
Abstract
Background
An Escherichia coli (E. coli) pathotype with invasive properties, first reported by Darfeuille-Michaud and termed adherent-invasive E. coli (AIEC), was shown to be prevalent in up to half the individuals with Crohn’s Disease (CD), suggesting that these bacteria could be involved in the pathophysiology of CD. Among the genes related to AIEC pathogenicity, fim has the potential to generate an inflammatory reaction from the intestinal epithelial cells and macrophages, as it interacts with TLR4, inducing the production of inflammatory cytokines independently of LPS. Therefore, targeting the bacterial adhesion of FimH-expressing bacteria seems a promising therapeutic approach, consisting of disarming bacteria without killing them, representing a selective strategy to suppress a potentially critical trigger of intestinal inflammation, without disturbing the intestinal microbiota.
Results
We analyzed the metagenomic composition of the gut microbiome of 358 patients with CD from two different cohorts and characterized the presence of FimH-expressing bacteria. To assess the pathogenic role of FimH, we used human intestinal explants and tested a specific FimH blocker to prevent bacterial adhesion and associated inflammation. We observed a significant and disease activity-dependent enrichment of Enterobacteriaceae in the gut microbiome of patients with CD. Bacterial FimH expression was functionally confirmed in ileal biopsies from 65% of the patients with CD. Using human intestinal explants, we further show that FimH is essential for adhesion and to trigger inflammation. Finally, a specific FimH-blocker, TAK-018, inhibits bacterial adhesion to the intestinal epithelium and prevents inflammation, thus preserving mucosal integrity.
Conclusions
We propose that TAK-018, which is safe and well tolerated in humans, is a promising candidate for the treatment of CD and in particular in preventing its recurrence.
Video abstract
Similar content being viewed by others


Background
Crohn’s disease (CD) is a chronic inflammatory disease of the gastrointestinal (GI) tract involving the dynamic interaction of host’s genetics, microbiome, and inflammatory responses [1]. Compelling evidence suggests that the microbiome plays a significant role in triggering an abnormal mucosal immune response in patients with CD [2]. An Escherichia coli (E. coli) pathotype with invasive properties, first reported by Darfeuille-Michaud [3] and termed adherent-invasive E. coli (AIEC) [4], was shown to be prevalent in up to half the individuals with CD [5,6,7,8,9], suggesting that these bacteria could be involved in the pathophysiology of CD.
Besides their ability to adhere and invade, AIECs can also survive and replicate in intestinal epithelial cells and macrophages, stimulating the production of inflammatory cytokines [2, 10, 11]. The presence of non-classic virulence factors of adherence and invasion distinguishes AIECs from other E. coli strains [12,13,14]. Interestingly, the genetic factors that are characteristic of the AIEC pathotype are still unknown, as the majority of genes related to their pathogenicity are present commonly in both commensal and pathogenic E. coli strains, suggesting that pathoadaptive evolution is an important determinant of E. coli pathogenicity. Among these genes, FimH codes for a mannose-binding adhesin presented at the tip of type 1 pili, expressed by pathogenic E.coli and members of the Enterobacteriaceae family [15,16,17,18], that allows them to recognize and binds terminal mannoses on epithelial glycoproteins [19,20,21], including carcinoembryonic antigen-related cell adhesion molecule 6 (CEACAM6) [19] and Toll-like receptor 4 (TLR4) [16, 17]. Even though not reported specifically, other mannosylated abundant components like mucus might also provide high-capacity substrates for FimH attachment [22], allowing for biofilm formation and bacteria-specific mucosal immune responses [12, 15, 19, 21, 23]. Despite their role as sources of pro-inflammatory cytokines [24], normal healthy intestinal epithelial cells (IECs), as well as intestinal macrophages lack the TLR4-accessory proteins CD14 or MD2, rendering these cell types resistant to lipopolysaccharides (LPS)-induced inflammation [25,26,27,28,29,30,31,32]. In particular, resident intestinal macrophages in non-inflamed mucosa have been described as being inflammation “adverse” or “anergic” and will receive the assistance of newly recruited circulating monocytes when needs arise [27]. In sharp contrast, the CD14/MD2-lacking TLR4 in IECs and intestinal macrophages remain sensitive to the action of FimH. In turn, FimH, which is expressed by the type of pathogenic E. coli reported to be found in the GI track of patients with CD, has the potential to generate an inflammatory reaction from the intestinal epithelial cells and macrophages residing in (so far) non-inflamed mucosa. Type 1 pili-mediated adhesion to host cells is a crucial step in the establishment of E. coli adherence and subsequent invasive process [33, 34]. These mechanisms set then the stage for a selective over colonization of the epithelium by AIECs, with subsequent biofilm formation and bacteria-specific mucosal immune responses [12, 15, 19, 21, 23]. FimH serves as a trigger of inflammation via its interaction with TLR4, inducing the production of tumor necrosis factor alpha (TNFα), interleukin 6 (IL-6) and IL-8 in the gut, independently of LPS [16]. The critical role of FimH as a pro-inflammatory mediator in CD stresses the importance of designing therapeutic strategies that can disrupt this pathogenic pathway [35].
Here, we report the metagenomic analyses of fecal samples from two different cohorts of patients with CD, representing 358 patients, compared with 43 healthy volunteers. Using unbiased shotgun sequencing, we show that E. coli, which represents a moderately abundant species in healthy individuals, becomes a quantitatively important species in CD fecal samples. Our data indicate that FimH-expressing bacteria might significantly contribute to the inflammatory process in CD and confer strong support for pharmacological inhibition of FimH in patients with CD. Accordingly, the FimH-blocker TAK-018, a safe and tolerated clinical candidate, might selectively help disarm and clear the harmful FimH-expressing bacteria without directly interfering with the other members of the microbial community. Its mode of action and its expected low impact on the microbiome will clearly avoid many of the generally unacceptable complications and limitations that chronic antibiotic treatments display.
Methods
Population for metagenomic study
The first cohort of patients with CD comes from the CrohnOmeter study (Enterome-sponsored) which was an exploratory longitudinal study, conducted at the Saint-Antoine and Saint-Louis Hospitals (Paris, France) and at the French patient association l’Association François Aupetit (AFA, Paris, France). Patients were followed longitudinally for about 9 months, providing monthly stool samples collected at home. For each sample, patients filled a questionnaire, and the disease activity was assessed by the Harvey-Bradshaw index (HBI) [36] and fecal calprotectin [37]. As the E. coli abundance and HBI remain stable throughout time (Supplementary Fig1a, b), for the sake of clarity, only the first time point available for each patient is depicted in figures.
The second cohort of patients with CD comes from the PREDICT study (AbbVie sponsored) which was a multicenter, global, cross-sectional, non-interventional study. A total of 305 patients with CD were enrolled into the study, of which 284 patients provided a stool sample before an ileocolonoscopy. Relevant clinical information such as HBI, calprotectin, C-reactive protein (CRP), and endoscopic scores were monitored.
The third cohort of patients with CD comes from the multicenter, international MOBIDIC descriptive study (Enterome-sponsored) which was a longitudinal study having enrolled 143 patients; 113 of them provided peri-ulcerative ileal biopsies and their associated stool DNAs. Conclusive results were obtained on 106/113 biopsies. Relevant clinical information such as HBI, calprotectin, CRP, and endoscopic scores were monitored.
The protocols for CrohnOmeter, PREDICT, and MOBIDIC studies were approved by the institutional medical ethics committees. Participants were given oral and written information prior to signing the informed consent form.
The HV cohort comes from the MICROLEAN study (Enterome sponsored with INRA MetaGenoPolis, Jouy-en-Josas, France and Universitätsspital Basel, Switzerland). Forty-three HV were enrolled, and no formal clinical information were collected. Each subject provided one stool sample collected at home.
Stool sample preparation and processing for sequencing
Stool samples were collected at home using Sarstedt tubes (Sarstedt, Nümbrect, Germany) filled with RNAlater. On reception, the tubes were stored at – 80 °C. Sample processing, including DNA isolation, library preparation, and shotgun sequencing were performed by GATC Biotech (Konstanz, Germany). A commercial extraction kit, the QIAamp Stool DNA mini kit (Qiagen, Hilden, Germany) was used after optimization by GATC. DNA concentrations were measured using Qubit fluorometric quantitation (Life Technologies, Carlsbad, CA, USA). DNA libraries were prepared following the manufacturer’s instruction (Illumina, San Diego, CA, USA) and sequenced on a HiSeq 2500 Illumina sequencer. The target of 40 million minimum paired-end reads was generated for each sample, and sequencing read length was 100 to 125 bp.
