage of the T cell immunoglobulin domain and mucin-containing protein 4, known as Tim4, and its adherence to the externalized plasma membrane phosphatidylserine [34]. In this method the extracellular domain of Tim4, normally expressed as a transmembrane protein on macrophages, is immobilized on magnetic beads. Since Tim4 strongly binds to phosphatidylserine it immobilizes the EV on the beads [35]. This binding is calcium dependent and thus EV can be released when calcium chelators are added. Tim4 affinity can also be used in ELISA and flow cytometry formats [34].
Post-translational modifications of EV proteins are under study [3, 36, 37]. Exosomes become more rigid increasing their stability after they are released from m into the extracellular space with its increased pH. They also undergo post-translational covalent attachment to fatty acids. Acetylation and myristoylation promote protein sorting and subsequent EV budding [36, 38].
Total mass of EVs in circulation
The total mass of EVs in the circulation at any given time can be represented by their rates of release into the circulation and their rates of clearance from the circulation. How long they survive in the circulation is unclear [33]. Clearance mechanisms include endocytosis, micropinocytosis, phagocytosis and membrane fusion [22, 39]. These processes are dependent upon exposed PSGL-1 on platelet-derived EV attaching to P-selectin expressed from the endothelial cell Weibel–Palade bodies and platelets alpha-granules; and upon the phosphatidylserine on their surface with tethering to Tim4 and lactadherin/MFG-E8 [25, 40]. The extent of surface phosphatidyl serine exposure in-vivo prior to ex vivo manipulation is unknown.
Their rates of clearance have acute and chronic patterns of change and appear to be influenced by their cell of origin [22, 41, 42]. The locales of the macrophage-mediated phagocytosis also differ based upon the organ in which the macrophages are anchored [43,44,45]. Endothelial receptors may also vary from site to site with different organ-specificities, such as the endothelial cell-rich lungs [46, 47].
EV activities
The total array of activities of the EVs is still under study. They are involved in cell-to-cell communications, transferring their contents to cell types that differ from their cell of origin [9,10,11,12]. This affects cell signaling, platelet and leukocyte adhesion to subendothelial matrix, vascular smooth muscle proliferation, inflammation, immune response, thrombosis, angiogenesis and angiocrine activities, and tumor niche formation leading to enhanced tumor implantation on endothelial surfaces [4, 47,48,49,50,51,52,53,54,55].
Contributions to thrombosis
An important and complex picture is emerging of how EVs participate in coagulation and fibrinolysis [8, 12, 49, 56,57,58].
EVs become anchored to endothelium in two ways. The PSGL-1 from platelet-derived EV attaches to the P-selectin released from Weibel–Palade bodies of the activated endothelial cell and from the platelet-derived α-granules [55, 59,60,61]. There also appears to be a phosphatidylserine receptor, at least on cultured activated microvascular endothelial cells derived from pulmonary and retinal tissues [62]. CD36, also known as platelet glycoprotein IIIB and IV among other names, is a scavenger receptor that binds several ligands including phosphatidylserine. It is a candidate cellular adhesion molecule for anchoring the EV. This pathway is blocked when the phosphatidylserine as pretreated with annexin V [63]. Subendothelial thrombospondin is another candidate cellular CAM for the exposed phosphatidylserine [64]. Other cellular adhesion molecules with ability to tether the phosphatidylserine exposed on the surface of the EV include Tim4 and lactadherin [25, 26].
EV trigger thrombosis via release of tissue factor they contain, supplementing the tissue factor released from perivascular tissue following endothelial disruption [9, 11, 12, 50, 65]. The released tissue factor is activated by thiol isomerases, with downstream platelet activation and thrombus formation [56, 57, 66, 67]. This has become a target of a new class of anticoagulants [68]. As an example, quercetin-3-rutinoside suppresses this tissue factor activation [56, 68]. Furthermore, the exposed phosphatidylserine is a catalytic surface for assembly of the prothrombinase complex [69].
Increased thrombin-generating activity has been recorded for EV derived from platelets and monocytes harvested from patients having recurrent thrombosis [70].
Suppression of tissue factor with anti-tissue factor antibody and the suppression of P-selectin in PSGL-1 knock out mice reduce clot formation [12, 56, 58]. Similarly, drugs with activity in this domain, such as the HMG-CoA reductase inhibitors, suppress the levels of soluble P-selectin and would consequently be expected to reduce the expression of tissue factor [71,72,73,74]. Also, the in vitro blockade of the phosphatidylserine on EVs with annexin V or lactadherin (also known as MFG-E8), both of which attach variably to phosphatidylserine, reduces the platelet-derived EVs contribution to thrombin generation [75,76,77,78]. These observations are the basis for new clinical studies.
EVs also accelerate fibrin polymerization, supporting the formation of denser clots that are more resistant to fibrinolysis by tissue plasminogen activator (tPA). Platelet-derived EV also attach to the fibrin fibers [79, 80]. When EVs are removed from the test sample the final clot formation is slowed, with reduced fibrin polymerization leading to increased fibrinolysis in response to added tPA [80].
Extracellular vesicles and malignancies
The complex relationship of EV and cancers is under investigation. These investigations point roles of EV in the metastatic process, cancer progression, drug- and radiotherapy-resistance and in cancer-related hypercoagulation [16, 48, 52,53,54, 81,82,83,84,85,86].
EVs derived from ovarian cancer, prostate cancer and fibrosarcoma paradoxically have the potential of potentiating fibrinolysis [8, 87,88,89,90,91,92]. The EV-associated urokinase plasminogen activator appears capable of promoting invasion of prostate cancer [89]. The EVs that are shed during blood storage loose their fibrinolytic capacity the longer the red cells are stored [92]. It is not yet known how these findings fit into in vivo clotting/fibrinolytic balance or for the metastatic process.
Activated platelets interact with cancer cells by way of P-selectin and the cancer PSGL-1 expressed on malignant cells [93]. Platelet-derived EV promotes tumor growth and tumor-induced angiogenesis [94, 95]. Melanoma cells release exosomes that change the local and systemic microenvironment so as to better support tumor growth and metastasis [52, 54]. They prepare the sentinel lymph node to trap and support growth of the incoming metastatic melanoma cells by inducing lymphangiogenesis in preparation of a premetastatic niche [53, 96]. This process is dependent upon transmembrane proteins. The tetraspanin-integrin complex contributes to the binding of exosomes to their target cells, possibly mediated by P-selectin/PSGL-1 complex [96,97,98,99].
