
Abstract
The skin constitutes the primary physical barrier between vertebrates and their external environment. Characterization of skin microorganisms is essential for understanding how a host evolves in association with its microbial symbionts, modeling immune system development, diagnosing illnesses, and exploring the origins of potential zoonoses that affect humans. Although many studies have characterized the human microbiome with culture-independent techniques, far less is known about the skin microbiome of other mammals, amphibians, birds, fish, and reptiles. The aim of this review is to summarize studies that have leveraged high-throughput sequencing to better understand the skin microorganisms that associate with members of classes within the subphylum Vertebrata. Specifically, links will be explored between the skin microbiome and vertebrate characteristics, including geographic location, biological sex, animal interactions, diet, captivity, maternal transfer, and disease. Recent literature on parallel patterns between host evolutionary history and their skin microbial communities, or phylosymbiosis, will also be analyzed. These factors must be considered when designing future microbiome studies to ensure that the conclusions drawn from basic research translate into useful applications, such as probiotics and successful conservation strategies for endangered and threatened animals.
Similar content being viewed by others

Introduction
Skin microbiome research seeks to better understand the largest organ of the body by providing information on the processes by which a host organism evolves in association with its diverse collection of fungi, bacteria, archaea, and viruses [1], characterizing the immune system and diagnosing illnesses [2, 3], and exploring the etiologies of diseases [4,5,6]. The advent of high-throughput sequencing has greatly expanded knowledge of the skin microbiome and its implications for health. For example, it is now recognized that humans are uniquely colonized by skin microbial communities that are linked to diet [7], age [8, 9], and the specific body region sampled [10, 11]. These baseline data are important for understanding how skin microbiota contribute to skin health and disease.
The majority of skin microbiome studies have focussed on humans, companion and domestic animals, and amphibians. Fish and birds have received substantially less attention, and many existing studies are cultivation-based. Few studies have explored the skin microbiome of reptiles [1]. The aim of this review is to summarize studies leveraging high-throughput sequencing to better understand the skin microorganisms that associate with members of classes within the subphylum Vertebrata (Additional file 1: Table S1). Specifically, links will be explored between the skin microbiome and vertebrate characteristics, including geographic location, biological sex, diet, captivity, maternal transfer, and phylosymbiosis (Fig. 1). This review will mainly focus on non-human animals because other reviews summarize human skin microbiome research [12,13,14,15].

Examples of factors that influence the vertebrate skin microbiome
Vertebrate skin physiology
The outermost layer of mammalian skin is the epidermis, which is frequently studied in part due to its non-invasive sampling protocols and direct contact with the surrounding environment. It is here that commensal microbiota protect the body from transient microorganisms [12] with the potential to cause disease by either producing inhibitory compounds [16] or competing for resources [17]. The epidermis is constantly shedding and it is considered to be a hostile environment, compared to the gut or mouth, because of its lower temperature, pH, and moisture levels, coupled with relatively high salt and antimicrobial concentrations [18]. Estimates suggest that between 106 and 109 microorganisms/cm2 are present on human skin [19, 20]. This difference of several orders of magnitude can be attributed to sampling different body locations. Although more invasive techniques, such as skin biopsies or scrapes using a surgical scalpel blade, collect a higher number of microorganisms than superficial skin swabs, there are no consistent depth-dependent differences in detected microbial communities [20]. A recent study comparing swabbing and tape-stripping techniques observed no differences between these techniques [21].
The Class Mammalia includes the closest evolutionary relatives of humans. Non-human mammals typically possess denser fur over a larger proportion of their bodies. Sebaceous glands and two types of sweat glands are present on mammals, including apocrine and eccrine, that may each select for distinct microbiota. Sebaceous glands produce oily viscous exudates. The large and spongy apocrine glands are associated with fur and hair [22]. In contrast, the eccrine glands, which are small and associated with pores, are predominant in human and non-human primate skin [22]. Other mammals have a wide distribution of apocrine glands throughout their bodies, with eccrine glands only on feet.