Bioinformatics processing for metagenomic stool samples sequencing
The resulting FASTQ files were processed using a customized version of the MOCAT pipeline [38]. Quality trimmed and filtered reads (PHRED quality cut-off 20) were then mapped against Enterome’s proprietary CD catalog including 4 million genes identified from the microbiome of healthy individuals and patients with CD. Sequence reads shorter than 45 bp, mapping to Illumina adapters, or to the human genome (GRCh37) were discarded. Bacterial genes were quantified using relative abundance measurements. Gene abundances for each sample were estimated as the sum of uniquely mapped reads per gene divided by the gene length and scaled by the sum of all the reads mapped on the microbiome gene catalogue [39]. The microbiome gene catalogue construction and annotation method were adapted from Li et al. [40]. Genes were annotated using the BLASTN alignment method against KEGG and RefSeq genomic databases [41] with different identity cut-offs for gene annotation at the species (95%), genus (80%), and phylum (65%) levels, requiring at least 80% of query sequence coverage. Genes with multiple hits deprived of any consensus (defined as 10% of hits having the same annotation) for their taxonomic associations were discarded. The relative abundances at each taxonomical level were computed by summing the relative abundances of all the genes belonging to the same species, family, genus or phylum. Species annotated to “others” and species annotated to “Homo Sapiens” were excluded from the analysis.
Quantitative PCR for FimS ON and FimS OFF
DNA was extracted from 1 g of stool using the QiAmp DNA stool kit (Qiagen, Hilden, Germany). Gene sequences were obtained from an internal database and LF82 sequencing and were used to design q-PCR primers for FimS ON (forward primer: CGGATTATGGGAAAGAAAT; reverse primer: CGATGCTTTCCTCTATGA) and FimS OFF (forward primer: CGATGCTTTCCTCTATGA; reverse primer: TTGTTTTGTCAACGAGTT). The q-PCR was performed with 1X SYBR Green master mix in a 10-μl reaction volume. The reactions were carried out in 96-well plates. Each reaction comprised 2 μl of DNA, 0.4 μl of primer (10 μM each), 2.6 µl of RNase free water, and 5 μl of 2X SYBR Green master mix. The initial denaturation time was 1 min at 95 °C, followed by 45 cycles at 95 °C for 5 s, 60 °C for 30 s (annealing), 55 °C for 30 s (elongation), 72 °C for 10 s (extension). Following PCR amplification, a dissociation curve was run to examine the amplification specificity. A portion of the DNA was diluted and was used for primer validation and determination of optimal template dilutions.
Cladogram representation
The cladogram (Supp Fig2) was built using the ggtree package [42]. We extracted from Ensembl database the list of species available in the human gut microbiome and those having a FimH gene. The clustering was done using 16S genes for each of these bacteria. The tree was built using MUSCLE software with its default parameters [43].
Isolated bacterial strains
A total of 105 isolated E. coli from intestinal mucosa and fecal samples were collected from Crohn’s disease patients at the Laboratory of Molecular Microbiology, Biology Department, Universitat de Girona, Girona, Spain [7] and College of Veterinary Medicine, Cornell University, Ithaca, NY, USA [6] (Supplementary Table 4).
DNA isolation of bacterial strains and full genome sequencing
DNA isolation for whole genome sequencing of bacterial strains was outsourced to GATC Biotech (Konstanz, Germany). A commercial extraction kit, the QIAamp Stool DNA mini kit (Qiagen, Hilden, Germany) was used to obtain at least 500 ng of DNA, as required for the library preparation. To control for contamination during DNA extraction, either a Tris–HCL buffer as negative control or an aliquot of the culturing media was used. The Qubit assay (Life Technologies, Carlsbad, CA, USA) was used to assess the DNA yield and purity using Abs260/280 nm. Fragmentation using gels was also performed on bacterial DNA to ensure that high molecular weight DNA was present. The genome sequencing was carried out as 300 bp paired-end with a MiSeq, using the V4 chemistry from Illumina, which did not vary for the duration of this project to ensure continuity. Negative controls were added to each of the sequencing runs, and a final quality control took place. A PhiX control was used to control the sequencing process.
Bioinformatics processing for E. coli sequencing data
The Escherichia coli strain LF82 sequence (BioSample SAMEA3138414 on NCBI) was downloaded from NCBI and was used as a reference for the mapping of the 105 sequences from the E. coli isolates. This strain contains 4 376 genes. Alignment of the LF82 genes with the 105 isolated E. coli genomes was done using 95% of identity and 90% of coverage as criteria of mapping using BLASTN.
TAK-018 stock solution preparation
Stock solutions of TAK-018 were prepared in dimethyl sulfoxide (DMSO) at a concentration of 10 mM and stored at – 20 °C until use. The TAK-018 concentrations tested were prepared in pure DMSO by tenfold serial dilution from the 10 mM stock solutions. Final DMSO concentration was 0.1%. Intermediate solutions were extemporaneously diluted in Ham’s F12 + Dulbecco’s modified Eagle’s medium (DMEM) (1:1) + 2 mM Glutamine + 10% Fetal bovine serum (FBS) + 1X Penicillin/streptomycin (used for T84 cell adhesion assays) and Luria–Bertani (LB) broth for the aggregation tests.
In vitro testing of TAK-018 in prevention of bacterial adhesion on T84 epithelial cells
T84 cell line culture
The T84 human carcinoma cell line (Sigma Aldrich) was cultured in Ham’s F12 + DMEM (1:1) + 2 mM Glutamine + 10% Fetal bovine serum (FBS) + 1X Penicillin/streptomycin. T84 cell lines were not used between 9 and 15 passages. Four days before infection, 40,000 cells/well were seeded on black 96-well plates coated with poly-D-lysine. Medium (without antibiotics) was changed every 2 days, and cells were grown until confluence.
Bacterial adhesion assay
The media from the confluent 96-well plate was discarded, and 50 μl of compound solutions (0.1 nM, 1 nM, 10 nM, and 100 nM), compound vehicle, or media were added. Bacterial adhesion was tested using 50 μl of AIEC for 40 min (LF82; multiplicity of infection (MOI) of 10) or 70 min (NRG857c; MOI of 100) incubation periods. The plate was centrifuged at 1200 rpm for 5 min before incubation at 37 °C, 5% CO2. Supernatants were removed, and the plate was washed to remove non-adherent bacteria and other debris. Cells were then fixed with 4% paraformaldehyde (PFA), incubated at room temperature (RT) for 30 min and washed with phosphate buffered saline (PBS). The plate was kept at 4 °C until immunostaining. PBS was discarded, cells were permeabilized using 0.2% triton in PBS, incubated 15 min at RT, washed before addition of 1:200 of goat anti-E. coli (anti-O and anti-K antigens) and incubated for a further 2 h at RT. Then, cells were washed again and incubated with 1:200 donkey anti-goat Alexa 488. Cells were washed, and 1:2 4’,6-diamidino-2-phenylindole (DAPI) was added, plates incubated 5 min at RT in the dark. Image acquisition was performed by high content imaging (HCI) technology. This methodology allows to visualize and quantify the bacterial adhesion to T84 cells and assess the TAK-018 efficacy. To study the ability of TAK-018 to dislodge bound AIEC, adhesion of bacteria to T84 cells was first run 45 min and, subsequently, unbound bacteria were washed. Increasing concentration (as above) of TAK-018 was added and plates incubated for 45 min. Thereafter, cells were washed again and subjected to the same procedure used in the adhesion assay to quantify bacteria.
Aggregation assay
The diverse bacterial suspensions (50 μl) were dispensed into 96-well plates, and 50 μl of the TAK-018 solutions (1 nM, 10 nM, 100 nM, 1 μM, 10 μM, and 100 μM) or LB with 0.2% DMSO were added. Plates were incubated for 5 h at 37 °C under slow agitation (200 rpm). Every hour, each well was observed with a microscope and the onset of aggregation was recorded. Bright field images were acquired to assess bacterial aggregation after 5 h of incubation.
No growth is indicative of samples for which, upon overnight culture, no bacterial growth could be observed. Therefore, no aggregation test could be performed. Negative is indicative of patients for which harvested bacteria grew but did not aggregate in the presence of TAK-018, indicative potentially of the lack of FimH expression.
