
Abstract
Background
Free-living and symbiotic chemosynthetic microbial communities support primary production and higher trophic levels in deep-sea hydrothermal vents. The shrimp Rimicaris exoculata, which dominates animal communities along the Mid-Atlantic Ridge, houses a complex bacterial community in its enlarged cephalothorax. The dominant bacteria present are from the taxonomic groups Campylobacteria, Desulfobulbia (formerly Deltaproteobacteria), Alphaproteobacteria, Gammaproteobacteria, and some recently discovered iron oxyhydroxide-coated Zetaproteobacteria. This epibiotic consortium uses iron, sulfide, methane, and hydrogen as energy sources. Here, we generated shotgun metagenomes from Rimicaris exoculata cephalothoracic epibiotic communities to reconstruct and investigate symbiotic genomes. We collected specimens from three geochemically contrasted vent fields, TAG, Rainbow, and Snake Pit, to unravel the specificity, variability, and adaptation of Rimicaris–microbe associations.
Results
Our data enabled us to reconstruct 49 metagenome-assembled genomes (MAGs) from the TAG and Rainbow vent fields, including 16 with more than 90% completion and less than 5% contamination based on single copy core genes. These MAGs belonged to the dominant Campylobacteria, Desulfobulbia, Thiotrichaceae, and some novel candidate phyla radiation (CPR) lineages. In addition, most importantly, two MAGs in our collection were affiliated to Zetaproteobacteria and had no close relatives (average nucleotide identity ANI < 77% with the closest relative Ghiorsea bivora isolated from TAG, and 88% with each other), suggesting potential novel species. Genes for Calvin-Benson Bassham (CBB) carbon fixation, iron, and sulfur oxidation, as well as nitrate reduction, occurred in both MAGs. However, genes for hydrogen oxidation and multicopper oxidases occurred in one MAG only, suggesting shared and specific potential functions for these two novel Zetaproteobacteria symbiotic lineages. Overall, we observed highly similar symbionts co-existing in a single shrimp at both the basaltic TAG and ultramafic Rainbow vent sites. Nevertheless, further examination of the seeming functional redundancy among these epibionts revealed important differences.
Conclusion
These data highlight microniche partitioning in the Rimicaris holobiont and support recent studies showing that functional diversity enables multiple symbiont strains to coexist in animals colonizing hydrothermal vents.
Video Abstract
Similar content being viewed by others
Background
Life in deep-sea hydrothermal vent ecosystems is sustained by both free-living and symbiotic microbial chemosynthetic primary production [1]. The shrimp Rimicaris exoculata dominates the faunal communities of several hydrothermal sites along the Mid-Atlantic Ridge (MAR) [2,3,4]. This species lives in dense aggregates in quite a warm part of the hydrothermal environment, 3–25°C, with nearly neutral pH [5], and slightly lower oxygen content than deep seawater [6]. It can be retrieved from geochemically contrasting environments. For example, hydrothermal vents such as those of Snake Pit and TAG, located at 3460 m depth, have developed on basaltic rocks, while Rainbow, located at 2400 m depth, is on ultramafic rock. Fluids emitted by basaltic sites are mainly enriched in sulfur and minerals [7]. In contrast, ultramafic fluids are relatively depleted in sulfur but enriched in methane, hydrogen, and ferrous iron [7], which may select for specific microbial chemoautotrophic communities. Whatever the site of sampling, adult Rimicaris harbor complex bacterial communities, one located in their hypertrophied cephalothoracic cavity and a second in their digestive system, where long microbial filaments have been identified whose role remains unknown [8,9,10]. The cephalothoracic cavity microbiota was first described as a single campylobacterial lineage (formerly called Epsilonproteobacteria [11,12,13]). It nevertheless proved to be much more diverse, including a secondary dominant lineage, Gammaproteobacteria [14,15,16,17], but also, to a lesser extent, Alpha and Deltaproteobacteria (renamed Desulfobulbia) [14, 18,19,20] and the more recently hypothesized Zetaproteobacteria [21]. In vivo experiments provided evidence for direct nutritional transfers from bacteria to the host across the cephalothoracic chamber integument and, therefore, indicating a true trophic association [22]. In addition, in silico and microscopy analyses suggest that four potential metabolic pathways (iron, sulfide, methane, and hydrogen oxidation) may co-occur within this community [14, 15, 18, 21]. This diversity of metabolisms indicates that the epibiotic community associated with R. exoculata is highly plastic, providing a potential adaptive advantage for the shrimp thriving in highly dynamic hydrothermal mixing zones. This diverse bacterial consortium could explain the clear success of the holobiont (i.e., the animal in interaction with its symbiotic partners) in colonizing these geochemically contrasted hydrothermal vents all along the Mid-Atlantic Ridge [4]. However, although previous studies identified diverse microbial populations, authors focused on dominant metabolisms and community members, leaving knowledge gaps regarding the overall functioning of co-occurring symbionts, some of which have been very little studied. Briefly, Jan and colleagues [21] described the dominant lineages, Campylobacteria and Gammaproteobacteria, and made a new hypothesis on the under-recovered Zetaproteobacteria. They focused on carbon fixation pathways (rTCA and CBB cycles for Campylobacteria and Gamma/Zetaproteobacteria, respectively), sulfur, hydrogen and nitrogen cycles, stress response, and interactions with the host. Still, authors identified taxobins as submetagenomes and did not undertake genome reconstruction [21]. Recently, Jian et al. [19] focused on Desulfobulbia and described a novel species, Candidatus Desulfobulbus rimicarensis, with specific symbiotic traits. However, the functional capacities and evolutionary relationships of the remaining epibionts, notably the Zetaproteobacteria, under-recovered compared with their free-living counterparts, remain unknown.
Biotic iron oxidation reactions were first hypothesized to occur in the Rimicaris cephalothoracic cavity based on observations of iron oxyhydroxide particles embedding microbial cells [6]. These mineral deposits are located in the upper part of the cavity where the outgoing seawater flow is expulsed. Here, they may be under microaerophilic conditions and partially enriched in carbon dioxide due to shrimp respiration. Two subsequent studies showed these iron particles have a stable mineral composition, with increasing deposits through the molt cycle, giving shrimps a rusty color by the end [23, 24]. An in vivo experiment also showed that chemoautotrophy was fueled by Fe2+ supply as the sole electron donor [22]. Still, only a very few genes affiliated to potential iron oxidizers, namely Zetaproteobacteria, have been identified in studies using molecular approaches [21], leading to major gaps in knowledge regarding potential symbiotic iron oxidation. It is possible that this coating with iron oxyhydroxides, closely attached to the bacterial cells [23, 24], impaired genomic DNA extraction from these specific epibionts.
In addition to these physical constraints, reconstructing and separating highly similar genomes from metagenomes, notably from complex host-associated microbial communities, remained a technical challenge until recently. The presence of closely related strains can increase assembly breaks, resulting in short orphan contigs that cannot be grouped together nor further analyzed. In addition, unlike for symbionts constrained in a specific organ such as tubeworm trophosomes [25], and despite precautions in the DNA extraction protocols, host contamination is unavoidable and can result in a high number of host-associated contiguous sequences. Therefore, efforts to understand the functioning of the epibiont community are still in their infancy. In particular, it remains unknown whether the highly diverse Rimicaris epibiont community is composed of specialists adapted to microniches that possibly complement or interact with each other through syntrophy, or rather composed of generalists able to perform a wide range of functions depending on the environmental conditions.
Here, we improved DNA extraction procedures to maximize DNA recovery from iron/mineral-embedded bacteria, followed by shotgun metagenomics and advanced binning strategies to reconstruct novel Rimicaris exoculata symbiotic genomes. We collected specimens from the three geochemically contrasted hydrothermal vents TAG, Snake Pit, and Rainbow to investigate functional diversity and assess potential niche differentiation at the genome level within and between sites. We aimed to decipher the functioning of the Rimicaris epibiont community as a whole, to better understand animal–epibiont interactions at a fine genomic scale and, for the first time, propose an overview of the capabilities of Zetaproteobacteria as symbionts.
Methods
Sample collection
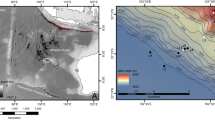
We collected Rimicaris exoculata specimens using a suction sampler manipulated by the remote operated vehicle (ROV) Victor 6000, controlled from on board the research vessel Pourquoi pas? Samples were taken from three hydrothermal sites on the Mid-Atlantic Ridge (Fig. 1): Rainbow (36° 13.760′ N, 33° 54.170′ W, 2292m depth) during the MoMAR (Leg2, August 25 to September 15, 2008, https://doi.org/10.17600/8010140) and BioBaz cruises (August 02 to 21, 2013, https://doi.org/10.17600/13030030), TAG (26° 8.237′ N, 44° 49.563′ W, 3625 m depth) and Snake Pit (23° 22.140′ N, 44° 57.054′ W, 3495 m depth) during the BICOSE cruise (January 11 to February 10, 2014, https://doi.org/10.17600/14000100), respectively. We rapidly froze the BioBaz and BICOSE samples at − 80°C on board for later genomic DNA extraction. We dissected branchiostegites and scaphognathites of some MoMAR specimens directly onboard under sterile conditions, fixed them for three hours in 3% formaldehyde seawater solution, and stored them frozen in PBS/Ethanol 1:1 until further use for FISH experiments.