The skin of avian reptiles (Class Aves, herein referred to as “birds” for clarity) has distinct physiological features from mammals. Although the most striking difference between birds and mammals is the presence of plumage, birds also have a thinner epidermis, no sebaceous glands, and a higher proportion of lipids in the transitional layer of the epidermis [23]. Birds are closer relatives of reptiles, especially modern-day crocodiles, than mammals. Their feathers are considered modified scales and a component of the integument, which is the outer protective organ system that includes the layers of skin, glands, hair, and nails in vertebrates [24]. Moreover, birds possess avian scales on their feet and only a single gland type [25]. The uropygial glands (“preen glands”), which are located in the dorsal region of most birds, exude an oily secretion that is used to coat feathers.
Non-avian reptiles (Class Reptilia, herein referred to as “reptiles” for clarity) include crocodiles, turtles, snakes, and lizards. This class of amniotes (“membrane surrounding the fetus”) represent the first animals to transition to land, which resulted in accompanying shifts to their integument. Reptiles were also the first organisms to evolve a stratum corneum (i.e., horny outer skin layer) with multiple layers and programmed cell death, coupled with additional lipids to prevent water loss on land [26]. A terrestrial lifestyle also led to the loss of gas exchange and mucous, which occurred approximately 340 million years ago [26]. The pleated-sheet beta-keratin polypeptides involved in creating sauropsid feathers, scales, and claws are distinct from the helical alpha-keratin polypeptides that form hair [25].
Amphibians, such as frogs and salamanders, possess a thin and persistently moist layer of skin that is water permeable and able to undergo gas exchange [27]. Unlike the other vertebrate classes, their skin contributes to respiration and osmoregulation while functioning as an innate immune organ [28]. Amphibian skin anatomy has been expertly reviewed elsewhere [28]. In brief, these tetrapods were the first vertebrates to evolve corneous cells [26], which form a protective external envelope around the organism and aid in terrestrial survival. An absence of protective integument layers, namely feathers or fur, makes them particularly susceptible to skin diseases [6]. Additionally, their skin is covered in a sugar-rich mucosal layer that can serve as a growth substrate for pathogenic bacteria and fungi. Consequently, many amphibian microbiome studies have focused on elucidating the differences between infected and uninfected animals in an attempt to create conservation strategies to prevent species extinctions. In particular, studies have focused on chytrid fungus, which was recently declared to have caused the most devastating recorded loss of biodiversity that can be attributed to disease [29]. As a result, the amphibian skin microbiome is better characterized than several of the other vertebrate classes.
Fish represent an evolutionarily diverse clade of six vertebrate classes. Their scales are formed in the mesoderm layer and they do not possess keratin and a corneous cell envelope, in contrast to keratinized reptilian scales that are formed in the epidermis [30]. Like amphibians, fish possess a layer of mucous that surrounds the epidermis [31] and represents an additional critical barrier between the animal and its aquatic environment. The mucous is a complex viscous mixture of immunogenic compounds, such as mucins, immunoglobins, lysozyme, antimicrobial peptides, and defensins that contribute to both innate and adaptive immunity [2, 32]. Despite these bactericidal compounds, the mucous layer also possesses numerous sugars and amino acids suitable for bacterial growth [33].
Microbial diversity and composition of vertebrate skin
Non-human terrestrial mammals
Despite the importance of the mammalian microbiota, only a few skin microbiome studies have been conducted on non-human mammals (key papers included in Additional file 1: Table S1). Initial culture-based studies of dogs and cats reported minimal skin bacterial diversity [34]. Other studies showed that squirrels, raccoons, cattle, pigs, sheep, and dogs were dominated by Micrococcus and Staphylococcus [35], with Staphylococcus being detected in 100% of pigs and cows, 90% of humans and horses, 77% of laboratory mice, and 40% of dogs [36].
Similar to human skin microbiome studies, the ability to use high-throughput sequencing has expanded our understanding of vertebrate skin microbial diversity. A large study using superficial skin swabs to evaluate the skin microbiome of wild, farm, zoo, and household animals found the majority of animals had higher diversity and distinct skin microbial communities, as compared to human samples [37]. The study evaluated skin samples from the back, torso, and inner thigh regions and found no significant variation among hair-covered body locations. The differences between human and animal skin were largely driven by a decreased relative abundance of Actinobacteria on mammalian skin, with corresponding increases in the abundances of Chloroflexi and Bacteroidetes. A study that compared human and primate axillae also found that human skin communities were unique compared to non-human primates, including gorillas, chimpanzees, rhesus macaques, and baboons [38]. Other 16S rRNA gene studies of healthy and allergic dogs [39] and cats [34, 40] also observed higher species richness and diversity on skin from animals compared to human studies, with relatively higher abundances in bacteria in the phyla Proteobacteria and Bacteroidetes. Mucosal surfaces of companion animals were inhabited by less diverse bacterial communities compared to haired skin. However, significant variations in community structure have also been observed among different haired anatomic regions in horses [41], suggesting that additional factors influence microbial communities on an animal, such as contact with other vertebrates and the environment.