Measurement of transepithelial electric resistance (TEER)
T84 cells at 80–90% of confluency were detached from 25 cm2 flasks in 2 ml of trypsin-ethylenediaminetetraacetic acid (EDTA) solution. After centrifugation (1200 rpm, 5 min), cells were resuspended in culture medium, cells were counted using Malassez’s cell, and the cell density was adjusted to approximately 200,000 cells/ml by dilution in culture medium. Cells were then plated onto 12-well inserts (ThinCert Greiner, diameter 1 cm2, pore size 3 µm, ref 665,630 Dominique Dutscher) at ~ 100,000 cells per well (500 µl of cell suspension). Cells were left to attach and differentiate for 21 days before bacterial infection with medium changes every 2–3 days. The day before the assay, tubes containing 3 ml of LB (from Sigma Aldrich, reference: L3522) were inoculated with glycerol stock of bacteria maintained at − 8 0 °C using sterile loops. Tubes were incubated overnight at 37 °C without shaking. The next day, optical density of the bacterial suspensions was read at 600 nm allowing estimation of the bacterial cell density. Bacterial suspensions were centrifuged at 3000 rpm, 10 min at 4 °C. Bacterial pellets were resuspended in DMEM without antibiotic or serum, and bacterial concentrations were adjusted at 109 bacteria/ml. Bacteria were then incubated 1 h at 37 °C with TAK-018 at three final concentrations (1 nM, 100 nM, 1 µM) or the vehicle DMSO (0.1% final concentration). In parallel, inserts of T84 cells were aspirated, and cells were washed 3 times with DMEM without antibiotic or serum. T84 cells were then exposed to 500 µl of bacteria suspensions exposed or not to TAK-018 or DMSO. Cells were also exposed to LPS or Flagellin pre-incubated or not with DMSO or TAK-018 at 1 nM, 100 nM, and 1 µM. LPS was from E. coli K12 (from Invivogen, ref: tlrl-eklps) and was used at 100 ng/ml as positive control of hTLR4 activation. Flagellin was from Salmonella typhimurium (from Invivogen, ref: tlrl-epstfla-5) and was used at 10 µg/ml as agonist of TLR5 receptor. Transepithelial electrical resistance (TEER) was measured before infection (at t0) using Millicell-ERS (electrical resistance system) Voltohmmeter (Millipore, ref: MERS00002). After 4 h at 37 °C, TEER was again measured, and basolateral compartments were collected and stored at – 80 °C before measurement of cytokine levels using enzyme-linked immunosorbent assay (ELISA) kits (detection kits for human IL-1β, IL-6, IL-8, or TNF-α; BD Biosciences ref: 557953, 555220, 555244, 555212, respectively).
In vitro experiments using HEK-TLR4+/+ and HEK-TLR4−/− cells
HEK cell culture
HEK-TLR4−/− (reference: hkb-null2) and HEK-hTLR4 (reference: hkb-hTLR4) cells were obtained from Invivogen and grown accordingly to manufacturer’s instructions. HEK-TLR4−/− cells were grown in the following medium: DMEM, 4.5 g/l glucose, 2–4 mM L-glutamine, 10% (v/v) fetal bovine serum, 50 U/ml penicillin, 50 μg/ml streptomycin, 100 μg/ml Normocin, and 100 μg/ml of the selection antibiotic Zeocin. HEK-hTLR4 cells were grown in the following medium: DMEM, 4.5 g/l glucose, 2–4 mM L-glutamine, 10% (v/v) fetal bovine serum, 50 U/ml penicillin, 50 μg/ml streptomycin, 100 μg/ml Normocin, and HEK-hTLR4 selection antibiotics diluted 1:250 (stock at 250X, reference hb-sel).
Cytokine secretion assay
HEK-TLR4−/− and HEK-hTLR4 cells at 80–90% of confluency were detached from 25 cm2 flasks in 2 ml of PBS using mechanical detachment technic with cell scraper (avoiding trypsin used accordingly to manufacturer’s recommendations). Cells were counted using Malassez’s cell, and the cell density was adjusted to approximately ~ 280,000 and ~ 140,000 cells/ml by dilution in culture medium. Cells were then seeded into 12-well plates (1 ml per well) and left to attach for 24 h before treatment. Cells were either left untreated or treated for 16 h at 37 °C with (i) increasing concentrations of purified E coli K12 FimH and (ii) LPS from E coli K12 (from Invivogen, reference: tlrl-eklps) used at 100 ng/ml as positive control of hTLR4 activation. After incubation, cell culture supernatants were collected and stored at – 80 °C before measurement of TNFa levels using ELISA kits (detection kits for human TNF alpha from BD Biosciences reference 555212).
In vitro experiments using human primary enterocytes
Primary enterocyte cell culture
Primary intestinal cells were isolated from a surgical piece of a patient with CD, by digestion of the mucosae using proteases. Briefly, mucosae were stripped from intestinal resection and incubated in lactated Ringer’s (LR) buffer containing dithiothreitol (DTT) to remove mucus. The mucosae was then transferred to new LR buffer containing collagenase and pronase and homogenized by pipetting. Cell suspension was filtered through sterile gauze and centrifuged. Pellet was resuspended in DMEM containing 10% FBS (referred as complete media) and antibiotics (penicillin–streptomycin solution and gentamycin at 50 µg/ml). Cells were centrifuged and resuspended in complete media to obtain a cell density of 200,000 cells/ml. Cells were seeded into 96-well plates at 20,000 cells per well and incubated at 37 °C for 16–24 h before adhesion assay.
Adhesion assay
After incubation, wells were aspirated and washed with DMEM without serum and without antibiotics. Bacteria were pre-incubated for 1 h at 37 °C with increasing concentration of TAK-018 or with vehicle (DMSO, 1:1000 dilution). Then, primary cells were exposed to 109 of AIEC and non-AIEC bacteria, exposed or not to TAK-018. In parallel to the bacteria, control wells were exposed to flagellin isolated from Salmonella, LPS from E. coli or human recombinant IL-1β for 4 h at 37 °C. Supernatant was collected and frozen at − 80 °C until determination of cytokines by ELISA. Cells were washed with PBS containing calcium and magnesium (PBS++) and fixed with 4% PFA diluted in PBS++ (100 µl per well). Wells were washed with PBS++ before determination of bacterial adhesion using ELISA assay.
Quantification of bacterial adhesion using ELISA assay
Primary antibodies directed against specific bacteria were incubated at 1:1000 in saturation buffer (PBS++ supplemented with 10% FBS). Primary antibodies used were Mycobacterium avium monoclonal antibody (ref. MA1-10788), Klebsiella pneumoniae rabbit polyclonal antibody (ref. PA1-7226), Salmonella rabbit polyclonal antibody (ref. PA1-7244), and E. coli serotypes O + K rabbit antibody (ref. PA1-7213) from ThermoFisher. After 1-h incubation at RT, wells were washed with PBS. Secondary antibodies goat anti-rabbit IgG (ref. 111-035-003) and goat anti-mouse IgG (ref. 115-035-003) from Jackson ImmunoResearch, diluted at 1:10,000 in saturation buffer and conjugated to horseradish peroxidase (HRP), were added (100 µl) and incubated for 1 h at RT. Wells were then washed with PBS, and HRP substrate (Sigma Fast-OPD, ref P9187) was added to reveal. Plates were incubated in the dark, before H2SO4 2 N was added and optical density was measured at 490 nm.
Ex vivo experiments on explants from patients with CD
Total adhesion measurements
Ex vivo experiments were performed on ileocecal resections from patients undergoing surgery and having agreed to use their tissues for research purposes. Only freshly prepared intestinal punches were used. TAK-018 compound was tested at 3 concentration levels on the adhesion of LF82-Green Fluorescent Protein (GFP) bacteria (109/ml, 4 h of incubation) for a total of 4 conditions. Human recombinant IL-1β (final concentration 2 ng/ml) was used as positive control of pro-inflammatory signal. At the end of the 4-h incubation period with LF82-GFP bacteria, cell culture media was collected for cytokine quantification. After 6 washes with PBS (1 ml), explants were transferred to matrix lysis tubes and subjected to mechanical lysis using bead-better apparatus. Green fluorescence was quantified using black 96-well plates and a spectrofluorometric plate reader.
Determination of inflammation marker by ELISA
Supernatant (SN) of LF82-GFP and/or TAK-018–treated explants were collected after 4 h and stored at – 80 °C until cytokine determination. Cytokine levels in tissues (“Tissue” samples) were also measured after mechanical lysis of the explants in 500 µl of PBS containing protease inhibitors (Sigma Aldrich; ref. P8340). For cytokine determination, commercial ELISA kits, allowing detection of human IL-1β, IL-6, IL-8, or TNF-α, were used according to manufacturer’s instructions (BD Biosciences kits; refs: 557953, 555220, 555244, 555212, respectively).