Geographic locations of Mid-Atlantic Ridge hydrothermal vent sites Rainbow, TAG, and Snake Pit where Rimicaris specimens were sampled
DNA extraction
We performed sterile dissections in the lab on thawed samples before total genomic DNA extraction of the six Rimicaris cephalothoracic appendages (2 branchiostegites + 2 scaphognathites + 2 exopodites). As mentioned above, symbionts have previously been found embedded in iron particles, which may impair DNA extraction. To maximize DNA recovery, we chose specimens in the middle of their molt cycles, to avoid over embedding in minerals, but with high levels of microbial colonization according to earlier color-based identification [23]. Then, we aseptically crushed each sample for few minutes in a sterile mortar in iced lysis buffer to release as many bacteria as possible from the shrimp cephalothorax. We then used a vortex step of 5 min on the crushed samples to help separate bacteria from minerals. The DNA was then extracted using the Nucleospin Soil extraction kit (Macherey-Nagel) following the manufacturer’s instructions.
Microbial community DNA preparation and sequencing
We analyzed six Rimicaris individuals (hereafter referred to as RE5 and RE6 from TAG, RE12, and RE13, from Rainbow, and RE3 and RE7 from Snake Pit) by shotgun sequencing of total community DNA. We quantified DNA using a NanoDrop 1000. We sheared DNA to 175 bp using a Covaris S-series sonicator and completed metagenomic library construction using the Ovation Ultralow Library DR multiplex system (Nugen) following the manufacturer’s instructions. We performed metagenomic sequencing on an Illumina HiSeq 1000 and a NextSeq at the W.M. Keck sequencing facility at the Marine Biological Laboratory (MBL, MA, USA). All libraries consisted of paired-end data, with a 30-bp overlap.
Metagenomic analysis
We trimmed adaptors using bbduk from bbmap 38.22 [26] after which we processed reads using a snakemake [27] workflow implemented in anvi’o v5 (http://merenlab.org/2018/07/09/anvio-snakemake-workflows/) [28]. Briefly, we used illumina-utils v1.4 [29] with the “iu-filter-quality-minoche” program with default parameters for sequence quality filtering. We co-assembled two metagenomic datasets per site (that is, RE5 and RE6 for TAG; RE12 and RE13 for Rainbow; and RE3 and RE7 for Snake Pit) using Megahit 1.1.3 [30] and the --metasensitive mode, discarding contigs smaller than 1000 bp. We performed read recruitment analyses with Bowtie v2.3.4 [31]. We used the option “all-against-all” to map reads from each of the six individual samples onto each of the three co-assemblies. The details and a scheme of the bioinformatics pipeline are provided in Additional File 1.
To estimate symbiont relative abundance, we retrieved the raw counts mapping to each MAG using samtools view [32]. We normalized data within and between samples with Gene length corrected Trimmed Mean of M-values (GeTMM, [33]) using genome length instead of gene length. We used DESeq2 [34] for differential MAG abundance analysis between sites. Differentially abundant MAGs with adjusted p-value of 0.01 (padj < 0.01) and absolute log2 fold change of 2 were regarded as significant in this study.
For phylogenomic analyses, we searched for and aligned 120 bacterial marker genes of the MAGs using the identity and align commands of GTDB-Tk v1.1.0 [35]. We filtered closely related GTDB taxa identified with the “classify_wf” workflow with the taxa-filter option during the alignment step. We trimmed multiple sequence alignments using trimAl v1.4.1 [36] with the setting “-gt 0.5” to remove positions with gaps in more than 50% of sequences. We reconstructed a Maximum Likelihood (ML) phylogenetic tree using IQ-TREE v1.6.12 [37] with the “WAG” general matrix model [38] and 1000 bootstrap replicates that we visualized using anvi’o and FigTree v1.4.4. Finally, we made a finer GTDB reference taxa filtration after a first visualization, providing a better representation of our MAG genera. For this purpose, we removed references from distant families and added some from the closest genera or families in a supervised manner.
We calculated average nucleotide identity (ANI) online using the ANI calculator (https://www.ezbiocloud.net/tools/ani) [39]. As for functional analyses, we extracted KO assignations from the anvi’o database for each MAG and used the KEGG Decoder v 1.0.8.2 (www.github.com/bjtully/BioData/tree/master/KEGGDecoder) [40] to determine the completeness of various metabolic pathways based on a key set of genes. In addition, we used the RAST platform to provide a more detailed analysis of the different Zetaproteobacteria genomes (from this study and their free-living counterparts). The schematic representation of the predicted metabolic potentials within Zetaproteobacteria MAGs was constructed based on the KEGG annotations and the Reconstruct Pathway tool (https://www.genome.jp/kegg/tool/map_pathway.html). To explore the taxonomic composition of each samples based on the small-subunit rRNA, the quality-filtered reads were analyzed using the phyloFlash v3.4 pipeline [41] with the option “almost everything” and the SILVA database release 138.1 [42].
Code and data availability
The metagenome raw reads are available in the European Nucleotide Archive under Bioproject Accession Number PRJEB37577. We also made the FASTA files available for individual metagenomic co-assemblies and for the 49 MAGs at DOI (10.12770/4186eeef-32b1-4ffb-9a40-61a02a3852b7), as well as the anvi’o merged profile databases for each co-assembly and MAG. The URL https://gitlab.ifremer.fr/rimicaris/rimicaris-exoculata-cephalothoracic-epibionts-metagenomes provides access to a detailed reproducible bioinformatics workflow for all the computational analyses.
Fluorescence in situ hybridization procedures
We carried out FISH procedures as described by Duran et al. [9]. Briefly, we hybridized 0.6-μm transverse sections using the Zeta123-Cy5 probe (5′-ACTGATGGGCAGGTAACCACG-3′, [43]) directed toward Zetaproteobacteria, and the EPSI549-Cy3 probe (5′-CAGTGATTCCGAGTAACG-3′, [44]) directed toward Campylobacteria, together with DAPI staining for cell nucleus observation. Hybridization parameters were set at 35°C and 55% formamide as in Jan et al. [21]. Nonsense probes were tested but gave no signal. For observations, we used an Apotome AxioImager Z2 equipped with a Colibri LED system (Zeiss, Göttingen).
Results and discussion
In this study, we used metagenomic data of Rimicaris exoculata shrimp epibiont communities from three contrasting hydrothermal sites along the Mid-Atlantic Ridge to reconstruct bacterial genomes and examine functional potential at the genome-resolved scale.
The reconstruction of 49 Rimicaris-associated MAGs
Shotgun sequencing of total community DNA recovered from the cephalothorax of six R. exoculata individuals (RE3, RE5, RE6, RE7, RE12, RE13) yielded 22–119 million paired-end sequences. Metagenomic assembly of each site yielded 34K–64K contigs longer than 1 kbp, which recruited 14–92% of the raw sequencing reads. Additional File 2 provides the statistics on the raw number of reads, quality trimming, filtering, assembly, and recruitment results for each sample.
We clustered contigs across the six Rimicaris specimens with respect to their sequence composition signatures and differential coverage patterns. This binning approach allowed us to efficiently segregate the metagenomic assembly into 29 genome bins for Rainbow and 21 for TAG. We retained bins with more than 2 Mbp or more than 60% completion and less than 10% contamination based on the single occurrence of 71 single-copy core genes (SCG) that represent a modified version of the HMM profiles published by Lee [45] implemented in anvi’o [28]. We were unable to reconstruct bacterial MAGs for Snake Pit using these criteria, due to a much lower sequencing depth for shrimp samples from this site compared with the Rainbow and TAG ones. Chemically contrasting site Snake Pit however proved informative because it provided differential coverage values that improved the binning strategy.
Dereplication (an alignment fraction between genomes of both 0.10 and 0.75%) generated a single cluster containing TAG_MAG_00006 and RB_MAG_00023 Desulfocapsa (Desulfocapsaceae), with lower scores for the latter, which was then excluded. This resulted in a final collection of 49 MAGs to be further analyzed (Fig. 2). We nevertheless note that the 49 MAGs reconstructed and described herein represent only part of the Rimicaris cephalothorax diversity and that some remaining groups might be misrepresented given the fact that strain diversity can result in assembly breaks and therefore impair binning and recruitment analyses [46]. MAG collections for TAG and Rainbow, with estimates of completion and redundancy, total bin length, and taxonomic affiliation determined by GTDB-Tk, are provided in Additional File 3. We used read mapping against these 49 newly reconstructed genomes and normalization with GeTMM to estimate symbiont relative abundance within and between samples (Additional File 4). We used DESeq2 to compare the differential abundance of the MAGs between sites (Additional File 5 and 6). We hereafter focus our distribution analyses according to vent origin on the most abundant genome bins.