As discussed above, tissue factor is activated by protein disulfide isomerases (PDI) [56, 100,101,102,103,104]. It is also known that many cancers are dependent upon protein disulfide isomerase for survival and metastasis [105]. Thus PDI inhibitors may serve both as anticoagulant and as tumor suppressant [68].
The EVs derived from cancer cells appear to carry some specific physiologic activities, including the increased tissue factor [6, 11, 106,107,108,109,110,111,112,113,114,115]. This blood-borne tissue factor may contribute to the hypercoagulation syndrome that accompanies many cancers [6, 110, 113,114,115,116,117]. The amount of tissue factor-positive EV correlates with venous thrombosis seen in patients with cancer [7, 113,114,115,116]. In another study epithelial cancer cells adopted mesenchymal features upon exposure to activated epithelial growth fact receptor coupled with blockade of E-cadherin, leading to a surge of released EV-containing epidermal growth factor receptor (EGFR) and tissue factor [117]. Upon transfer of the tissue factor to cultured endothelial cells, they become procoagulant [11, 117]. Cancer cell-derived EV carrying PSGL-1 can accelerate thrombus formation in vivo by aggregating platelets via the tissue factor-dependent pathway. Their intact parent cells do not have the same capacity [113, 114]. When microparticles derived from tumor cell are injected into mice acute thrombocytopenia and signs of shock follow. This was prevented by prior heparinization [42]. When exposed to hypoxic conditions tumor cell-derived EVs demonstrate the potential for increasing angiogenesis and facilitating metastasis, leading to changes in cell–cell and cell-extracellular matrix interaction allowing for increased invasiveness [118].
Further exploration of extracellular vesicle activity on vascular endothelium
Endothelial cells are involved with tumor growth, tumor-induced angiogenesis and angiocrine functions for self-renewal and differentiation following trauma and thrombosis [119, 120].
As demonstrated with phage display, endothelium expresses different receptors depending upon their organ of origin and their functional status [121,122,123,124,125]. This phenomenon has been nicknamed endothelial ZIP codes [126]. This may have therapeutic implications.
Endothelial dysfunction occurs with many pathological states, including sepsis, thrombotic thrombocytopenia purpura, pulmonary hypertension, sickle cell diseases, activation of the complement C5-9 membrane attack complex and exposure to cytotoxic chemotherapy among others [108,109,110]. These result in changes of phenotype for thrombo-resistance including decreased production of thrombomodulin, tissue-derived plasminogen activator (tPA), heparan sulfate, plasminogen activator inhibitor-1 (PAI-1), but also increased expression of selectins, including over expression of P-selectin from the Weibel–Palade bodies thus leading increased production of procoagulant activity [126]. The over expression of P-selectin facilitates further attachment of EV allowing for further expression of its tissue factor.
The family of selectins and their ligands play roles in the metastatic process and possibly organ selectivity for metastasis. P-selectin facilitates the initial attachment, with subsequent other cellular adhesion molecules (CAMs) furthering the process, including interaction with platelet- and fibrinogen-causing clots with further anchoring of circulating malignant cells [127,128,129,130,131]. Increased adhesion for monocytes and leukocytes further accelerate the clotting process, leading to increased endothelial cell apoptosis and shear-induced endothelial cell loss causing exposure of subendothelial substances [132].
The angiocrinic function of endothelial cells has been reviewed describing its influence upon tumor growth, as well as organ regeneration and differentiation [121]. The endothelial cells preserve specific niches useful for selective stem cell implantation. This process is associated with inhibitory or stimulatory activities that effect downstream trophagens and cytokines that in turn regulate adoptive healing and metastatic processes [52, 54, 95, 119, 120, 133].
Annexin I and annexin V are cell-specific receptors for the exposed phosphatidyl serine on the EV surfaces, working synergistically with its PSGL-1 ligand for P-selectin [24, 41, 55, 134,135,136].
Vascular ligand–receptor mapping
Combinatorial screenings with phage display can be used to identify peptides and proteins with high affinity and specificity for EV [137, 138]. The technique has been used to examine ligand–receptor interactions on the endothelial cells of blood vessels allowing selection of peptides that bind specifically to different vascular beds [123, 138,139,140,141]. This technique has been used for unbiased mapping of vascular diversity, the vascular ZIP codes [142, 143]. These ZIP codes are conceived to be the basis for specific ligand delivery on intravascular endothelium. Furthermore, it is conceived that these ZIP codes may vary in certain diseases, including cancers [142, 143]. They are evolving into site-specific targets for drug delivery for prevention of metastatic cell deposits and for the treatment of cancers [141, 142].
It appears possible that phosphatidyl serine receptor-mediated actions as assayed using the ZIP code identification may allow detection of differences in the receptor density based upon source of the endothelial cells and thus differences of EV adherence to those cells. This may add to our understanding of why certain veins are more likely to anchor circulating malignant cells and/or serve as a more effective platform have thrombosis.
Blocking EV function
Creating agents that specifically interfere with the activity of selectin or their ligands is a major area of pharmacologic study [144]. Among already available agents are the HMG-CoA reductase inhibitors that reduce expression of P-selectin so as to reduce anchoring of EVs to the venous endothelium [76,77,78,79,80,81, 145]. Therapy with statin agents interferes with platelet microparticle (EV) attachment, seemingly causing a fall in the tissue factor expression and subsequent thrombin generation [79, 81, 145]. This may become a model for therapies in other diseases wherein EV play facilitating roles. Another example is a novel recombinant homodimer of annexin V, Diannexin, which binds to and shields phosphatidylserine thereby suppressing phosphatidylserine-mediated leukocyte and platelet attachments [146]. It has been used with success in animal models for preventing reperfusion injury after transplants of lung, muscle, kidney, liver, and islet cell transplants, and following myocardial infarct [147,148,149,150,151]. Quercetin-3-rutinoside blocks EV-related tissue factor activation and platelet aggregation. This latter agent may become a new class of anticoagulant [56, 105]. These same agents may reduce the potential of other EV functions, thus suppressing metastatic cell anchorage and tumor-related angiogenesis. Agents that block Tim4 activity, such as anti-Tim4 antibody, may have future roles, as may anti-P-selectin and P-selectin glycoprotein ligand -1 antibodies [35].