Microscopy
For microscopy analysis, at the end of the 4-h incubation, the punches of tissue used were fixed in 1 ml of PFA 4% for 24 h. Punches were washed twice with 1 ml of PBS and cut in by half and included in inclusion medium (TFM), in transverse position to allow sectioning in the crypt–villosity axis. Then, 3 sections of 5-µm thickness were obtained per piece of punch, each section being separated by 60 µm from the next to cover all the tissue. Sections were stained with Phalloidin, conjugated to Alexa-547 to microscopically assess bacterial adhesion or stained with hematoxylin and eosin (H&E) to analyze histopathology damage.
Statistical analysis
For the Cohort 1 from the CrohnOmeter study, data studied corresponds to the first timepoint available for each patient. For both cohorts of patients with CD, patients were considered “Active CD patients” if their HBI was > 4, “Quiet CD patients” otherwise. Clinical variables were summarized as medians with interquartile ranges (IQRs) or as frequencies with percentages. For non-paired samples, the nonparametric Mann–Whitney U test was performed to compare continuous variables between 2 groups and the nonparametric Kruskal–Wallis test was performed to compare continuous variables between more than 2 groups. For paired samples, nonparametric Wilcoxon’s signed-rank test for paired samples was performed to compare continuous variables between 2 groups/timepoints, and linear mixed models were performed to compare continuous variables between more than 2 groups/timepoints. Additionally, we studied only species detected (relative abundance > 0) in at least 70% of samples from at least one cohort. Additionally, for all metagenomics data analyses except the gene diversity, a pseudo-count of 1e-10 was added. ANOVA was performed for each group on the aggregation data from Fig. 4a and b. A P value lower than 0.05 was considered statistically significant. All P values presented come from two-sided tests. All statistical representations were added manually on figures. All statistical analyses were performed using R software (version 3.6.1).
Data availability
The datasets analyzed during the current study are available (“Blockage of bacterial FimH prevents mucosal inflammation associated with Crohn’s disease”, Mendeley Data, V1, https://doi.org/10.17632/4s3f4dv59g.1). The informed consent does not allow us to share on a public domain the metagenomic data. However, these data are available from the corresponding author on reasonable request.
Results
Characteristics of study participants
We assessed the intestinal microbial communities of 401 individuals, including 358 patients with CD (from two distinct cohorts) and 43 healthy volunteers (HV), using shotgun metagenomic sequencing in fecal samples. All samples collected were analyzed using shotgun sequencing, and reads corresponding to different bacterial species were quantified and normalized relative to the total stool DNA in order to generate unbiased quantification of the different bacterial DNA species studied. Demographic data and history of CD and disease activity for each of the studies are described in Supplementary Table 1. Demographic data for the HV is presented in Supplementary Table 2. The first cohort of patients with CD, from the CrohnOmeter study, was followed longitudinally, and we did not observe statistically significant differences in terms of HBI (Supplementary Fig1a) and E. coli relative abundance (Supplementary Fig1b) across the different time points. Therefore, we chose to focus our analysis on the data corresponding to the first time point available, in order to compare this cohort with the second cohort, from the PREDICT study.
Patients with CD show altered microbiota profiles characterized by a blooming of Enterobacteriaceae
As expected, the overall alpha diversity, measured according to the Shannon and Simpson indices, was significantly reduced in samples from patients with CD when compared with those from HV, in both patient cohorts (Fig. 1a, b). The distinctiveness of the microbiota from patients with CD was confirmed by beta diversity analysis, demonstrating a clustering of samples according to CD diagnosis for both cohorts with the same homogeneity by Bray–Curtis dissimilarity metrics (Fig. 1c). We also observed significant differences between Cohort 1 and Cohort 2 regarding the Shannon and Simpson indices (Fig. 1a, b) as well as the Bray–Curtis dissimilarity metrics (Fig. 1c), likely because of differences in the inflammatory status of the patients between the two cohorts, illustrated by higher Harvey-Bradshaw index and higher calprotectin levels in cohort 2 (Supplementary Table 1). Determining the relative abundance of E. coli and other Enterobacteriaceae spp, we observed that Enterobacteriaceae were strongly enriched in patients with CD compared with HV: indeed, 30 to 55% of samples showed a greater-than tenfold increase in different Enterobacteriaceae spp including Shigella flexneri, Klebsiella pneumoniae, Salmonella enterica, and Escherichia coli (Fig. 2a, b and Supplementary Table 3), illustrating the blooming of Enterobacteriaceae observed in CD (species for which a tenfold increase has been observed in patients with CD compared with mean abundance in HV). Moreover, we observed a different clustering of the patients from Cohort 2 in the PC1 scores in the PCoA analysis (Fig. 1c). Indeed, some patient samples clustered with HV, but a larger fraction was very different from healthy controls (Supplementary Fig3a). This dichotomy of patient samples in Cohort 2 has been further investigated at the levels of Enterobacteriaceae spp abundances and we observed striking differences, with the population of patients from Cohort 2 overlapping HV on the PCoA having highly significantly lower Enterobacteriaceae spp abundance compared with the other patients from Cohort 2 (Supplementary Fig3b). In terms of relative abundances, E. coli species which in HV account for less than 0.02% of all fecal bacterial DNA, becomes a major species accounting for 1 up to 80% of all stool DNA in > 20% of all our samples from patients with CD. We next separated the samples according to the disease status of the donor (considered in an active phase of the disease when HBI > 4 and in a quiet phase when HBI ≤ 4) and observed an association between levels of Enterobacteriaceae spp and disease activity (Fig. 2c, d).

Altered microbiota profiles in patients with CD from 2 different cohorts. a, b Species diversity in patients with CD and healthy volunteers were calculated using alpha diversity: Shannon (a) and Simpson indices (b). c Microbial clustering is shown based on Bray–Curtis dissimilarity principal coordinate analysis (PCoA) metrics for Cohort 1, Cohort 2, and HV. Ellipsoids represent a 95% confidence interval surrounding each group. There were 74 patients with CD in Cohort 1, 284 patients with CD in Cohort 2, and 43 healthy volunteers. Bars represent the median of all points. Nonparametric Mann–Whitney U test was used to identify the statistically significant differences between groups. (*P < 0.05, **P < 0.005, ***P < 0.0005, ****P < 0.0001)

Fecal microbial composition of patients with CD is characterized by a blooming of Enterobacteriaceae. a, b Top blooming species by percentage of patients with CD in Cohort 1 (a) and Cohort 2 (b), for whom a tenfold increase has been observed compared with mean relative abundance in HV. Are represented only the species with at least 20% of patients in blooming either in Cohort 1 or in Cohort 2. Note the presence of 4 Enterobacteriaceae among the top 5 species. c Relative abundance of Enterobacteriaceae spp in HV and patients with CD from Cohort 1 and Cohort 2. d Relative abundance of Enterobacteriaceae spp in HV, active and quiet patients with CD from Cohort 1 and Cohort 2. Patients with CD were considered in an active phase of the disease when HBI > 4 and in a quiet phase when HBI ≤ 4. Bars represent the median of all points. Nonparametric Mann–Whitney U test was used to identify the statistically significant differences between groups. (*P < 0.05, ***P < 0.0005, ****P < 0.0001)
FimH adhesin is expressed by bacteria in ileal biopsies from patients with CD
At the metagenomic level, we confirmed that FimH presence was restricted to a family of Proteobacteria, all species of the Enterobacteriaceae family expressing FimH (Supplementary Fig2) [18]. We also tested for the presence of FimH-expressing bacteria in ileal biopsies in a third cohort of patients with CD from the MOBIDIC study (Supplementary Table 1). In order to do so, we used our designed bi-mannosylated compound TAK-018 (EB8018/Sibofimloc) that sensitively identifies FimH expression [44, 45] (Supplementary Fig4). We showed that TAK-018 aggregates the reference FimH-expressing AIEC E. coli LF82 strain, whereas a FimH negative mutant of E. coli LF82 did not aggregate upon incubation with TAK-018, allowing for the detection of FimH-expressing bacteria with TAK-018 (Fig. 3a). We observed that 65% of patients (69/106 patients) did display adherent colonies made up of FimH-expressing bacteria, as measured by aggregation to TAK-018 (Fig. 3b). Moreover, we characterized the relationship between FimH and the “AIEC” phenotype by sequencing a total of 98 isolated E. coli strains described as “AIECs” (Supplementary Table 4). We correlated sequencing data with results from the aggregation assay and mapped sequence reads against the E. coli LF82 strain used as reference, and specifically focused on the fim operon. The mapping against the fimS region revealed a strong association between the percentage of reads in the “ON” position, indicative of expression of the entire fim operon, and the aggregation to TAK-018 [46] (Supplementary Fig5). When applied to the samples from patients with CD, we also observed a strong relationship between the level of fimS-ON analyzed by qPCR in feces and functional expression of FimH in ileal biopsies from these samples (Fig. 3c).