Static image from the anvi’o interactive display for the Rimicaris datasets with the 49 genome bins retained for this study. From inner to outer layers: phylogenomic tree based on concatenated marker proteins according to GTDB-Tk, length layer (shows the actual length of a genome), auxiliary layer with information about contigs stored in the contig database (GC-content), four view layers with information about MAGs across samples stored in the profile database (mean coverage), completion, redundancy, genome class based on GTDB-Tk, and bacterial genome bin layers. The horizontal layers show genome bin taxonomy based on GTDB-Tk for the ten most abundant families, total number of reads mapped and corresponding mapping percentage of reads, and total number of reads for each sample. Relative abundances of families are noted as the percentages of reads recruited to the bins for each sample
Abundance of Campylobacteria, Desulfobulbia (formerly Deltaproteobacteria), Gammaproteobacteria, Alphaproteobacteria, and newly recovered candidate phyla radiation (CPR) lineages
We observed a small number of symbionts that seemed to dominate at the different hydrothermal vent sites (Additional Files 4, 5, 6). The two most abundant MAGs at ultramafic Rainbow, RB_MAG_00025 Gammaproteobacteria (Thiotrichaceae family) and RB_MAG_00022 Patescibacteria (GCA-2747955 family), were not differentially abundant at any site. On the other hand, the third most abundant MAG at Rainbow RB_MAG_00015, Campylobacteria (Sulfurovaceae family), was significantly more abundant at Rainbow than at TAG (log2FoldChange>3). Similarly, we observed RB_MAG_00011, Campylobacteria (Sulfurimonadaceae family), in much higher abundance in the shrimps from Rainbow than TAG (log2FoldChange>7). Both lineages were retrieved from moderate temperature fluids with quite elevated sulfide concentration and in the presence of oxygen. Sulfurovaceae are well known to occur as symbionts associated with hydrothermal fauna, but this is less the case for Sulfurimonadaceae, whose genomes have been recently sequenced from Alviniconcha snails colonizing almost the same biotope as Rimicaris in Pacific hydrothermal vents [47]. The seemingly absence of dominant Campylobacteria MAGs at TAG, although they had been observed in previous studies [48], could be due to the presence of closely related Campylobacter strains impairing genome reconstruction of these populations at this site. Diverse Campylobacter were actually observed based on small-subunit rRNA reconstruction using phyloFlash (Additional File 7). The ultramafic Rainbow fluids partly depleted in sulfur and enriched in hydrogen may be more selective for a few Campylobacter lineages able to cope with hydrogen, while many closely related species could be present at TAG. Deep-sea vent Campylobacteria have also been shown to lack many DNA-repair genes, which could lead to numerous recombination, mutation, gene loss, and horizontal gene transfer events increasing microdiversity and conferring genomic plasticity on these taxa [49].
On the contrary, TAG most abundant MAGs TAG_MAG_00015 and RB_MAG_00014, Alphaproteobacteria Marinosulfonomonas sp. (Rhodobacteraceae family) were significantly more abundant at TAG than Rainbow (log2FoldChange =−5.5 and −3.13, respectively). In addition, abundant TAG_MAG_00007 (Candidatus Patescibacteria phylum, Paceibacteria class, from the candidate phyla radiation (CPR)) [50] showed a higher abundance at TAG as compared to Rainbow (log2FoldChange =−9.7). The latter contrasted with MAG RB_MAG_00022 from the same phylum and class, which did not show any significant differential abundance at Rainbow and TAG shrimps. TAG_MAG_00018, from the same phylum (Gracilibacteria class), was also significantly more abundant at TAG than Rainbow (log2FoldChange=−2.8). The predominance of these groups in the Rimicaris holobiont, associated with small genome size for TAG_MAG_00007, TAG_MAG_00018, and RB_MAG_00022 (1,201,735 bp, 841,694 bp, and 624,325 bp, respectively), adds to recent studies showing a wide distribution of CPR organisms. These lineages are often reported in association with abundant autotrophic taxa involved in nitrogen, sulfur, and iron cycling [51, 52] as well as arsenic in contaminated sediments [53].
We also observed several MAGs which abundance was not significantly different at both sites (abs(log2FoldChange)<2) like TAG_MAG_00006, Desulfobulbia (Desulfocapsaceae family), and RB_MAG_00001, Bacteroidia (Flavobacteriaceae family). Like for its counterpart RB_MAG_00025, TAG_MAG_00019 Gammaproteobacteria (Thiotrichaceae family) was not differentially abundant at both Rainbow and TAG shrimps.
These data provide further evidence for the occurrence of similar genomes in each host individual regardless of the site of sampling, yet with varying abundance partly reflecting contrasting environmental conditions. This may indicate that the cephalothoracic cavity, a nearly closed environment, protects symbionts from steep environmental modifications, leading to an overall similar symbiotic community whatever the site. Alternatively, a second hypothesis, not exclusive from the previous one, is that the shrimp have a pool of diverse symbiotic bacteria that can express their potential according to the chemical signature, thus explaining the colonization success of this holobiont.
Dual Zetaproteobacteria symbiosis in Rimicaris: Candidatus Ghiorsea rimicarensis and Candidatus Ghiorsea crypta
Iron-oxidizing Zetaproteobacteria Mariprofundus ferrooxydans PV-1 and JV-1 isolates were first described from the Lō’ihi hydrothermal systems in Hawaii [54, 55]. Zetaproteobacteria have since been described from very distinct habitats showing high ferrous iron concentrations, including coastal sediments, steel corrosion biofilms, and saline terrestrial springs [56, 57]. They are also reported at the MAR hydrothermal sites of the present study: TAG, Rainbow, and Snake Pit, where they dominate iron rich microbial mat communities [58]. These specialized, yet diverse, bacteria are well adapted to microaerophilic growth on Fe(II) and play an important role in the biogeochemical iron cycle within these diverse ecosystems, where abiotic iron oxidation was first thought to be the rule [43, 59]. Two distinct Zetaproteobacteria symbiotic genomes with high completeness, TAG_MAG_00014 and RB_MAG_00008, were recovered for the first time at both TAG and Rainbow sites. Zetaproteobacteria TAG_MAG_00014 was significantly more represented at TAG (log2FoldChange=−5.36, Additional File 5). However, Zetaproteobacteria RB_MAG_00008 was not significantly more abundant at Rainbow or TAG (abs(log2 Fold Change)<2). Differential abundance for the two lineages could suggest that specific Zetaproteobacteria lineages dominate at distinct or within a single hydrothermal vent site, depending on the environmental conditions. Surprisingly, the number of Zetaproteobacteria was higher at TAG, where G. bivora was also isolated [60], than at the iron-richer Rainbow site. This may be due to more robust iron incrustations on Rainbow specimens, despite the use of a thorough DNA extraction protocol.
Fluorescence in situ hybridization revealed Zetaproteobacteria in previously sampled Rainbow R. exoculata individuals [21], yet their genomic potential and potential microdiversity remained hidden. Here, we confirmed these findings using a similar FISH procedure, showing cells as small, curved rods, closely attached to the host cuticle under the long filamentous Campylobacteria symbionts (Fig. 3a, b). These observations are in agreement with previous TEM observations [24]. Unfortunately, we were able to retrieve one 16S rRNA gene for Zetaproteobacteria TAG_MAG_00014 only, and not for RB_MAG_00008. A blast analysis between the 16S rRNA gene full-length sequence extracted from TAG_MAG_0014 and the ones retrieved using PhyloFlash from TAG (RE5 and RE6) and Rainbow (RE12 and RE13) showed 100% and 97.7% identity, respectively (Additional File 8). Overall, data did not allow designing specific probes for each symbiont, nor did previous studies, which led to a single lineage sequence [16, 21] or unpublished data. Therefore, we cannot yet fully describe the distribution of either recovered Zetaproteobacteria-related MAGs.