Conclusions
While some details are known about the roles of circulating EV’s in the complex physiologic and pathophysiologic activities of endothelium there is much yet to be examined. It may be instructive to determine if EV from different source cells adhere to different organ-sourced endothelial cells with different densities and with differing endothelial cell response. It is unknown whether there is a steady state of microparticle attachment to endothelial surfaces in normal circumstances. Are there differences of any such density of adherence depending upon the site of origin of these veins? Is there an accelerated adherence of these particles in disease states that effect endothelial function and integrity? Is there enough of a difference in this density to explain difference in the rates of deep vein thrombosis in certain vessels and the placement of malignant masses in certain vessels but not others? Whether there are specific receptors that facilitate the incorporation of the EV’s membranous and submembranous contents into the interior of the adherent cells is another area in need of further study. Can cancer-derived EV facilitate the initiation of tumor metastasis cascade more efficiently than do intact circulating tumor cells?
References
Lötvall J, Hill AF, Hochberg F, Buzás EI, Di Vizio D, Gardiner C et al (2014) Minimal experimental requirements for definition of extracellular vesicles and their functions: a position statement from the International Society for Extracellular Vesicles. J Extracell Vesicles 3:26913. doi:10.3402/jev.v3.26913
Mathivanan S, Ji H, Simpson RJ (2010) Exosomes: extracellular organelles important in intercellular communication. J Proteom 10(73):1907–1920. doi:10.1016/j.jprot.2010.06.006
Kreimer S, Belov AM, Ghiran I, Murthy SK, Frank DA, Ivanov AR (2015) Mass-spectrometry-based molecular characterization of extracellular vesicles: lipidomics and proteomics. J Proteome Res 14:2367–2384. doi:10.1021/pr501279t
Vlassov AV, Magdaleno S, Setterquist R, Conrad R (2012) Exosomes: current knowledge of their composition, biological functions, and diagnostic and therapeutic potentials. Biochim Biophys Acta 1820:940–948. doi:10.1016/j.bbagen.2012.03.017
van der Pol E, Böing AN, Gool EL, Nieuwland R (2016) Recent developments in the nomenclature, presence, isolation, detection and clinical impact of extracellular vesicles. J Thromb Haemost 14:48–56. doi:10.1111/jth.13190
Zahra S, Anderson JA, Stirling D, Ludlam CA (2011) Microparticles, malignancy and thrombosis. Br J Haematol 152:688–700. doi:10.1111/j.1365-2141.2010.08452.x
Zwicker JI, Trenor CC 3rd, Furie BC, Furie B (2011) Tissue factor-bearing microparticles and thrombus formation. Arterioscler Thromb Vasc Biol 31:728–733. doi:10.1161/ATVBAHA.109.200964
Lacroix R, Dignat-George F (2012) Microparticles as a circulating source of procoagulant and fibrinolytic activities in the circulation. Thromb Res 129(Suppl):27–29. doi:10.1016/j.thromres.2012.02.025
Hussein MA, Böing AN, Biró E, Hoek FJ, Vogel GM, Meuleman DG et al (2008) Phospholipid composition of in vitro endothelial microparticles and their in vivo thrombogenic properties. Thromb Res 121:865–871
Vickers KC, Remaley AT (2012) Lipid-based carriers of microRNAs and intercellular communication. Curr Opin Lipidol 23:91–97. doi:10.1097/MOL.0b013e328350a425
Collier ME, Mah PM, Xiao Y, Maraveyas A, Ettelair C (2013) Microparticle-associated tissue factor is recycled by endothelial cells resulting in enhanced surface tissue factor activity. Thromb Haemost 110:966–976. doi:10.1160/TH13-01-0055
Falati S, Liu Q, Gross P, Merrill-Skoloff G, Chou J, Vandendries E et al (2003) Accumulation of tissue factor into developing thrombi in vivo is dependent upon microparticle P-selectin glycoprotein ligand 1 and platelet P-selectin. J Exp Med 197:1585–1598
Hrachovinová I, Cambien B, Hafezi-Moghadam A, Kappelmayer J, Camphausen RT, Widom A et al (2003) Interaction of P-selectin and PSGL-1 generates microparticles that correct hemostasis in a mouse model of hemophilia A. Nat Med 9:1020–1025
Hannun YA, Obeid LM (2011) Many ceramides. J Biol Chem 286:27855–27862. doi:10.1074/jbc.R111.254359
Valadi H, Ekström K, Bossios A, Sjöstrand M, Lee JJ, Lötvall JO (2007) Exosome-mediated transfer of mRNAs and microRNA is a novel mechanism of genetic exchange between cells. Nat Cell Biol 9:654–659. doi:10.1038/ncb1596
Thakur BK, Zhang H, Becker A, Matei I, Huang Y, Costa-Silva B et al (2014) Double-stranded DNA in exosomes: a novel biomarker in cancer detection. Cell Res 24:766–769. doi:10.1038/cr.2014.44
Zomer A, Vendrig T, Hopmans ES, von Eijndhoven M, Middeldorp JM, Pegtel DM (2010) Exosomes: fit to deliver small RNA. Commun Integr Biol 3:447–450. doi:10.4161/cib.3.5.12339
Gustafson CM, Shepherd AJ, Miller VM, Jayachandran M (2015) Age- and sex-specific differences in blood-borne microvesicles from apparently healthy humans. Biol Sex Differ 6:10. doi:10.1186/s13293-015-0028-8
Rautou PE, Mackman N (2013) Microvesicles as risk markers for venous thrombosis. Exp Rev Hematol 6:91–101. doi:10.1586/ehm.12.74
Herring JM, McMichael MA, Smith SA (2013) Microparticles in health and disease. J Vet Intern Med 27:1020–1033. doi:10.1111/jvim.12128
Cocucci E, Racchetti J, Meldolesi J (2009) Shedding microvesicles: artifacts no more. Trends Cell Biol 19:43–51. doi:10.1016/j.tcb.2008.11.003