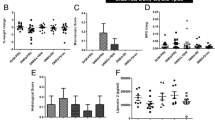
FimH adhesin is expressed by bacteria in ileal biopsies from patients with CD and induces inflammatory response in a TLR4-dependent manner. a Aggregation of LF82 E. coli upon incubation with TAK-018. Note that FimH−/− LF82 E. coli did not aggregate upon incubation with E. coli illustrating that aggregation is a FimH-dependent phenomenon. b Presence of FimH-expressing bacteria in patients with CD as measured by aggregation of bacteria to TAK-018 in ileal biopsies. One to two biopsies were analyzed per patient. c FimS-ON expression in fecal samples from patients with CD, as measured by qPCR. Bars represent the median of all points. Nonparametric Mann–Whitney U test was used to identify the statistically significant differences between groups (*P < 0.05; n.s. nonsignificant). d Secretion of TNF-α of HEK-hTLR4+/+ and HEK-TLR4−/− cells upon incubation with increased concentration of FimH. LPS was used as a positive control at a concentration of 100 ng/ml. FimH was produced in yeast in order to be LPS-free. The data are centered on the mean value at 0. Bars represent the mean of all points. An analysis of covariance (ANCOVA) was used to identify the statistically significant difference between TLR4−/− and TLR4+/+ groups upon FimH treatment (****P < 0.0001; 0 nM and LPS conditions were excluded from the ANCOVA)
FimH blocker TAK-018 prevents Enterobacteriaceae adhesion and inflammation in gut explant in a TLR4-dependent manner
FimH has been described as a potent TLR4 agonist in the gut which can trigger inflammation independently of the presence of lipopolysaccharides (LPS) [16], even though TLR4 expression is upregulated in ileal biopsies from patients with CD [23, 28, 47]. As previously reported [15,16,17], we confirmed a dose-dependent increased secretion of tumor necrosis factor alpha (TNFα) by epithelial human embryonic kidney (HEK) cells upon incubation with purified FimH, which was completely abrogated in TLR4−/− HEK cells (Fig. 3d). Considering that TLR4 expression is upregulated by inflammatory cytokines in ileal biopsies from patients with CD [23, 28, 47] and that an inflamed environment is conducive to blooms of Enterobacteriaceae [48], we hypothesized a critical role for FimH as a pro-inflammatory trigger leading to a vicious circle in CD, stressing the importance of designing therapeutic strategies that can disrupt this pathogenic loop.
Accordingly, we studied the effects of blocking bacterial adhesin FimH with TAK-018 by performing adhesion assays on AIEC and non-AIEC strains to epithelial cells. We observed that different bacteria described as AIECs (most notably E. coli LF82) are able to adhere to human primary intestinal cells in a FimH-dependent manner, except the 41CB2 strain that does not express FimH (Fig. 4a and Supplementary Table 4). Moreover, we demonstrated a concentration-dependent decrease in bacterial adhesion by TAK-018 with full blocking activity observed at 1 μM TAK-018 for all strains (Fig. 4a), also confirmed on T84 epithelial cell line (Supplementary Fig6) and primary ileal cells (Supplementary Fig7). This blocking of bacterial adhesion further prevented intracellular infection of epithelial cells by LF82 E. coli (Fig. 4b).

FimH blocker TAK-018 prevents Enterobacteriaceae adhesion and inflammation of gut explant. a Adhesion of different strains of E. coli to human primary intestinal epithelial cells, in the presence of increasing concentrations of TAK-018. Note that the 41CB2 strain did not express FimH. Bars represent the mean value of individual points which themselves correspond to biological replicates. ANOVA was performed for each group to identify the statistically significant effect of TAK-018 (***P < 0.001). b Number of intracellular colonies in epithelial cells from human ileal explant incubated with LF82 E. coli, in the presence of increasing concentrations of TAK-018. Bars represent the mean value of individual points which themselves correspond to biological replicates. ANOVA was performed to identify the statistically significant effect of TAK-018 (*P < 0.05). c TNF-α secretion of human ileal explant incubated for 4 h with 109 LF82 E. coli, in the presence of increasing concentrations of TAK-018. IL-1β was used as a positive control to trigger inflammation. Bars represent the mean value of individual points which themselves correspond to biological replicates. A nonparametric Wilcoxon signed-rank paired test was used to identify the statistically significant differences between the LF82 and LF82 + TAK-018 1 µM groups (P = 0.031). A linear mixed model was used to identify the statistically significant differences between the groups LF82, LF82 + 500 nM, and LF82 + 1 µM (P = 0.07). d Variation of transepithelial electric resistance (TEER) of T84 cells incubated for 4 h with 109 LF82 E. coli, in the presence of increasing concentrations of TAK-018. Note that FimH−/− LF82 E. coli does not adhere to T84 cells and therefore does not affect TEER. Bars represent the mean value of individual points which themselves correspond to biological replicates. A nonparametric Kruskal–Wallis test was used to identify the statistically significant differences between the LF82 groups (P = 0.02). e Histopathologic effects of LF82 E. coli adhesion on human ileal explants from a patient with CD and respective countereffects in the presence of increasing concentrations of TAK-018. Note the characteristic mucosa desquamation upon 4-h incubation with LF82 E. coli, visibly reduced in the presence of increased concentrations of TAK-018
We also determined the effect of preventing FimH sensing by TLR4 with TAK-018 on cytokine secretion. E. coli LF82 induced a robust secretion of TNFα, IL-6, and IL-8, TAK-018 inhibiting dose-dependently the secretion of all three cytokines, with 1 μM retaining levels close to or below basal ones (Fig. 4c and Supplementary Fig8). We further assessed the functional implications in terms of tight junction dynamics and gut permeability by determining transepithelial electrical resistance (TEER) in T84 cells. E. coli LF82 induced a robust decrease in TEER in a FimH-dependent manner; as a FimH-deleted LF82 mutant did not affect TEER [49]. These effects were dose-dependently counteracted by TAK-018, with full efficacy at 1 µM (Fig. 4d). Similarly, histological analyses of the human gut explants incubated with E. coli LF82 showed an extremely pronounced inflammatory effect on the intestinal tissue with mucosa desquamation (Fig. 4e). Incubation with increasing amount of TAK-018 showed again gradual decrease in the inflammatory reaction and a general and clear improvement of gut explant integrity, with a concentration of 1 μM TAK-018 being able to maintain normal tissue integrity (Fig. 4e).
Discussion
The present study documents and quantifies an enrichment of E. coli and other Enterobacteriaceae spp in the gut of patients with CD. Our data suggest that a blooming of FimH-expressing bacteria could participate in initiating inflammation patients with CD. We observed potentially pathogenic levels of Enterobacteriaceae in over 75% of patients with CD when followed longitudinally. On human ileal resections, FimH contributes to mucus colonization and decreased mucus thickness, epithelial adherence and inflammation, and desquamation. Moreover, in animal models of colitis, FimH has been shown to be required to induce severe inflammatory damages since genetic deletion of FimH led to less engraftment and inflammation upon gavage with FimH-mutant LF82 AIEC [49]. The pathogenesis of AIECs has been linked to the overexpression of the fim operon [50]. The clinical cohorts reported here show an increased number of FimH-expressing bacteria associated with CD and with disease activity that account for the top 4 bloomers. The gene fimH is under strict control of a “switch” mechanism of the invertible fimS promoter [34, 51]. A qPCR strategy was designed using primers that amplify the level of fimS-ON in stool samples. The results showed that fimS-ON in stools is statistically linked to aggregation in ileal biopsies, suggesting that this might be a reliable and noninvasive tool to diagnose patients with CD hosting FimH-expressing bacteria. However, we did not perform metagenomic analysis of fecal samples and cannot infer whether AIEC levels would correlate between fecal and ileal samples. To date, among human gut bacteria, only FimH proteins and in particular E. coli FimH proteins, are annotated with the gene ontology term “mannose binding” (GO:0005537) on the UniProt database. To our knowledge, there is no other gene in Enterobacteriaceae capable of recognizing mannose. These bacteria interact with TLR4 receptors, insensitive to LPS in the gut, through its FimH, inducing large release of TNFα [16]. The gene fimH is not constitutively expressed in non-inflammatory conditions, and therefore, these commensals are not harmful for the host. Once the biofilm is formed, FimH-expressing bacteria can invade the mucosa by transcytosis of the epithelial cells and are also phagocyted by M cells in Peyer’s patches [52].