Eukaryotic microscopic fungi are an additional group of microorganisms within the skin microbiome (i.e., the “mycobiome”). Fungi are less abundant than bacteria according to human skin metagenomic analysis [42]. Dogs and cats [43, 44] are also colonized by diverse fungal communities, which often vary across haired and mucosal surfaces and disease status. Their mycobiome seems to be more diverse than the human mycobiome and dominated by fungal genera including Cladosporium, Alternaria, and Epicoccum, whereas the genus Malassezia was recorded at > 90% relative abundance in human skin, except the feet [45].
Host species is an important predictor of skin microbial communities. Indeed, a survey of 38 mammalian species determined that host order and species were the most significant influences on skin microbial communities [37]. This has been also observed in North American bats [46] and early culture-based studies of mammalian skin determined that non-human animals had distinct dominant staphylococci from humans [36]. Even breed can influence the skin microbiome of mammals, as has been recently demonstrated cats [44].
In humans, overall microbial community composition is influenced by biological sex [47], age [8, 48], diet [7, 49, 50], use of hygiene products [10], ethnicity [51], cohabitation [52], habitats, and geographic location [53, 54] (Fig. 1). In terms of biological sex, only a few studies have identified significant differences between male and female mammals, including captive red kangaroos in Canada [37] and wild bank voles in Ukraine [55]. Although the link between diet and skin microbiota has not been established, diet has been linked to the composition of the gut microbiome in healthy mammals [56], including carnivores, omnivores, and herbivores. Thus, diet is presumed to also impact the skin microbiome and influence skin diseases. Canine odor is another factor associated with changes in microbial communities, with bloodhound dogs with malodor having lower diversity than controls, mainly due to higher abundances of the genera Psychrobacter and Pseudomonas [57].
Similar to humans, cutaneous microorganisms are transferred maternally to non-human vertebrates (Fig. 1). The pouch of the Tasmanian devil (Sarcophilus harrisii), where the marsupial protects its developing offspring, is associated with similar microbial community composition to its skin, in terms of phylotype richness and the number of operational taxonomic units (OTUs) present. Skin samples were clustered with pouch samples instead of mouth and gut samples [58]. Significant differences were also observed among these specimens, with an increase in Clostridia and decrease in Bacilli for pouch samples.
Geographic location is an important factor influencing the skin microbiome of mammals (Fig. 1). Two studies of North American bats concluded that geographic location was an important predictor of microbial community composition [46, 59]. Within Southwestern Ontario (Canada), the source location for sampled mammals was a significant factor influencing skin microbial communities, albeit exhibiting less influence than host taxonomic order [37]. A recent study identified that the skin microbiome of humans and three species of pigs differed among inhabitants from high and low altitudes [60]. In particular, Arthrobacter, Carnobacterium, Cellulomonadaceae, Paenibacillus, and Xanthomonadaceae were five taxa significantly increased in individuals from high altitudes [60]. Wild bank voles in Ukraine had shifts in their skin communities over large spatial distances between Kyiv and the Chernobyl Exclusion Zone, irrespective of skin radionuclide contamination [55]. One study also described seasonality to have an effect on the skin microbiota of dogs [61]; however, sampling across breeds was performed in a single year. The same study described that cohabiting individuals shared their microbiota, as previously demonstrated in humans [52].