Ayers L, Nieuwland R, Kohler M, Kraenkel N, Ferry B, Leeson P (2015) Dynamic microvesicle release and clearance within the cardiovascular system: triggers and mechanisms. Clin Sci 129:915–931. doi:10.1016/j.tcb.2008.11.003
Johnstone RM, Mathew A, Mason AB, Teng K (1991) Exosome formation during maturation of mammalian and avian reticulocytes: evidence that exosome release is a major route for externalization of obsolete membrane proteins. J Cell Physiol 147:27–36
Stace CL, Ktistakis NT (2006) Phosphatidic acid- and phosphatidylserine-binding proteins. Biochem Biophys Acta 1761:913–926
Ye H, Li B, Subramanian V, Choi BH, Liang Y, Harikishore A et al (2013) NMR solution structure of C2 domain of MFG-E8 and insights into its molecular recognition with phosphatidylserine. Biochim Biophys Acta 1828:1083–1093. doi:10.1016/j.bbamem.2012.12.009
Tietjen GT, Gong Z, Chen CH, Vargas E, Crooks JE, Cao KD et al (2014) Molecular mechanism for differential recognition of membrane phosphatidylserine by the immune regulatory receptor Tim4. Proc Natl Acad Sci USA 111:E1463–E1472. doi:10.1073/pnas.1320174111
Ludwig A, Giebel B (2012) Exosomes: small vesicles participating in intercellular communication. Int J Biochem Cell Biol 44:11–15. doi:10.1016/j.biocel.2011.10.005
Li XB, Zhang ZR, Schluesener HJ, Xu SQ (2006) Role of exosomes in immune regulation. J Cell Mol Med 10:364–375. doi:10.1111/j.1582-4934.2006.tb00405.x
Hauser P, Wang S, Didenka W (2017) Apoptotic bodies: selective detection in extracellular vesicles. Methods Mol Biol 1554:193–200. doi:10.1007/978-1-4939-6759-9_12
Muller L, Hong CS, Stolz DB, Watkins SC, Whiteside TL (2014) Isolation of biologically-active exosomes from human plasma. J Immunol Methods 411:55–65. doi:10.1016/j.jim.2014.06.007
Lacroix R, Judicone C, Poncelet P, Rovert S, Arnaud L, Sampol J et al (2012) Impact of pre-analytical parameters on the measurement of circulating microparticles: towards standardization of protocol. J Thromb Haemost 10:437–446. doi:10.1111/j.1538-7836.2011.04610.x
Januszyk K, Lima CD (2010) Structural components and architectures of RNA exosomes. Adv Exp Med Biol 702:9–28
Poncelet P, Robert S, Bailly N, Garnache-Ottou F, Bouriche T, Devalet B, Segatchian JH et al (2015) Tips and tricks for flow cytometry-based analysis and counting of microparticles. Transfus Apher Sci 53:110–126. doi:10.1016/j.transci.2015.10.008
Nakai W, Yoshida T, Diez D, Miyatake Y, Nishibu T, Imawaka N et al (2016) A novel affinity-based method for the isolation of highly purified extracellular vesicles. Sci Rep 6(33935):1–11. doi:10.1038/srep33935
Miyanishi M, Tada K, Koike M, Uchiyama Y, Kitamura T, Nagata S et al (2007) Identification of Tim4 as a phosphatidylserine receptor. Nature 450:435–439
Moreno-Gonzalo O, Villarroya-Beltri C, Sánchez-Madrid F (2014) Post-translational modifications of exosomal proteins. Front Immunol 5:383. doi:10.3389/fimmu.2014.00383
Szabó-Taylor K, Ryan B, Osteikoetxea X, Szabó TG, Sódar B, Holub M et al (2015) Oxidative and other posttranslational modifications in extracellular vesicle biology. Semin Cell Dev Biol 40:8–16. doi:10.1016/j.semcdb.2015.02.012
Charrin S, Manie S, Oualid M, Billard M, Boucheix C, Rubinstein E (2002) Differential stability of tetraspanin–tetraspanin interactions: role of palmitoylation. FEBS Lett 516:139–144
Henson PM, Bratton DL, Fadok VA (2001) Apoptotic cell removal. Curr Biol 11:R795–R805
Zhang Y, Meng H, Ma R, He Z, Wu X, Cao M et al (2016) Circulating microparticles, blood cells, and endothelium induce procoagulant activity in sepsis through phosphatidylserine exposure. Shock 45:299–307. doi:10.1097/SHK.0000000000000509
Jansen F, Yang X, Hoyer FF, Paul K, Heiermann N, Becher MU et al (2012) Endothelial microparticle uptake in target cells in annexin 1/phosphatidylserine receptor dependent and prevents apoptosis. Arterioscler Thromb Vasc Biol 32:1925–1935. doi:10.1161/ATVBAHA.112.253229
Brown S, Heinisch I, Ross E, Shaw K, Buckley CD, Savill J (2002) Apoptosis disables CD31-mediated cell detachment from phagocytes promoting binding and engulfment. Nature 418:200–203
Dasgupta SK, Abdel-Monem H, Niravath P, Le A, Bellera RV, Langlois K (2009) Lactadherin and clearance of platelet-derived microvesicles. Blood 113:1332–1339. doi:10.1182/blood-2008-07-167148
Willekens F, Werre JM, Kruijt JK, Roerdinkholder-Stoelwinder B, Groenen-Döpp YA, van den Bos AG et al (2005) Liver Kupffer cells rapidly remove red blood cell-derived vesicles from the circulation by scavenger receptors. Blood 105:2141–2145
Al Faraj A, Gazeau F, Wilhelm C, Devue C, Guérin CL, Péchoux C et al (2012) Endothelial cell-derived microparticles loaded with iron oxide nanoparticles: feasibility of MR imaging monitoring in mice. Radiology 263:169–178. doi:10.1148/radiol.11111329
Qiu Q, Xiong W, Yang C, Gagnon C, Hardy P (2013) Lymphocyte-derived microparticles induce bronchial epithelial cells’ pro-inflammatory cytokine production and apoptosis. Mol Immunol 55:220–230. doi:10.1016/j.molimm.2013.01.017
Bastarache JA, Fremont RD, Kropski JA, Bossert FR, Ware LB (2009) Procoagulant alveolar microparticles in the lung of patients with acute respiratory distress syndrome. Am J Physiol Lung Cell Mol Physiol 297:L1035–L1041. doi:10.1152/ajplung.00214.2009
Janowska-Wieczorek A, Wysoczynski M, Kijowski J, Marquez-Curtis L, Machalinski B, Ratajczak J et al (2005) Microvesicles derived from activated platelets induce metastasis and angiogenesis in lung cancer. Int J Cancer 113:752–760