In the current status of drug-resistant bacterial pathogens, the availability of antibiotics to effectively treat patients is limited and can even further aggravate dysbiosis by killing the beneficial bacteria and allow overgrowth of the resistant species. Traditional treatments and novel therapies for CD include biologic agents that target various mechanisms of action in the inflammatory pathways [53], but they do not target the source of the inflammation that originates from an imbalanced gut microbiota and the emergence of pro-inflammatory pathogenic bacteria. Elimination of pathogenic FimH-carrying bacteria from the gut would thus represent a selective strategy to suppress a potentially critical trigger of intestinal inflammation.
We have shown using the AIEC reference strain LF82 expressing FimH that targeting FimH could prevent bacterial adhesion and alleviate inflammation in ileal explants. Considering that all members of the Enterobacteriaceae family express FimH [18], we assume that this strategy would also be relevant for other Enterobacteriaceae species. However, we did not test our FimH blocker in animal models and chose to move forward into the clinic, based on these results. Therefore, even though our data suggest that FimH is an inflammatory trigger in CD, the initiation of a phase Ib clinical study in patients with CD (NCT03709628) will bring the expected information whether FimH blockage represent a realistic strategy for treating patients with CD. Targeting the bacterial adhesion of FimH-expressing bacteria is a promising therapeutic method that consists of ‘disarming’ bacteria without killing them, representing a selective strategy to suppress a potentially critical trigger of intestinal inflammation, without disturbing the intestinal microbiota [54,55,56,57,58]. In addition, this therapeutic approach exerts weaker selective pressure compared with other antibacterial agents and thus the emergence of bacterial resistance is expected to be residual. Accordingly, to interfere with AIEC adhesion to host cells, the development of drugs rationally designed to saturate the carbohydrate recognition domain (CRD) of FimH, by mimicking its natural ligand, appears of great interest for a better personalized therapeutic strategy to manage CD [35].
Conclusions
Considering the limits of other therapies, TAK-018 has been designed as a novel, first-in-class small molecule FimH blocker, orally administered and gut-restricted with minimal systemic absorption [58]. We have shown here that the microbiome of patients with CD was enriched with Enterobacteriaceae spp, this enrichment being even more pronounced in patients with active CD. As Enterobacteriaceae express the bacterial adhesin FimH, we confirmed the presence of bacterial FimH expression in ileal biopsies from patients with CD, suggesting a pathophysiological role for FimH-expressing bacteria in CD. Using human intestinal explants, we further showed that FimH is essential for adhesion and to trigger inflammation. Finally, a specific FimH blocker, TAK-018, was shown to reduce bacterial adhesion in a dose-dependent manner, totally blunting inflammation and preserving tissue integrity. Moreover, we have conducted a 2-week phase Ia clinical study where TAK-018 has been shown to be well tolerated in healthy volunteers with a maximum daily dose of 3000 mg/day (NCT02998190) [58]. The benefit–risk balance of TAK-018 enabled the initiation of a phase Ib clinical study in patients with CD to assess the pharmacokinetics, under TAK-018 designation (NCT03709628) [59]. Because of its properties and mode of action, TAK-018 can be especially useful in maintenance therapy and be complementary to other more symptomatic therapeutics. Accordingly, TAK-018 is currently evaluated in postoperative CD in a phase II study.
Availability of data and materials
The datasets analyzed during the current study are available (“Blockage of bacterial FimH prevents mucosal inflammation associated with Crohn’s disease”, Mendeley Data, V1, https://doi.org/10.17632/4s3f4dv59g.1). The informed consent does not allow us to share on a public domain the metagenomic data. However, these data are available from the corresponding author on reasonable request.
References
Sartor RB, Wu GD. Roles for intestinal bacteria, viruses, and fungi in pathogenesis of inflammatory bowel diseases and therapeutic approaches. Gastroenterology. 2017;152(2):327-339.e4.
Buisson A, Bringer MA, Barnich N, Vazeille E. Macrophages versus Escherichia coli: a decisive fight in Crohn’s disease. Inflamm Bowel Dis. 2016;22(12):2943–55.
Darfeuille-Michaud A, Neut C, Barnich N, Lederman E, Di Martino P, Desreumaux P, et al. Presence of adherent Escherichia coli strains in ileal mucosa of patients with Crohn’s disease. Gastroenterology. 1998;115(6):1405–13.
Boudeau J, Glasser AL, Masseret E, Joly B, Darfeuille-Michaud A. Invasive ability of an Escherichia coli strain isolated from the ileal mucosa of a patient with Crohn’s disease. Infect Immun. 1999;67(9):4499–509.
Darfeuille-Michaud A, Boudeau J, Bulois P, Neut C, Glasser AL, Barnich N, et al. High prevalence of adherent-invasive Escherichia coli associated with ileal mucosa in Crohn’s disease. Gastroenterology. 2004;127(2):412–21.
Baumgart M, Dogan B, Rishniw M, Weitzman G, Bosworth B, Yantiss R, et al. Culture independent analysis of ileal mucosa reveals a selective increase in invasive Escherichia coli of novel phylogeny relative to depletion of Clostridiales in Crohn’s disease involving the ileum. ISME J. 2007;1(5):403–18.
Martinez-Medina M, Aldeguer X, Lopez-Siles M, González-Huix F, López-Oliu C, Dahbi G, et al. Molecular diversity of Escherichia coli in the human gut: new ecological evidence supporting the role of adherent-invasive E. coli (AIEC) in Crohn’s disease. Inflamm Bowel Dis. 2009;15(6):872–82.
Elliott TR, Hudspith BN, Wu G, Cooley M, Parkes G, Quiñones B, et al. Quantification and characterization of mucosa-associated and intracellular Escherichia coli in inflammatory bowel disease. Inflamm Bowel Dis. 2013;19(11):2326–38.
Martinez-Medina M, Garcia-Gil LJ. Escherichia coli in chronic inflammatory bowel diseases: an update on adherent invasive Escherichia coli pathogenicity. World J Gastrointest Pathophysiol. 2014;5(3):213–27.
Glasser AL, Boudeau J, Barnich N, Perruchot MH, Colombel JF, Darfeuille-Michaud A. Adherent invasive Escherichia coli strains from patients with Crohn’s disease survive and replicate within macrophages without inducing host cell death. Infect Immun. 2001;69(9):5529–37.
Eaves-Pyles T, Allen CA, Taormina J, Swidsinski A, Tutt CB, Eric Jezek G, et al. Escherichia coli isolated from a Crohn’s disease patient adheres, invades, and induces inflammatory responses in polarized intestinal epithelial cells. Int J Med Microbiol. 2008;298(5–6):397–409.
Martinez-Medina M, Mora A, Blanco M, López C, Alonso MP, Bonacorsi S, et al. Similarity and divergence among adherent-invasive Escherichia coli and extraintestinal pathogenic E. coli strains. J Clin Microbiol. 2009;47(12):3968–79.
Nash JHE, Villegas A, Kropinski AM, Aguilar-Valenzuela R, Konczy P, Mascarenhas M, et al. Genome sequence of adherent-invasive Escherichia coli and comparative genomic analysis with other E. coli pathotypes. BMC Genomics. 2010;11:667.
Céspedes S, Saitz W, Del Canto F, De la Fuente M, Quera R, Hermoso M, et al. Genetic diversity and virulence determinants of Escherichia coli strains isolated from patients with Crohn’s disease in Spain and Chile. Front Microbiol. 2017;8:639.
Ashkar AA, Mossman KL, Coombes BK, Gyles CL, Mackenzie R. FimH adhesin of type 1 fimbriae is a potent inducer of innate antimicrobial responses which requires TLR4 and type 1 interferon signalling. PLoS Pathog. 2008;4(12):e1000233.
Mossman KL, Mian MF, Lauzon NM, Gyles CL, Lichty B, Mackenzie R, et al. Cutting edge: FimH adhesin of type 1 fimbriae is a novel TLR4 ligand. J Immunol. 2008;181(10):6702–6.
Fischer H, Yamamoto M, Akira S, Beutler B, Svanborg C. Mechanism of pathogen-specific TLR4 activation in the mucosa: fimbriae, recognition receptors and adaptor protein selection. Eur J Immunol. 2006;36(2):267–77.
Jones CH, Pinkner JS, Roth R, Heuser J, Nicholes AV, Abraham SN, et al. FimH adhesin of type 1 pili is assembled into a fibrillar tip structure in the Enterobacteriaceae. Proc Natl Acad Sci U S A. 1995;92(6):2081–5.