FISH observations of a Rainbow R. exoculata branchiostegite with epibionts. All cells are labeled with DAPI (blue). Zetaproteobacteria are hybridized with the Zeta123 probe (red) [43]. a Zetaproteobacteria are closely attached to the cuticle. Dominant large filamentous bacteria (Campylobacteria) and thinner Gammaproteobacteria filaments can be seen. b Co-hybridization of filamentous Campylobacteria with the EPSI 549 probe (orange) [44] and small Zetaproteobacteria rods (red), highlighting the co-occurrence of both lineages
We were nevertheless able to perform a phylogenomic analysis using 120 bacterial marker genes from our 49 MAGs, as well as 600 closely related genomes from GTDB, to infer how Zetaproteobacteria and other MAGs reconstructed in this study are distributed relative to known taxa (Additional File 9A). Both RB-MAG-00008 and TAG-MAG-00014 were found to be divergent, yet close relatives of Ghiorsea bivora isolated from TAG (Additional File 9B) [60]. RB_MAG_00008, estimated to have a genome size of 1,853,475 bp and 92.96% completion based on the presence of SCG genes [45], suggests an actual genome size of around 2 Mbp. TAG_MAG_00014, on the contrary, had a reconstructed genome size of 1,625,457 bp and 94.37% completion, suggesting an actual genome size of around 1.7 Mbp, that is, a slightly reduced and presumably streamlined genome. Nevertheless, this is to our knowledge the first study to report symbiotic Zetaproteobacteria genomes. Based on average nucleotide identity (ANI) values of 88.74% between the two newly reconstructed Zetaproteobacteria MAGs and less than 77% with their closest relatives Ghiorsea bivora from TAG, we propose that they belong to potentially novel species [61]. We suggest the names Candidatus Ghiorsea rimicarensis for RB_MAG_00008 and Candidatus Ghiorsea crypta for TAG_MAG_00014.
Like their closest cultivated counterpart, both of these Zetaproteobacteria MAGs have the complete set of genes to fix carbon though the CBB cycle ([57, 60], Fig. 4, Additional File 10), confirming the possibility of autotrophic carbon assimilation in these host-associated lineages. Both have the form II of ribulose-1,5-bisphosphate carboxylase/oxygenase (cbbM gene, Additional File 10), which is in agreement with their symbiotic life under external seawater flow with relatively low oxygen and high carbon dioxide content [14, 57]. In addition, although they are described as fully autotrophic bacteria under culturing approaches [55, 60], the presence of genes for putative heterotrophic behavior may suggest they are capable of mixotrophy, as already proposed by Singer and colleagues [62] for M. profundus PV1. These two Zetaproteobacteria MAGs share genes for formate/nitrite transporter (focA), di- and tricarboxylate transporters (trk), as well as putative tricarboxylic transport membrane protein (tctA) in RB_MAG_00018 (Additional File 11). These carbon compound transporters could be used by the symbionts to import small organic molecules. Notably, focA encodes the formate/nitrite transporter (FNT) family of integral membrane proteins that show a great specificity for small anions, formate, nitrite, hydrosulfide, and also larger organic acids [63, 64].

Schematic representation of the predicted metabolic potentials within Zetaproteobacteria MAGs: RB_MAG_00008 and TAG_MAG_00014. The potential metabolic traits are focused on carbon metabolism, nitrogen uptake, sulfur and iron oxidation, hydrogen utilization, flagellum, chemotaxis, and detoxification. Genes present in both MAGs are indicated in red; those present only in RB_MAG_00008 or TAG_MAG_00014 are indicated in blue and green, respectively. CBB, Calvin–Benson–Bassham; H2ase, Hydrogenase; PEP, phosphoenolpyruvate; SDH, succinate dehydrogenase
Both strains seem capable of using several electron donors. The detection, for the first time, of canonical cyc2 iron oxidizing genes (Additional File 12) in both MAGs strengthens the possibility that these epibionts oxidize iron, as suggested by several authors [5, 8, 14, 21, 23, 24]. It is noteworthy that cyc2 genes, and splits containing them, showed a slightly lower average coverage compared with the remaining Zetaproteobacteria genes for RB_MAG_00008 (Additional File 13A). These data could imply environmental populations with high levels of strain heterogeneity, as described in [65]. Furthermore, Zetaproteobacteria RB-MAG-0008 showed genes coding for multicopper oxidase (MCO), which have been shown as potential homologs for iron oxidation in several other taxa oxidizing iron [66].
Detection of sulfide:quinone oxidoreductase (sqr) in both bins could suggest the ability to use sulfur in addition to H2 and Fe(II), as shown for G. bivora, and further extend the potential metabolic repertoire of the Zetaproteobacteria. It is however of note that no other sulfur oxidation genes were retrieved. Cbb3-type cytochrome c oxidase encoding genes, involved in respiration, oxygen sensing, and detoxification [62], but also in aerobic neutrophilic Fe oxidation, and which are highly expressed in cultivated Mariprofundus ferrooxydans PV-1, were also found in both bins [67].
Nevertheless, we also observed striking differences between the two MAGs, such as the capacity for hydrogen oxidation using at least three NiFe hydrogenase coding genes (Hydrogenase I cytochrome b subunit (hya), NiFe bidirectional hydrogenase (hox), and NiFe- hydrogen uptake hydrogenase (hup) genes) that were detected in RB_MAG_00008 only (Additional File 10). This may contribute to niche partitioning between these symbionts, giving them more flexibility with regard to variable environmental conditions and avoiding potential competition.
Both MAGs show genes implied in dissimilatory nitrate reduction to ammonium (DNRA, using nir genes; Fig. 4 and Additional File 10), suggesting the potential ability for nitrogen acquisition, like their closest relative [60]. Glutamine synthetase and glutamate synthase enzymes were also encoded in both bins, suggesting that ammonia could be incorporated into amino acids, whether it comes from nitrate reduction or environmental uptake, and that the symbionts may, therefore, have the potential to recycle their host’s ammonium waste. Together with genes coding for other amino acids like threonine, these findings add to previous data showing that Rimicaris epibionts can synthesize amino acids. The recently reconstructed Candidatus Desulfobulbus rimicarensis from Rimicaris proved capable of synthesizing all 20 amino acids, with all the genes essential for amino acid biosynthesis present in the genome and expressed [19]. The presence of genes coding for thiamin (thi), biotin (bio), riboflavin (rib), and cobalamin (cob) biosynthesis also indicates that both MAGs probably have the ability to synthesize vitamins (Additional File 10 and 11).
Finally, we observed that numerous flagella biosynthesis encoding genes including hook-associated genes (flgBCGIJK), possibly from the same operon [68], as well as flagellar rotation genes (motAB), together with chemotaxis genes (che), were detected in both symbiotic bins. The presence of these genes suggests Zetaproteobacteria are able to actively move toward favorable environments, in agreement with host–symbiont recognition and colonization. The squid-vibrio symbiosis shows distinct flagellar functions ranging from swimming capacity to chemotaxis and host signaling and communication, which suggest essential and constitutive roles for these structures [69]. It is possible that flagella identified in free-living closest relative G. bivora [60] are also used for host–symbiont colonization.
Occurrence of genes coding for arsenic resistance (arsBC) proteins suggests the Zetaproteobacteria symbionts have the genetic potential for arsenic detoxification that might benefit the shrimps. Arsenic is considered as a toxic metalloid acting as a molecular analog of phosphate and glycerol, leading to metabolic damage (proteins, lipids, DNA breaks, and inhibition of DNA repair; for review, see [70]). In animals, intoxication leads to organ necrosis and brain damage.
We observed genes coding for twin-arginine translocation protein (tat), phosphate (pst), and copper transporters (cop) in each MAG, suggesting they can also transport proteins (including potential bacterial toxins and degradative enzymes such as proteases and lipases), ions, and metals. In addition, the presence of exopolyphosphatases (ppx) and polyphosphate kinase (ppk) in both MAGs suggests they are capable of producing polyphosphate, in agreement with previous observations [14, 21, 62]. In Fig. 4, we present a metabolic model highlighting the predicted metabolic potentials together with host–symbiont patterns for the zetaproteobacterial symbionts.
Metabolically diverse Rimicaris epibionts
Overall, we observed Rimicaris symbionts to have genomes with considerable functional diversity (Fig. 5; for details, see Additional File 14). The potential for autotrophic growth was observed in ten out of the 49 MAGs. Eight of those ten MAGs, affiliated to Gamma, Zeta, and Alphaproteobacteria, contained the complete set of genes for the CBB cycle. Our data confirmed that Campylobacteria MAGs were the only ones capable of mediating chemoautotrophy through the rTCA cycle. The presence of both rTCA and CBB cycles in the Rimicaris epibiont community has already been reported [16, 21, 47] and suggests that it might allow the consortium to switch cycles depending on the oxygen and carbon dioxide balance in the environment. The rTCA cycle, harboring oxygen-sensitive enzymes, is supposedly better adapted to more anoxic conditions combined with higher temperatures and is energetically more efficient than the CBB cycle [18, 71]. This property might partly explain the success of Campylobacteria in these environments. In addition, the Desulfocapsaceae MAGs from both Rainbow and TAG sites were shown to possess genes for the Wood-Ljungdahl pathway, allowing the use of hydrogen as an electron donor and carbon dioxide as both an electron acceptor and for biosynthesis, confirming the recent findings of [19].