Furie B, Furie BC (2005) Thrombosis formation in vivo. J Clin Investig 115:3355–3362
Chou J, Mackman N, Merrill-Skoloff G, Pederson B, Furie BC, Furie B (2004) Hematopoietic cell-derived microparticle tissue factor contributes to fibrin formation during thrombus propagation. Blood 104:3190–3197
Simoncini S, Njock MS, Robert S, Camoin-Jau L, Sampol J, Harlé JR et al (2009) TRAIL/Apo2L mediates the release of procoagulant endothelial microparticles induced by thrombin in vitro. A potential mechanism linking inflammation and coagulation. Circ Res 104:943–951. doi:10.1161/CIRCRESAHA.108.183285
Kahlert C, Kalluri R (2013) Exosomes in tumor microenvironment influence cancer progression and metastasis. J Mol Med 91:431–437. doi:10.1007/s00109-013-1020-6
Hood JL, San RS, Wickline SA (2011) Exosomes released by melanoma cells prepare sentinel lymph nodes for tumor metastasis. Cancer Res 71:3792–3801. doi:10.1158/0008-5472
Rinderknecht M, Detmar M (2008) Tumor lymphangiogenesis and melanoma metastasis. J Cell Physiol 216:347–354. doi:10.1002/jcp.21494
Cambien B, Wagner DD (2004) A new role in hemostasis for the adhesion receptor P-selectin. Trends Mol Med 10:179–186
Furie B, Flaumenhaft R (2014) Thiol isomerases in thrombus formation. Clin Res 114:1162–1173. doi:10.1161/CIRCRESAHA.114.301808
Zhou L, Qi XL, Xu MX, Mao Y, Liu ML, Song HM (2014) Microparticles: new light shed on the understanding of venous thromboembolism. Acta Pharmacol Sin 35:1103–1110. doi:10.1038/aps.2014.73
Furie B, Furie BC (2004) Role of platelet P-selectin and microparticle PSGL-1 in thrombus formation. Trends Mol Med 10:171–178
Vandendries ER, Furie BC, Furie B (2004) Role of P-selectin and PSGL-I in coagulation and thrombosis. Thromb Haemost 92:459–466
McEver RP, Beckstead JH, Moore KL, Marshall-Carlson L, Bainton DF (1989) GMP-140, a platelet alpha-granule membrane protein, is also synthesized by vascular endothelial cells and is localized in Weibel–Palade bodies. J Clin Investig 84:92–99
Montoro-Garcia S, Shantsila E, Hermández-Romero D, Jover E, Valdés M, Marin F et al (2014) Small-size platelet microparticles trigger platelet and monocyte functionality and modulate thrombogenesis via P-selectin. Br J Haematol 166:571–580. doi:10.1111/bjh.12913
Setty BN, Betal SG (2008) Microvascular endothelial cells express a phosphatidylserine receptor: a functionally active receptor for phosphatidylserine-positive erythrocytes. Blood 111:905–914
Lopez-Vilchez I, Diaz-Ricart M, Galan AM, Roque M, Caballo C, Molina P et al (2016) Internalization of tissue factor-rich microvesicles by platelets occurs independently of GPIIb-IIIa, and involves CD36 receptor, serotonin transporter and cytoskeletal assembly. J Cell Biochem 117:448–457. doi:10.1002/jcb.25293
Manodori AB, Barabino GA, Lubin BH, Kuypers FA (2000) Adherence of phosphatidylserine-exposing erythrocytes to endothelial matrix thrombospondin. Blood 95:1293–1300
Sabatier F, Roux V, Anfosso F, Camoin L, Sampol J, Dignat-George F (2002) Interaction of endothelial microparticles with monocytic cells in vitro induces tissue factor-dependent procoagulant activity. Blood 99:3962–3970
Furie B, Furie BC, Flaumenhaft R, Zwicker JI (2014) Animal models of arterial and venous thrombosis. Blood 124:SCI 2
Reinhardt C, von Brühl ML, Manukyan D, Grahl L, Lorenz M, Altmann B et al (2008) Protein disulfide isomerase acts as an injury response signal that enhances fibrin generation via tissue factor activation. J Clin Investig 118:1110–1122. doi:10.1172/JCI32376
Jasuja R, Passam FH, Kennedy DR, Kim SH, van Hessem L, Lin L et al (2012) Protein disulfide isomerase inhibitors constitute a new class of antithrombotic agents. J Clin Investig 122:2104–2113. doi:10.1172/JCI61228
Hamilton KK, Hattori R, Esmon CT, Sims PJ (1990) Complement proteins C5b-9 induce vesiculation of the endothelial plasma membrane and expose catalytic surface for the assembly of the prothrombinase enzyme complex. J Biol Chem 265:3809–3814
Bidot L, Jy W, Bidot C Jr, Jimenez JJ, Fontana V, Horstman LL et al (2008) Microparticle-mediated thrombin generation assay: increased activity in patients with recurrent thrombosis. J Thromb Haemost 6:913–919. doi:10.1111/j.1538-7836.2008.02963.x
Mobarrez F, He S, Bröijersen A, Wiklund B, Antovic A, Antovic J et al (2011) Atrovastatin reduces thrombin generation and expression of tissue factor, P-selectin and GPIIIa on platelet-derived microparticles in patients with peripheral arterial occlusive disease. Thromb Haemost 106:344–352. doi:10.1160/TH10-12-0810
Romano M, Mezzetti A, Marulli C, Ciabattoni G, Febo F, Di Ienno S et al (2000) Fluvastatin reduces soluble P-selectin and ICAM-1 levels on hypercholesterolemic patients: role of nitric oxide. J Investig Med 48:183–189
Berkan O, Katrancioglu N, Ozker E, Ozerdem G, Bakici Z, Yilmaz MB (2009) Reduced P-selectin in hearts pretreated with fluvastatin: a novel benefit for patients undergoing open heart surgery. Thorac Cardiovasc Surg 57:91–95. doi:10.1055/s-2008-1039107
Mobarrez F, Egberg N, Antovic J, Bröijersen A, Jörenkog G, Wallén H (2012) Release of endothelial microparticles in vivo during atorvastatin treatment; a randomized double blind placebo-controlled study. Thromb Res 129:95–97. doi:10.1016/j.thromres.2011.09.027