Barnich N, Carvalho FA, Glasser AL, Darcha C, Jantscheff P, Allez M, et al. CEACAM6 acts as a receptor for adherent-invasive E. coli, supporting ileal mucosa colonization in Crohn disease. J Clin Invest. 2007;117(6):1566–74.
Hase K, Kawano K, Nochi T, Pontes GS, Fukuda S, Ebisawa M, et al. Uptake through glycoprotein 2 of FimH + bacteria by M cells initiates mucosal immune response. Nature. 2009;462(7270):226–30.
Ohno H, Hase K. Glycoprotein 2 (GP2) grabbing the FimH+ bacteria into M cells for mucosal immunity. Gut Microbes. 2010;1(6):407–10.
Teague RH, Fraser D, Clamp JR. Changes in monosaccharide content of mucous glycoproteins in ulcerative colitis. Br Med J. 1973;2(5867):645–6.
Barnich N, Darfeuille-Michaud A. Abnormal CEACAM6 expression in Crohn disease patients favors gut colonization and inflammation by adherent-invasive E. coli. Virulence. 2010;1(4):281–2.
Roulis M, Armaka M, Manoloukos M, Apostolaki M, Kollias G. Intestinal epithelial cells as producers but not targets of chronic TNF suffice to cause murine Crohn-like pathology. Proc Natl Acad Sci U S A. 2011;108(13):5396–401.
Smith PD, Janoff EN, Mosteller-Barnum M, Merger M, Orenstein JM, Kearney JF, et al. Isolation and purification of CD14-negative mucosal macrophages from normal human small intestine. J Immunol Methods. 1997;202(1):1–11.
Smith PD, Smythies LE, Mosteller-Barnum M, Sibley DA, Russell MW, Merger M, et al. Intestinal macrophages lack CD14 and CD89 and consequently are down-regulated for LPS- and IgA-mediated activities. J Immunol. 2001;167(5):2651–6.
Smith PD, Smythies LE, Shen R, Greenwell-Wild T, Gliozzi M, Wahl SM. Intestinal macrophages and response to microbial encroachment. Mucosal Immunol. 2011;4(1):31–42.
Cario E, Podolsky DK. Differential alteration in intestinal epithelial cell expression of Toll-like receptor 3 (TLR3) and TLR4 in inflammatory bowel disease. Infect Immun. 2000;68(12):7010–7.
Abreu MT, Vora P, Faure E, Thomas LS, Arnold ET, Arditi M. Decreased expression of Toll-like receptor-4 and MD-2 correlates with intestinal epithelial cell protection against dysregulated proinflammatory gene expression in response to bacterial lipopolysaccharide. J Immunol. 2001;167(3):1609–16.
Naik S, Kelly EJ, Meijer L, Pettersson S, Sanderson IR. Absence of Toll-like receptor 4 explains endotoxin hyporesponsiveness in human intestinal epithelium. J Pediatr Gastroenterol Nutr. 2001;32(4):449–53.
Suzuki M, Hisamatsu T, Podolsky DK. Gamma interferon augments the intracellular pathway for lipopolysaccharide (LPS) recognition in human intestinal epithelial cells through coordinated up-regulation of LPS uptake and expression of the intracellular Toll-like receptor 4-MD-2 complex. Infect Immun. 2003;71(6):3503–11.
He YY, Liu SB, Kling DE, Leone S, Lawlor NT, Huang Y, et al. The human milk oligosaccharide 2′-fucosyllactose modulates CD14 expression in human enterocytes, thereby attenuating LPS-induced inflammation. Gut. 2016;65(1):33–46.
Boudeau J, Barnich N, Darfeuille-Michaud A. Type 1 pili-mediated adherence of Escherichia coli strain LF82 isolated from Crohn’s disease is involved in bacterial invasion of intestinal epithelial cells. Mol Microbiol. 2001;39(5):1272–84.
Barnich N, Boudeau J, Claret L, Darfeuille-Michaud A. Regulatory and functional co-operation of flagella and type 1 pill in adhesive and invasive abilities of AIEC strain LF82 isolated from a patient with Crohn’s disease. Mol Microbiol. 2003;48(3):781–94.
Sivignon A, Bouckaert J, Bernard J, Gouin SG, Barnich N. The potential of FimH as a novel therapeutic target for the treatment of Crohn’s disease. Expert Opin Ther Targets. 2017;21(9):837–47.
Harvey RF, Bradshaw JM. A simple index of Crohn’s Disease activity. Lancet. 1980;1(8167):514.
Gaya DR, Lyon TDB, Duncan A, Neilly JB, Han S, Howell J, et al. Faecal calprotectin in the assessment of Crohn’s disease activity. QJM. 2005;98(6):435–41.
Kultima JR, Sunagawa S, Li J, Chen W, Chen H, Mende DR, et al. MOCAT: a metagenomics assembly and gene prediction toolkit. PLoS One. 2012;7(10):e47656.
Wang J, Qin J, Li Y, Cai Z, Li S, Zhu J, et al. A metagenome-wide association study of gut microbiota in type 2 diabetes. Nature. 2012;490(7418):55–60.
Li J, Wang J, Jia H, Cai X, Zhong H, Feng Q, et al. An integrated catalog of reference genes in the human gut microbiome. Nat Biotechnol. 2014;32(8):834–41.
Tatusova T, Ciufo S, Fedorov B, O’Neill K, Tolstoy I. RefSeq microbial genomes database: new representation and annotation strategy. Nucleic Acids Res. 2014;42(Database issue):D553-9.
Yu G, Smith DK, Zhu H, Guan Y, Lam TTY. ggtree: an r package for visualization and annotation of phylogenetic trees with their covariates and other associated data. Methods Ecol Evol. 2017;8(1):28–36.
Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32(5):1792–7.
Moor K, Diard M, Sellin ME, Felmy B, Wotzka SY, Toska A, et al. High-avidity IgA protects the intestine by enchaining growing bacteria. Nature. 2017;544(7651):498–502.
Spaulding CN, Klein RD, Ruer S, Kau AL, Schreiber HL, Cusumano ZT, et al. Selective depletion of uropathogenic E. coli from the gut by a FimH antagonist. Nature. 2017;546(7659):528–32.
Burns LS, Smith SGJ, Dorman CJ. Interaction of the FimB integrase with the fimS invertible DNA element in Escherichia coli in vivo and in vitro. J Bacteriol. 2000;182(10):2953–9.
Abraham C, Cho JH. Bugging of the intestinal mucosa. N Engl J Med. 2007;357(7):708–10.
Zeng MY, Inohara N, Nuñez G. Mechanisms of inflammation-driven bacterial dysbiosis in the gut. Mucosal Immunol. 2017;10(1):18–26.
Carvalho FA, Barnich N, Sivignon A, Darcha C, Chan CHF, Stanners CP, et al. Crohn’s disease adherent-invasive Escherichia coli colonize and induce strong gut inflammation in transgenic mice expressing human CEACAM. J Exp Med. 2009;206(10):2179–89.
Dreux N, Denizot J, Martinez-Medina M, Mellmann A, Billig M, Kisiela D, et al. Point mutations in FimH adhesin of Crohn’s disease-associated adherent-invasive Escherichia coli enhance intestinal inflammatory response. PLoS Pathog. 2013;9(1):e1003141.
Eisenstein BI. Phase variation of type 1 fimbriae in Escherichia coli is under transcriptional control. Science (80-). 1981;214(4518):337–9.
Bennett KM, Parnell EA, Sanscartier C, Parks S, Chen G, Nair MG, et al. Induction of colonic M cells during intestinal inflammation. Am J Pathol. 2016;186(5):1166–79.
Katsanos KH, Papadakis KA. Inflammatory bowel disease: updates on molecular targets for biologics. Gut Liver. 2017;11(4):455–63.
Brument S, Sivignon A, Dumych TI, Moreau N, Roos G, Guérardel Y, et al. Thiazolylaminomannosides as potent antiadhesives of type 1 piliated escherichia coli isolated from Crohn’s disease patients. J Med Chem. 2013;56(13):5395–406.
Sivignon A, Yan X, Dorta DA, Bonnet R, Bouckaert J, Fleury E, et al. Development of heptylmannoside-based glycoconjugate antiadhesive compounds against adherent-invasive Escherichia coli bacteria associated with Crohn’s disease. MBio. 2015;6(6):e01298-15.
Alvarez Dorta D, Sivignon A, Chalopin T, Dumych TI, Roos G, Bilyy RO, et al. The antiadhesive strategy in Crohn’s disease: orally active mannosides to decolonize pathogenic Escherichia coli from the gut. ChemBioChem. 2016;17(10):936–52.