KEGG Decoder heat map based on KEGG annotation. The heat map represents metabolic pathway completeness of the MAGs based on the presence or absence of genes as determined by KEGG Decoder. The data represent the discussed pathways, all of which are shown in Additional File 15. The dendrogram at the top represents the similarity between the MAGs based on their metabolic pathways, using Euclidean distance and complete linkage clustering. Taxonomic affiliations at the class and phylum levels are indicated and represented by the different colors. NAD-reducing hydrogenase, hoxHFUY; NiFe hydrogenase Hyd−1, hyaABC
Diverse energy sources potentially powering those symbioses were identified (Additional File 14). Genes related to sulfur metabolism, including sox, sat, apr, dsr, and sqr, were shown in 32 MAGs (that is, more than half of the population). Genes indicating the potential for hydrogen oxidation, including hox and hyd genes, were shown in 17 MAGs. This confirms the importance of H2 as an electron donor for the Rimicaris symbiosis, which probably contributes to its success at Rainbow where sulfide is lower. Finally, two Zetaproteobacteria MAGs may oxidize iron, as mentioned above. We observed 29 MAGs encoding the cbb3-type cytochrome c oxidase enzyme, used for sensing and respiration with oxygen as electron acceptor, but also for protection against oxidative stress. Four MAGs (belonging to Rhodobacteraceae and Marinicellaceae families) showed the possible use of an alternative electron acceptor to oxygen, such as nitrate as they encoded all the genes required for the complete reduction of nitrate to dinitrogen. Although the gene coding for nitrate reductase was lacking in Campylobacteria, possibly due to incomplete genome bins, the presence of three other denitrification genes suggests that they could also potentially use nitrate, as described in Jan et al. [21].
In addition, 30 other MAGs encoded some of the enzymes involved in nitrogen metabolism. Members of families like Rhodobacteraceae seemed to possess the capacity for denitrification, DNRA, and nitrification, which may provide them with greater metabolic flexibility compared with more specialist taxa. This seemingly redundant genetic potential for each cycle might indicate microniches, where each symbiont could perform part of a cycle relayed by another so as to perform complete pathways together. This is the case for carbon, as both autotrophy and heterotrophy fuel the host with diverse organic carbon [22], but also for nitrogen uptake and the sulfur cycle.
In addition, because the epibionts have the potential to synthesize and transport vitamins, such as thiamin, riboflavin, and cobalamin, the nutritional advantages for the host may go beyond a rich source of carbon and energy. The presence of genes for the biosynthesis of energy storage compounds, such as polyphosphate, supports previous results showing polyphosphate granules in the R. exoculata holobiont as well as in M. ferrooxydans cells [14, 62] and the genomic potential for their synthesis [21].
A total of 41 MAGs showed genes involved in arsenic reduction (arsRABC), which may be an environmental adaptation with the ars operon possibly being transferred among bacterial lineages. The presence of high concentrations of arsenic, highly correlated with zinc, was reported along the Mid-Atlantic Ridge [72]. Microorganisms having the ars operon, which can be extrachromosomal and subject to lateral gene transfer, are resistant and can cope with arsenic by reduction (arsC) and export (arsB, helped by arsA), which is regulated by arsenic level (arsR). Biofilms, chemotactism, flagellar synthesis, and quorum sensing are also enhanced by arsenic, which can be chelated by polysaccharides embedding cells in biofilms [73]. It is noteworthy that polymers were observed on MET on several Rimicaris samples [24]. In our model, Campylobacteria, Gammaproteobacteria, and now Zetaproteobacteria produce a biofilm, synthetize polysaccharides, and share ars genes. These lineages probably enter an arsenic cycle leading to chelation, which lowers potential impact on the host.
Finally, every 10 days, adult shrimp undergo a molt event, which necessitates continual and controlled microbiota colonization processes. Among them, secretion systems and biofilm formation are considered as central to host–symbiont recognition. In line with this, the majority of the MAGs [43] share characteristics of pathogens and beneficial microbes through genes encoding secretion systems (the general types II and IV and a striking number of twin targeting or Tat systems) and biofilm formation that probably facilitate their success at colonizing the host (detailed in Additional File 14).
Niche partitioning in the Rimicaris holobiont
As for the Zetaproteobacteria, we observed genomic differences between closely related epibiotic strains that suggest niche partitioning is widespread in the Rimicaris holobiont. Genes for glycolysis, dissimilatory nitrate reduction, and thiosulfate oxidation were present in both RB_MAG_00025 and TAG_MAG_00019 abundant Thiotrichaceae symbiont populations. Nevertheless, enzymes for nitric oxide reduction, NiFe hydrogenase Hyd-1, and dissimilatory sulfate reduction were encoded only in TAG_MAG_00019, while those for dissimilatory sulfite reduction and sulfide oxidation was solely encoded in RB_MAG_00025. The observed functional differences between the closely related Thiotrichaceae MAGs could suggest some complementary rather than competitive strains and may explain their co-occurrence. It is also possible that one Thiotrichaceae strain performs some of the dissimilatory sulfate metabolic steps while the other covers the remaining ones. Likewise, both Marinosulfonomonas sp. RB_MAG_00014 and TAG_MAG_00015 have the potential for glycolysis, carbon fixation via the CBB cycle, and partial to complete reduction of nitrate to dinitrogen (N2), as well as sulfide and hydrogen oxidation (Fig. 5). Nevertheless, only TAG_MAG_00015 showed genes for both the complete oxidation of nitrate to N2 and DNRA. It should be noted that no Marinosulfonomonas sp. seemed able to utilize CH4, suggesting strains with different metabolic pathways than the ones described by Holmes et al. [74], putatively constrained by their association with animal hosts. Overall, we observed behind an apparent functional redundancy, a high symbiont strain diversity that possibly has important implications for the functioning of the complex Rimicaris symbiosis. These data are congruent with recent studies showing genomic heterogeneity in vent mussel symbiont populations that either possess or lack a key gene cluster, suggesting specialized rather than generalist symbionts [75, 76]. The high diversity of Rimicaris MAGs capable of sulfur oxidation is in agreement with previous work describing more than 16 different sulfur oxidizing strains in four Bathymodiolus species from the Mid-Atlantic Ridge, which showed a large adaptability to the holobiont in the vents [75]. These seemingly closely related strains were suggested to differ in key functions including the use of energy and nutrient sources, viral defense genes, and electron acceptors. These different studies posit that the costs for the maintenance of such symbiont diversity may be counterbalanced by the plasticity it offers, i.e., a larger adaptability and resilience, especially in these unstable environments. In addition to obvious functional differences, it is likely that the symbionts have more subtle phenetic differences such as a better adaptation to temperature, chemicals, or pressure, allowing each of these strains to occupy and be adapted to different microniches. High levels of strain variability and numerous ortholog key proteins in vent-associated polychaete worm Alvinella pompejana were hypothesized as being each optimally adapted to thermal fluctuations within the worm’s habitat [77]. Similarly, Alcaide et al. [78] suggested that diverse carboxyl esterases of the gill-associated microbiota from Rimicaris may reflect distinct habitat-specific adaptations. Although it was not possible to determine whether geochemical or thermal fluctuations impose selection pressures on the epibiont community, this study adds to previous work showing that symbiont genetic diversity is more widespread than currently appreciated and that it might underpin ecosystem functioning and resilience in the highly dynamic hydrothermal vents.
Conclusions
These data reveal a much more complex microbial consortium associated within R. exoculata than previously appreciated and highlight some generalized niche partitioning between symbionts. Our study stresses that the apparent functional redundancy at the genome-wide level between co-existing strains hides differences that may reflect distinct history traits. This may be the key to the success of this holobiont along the Mid-Atlantic Ridge, where it encounters contrasted habitats and shows itself capable of a great degree of connectivity [79]. Two zetaproteobacterial symbionts add to the metabolic catalog shared by the Rimicaris holobiont and to the repertoire of metabolic diversity and lifestyle of the Zetaproteobacteria. Taken together, our results reveal highly complex symbioses providing new insights into deep-sea ecosystem functioning and resilience to potential threats of anthropogenic or natural origin.
Availability of data and materials
All data generated or analyzed during this study are included in this published article (and its supplementary information files).
Change history
18 March 2022
A Correction to this paper has been published: https://doi.org/10.1186/s40168-022-01257-4
References
Dubilier N, Bergin C, Lott C. Symbiotic diversity in marine animals: The art of harnessing chemosynthesis. Nat Rev Microbiol. 2008;6(10):725–40. https://doi.org/10.1038/nrmicro1992.
Williams A, Rona P. Two new caridean shrimps (bresiliidae) from a hydrothermal field on the Mid- Atlantic Ridge. J Crustac Biol. 1986;6(3):446–62. https://doi.org/10.2307/1548184.
Segonzac M, de Saint Laurent M, Casanova B. L'énigme du comportement trophique des crevettes Alviocarididae des sites hydrothermaux de la dorsale médio-atlantique. Cah Biol Mar. 1993;34:535−71.
Zbinden M, Cambon-Bonavita M. Rimicaris exoculata: biology and ecology of a shrimp from deep-sea hydrothermal vents associated with ectosymbiotic bacteria. Mar Ecol Prog Ser Inter-Res Sci Center. 2020;652:187–222.