Connor DE, Exner T, Ma DD, Josepth JE (2010) The majority of circulating platelet-derived microparticles fail to bind annexin V, lack phospholipid-dependent procoagulant activity and demonstrate greater expression of glycoprotein 1B. Thromb Haemost 103:1044–1052. doi:10.1160/TH09-09-0644
Ravanat C, Archipoff G, Beretz A, Freund G, Cazenave JP, Freyssinet JM (1992) Use of annexin-V to demonstrate the role of phosphatidylserine exposure in the maintenance of haemostatic balance by endothelial cells. Biochem J 282:7–13
Albanyan AM, Murphy MF, Rasmussen JT, Heegaard CW, Harrison P (2009) Measurement of phosphatidylserine exposure during storage of platelet concentrates using the novel probe lactadherin: a comparison study with annexin V. Transfusion 49:99–107. doi:10.1111/j.1537-2995.2008.01933.x
Ravassa S, Bennaghmouch A, Kenis H, Lindhout T, Hackeng T, Narula J et al (2005) Annexin A5 down-regulates surface expression of tissue factor: a novel mechanism of regulating the membrane receptor repertoir. J Biol Chem 280:6028–6035
Zubairova LD, Nabiulina RM, Nagaswami C, Zuev YE, Mustafin IG, Litvinov RI et al (2015) Circulating microparticles alter formation, structure, and properties of fibrin clots. Sci Rep 5:17611. doi:10.1038/srep17611
Nabiullina RM, Mustafin IG, Zuev YF, Faizullin DA, Litvinov RI, Zubairova LD (2015) Effect of blood microparticles on the kinetics of polymerization and enzymatic hydrolysis of fibrin. Dokl Biochem Biophys 462:151–154. doi:10.1134/S1607672915030035
Milane L, Singh A, Mattheolabakis G, Suresh M, Amiji MM (2015) Exosome mediated communication within the tumor microenvironment. J Control Release 219:278–294. doi:10.1016/j.jconrel.2015.06.029
Brinton LT, Sloane HS, Kester M, Kelly KA (2015) Formation and role of exosomes in cancer. Cell Mol Life Sci 72:659–671. doi:10.1007/s00018-014-1764-3
Chin AR, Wang SE (2016) Cancer-derived extracellular vesicles: the ‘soil conditioner’ in breast cancer metastasis. Cancer Metastasis Rev 35:669–676. doi:10.1007/s10555-016-9639-8
Suchorska WM, Lach MS (2016) The role of exosomes in tumor progression and metastasis. Oncol Rep 25:1237–1244. doi:10.3892/or.2015.4507
Pultz BDA, Luz FAC, Faria SS, Souza LPF, Tavares PCB, Goulart VA et al (2017) The multifaceted role of extracellular vesicles in metastasis: priming the soil for seeding. Int J Cancer 140:2397–2407. doi:10.1002/ijc.30595
Lobb RJ, van Amerongen R, Wiegmans A, Ham S, Larsen JE, Möller A (2017) Exosomes derived from mesenchymal non-small cell lung cancer cells promote chemoresistance. Int J Cancer 141:614–620. doi:10.1002/ijc.30752
Dolo V, D’Ascenzo S, Violini S, Pompucci L, Festuccia C, Ginestra A et al (1999) Matrix-degrading proteinases are shed in membrane vesicles by ovarian cancer cells in vivo and in vitro. Clin Exp Metastasis 17:131–140
Ginestra A, Monea S, Seghezzi G, Dolo V, Nagase H, Mignatti P et al (1997) Urokinase plasminogen activator and gelatinases are associated with membrane vesicles shed by human HT1080 fibrosarcoma cells. J Biol Chem 272:17216–17222
Angelucci A, D’Ascenzo S, Festuccia C, Gravina GL, Bologa M, Dolo V et al (2000) Vesicle-associated urokinase plasminogen activator promotes invasion in prostate cancer cells lines. Clin Exp Metastasis 18:163–170
Brodsky SV, Malinowski K, Golightly M, Jesty J, Goligorsky MS (2002) Plasminogen activator inhibitor-I promotes formation of endothelial microparticles with procoagulant potential. Circulation 106:2372–2378
Lacroix R, Sabatier F, Mialhe A, Basire A, Pannell R, Borghi H et al (2007) Activation of plasminogen into plasmin at the surface of endothelial microparticles: a mechanism that modulates angiogenic properties of endothelial progenitor cells in vitro. Blood 110:2432–2439
Levin G, Sukhareva E, Lavrentieva A (2016) Impact of microparticles derived from erythrocytes on fibrinolysis. J Thromb Thrombolysis 41:452–458. doi:10.1007/s11239-015-1299-y
Gong L, Cai Y, Zhou X, Yang H (2012) Activated platelets interact with lung cancer cells through P-selectin glycoprotein ligand-1. Pathol Oncol Res 18:989–996. doi:10.1007/s12253-012-9531-y
Goubran H, Sabry W, Kolb R, Seghatchian J, Burnouf T (2015) Platelet microparticles and cancer: an intimate cross-talk. Transfus Apher Sci 53:168–172. doi:10.1016/j.transci.2015.10.014
Sierko E, Wojtukiewicz MZ (2004) Platelets and angiogenesis in malignancy. Semin Thromb Hemost 30:95–108
Hood JL, Pan H, Lanza GM, Wickline SA (2009) Consortium for translational research in advanced imaging and nanomedicine (C-TRAIN) paracrine induction of endothelium by tumor exosomes. Lab Investig 89:1317–1328. doi:10.1038/labinvest.2009.94
Nazarenko I, Rana S, Baumann A, McAlear J, Hellwig A, Trendelenburg M et al (2010) Cell surface tetraspanin Tspan8 contributes to molecular pathways of exosome-induced endothelial cell activation. Cancer Res 70:1668–1678. doi:10.1158/0008-5472.CAN-09-2470
Rana S, Yue S, Stadel D, Zoller M (2012) Toward tailored exosomes: the exosomal tetraspanin web contributes to target cell selection. Int J Biochem Cell Biol 44:1574–1584. doi:10.1016/jbiocel.2012.06.018
Chen M, Geng JG (2006) P-selectin mediates adhesion of leukocytes, platelets, and cancer cells in inflammation, thrombosis, and cancer growth and metastasis. Arch Immunol Ther Exp 54:75–84. doi:10.1007/s00005-006-0010-6
Cho J, Furie BC, Coughlin SR, Furie B (2008) A critical role for extracellular protein disulfide isomerase during thrombus formation in mice. J Clin Investig 118:1123–1131. doi:10.1172/JCI34134