Chalopin T, Alvarez Dorta D, Sivignon A, Caudan M, Dumych TI, Bilyy RO, et al. Second generation of thiazolylmannosides, FimH antagonists for E. coli-induced Crohn’s disease. Org Biomol Chem. 2016;14(16):3913–25.
Patel J, Paillarse J-M, Françon P, Laveissière A, Bonny C, Ostos M. Sa1767 - Eb8018, a novel FimH blocker is well tolerated in a randomized, double-blind, placebo-controlled phase I study in healthy volunteers. Gastroenterology. 2018;154(6):S-387.
Reinisch W, Hébuterne X, Buisson A, Schreiber S, Desreumaux P, Paillarse JM, et al. P568 An open-label, multicenter, phase Ib, pharmacokinetic (pk) and safety study of a FimH blocker, Sibofimloc (TAK-018/EB8018), in patients with Crohn’s disease (CD). J Crohn’s Colitis. 2020;14(Supplement_1):S479–80.
Acknowledgements
We would like to thank Dr. Christoph Beglinger and Dr. Bettina Woelnerhanssen from the Claraspital, Basel, Switzerland and Dr. Joël Doré from INRA Metagenopolis, France for the recruitment of healthy volunteers. We want to thank the French association of Crohn’s Disease patients named AFA (Association François Aupetit) and the patients involved in the CrohnOmeter study. We would like to thank Abbvie for authorizing additional analyses of its PREDICT cohort.
MOBIDIC Study Investigators
Temitayo Adegbamigbe11, Tariq Ahmad12, Ian Arnott13, Yoram Bouhnik14, Franck Carbonnel15, Jean-Frédéric Colombel16, Glen Doherty17, J. R. Fraser Cummings18, Xavier Hébuterne19, Hans Herfarth20, David Kevans21, Guillaume Pineton de Chambrun22, Maria Nachury23, Stéphane Nancey24, Xavier Roblin25 and Mark A. W. Tremelling26
11Carolinas Medical Center, Charlotte, North Carolina, United States. 12Department of Gastroenterology, Royal Devon and Exeter Hospital National Health Service Foundation Trust, Exeter, UK. 13Gastrointestinal Unit, Western General Hospital, Edinburgh, UK. 14Beaujon Hospital, University Paris 7 Denis Diderot, Clichy, France. 15Department of Gastroenterology, Kremlin-Bicêtre Hospital, Assistance Publique-Hôpitaux de Paris, Le Kremlin-Bicêtre, France. 16Division of Gastroenterology, Department of Medicine, Icahn School of Medicine at Mount Sinai, New York, NY, United States. 17Center for Colorectal Disease, St. Vincent’s University Hospital & School of Medicine, University College Dublin, Ireland. 18Department of Gastroenterology, University Hospital Southampton National Health Service Foundation Trust, Southampton, UK. 19Nice University Hospital, University of Nice Sophia-Antipolis, Nice, France. 20Division of Gastroenterology and Hepatology, University of North Carolina, Chapel Hill, North Carolina, United States. 21Gastroenterologist at St James’s Hospital, Dublin, Ireland. 22Hôpital Saint-Eloi, University Hospital of Montpellier, Montpellier, France. 23Huriez Hospital, Université of Lille, Lille, France. 24Hospices Civils de Lyon and University Claude Bernard Lyon 1, Pierre-Benite, France. 25Saint-Etienne University Hospital, Saint-Etienne, France. 26Department of Gastroenterology, Norfolk and Norwich University Hospital, Colney Lane, Norwich, NR4 7UY, UK.
Funding
This work was supported by Enterome.
Author information
Authors and Affiliations
Consortia
Contributions
AL, RM, AC, and CB conceived the study and designed the experiments. NB, AS, MM, CN, and ED performed the experiments. AL, RM, JP, CB, GC, and GD analyzed and interpreted the data and supervised the overall research. MM-M and KWS provided AIEC strains. GC, GD, RM, AL, and CB wrote the manuscript. The author(s) read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The protocols for CrohnOmeter, PREDICT, and MOBIDIC studies were approved by the institutional medical ethics committees:
• Program for Protection of Human Subjects from Mount Sinai Hospital, HS#: 17-00029 (GCO#1: 17-0114(0001)).
• New England Independent Review Board (NEIRB) (Sponsor Protocol Number: IBD210 / OP097015.ENT).
• Office for Research Ethics Committees Northern Ireland (IBD210/OP097015.ENT).
• Comité de Protection des Personnes de la Pitié Salpêtrière.
Participants were given oral and written information prior to signing the informed consent form.
Consent for publication
Not applicable.
Competing interests
Jonathan Plassais, Alessandra Cervino, and Christophe Bonny are among the inventors of a patent protecting tools for assessing FimH blockers’ therapeutic efficiency (WO2019076931). All other authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Supplementary Table 1.
Demographic data and history of Crohn’s Disease of the patients with CD included in each of the presented studies. *: Montreal classification for Cohort 1 and 3. **: MOBIDIC: QUANTA Lite/LLOD=15.6mg/kg. CrohnOmeter: Bühlmann/LLOD=50mg/kg. PREDICT: Bühlmann/LLOD=30mg/kg. NA=Not available. Supplementary Table 2. Demographic data of the healthy volunteers included in the presented study. Supplementary Table 3. Median values for Enterobacteriaceae species in Fig. 2c (top table) and d (bottom table). Supplementary Table 4. List of isolated E. coli strains and associated information. Supplementary Figure 1. Evolution over time of the first cohort regarding HBI (left) and E. coli relative abundance (right). A linear mixed model was used to identify statistically significant differences between time points (visits) and did not reach significance for E. coli abundance (P = 0.51) nor for HBI (P = 0.41). Supplementary Figure 2. Some Proteobacteria express FimH adhesin. Cladogram representing the bacteria phyla detected in the human gut microbiome (left) and FimH presence with a focus on Enterobacteriaceae spp (right). Supplementary Figure 3. Dichotomy in patient with CD from Cohort 2. a, Microbial clustering as shown based on Bray–Curtis dissimilarity principal Coordinate Analysis (PCoA) metrics for HV and patients with CD from Cohort2 with PC1 ≤ 0.1 and PC1 > 0.1. Ellipsoids represent a 95% confidence interval surrounding each group. b, Relative abundance of Enterobacteriaceae spp in HV and patients with CD from Cohort2 with PC1 ≤ 0.1 and PC1 > 0.1. Non-parametric Mann-Whitney U test was used to identify the statistically significant differences between groups. (* P < 0.05, ** P < 0.005, **** P < 0.0001). Supplementary Figure 4. Structure of the bi-mannosylated FimH-blocker TAK-018. Supplementary Figure 5. Association between percentage of FimS-ON expression and aggregation to TAK-018 of different AIEC strains. The mapping against the fimS region revealed a strong association between the percentage of reads in the “ON” position, indicative of expression of the entire fim operon, and the aggregation to TAK-018. Median are represented. Non-parametric Mann-Whitney U test was used to identify the statistically significant differences between groups (n.s. non-significant, *** P < 0.0005, **** P < 0.0001). Supplementary Figure 6. TAK-018 prevents adhesion of LF82 E. coli to T84 intestinal epithelial cells in a FimH-dependent manner. Bars represent the mean value of individual points which themselves correspond to biological replicates. A non-parametric Kruskal-Wallis test was used to identify the statistically significant differences between the LF82 groups (P = 0.0002). Supplementary Figure 7. TAK-018 prevents pro-inflammatory cytokine secretion of human ileal explants upon incubation with LF82 E. coli. IL-6 and IL-8 secretion of human ileal explants incubated for 4 hours with 109 LF82 E. coli, in the presence of increasing concentrations of TAK-018. IL-1β was used as a positive control to trigger inflammation. Bars represent the mean value of individual points which themselves correspond to biological replicates. A linear mixed model was used to identify statistically significant differences between the groups No TAK-018, TAK-018 500nM and TAK-018 1µM (IL-6, P = 0.1; IL-8, P = 0.0006). Supplementary Figure 8. TAK-018 prevents adhesion of LF82 E. coli to primary human intestinal cells isolated from patient with Crohn’s disease. Microscopy images of GFP LF82 E. coli (green) on primary human ileal cells stained with phalloidin Alexa-547 (red) in presence of TAK-018 (bottom) or not (top).
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Chevalier, G., Laveissière, A., Desachy, G. et al. Blockage of bacterial FimH prevents mucosal inflammation associated with Crohn’s disease. Microbiome 9, 176 (2021). https://doi.org/10.1186/s40168-021-01135-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40168-021-01135-5