Schmidt C, Le Bris N, Gaill F. Interactions of deep-sea vent invertebrates with their environment: the case of Rimicaris exoculata. J Shellfish Res. 2008;27(1):79–90. https://doi.org/10.2983/0730-8000(2008)27[79:IODVIW]2.0.CO;2.
Zbinden M, Le Bris N, Gaill F, Compère P. Distribution of bacteria and associated minerals in the gill chamber of the vent shrimp Rimicaris exoculata and related biogeochemical processes. Mar Ecol Prog Ser. 2004;284:237–51. https://doi.org/10.3354/meps284237.
Charlou JL, Donval JP, Konn C, Ondréas H, Fouquet Y, Jean-Baptiste P, et al. High production and fluxes of H2 and CH4 and evidence of abiotic hydrocarbon synthesis by serpentinization in ultramafic-hosted hydrothermal systems on the Mid-Atlantic Ridge. Divers Hydrothermal Syst Slow Spreading Ocean Ridges. 2010:265–96. https://doi.org/10.1029/2008GM000752.
Zbinden M, Cambon-Bonavita M-A. Occurrence of Deferribacterales and Entomoplasmatales in the deep-sea Alvinocarid shrimp Rimicaris exoculata gut. FEMS Microbiol Ecol. 2003;46(1):23–30. https://doi.org/10.1016/S0168-6496(03)00176-4.
Durand L, Zbinden M, Cueff-Gauchard V, Duperron S, Roussel EG, Shillito B, et al. Microbial diversity associated with the hydrothermal shrimp Rimicaris exoculata gut and occurrence of a resident microbial community. FEMS Microbiol Ecol. 2010;71(2):291–303. https://doi.org/10.1111/j.1574-6941.2009.00806.x.
Durand L, Roumagnac M, Cueff-Gauchard V, Jan C, Guri M, Tessier C, et al. Biogeographical distribution of Rimicaris exoculata resident gut epibiont communities along the Mid-Atlantic Ridge hydrothermal vent sites. FEMS Microbiol Ecol. 2015;91:1–15.
Polz MF, Cavanaugh CM. Dominance of one bacterial phylotype at a Mid-Atlantic Ridge hydrothermal vent site. Proc Natl Acad Sci U S A. 1995;92(16):7232–6. https://doi.org/10.1073/pnas.92.16.7232.
Waite DW, Vanwonterghem I, Rinke C, Parks DH, Zhang Y, Takai K, et al. Comparative genomic analysis of the class Epsilonproteobacteria and proposed reclassification to epsilonbacteraeota (phyl. nov.). Front Microbiol. 2017;8:682.
Waite DW, Vanwonterghem I, Rinke C, Parks DH, Zhang Y, Takai K, et al. Addendum: Comparative genomic analysis of the class Epsilonproteobacteria and proposed reclassification to Epsilonbacteraeota (phyl. nov.). Front Microbiol. 2018;9:1–2.
Zbinden M, Shillito B, Le Bris N, de Villardi de Montlaur C, Roussel E, Guyot F, et al. New insights in metabolic diversity among the epibiotic microbial communities of the hydrothermal shrimp Rimicaris exoculata. J Exp Mar Biol Ecol. 2008;159:131–40.
Petersen JM, Zielinski FU, Pape T, Seifert R, Moraru C, Amann R, et al. Hydrogen is an energy source for hydrothermal vent symbioses. Nature. 2011;476(7359):176–80. https://doi.org/10.1038/nature10325.
Guri M, Durand L, Cueff-Gauchard V, Zbinden M, Crassous P, Shillito B, et al. Acquisition of epibiotic bacteria along the life cycle of the hydrothermal shrimp Rimicaris exoculata. ISME J. 2012;6(3):597–609. https://doi.org/10.1038/ismej.2011.133.
Cowart DA, Durand L, Cambon-Bonavita MA, Arnaud-Haond S. Investigation of bacterial communities within the digestive organs of the hydrothermal vent shrimp Rimicaris exoculata provide insights into holobiont geographic clustering. PLoS One. 2017;12:1–22.
Hügler M, Sievert SM. Beyond the calvin cycle: autotrophic carbon fixation in the ocean. Annu Rev Mar Sci. 2011;3(1):261–89. https://doi.org/10.1146/annurev-marine-120709-142712.
Jiang L, Liu X, Dong C, Huang Z, Cambon-Bonavita M-A, Alain K, et al. Candidatus Desulfobulbus rimicarensis, an uncultivated deltaproteobacterial epibionts from the deep-sea hydrothermal vent shrimp Rimicaris exoculata. Appl Environ Microbiol. 2020;86(8):e02549–19.
Parks DH, Chuvochina M, Waite DW, Rinke C, Skarshewski A, Chaumeil PA, et al. A standardized bacterial taxonomy based on genome phylogeny substantially revises the tree of life. Nat Biotechnol. 2018;36(10):996–1004. https://doi.org/10.1038/nbt.4229.
Jan C, Petersen JM, Werner J, Teeling H, Huang S, Glöckner FO, et al. The gill chamber epibiosis of deep-sea shrimp Rimicaris exoculata: an in-depth metagenomic investigation and discovery of Zetaproteobacteria. Environ Microbiol. 2014;16(9):2723–38. https://doi.org/10.1111/1462-2920.12406.
Ponsard J, Cambon-Bonavita M-A, Zbinden M, Lepoint G, Joassin A, Corbari L, et al. Inorganic carbon fixation by chemosynthetic ectosymbionts and nutritional transfers to the hydrothermal vent host-shrimp Rimicaris exoculata. ISME J. 2013;7(1):96–109. https://doi.org/10.1038/ismej.2012.87.
Corbari L, Zbinden M, Cambon-Bonavita MA, Gaill F, Compère P. Bacterial symbionts and mineral deposits in the branchial chamber of the hydrothermal vent shrimp Rimicaris exoculata: relationship to moult cycle. Aquat Biol. 2008;1:225–38. https://doi.org/10.3354/ab00024.
Corbari L, Cambon-Bonavita MA, Long GJ, Grandjean F, Zbinden M, Gaill F, et al. Iron oxide deposits associated with the ectosymbiotic bacteria in the hydrothermal vent shrimp Rimicaris exoculata. Biogeosciences. 2008;5(5):1295–310. https://doi.org/10.5194/bg-5-1295-2008.
Reveillaud J, Anderson R, Reves-Sohn S, Cavanaugh C, Huber JA. Metagenomic investigation of vestimentiferan tubeworm endosymbionts from Mid-Cayman Rise reveals new insights into metabolism and diversity. Microbiome. 2018;6(1):19. https://doi.org/10.1186/s40168-018-0411-x.
Bushnell B, Rood J, Singer E. BBMerge – Accurate paired shotgun read merging via overlap. PLoS One. 2017;12(10):e0185056. https://doi.org/10.1371/journal.pone.0185056.
Köster J, Rahmann S. Snakemake—a scalable bioinformatics workflow engine. Bioinformatics. 2012;28(19):2520–2. https://doi.org/10.1093/bioinformatics/bts480.
Eren AM, Esen ÖC, Quince C, Vineis JH, Morrison HG, Sogin ML, et al. Anvi’o: an advanced analysis and visualization platform for ‘omics data. PeerJ. 2015;3:e1319. https://doi.org/10.7717/peerj.1319.
Eren AM, Vineis JH, Morrison HG, Sogin ML. A filtering method to generate high quality short reads using Illumina paired-end technology. PLoS One. 2013;8:6–11.
Li D, Liu CM, Luo R, Sadakane K, Lam TW. MEGAHIT: An ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics. 2015;31(10):1674–6. https://doi.org/10.1093/bioinformatics/btv033.
Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9(4):357–9. https://doi.org/10.1038/nmeth.1923.
Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25(16):2078–9. https://doi.org/10.1093/bioinformatics/btp352.
Smid M, van den Braak RRJ C, van de Werken HJG, van Riet J, van Galen A, de Weerd V, et al. Gene length corrected trimmed mean of M-values (GeTMM) processing of RNA-seq data performs similarly in intersample analyses while improving intrasample comparisons. BMC Bioinformatics. 2018;19:1–13.
Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:1–21.
Chaumeil P-A, Mussig AJ, Hugenholtz P, Parks DH. GTDB-Tk: a toolkit to classify genomes with the Genome Taxonomy Database. Bioinformatics. 2019;36:1925–7.
Capella-Gutiérrez S, Silla-Martínez JM, Gabaldón T. trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics. 2009;25(15):1972–3. https://doi.org/10.1093/bioinformatics/btp348.
Nguyen LT, Schmidt HA, Von Haeseler A, Minh BQ. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol Biol Evol. 2015;32(1):268–74. https://doi.org/10.1093/molbev/msu300.
Whelan S, Goldman N. A general empirical model of protein evolution derived from multiple protein families using a maximum-likelihood approach. Mol Biol Evol. 2001;18(5):691–9. https://doi.org/10.1093/oxfordjournals.molbev.a003851.