Flaumenhaft R, Furie B (2016) Vascular thiol isomerases. Blood 128:893–901. doi:10.1182/blood-2016-04-636456
Chen VM (2013) Tissue factor de-encryption, thrombus formation, and thiol-disulfide exchange. Semin Thromb Hemost 39:40–47. doi:10.1055/s-0032-1333311
Schulman S, Bendapudi P, Sharda A, Chen V, Bellido-Martin L, Jasuja R et al (2016) Extracellular thiol isomerases and their role in thrombus formation. Antioxid Redox Signal 24:1–15. doi:10.1089/ars.2015.6530
Ruf W (2012) Role of thiol pathways in TF procoagulant regulation. Thromb Res 129(Suppl 2):11–12. doi:10.1016/j.thromres.2012.02.020
Xu S, Sankar S, Neamati N (2014) Protein disulfide isomerase: a promising target for cancer therapy. Drug Discov Today 19:222–240. doi:10.1016/j.drudis.2013.10.017
Tesselaar ME, Romijn FP, Van Der Linden IK, Prins FA, Bertina RM, Osanto S (2007) Microparticle-associated tissue factor activity: a link between cancer and thrombosis? J Thromb Haemost 5:520–527
Hron G, Kollars M, Weber H, Sagaster V, Quehenberger P, Eichinger S et al (2007) Tissue factor-positive microparticles: cellular origin and association with coagulation activation in patients with colorectal cancer. Thromb Haemost 97:119–123
Simák J, Holada K, Vostal JG (2002) Release of annexin V-binding membrane microparticles from cultured human umbilical endothelial cells after treatment with camptothecin. BMC Cell Biol 3:11–21. doi:10.1186/1471-2121-3-11
Brunetta DM, De Santis GC, Silva-Pinto AC, Oliveira de Oliveira LC, Covas DT (2015) Hydroxyurea increases plasma concentrations of microparticles and reduces coagulation activation and fibrinolysis in patients with sickle cell disease. Acta Haematol 133:287–294. doi:10.1159/000362148
Lysov Z, Dwivedi D, Gould TJ, Liaw PC (2017) Procoagulant effects of lung cancer chemotherapy: impact on microparticles and cell-free DNA. Blood Coagul Fibrinolysis 28:72–82. doi:10.1097/MBC.0000000000000546
Fernandez PM, Rickles FR (2002) Tissue factor and angiogenesis in cancer. Curr Opin Hematol 9:401–406
Bromberg ME, Konigsberg WH, Madison JF, Pawashe A, Garen A (1995) Tissue factor promotes melanoma metastasis by a pathway independent of blood coagulation. Proc Natl Acad Sci USA 92:8205–8209
Thomas GM, Panicot-Dubois L, Lacroix R, Dignat-George F, Lombardo D, Dubois C (2009) Cancer cell-derived microparticles bearing P-selectin glycoprotein ligand 1 accelerate thrombus formation in vivo. J Exp Med 206:1913–1927. doi:10.1084/jem.20082297
Thomas GM, Brill A, Mezouar S, Crescence L, Gallant M, Dubois C et al (2015) Tissue factor expressed by circulating cancer cell-derived microparticles drastically increases the incidence of deep vein thrombosis in mice. J Thromb Haemost 13:1310–1319. doi:10.1111/jth.13002
Geddings JE, Mackman N (2013) Tumor-derived tissue factor-positive microparticles and venous thrombosis in cancer patients. Blood 122:1873–1880. doi:10.1182/blood-2013-04-460139
Khorana AA, Francis CW, Menzies KE, Wang JG, Hyrien O, Hathcock J et al (2008) Plasma tissue factor may be predictive of venous thromboembolism in pancreatic cancer. J Thromb Haemost 6:1983–1985. doi:10.1111/j.1538-7836.2008.03156.x
Garnier D, Magnus N, Lee TH, Bentley V, Meehan B, Milsom C et al (2012) Cancer cells induced to express mesenchymal phenotype release exosome-like extracellular vesicles carrying tissue factor. J Biol Chem 287:43565–43572. doi:10.1074/jbc.M112.401760
Park JE, Tan HS, Datta A, Lai RC, Zhang H, Meng W et al (2010) Hypoxic tumor cell modulates its microenvironment to enhance angiogenic and metastatic potential by secretion of proteins and exosomes. Mol Cell Proteom 9:1085–1099. doi:10.1074/mcp.M900381-MCP200
Butler JM, Kobayashi H, Rafii S (2010) Instructive role of the vascular niche in promoting tumour growth and tissue repair by angiocrine factors. Nat Rev Cancer 10:138–146. doi:10.1038/nrc2791
Rafii S, Butler JM, Ding BS (2016) Angiocrine functions of organ-specific endothelial cells. Nature 529:316–325. doi:10.1038/nature17040
Pasqualini R, Ruoslahti E (1996) Organ targeting in vivo using phage display peptide libraries. Nature 380:364–366
Rajotte D, Arap W, Hagedorn M, Koivunen E, Pasqualini R, Ruoslahti E (1998) Molecular heterogeneity of the vascular endothelium revealed by in vivo phage display. J Clin Investig 102:430–437
Trepel M, Pasqualini R, Arap W (2008) Screening phage-display peptide libraries for vascular targeted peptides. Methods Enzymol 445:83–106. doi:10.1016/S0076-6879(08)03004-8
Bredesen DE, Pasqualini R, Ruoslahti E (2002) Targeting the prostate for destruction through vascular address. Proc Natl Acad Sci USA 99:1527–1531
Arap W, Kolonin MG, Trepel M, Lahdenranta J, Cardó-Vila M, Giordano RJ et al (2002) Steps toward mapping the human vasculature by phage display. Nat Med 8:121–127
Ruoslahti E (2004) Vascular zip codes in angiogenesis and metastasis. Biochem Soc Trans 32:397–402. doi:10.1042/BST0320397
Vallet B, Wiel E (2001) Endothelial cell dysfunction and coagulation. Crit Care Med 29(Suppl 7):36–41
Borsig L, Wong R, Feramisco J, Nadeau DR, Varki NM (2001) Varki A Heparin and cancer revisited: mechanistic connections involving platelets, P-selectin, carcinoma mucins, and tumor metastasis. Proc Natl Acad Sci USA 98:3352–3357. doi:10.1073/pnas.061615598
Borsig L, Wong R, Hynes RO, Varki NM, Varki A (2002) Synergistic effects of L- and P-selectin in facilitating tumor metastasis can involve non-mucin ligands and implicate leukocytes as enhancers of metastasis. Proc Natl Acad Sci USA 99:2193–2198. doi:10.1073/pnas.261704098
Im JH, Fu W, Wang H, Bhatia SK, Hammer DA, Kowalska MA et al (2004) Coagulation facilitates tumor cell spreading in the pulmonary vasculature during early metastatic colony formation. Cancer Res 64:8613–8619
Biggerstaff JP, Seth N, Amirkhosravi A, Amaya M, Fogarty S, Meyer TV et al (1999) Soluble fibrin augments platelet/tumor cell adherence in vitro and in vivo, and enhances experimental metastasis. Clin Exp Metastasis 17:723–730
Bombeli T, Karsan A, Tait JF, Harlan JM (1997) Apoptotic vascular endothelial cells become procoagulant. Blood 89:2429–2442
Curtis AM, Edelberg J, Jonas R, Rogers WT, Moore JS, Syed W et al (2013) Endothelial microparticles: sophisticated vesicle modulating vascular function. Vasc Med 18:204–214. doi:10.1177/1358863X13499773
Walker B, Towhid ST, Schmid E, Hoffmann SM, Abed M, Münzer P et al (2014) Dynamic adhesion of eryptotic erythrocytes to immobilized platelets via platelet phosphatidylserine receptors. Am J Physiol Cell Physiol 306:C291–C297. doi:10.1152/ajpcell.00318.2013
van Engeland M, Nieland LJ, Ramaekers FC, Schutte B, Reutelingsperger CP (1998) Annexin V-affinity assay: a review on an apoptosis detection system based on phosphatidylserine exposure. Cytometry 31:1–9
van Genderen HO, Kenis H, Hostra L, Narula J, Reutelingsperger CP (2008) Extracellular annexin A5: functions of phosphatidylserine-binding and two-dimensional crystallization. Biochim Biophys Acta 1783:953–963. doi:10.1016/j.bbamcr.2008.01.030
Christianson DR, Ozawa MG, Pasqualini R, Arap W (2007) Techniques to decipher molecular diversity by phage display. Methods Mol Biol 357:385–406
Azzazy HM, Highsmith WE Jr (2002) Phage display technology: clinical applications and recent innovations. Clin Biochem 35:425–445
Staquicini FI, Moeller BJ, Arap W, Pasqualini R (2010) Combinatorial vascular targeting in translational medicine. Proteom Clin Appl 4:626–632. doi:10.1002/prca.200900213
Giordano RJ, Edwards JK, Tuder RM, Arap W, Pasqualini R (2009) Combinatorial ligand-directed lung targeting. Proc Am Thorac Soc 6:411–415. doi:10.1513/pats.200903-014AW
Pasqualini R, Moeller BJ, Arap W (2010) Leveraging molecular heterogeneity of the vascular endothelium for targeted drug delivery and imaging. Semin Thromb Hemost 36:343–351. doi:10.1055/s-0030-1253456
Li XB, Schluesener HJ, Xu SQ (2006) Molecular addresses of tumors: selection by in vivo phage display. Arch Immunol Ther Exp 54:177–181
Staquicini FI, Cardó-Vila M, Kolonin MG, Trepel M, Edwards JK, Nunes DR et al (2011) Vascular ligand-receptor mapping by direct combinatorial selection in cancer patients. Proc Natl Acad Sci 108:18637–18642. doi:10.1073/pnas.1114503108
Kneuer C, Ehrhardt C, Radomski MW, Bakowsky U (2006) Selectins-potential pharmacological targets? Drug Discov Today 11:1034–1040. doi:10.1016/j.drudis.2006.09.004
Hwang YS, Tsai WC, Lu YH, Lin CC, Chen YF (2004) Effect of atorvastatin on the expression of CD40 ligand and P-selectin on platelets in patients with hypercholesterolemia. Am J Cardiol 94:364–366. doi:10.1016/j.amjcard.2004.04.037
Kuypers FA, Larkin SK, Emeis JJ, Allison AC (2007) Interaction of an annexin V homodimer (Diannexin) with phosphatidylserine on cell surfaces and consequent antithrombotic activity. Thromb Haemost 97:478–486
Hale SL, Allison AC, Kloner RA (2011) Diannexin reduces no-reflow after reperfusion in rabbits with large ischemic myocardial risk zones. Cardiovasc Ther 29:e42–e52. doi:10.1111/j.1755-5922.2010.00223.x
Molski M, Groth A, Allison AC, Hendrickson M, Siemionow M (2009) Diannexin treatment decreases ischemia-reperfusion injury at the endothelial cell level of the microvascular bed in muscle flaps. Ann Plast Surg 63:564–571. doi:10.1097/SAP.0b013e3181935a4e
Wever KE, Wagener FA, Frielink C, Boerman OC, Scheffer GJ, Allison A et al (2011) Diannexin protects against renal ischemia reperfusion injury and targets phosphatidylserines in ischemic tissue. PLoS ONE 6:e24276. doi:10.1371/journal.pone.0024276
Shen XD, Ke B, Zhai Y, Tsuchihashi SI, Gao F, Duarte S et al (2007) Diannexin, a novel annexin V homodimer, protects rat liver transplants against cold ischemia-reperfusion injury. Am J Transplant 7:2463–2471
Hashimoto K, Kim H, Oishi H, Chen M, Iskender I, Sakamoto J et al (2016) Annexin V homodimer protects against ischemia reperfusion-induced acute lung injury in lung transplantation. J Thorac Cardiovasc Surg 151:861–868. doi:10.1016/j.jtcvs.2015.10.112
Acknowledgements
Diane Stanley prepared the final renditions of Figs. 1 and 2.
Competing interests
The author declares no competing interests.
Availability of data and materials
All of the data and material discussed in this review manuscript is available in the primary published articles listed in the references.
Consent for publication
Not applicable.
Ethics approval and consent to participate
Not applicable.
Funding
The Cancer Center Research Foundation.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Bern, M.M. Extracellular vesicles: how they interact with endothelium, potentially contributing to metastatic cancer cell implants. Clin Trans Med 6, 33 (2017). https://doi.org/10.1186/s40169-017-0165-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40169-017-0165-2