Yoon S-H, Ha S, Lim J, Kwon S, Chun J. A large-scale evaluation of algorithms to calculate average nucleotide identity. Antonie Van Leeuwenhoek. 2017;110(10):1281–6. https://doi.org/10.1007/s10482-017-0844-4.
Graham ED, Heidelberg JF, Tully BJ. Potential for primary productivity in a globally-distributed bacterial phototroph. ISME J. 2018;12(7):1861–6. https://doi.org/10.1038/s41396-018-0091-3.
Gruber-Vodicka HR, Seah BKB, Pruesse E. phyloFlash: Rapid small-subunit rRNA profiling and targeted assembly from metagenomes. mSystems. 2020;5(5):e00920.
Gurevich A, Saveliev V, Vyahhi N, Tesler G. QUAST: Quality assessment tool for genome assemblies. Bioinformatics. 2013;29(8):1072–5. https://doi.org/10.1093/bioinformatics/btt086.
Kato S, Yanagawa K, Sunamura M, Takano Y, Ishibashi JI, Kakegawa T, et al. Abundance of Zetaproteobacteria within crustal fluids in back-arc hydrothermal fields of the Southern Mariana Trough. Environ Microbiol. 2009;11(12):3210–22. https://doi.org/10.1111/j.1462-2920.2009.02031.x.
Lin X, Wakeham SG, Putnam IF, Astor YM, Scranton MI, Chistoserdov AY, et al. Comparison of vertical distributions of prokaryotic assemblages in the anoxic cariaco basin and black sea by use of fluorescence in situ hybridization. Appl Environ Microbiol. 2006;72(4):2679–90. https://doi.org/10.1128/AEM.72.4.2679-2690.2006.
Lee MD. GToTree: a user-friendly workflow for phylogenomics. Bioinformatics. 2019;35:4162–4. https://doi.org/10.1093/bioinformatics/btz188.
Olm MR, Brown CT, Brooks B, Banfield JF. DRep: A tool for fast and accurate genomic comparisons that enables improved genome recovery from metagenomes through de-replication. ISME J. 2017;11(12):2864–8. https://doi.org/10.1038/ismej.2017.126.
Beinart RA, Luo C, Konstantinidis KT, Stewart FJ, Girguis PR. The bacterial symbionts of closely related hydrothermal vent snails with distinct geochemical habitats show broad similarity in chemoautotrophic gene content. Front Microbiol. 2019;10:1–13.
Le Bloa S, Durand L, Cueff-Gauchard V, Le Bars J, Taupin L, Marteau C, et al. Highlighting of quorum sensing lux genes and their expression in the hydrothermal vent shrimp Rimicaris exoculata ectosymbiontic community. Possible use as biogeographic markers. PLoS One. 2017;12:1–19.
Nakagawa S, Takaki Y, Shimamura S, Reysenbach AL, Takai K, Horikoshi K. Deep-sea vent ε-proteobacterial genomes provide insights into emergence of pathogens. Proc Natl Acad Sci U S A. 2007;104(29):12146–50. https://doi.org/10.1073/pnas.0700687104.
Hug LA, Baker BJ, Anantharaman K, Brown CT, Probst AJ, Castelle CJ, et al. A new view of the tree of life. Nat Microbiol. 2016;1:1–6.
Herrmann M, Wegner CE, Taubert M, Geesink P, Lehmann K, Yan L, et al. Predominance of Cand. Patescibacteria in groundwater is caused by their preferential mobilization from soils and flourishing under oligotrophic conditions. Front Microbiol. 2019;10:1–15.
Sieber CMK, Paul BG, Castelle CJ, Hu P, Tringe SG, Valentine DL, et al. Unusual metabolism and hypervariation in the genome of a gracilibacterium (Bd1-5) from an oil-degrading community. MBio. 2019;10:1–14.
Suhadolnik MLS, Salgado APC, Scholte LLS, Bleicher L, Costa PS, Reis MP, et al. Novel arsenic-transforming bacteria and the diversity of their arsenic-related genes and enzymes arising from arsenic-polluted freshwater sediment. Sci Rep. 2017;7:1–17.
Emerson D, Moyer CL. Neutrophilic Fe-oxidizing bacteria are abundant at the Loihi seamount hydrothermal vents and play a major role in Fe oxide deposition. Appl Environ Microbiol. 2002;68(6):3085–93. https://doi.org/10.1128/AEM.68.6.3085-3093.2002.
Emerson D, Rentz JA, Lilburn TG, Davis RE, Aldrich H, Chan C, et al. A novel lineage of proteobacteria involved in formation of marine Fe-oxidizing microbial mat communities. PLoS One. 2007;2(8):e667. https://doi.org/10.1371/journal.pone.0000667.
Field EK, Sczyrba A, Lyman AE, Harris CC, Woyke T, Stepanauskas R, et al. Genomic insights into the uncultivated marine Zetaproteobacteria at Loihi Seamount. ISME J. 2015;9(4):857–70. https://doi.org/10.1038/ismej.2014.183.
McAllister SM, Moore RM, Gartman A, Luther GW, Emerson D, Chan CS. The Fe(II)-oxidizing Zetaproteobacteria: historical, ecological and genomic perspectives. FEMS Microbiol Ecol. 2019;95:1–18.
Scott JJ, Breier JA, Luther GW, Emerson D. Microbial iron mats at the Mid-Atlantic Ridge and evidence that Zetaproteobacteria may be restricted to iron-oxidizing marine systems. PLoS One. 2015;10:1–19.
Hoshino T, Kuratomi T, Morono Y, Hori T, Oiwane H, Kiyokawa S, et al. Ecophysiology of Zetaproteobacteria associated with shallow hydrothermal iron-oxyhydroxide deposits in Nagahama Bay of Satsuma Iwo-Jima, Japan. Front Microbiol. 2016;6:1–11.
Mori JF, Scott JJ, Hager KW, Moyer CL, Küsel K, Emerson D. Physiological and ecological implications of an iron- or hydrogen-oxidizing member of the Zetaproteobacteria, Ghiorsea bivora, gen. nov., sp. Nov. ISME J. 2017;11(11):2624–36. https://doi.org/10.1038/ismej.2017.132.
Chun J, Oren A, Ventosa A, Christensen H, Arahal DR, da Costa MS, et al. Proposed minimal standards for the use of genome data for the taxonomy of prokaryotes. Int J Syst Evol Microbiol. 2018;68(1):461–6. https://doi.org/10.1099/ijsem.0.002516.
Singer E, Emerson D, Webb EA, Barco RA, Kuenen JG, Nelson WC, et al. Mariprofundus ferrooxydans PV-1 the first genome of a marine Fe(II) oxidizing Zetaproteobacterium. PLoS One. 2011;6(9):e25386. https://doi.org/10.1371/journal.pone.0025386.
Lü W, Du J, Schwarzer NJ, Wacker T, Andrade SLA, Einsle O. The formate/nitrite transporter family of anion channels. Biol Chem Germany. 2013;394(6):715–27. https://doi.org/10.1515/hsz-2012-0339.
Sun P, Li J, Zhang X, Guan Z, Xiao Q, Zhao C, et al. Crystal structure of the bacterial acetate transporter SatP reveals that it forms a hexameric channel. J Biol Chem. 2018;293(50):19492–500. https://doi.org/10.1074/jbc.RA118.003876.
McKay LJ, Dlakić M, Fields MW, Delmont TO, Eren AM, Jay ZJ, et al. Co-occurring genomic capacity for anaerobic methane and dissimilatory sulfur metabolisms discovered in the Korarchaeota. Nat Microbiol. 2019;4(4):614–22. https://doi.org/10.1038/s41564-019-0362-4.
He S, Barco RA, Emerson D, Roden EE. Comparative genomic analysis of neutrophilic iron(II) oxidizer genomes for candidate genes in extracellular electron transfer. Front Microbiol. 2017;8:1–17.
Barco RA, Emerson D, Sylvan JB, Orcutt BN, Jacobson Meyers ME, Ramírez GA, et al. New insight into microbial iron oxidation as revealed by the proteomic profile of an obligate iron-oxidizing chemolithoautotroph. Appl Environ Microbiol. 2015;81(17):5927–37. https://doi.org/10.1128/AEM.01374-15.
Waters RC, O’Toole PW, Ryan KA. The FliK protein and flagellar hook-length control. Protein Sci. 2007;16(5):769–80. https://doi.org/10.1110/ps.072785407.
Aschtgen MS, Brennan CA, Nikolakakis K, Cohen S, McFall-Ngai M, Ruby EG. Insights into flagellar function and mechanism from the squid–vibrio symbiosis. NPJ Biofilms Microbiomes. 2019;5(1):32. https://doi.org/10.1038/s41522-019-0106-5.
Andres J, Bertin PN. The microbial genomics of arsenic. FEMS Microbiol Rev. 2016;40(2):299–322. https://doi.org/10.1093/femsre/fuv050.
Markert S, Arndt C, Felbeck H, Becher D, Sievert SM, Hügler M, et al. Physiological proteomics of the uncultured endosymbiont of Riftia pachyptila. Science. 2007;315:247–50.
Fouquet Y, Cambon P, Etoubleau J, Charlou JL, Ondréas H, Barriga FJAS, et al. Geodiversity of hydrothermal processes along the Mid-Atlantic Ridge and ultramafic-hosted mineralization: a new type of oceanic Cu-Zn-Co-Au volcanogenic massive sulfide deposit. Geophys Monogr Ser. 2013;188:321–67.
Gupta P, Diwan B. Bacterial Exopolysaccharide mediated heavy metal removal: a Review on biosynthesis, mechanism and remediation strategies. Biotechnol Reports. 2017;13:58–71. https://doi.org/10.1016/j.btre.2016.12.006.
Holmes AJ, Kelly DP, Baker SC, Thompson AS, De Marco P, Kenna EM, et al. Methylosulfonomonas methylovora gen. nov., sp. nov., and Marinosulfonomonas methylotropha gen. nov., sp. nov.: novel methylotrophs able to grow on methanesulfonic acid. Arch Microbiol. 1997;167(1):46–53. https://doi.org/10.1007/s002030050415.
Ansorge R, Romano S, Sayavedra L, Porras MÁG, Kupczok A, Tegetmeyer HE, et al. Functional diversity enables multiple symbiont strains to coexist in deep-sea mussels. Nat Microbiol. 2019;4(12):2487–97. https://doi.org/10.1038/s41564-019-0572-9.
Ikuta T, Takaki Y, Nagai Y, Shimamura S, Tsuda M, Kawagucci S, et al. Heterogeneous composition of key metabolic gene clusters in a vent mussel symbiont population. ISME J. 2016;10(4):990–1001. https://doi.org/10.1038/ismej.2015.176.
Grzymski JJ, Murray AE, Campbell BJ, Kaplarevic M, Gao GR, Lee C, et al. Metagenome analysis of an extreme microbial symbiosis reveals eurythermal adaptation and metabolic flexibility. Proc Natl Acad Sci U S A. 2008;105(45):17516–21. https://doi.org/10.1073/pnas.0802782105.
Alcaide M, Tchigvintsev A, Martínez-Martínez M, Popovic A, Reva ON, Lafraya Á, et al. Identification and characterization of carboxyl esterases of gill chamber-associated microbiota in the deep-sea shrimp Rimicaris exoculata by using functional metagenomics. Appl Environ Microbiol. 2015;81(6):2125–36. https://doi.org/10.1128/AEM.03387-14.
Teixeira S, Cambon-Bonavita M-A, Serrão EA, Desbruyéres D, Arnaud-Haond S. Recent population expansion and connectivity in the hydrothermal shrimp Rimicaris exoculata along the Mid-Atlantic Ridge. J Biogeogr. 2011;38(3):564–74. https://doi.org/10.1111/j.1365-2699.2010.02408.x.
Acknowledgements
We thank chief scientists of the MOMAR2008-Leg2, BioBaz 2013, and BICOSE 2014 cruises (J. Dyment, F. Lallier, and M.A. Cambon-Bonavita) and the captains and crew of R/Vs Pourquoi pas? and Atalante and ROV Victor for logistic assistance in collecting samples. We also thank Cyrielle Jan for FISH experiments and pictures, Hilary Morrison for assistance with sequencing, and A. Murat Eren, Florian Trigodet, Cyril Noël, and Loïs Maignien for their insights on the manuscript and on bioinformatics and statistical analyses. We are really grateful to two anonymous reviewers for their constructive comments, which helped us to improve the manuscript.
Funding
Funding was provided by the Ifremer REMIMA program and LabexMer post-doctoral fellowship from the University of Brest.
Author information
Authors and Affiliations
Contributions
MAC interpreted the data and wrote the manuscript. JA analyzed the data, prepared the figures, and wrote the manuscript. VCG organized the samples and performed the laboratory assays. JR performed the laboratory assays, analyzed the data, and wrote the manuscript. All authors read and approved the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable
Consent for publication
Not applicable
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1.
Details and scheme of the bioinformatics pipeline
Additional file 2.
Quality trimming and filtering statistics for each individual, number of assembled contigs longer than 1 kbp for each site, and number of reads recruited by these contigs for each metagenome
Additional file 3.
MAG collection, estimates of completion and redundancy calculated based on the occurrence of Single-copy Core Genes (SCG), number of contigs, total bin length, and taxonomic affiliation from TAG (above) and Rainbow (below) determined by GTDB-Tk
Additional file 4.
GeTMM normalized counts of each MAG
Additional file 5.
DESeq2 results table with baseMean, Log2FoldChange, lfcSE, stat, pvalue and padj for each of the 49 studied MAGs
Additional file 6.
Differentially abundants MAGs between site identified through DESeq2. MAGs are indicated with the family they belong when they are affiliated until this level. Positive log2FoldChange indicate abundant taxa in Rainbow, negative log2FoldChange indicate abundant taxa in TAG. MAG are colored according to the phyla they belong. Cut off value for inclusion in the plot was 0.01 for padj and 2 for log2FoldChange absolute value. Dot sizes correspond to mean counts after normalization with GeTMM and DESeq2 (corresponding to baseMean in Additional file 5)
Additional file 7.
PhyloFlash analysis results. Heatmap of taxonomic assignments (rows) for small-subunit rRNA reads in the six individual metagenomes (columns). The plot was generated using with the comparison script provided with phyloFlash. Color intensities represent the percentage of reads mapping to a given taxon, separated by prokaryotes (blue) and eukaryotes (red). Samples are clustered by their similarity in terms of taxonomic content and taxa are clustered by their co-occurrence across samples. Clustering are based on the euclidean distance and on the Ward's minimum variance method
Additional file 8
BLAST of 16S rRNA genes for Zetaproteobacteria. BLASTN search were performed between the 16S rRNA sequences affiliated to the Zetaproteobacteria assembled using SPAdes in phyloFlash and the 16S rRNA sequence retrieved from TAG_MAG_00014
Additional file 9 A
. Maximum-likelihood tree based on concatenated marker proteins according to the GTDB-Tk genome phylogeny visualized using anvi’o. Tree includes 600 genomes from GTDB and 49 MAGs covering mostly unknown genera, highlighting the importance of lineages lacking representatives. A single Firmicutes was used to root the tree. The bars in the innermost circular layer show the phylum affiliation of each genome. The second layer represents the family affiliation. The third layer marks genomes as either MAGs from our study (49, black) or genomes from GTDB (grey). The outermost layer shows the genus affiliation (10) or the lack thereof (19) of our MAGs. Only the families and genera observed in the MAGs are shown. B. Zoom inset of the Zetaproteobacteria phylogenetic relationships visualized using FigTree. Nodes represented by a dot indicate a bootstrap value of 100; lower values are specified
Additional file 10
Key gene predictions from Zetaproteobacterial MAGs RB_MAG_00008 and TAG_MAG_00014 and free-living Ghiorsea bivora reference genome (NCBI accession number GCF_000744415.1) using RAST and FeGenie (indicated by a star). NA: “Not Available’
Additional file 11.
Table containing the names of all genes found per MAG using KEGG and COG annotation
Additional file 12
Iron genes and gene clusters identified by FeGenie for the 49 MAGs and reference genomes Ghiorsea bivora and Mariprofundus ferrooxydans PV-1 (NCBI accession number GCF_000744415.1 and GCF_000153765.1). Cyc2 genes were retrieved in both MAGs, confirming they have the potential to oxidize Fe (II)
Additional file 13
Differential coverage of contigs within Zetaproteobacteria bins. Static image from the anvi’o refine display for A. RB_MAG_00008 and B. TAG_MAG_00014. From inner to outer layers: clustering based on sequence composition and differential coverage with Euclidian distance and Ward clustering method, length layer (shows the actual length of a split), auxiliary layer with information about contigs stored in the contig database (GC-content), four view layers with information about MAGs across samples stored in the profile database (mean coverage), and Ribosomal RNA presence. Splits containing the cyc2 genes are highlighted in red
Additional file 14
Analysis supplement: analysis details of metabolic potential among Rimicaris epibionts as depicted in Fig. 5
Additional file 15
KEGG Decoder heat map representing metabolic pathway completeness of the MAGs based on the presence or absence of genes as determined by KEGG Decoder. The dendrogram at the top represents the similarity between the MAGs based on their metabolic pathways, using Euclidean distance and complete linkage clustering. Taxonomic affiliations at the class and phylum levels are indicated and represented by the different colors. NAD-reducing hydrogenase: hoxHFUY. NiFe hydrogenase Hyd−1: hyaABC
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Cambon-Bonavita, MA., Aubé, J., Cueff-Gauchard, V. et al. Niche partitioning in the Rimicaris exoculata holobiont: the case of the first symbiotic Zetaproteobacteria. Microbiome 9, 87 (2021). https://doi.org/10.1186/s40168-021-01045-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40168-021-01045-6