Evidence suggests that companion animals and their owners transfer microorganisms to each other, and in turn, impact the detected human skin microbiome [62] (Fig. 1). Such evidence demonstrates that shedding of the skin microbiome impacts both the microbial community composition of inanimate objects and living macroorganisms alike. Indeed, exclusively indoor cats were shown to have similar microbial communities to their owners, compared to outdoor barn cats [37]. Animals that inhabit the same enclosed habitat, such as humans and their pets in a house [63], companion animals in a barn, or zoo animals in a cage, likely alter each other’s respective microbiomes. Built environment studies demonstrate that household surfaces are rapidly colonized with the microbiome of their inhabitants [64]. Therefore, the transfer of skin microbiota between animals can occur from either direct skin-to-skin contact or via indirect contact with shared surfaces. Although the direction of transfer can be difficult to determine in uncontrolled and complex environments, these transmission routes have important implications for the spread of infectious and zoonotic diseases. In turn, the environment is also likely an important source for new microorganisms to inhabit the skin, which can occur from direct contact with water, soil, or household surfaces. Within non-human mammals, Tasmanian devils had significant differences in skin microbial communities between wild and captive specimens, although larger differences were observed between gut microbiota [58]. Captive devils had elevated levels of Mycobacterium, a common cause of skin infections in captive facilities. Indoor and outdoor environments can also affect the skin microbiome of companion animals. Although cats primarily kept outdoors did not present higher microbial diversity as compared to indoor cats, their microbial structure and composition varied across these animals, with Corynebacterium spp., common bacteria on human skin, being more common on indoor cats [44].
Dysbiosis, defined as a shift from a normal microbiome, is associated with numerous skin diseases [4] and has been reviewed elsewhere for humans and domestic animals [65]. These polymicrobial diseases are complex and can involve the interactions of numerous microorganisms. Many pathogenic microorganisms compete directly for physical space and sources of food on human skin including sugars, ammonia, and amino acids [66], but possess virulence factors that harm the host in comparison to commensals. Commensal skin bacteria, such as Staphylococcus epidermidis, produce antimicrobial compounds to limit transient microorganisms from colonizing and appropriating resources. However, pathogens with pathogenicity islands are capable of outcompeting abundant commensals for resources [66], evading the host immune system, and subsequently lowering the abundance of typical healthy skin populations. Moreover, defects in the skin barrier may lead to penetration of pathogenic microorganisms and subsequent cutaneous inflammation, as has been shown in patients with atopic dermatitis [67,68,69]. Filaggrin is an important component of the skin barrier. Mutations in its encoding FLG gene results in a thickened, dehydrated stratum corneum and more clinically severe signs of disease [68]. Defects in the lipid bilayer, tight junctions, and proteases have also been associated with increased atopic dermatitis severity [69].
Microbial communities are typically more diverse on healthy skin, and there is evidence that microbial community composition affects several skin conditions in companion animals with atopic dermatitis and allergic skin diseases [39, 40, 43, 70, 71], bovine digital dermatitis [72], demodectic mange [73], white nose syndrome in bats [74, 75], and camel dermatophilosis [76] in other vertebrates. Dogs with skin allergies and atopic dermatitis exhibit lower bacterial richness [39] and diversity on their skin than their healthy counterparts [70], due to increases in proportions of Staphylococcus pseudintermedius. Although changes in diversity have not been observed in allergic cats, their skin is also inhabited by higher proportions of Staphylococcus spp. [40]. Horses have a stable skin microbiome that is able to return to its initial composition once a wound has healed [41]. During an experimentally induced wound experiment, the abundance of Fusobacteria and Actinobacillus increased during the early stages after wound formation. Unbandaged wounds had greater microbial diversity. This study recorded key information on temporal changes to the mammalian skin community throughout an ~80 day wound healing process and provided data that may inform veterinary practices for successful treatment of wounds. The aforementioned studies focused on reporting diversity as a proxy for health. These comparisons are based upon the community ecology perspective that diverse communities are more stable and resilient to external disturbances. Bioreactor experiments demonstrate that dynamically shifting communities can still be capable of maintaining stable ecosystem functions [77]. Subsequent human microbiome research shows that the healthy human skin microbiome is relatively stable over time due to fixed abundant species [78] and that subsequent decreases in diversity can result in disease. Atopic dermatitis treatments that increase microbial diversity ameliorate the condition [79] and provide a prime example of why researchers should continue to test for diversity when studying dysbiosis. Diversity should ideally not just be reported but should be further explored to determine how skin ecosystem diversity influences disease severity and response to treatment.
Digital dermatitis affects the hooves of cattle and results in lameness, corresponding to major economic losses to the agricultural industry [80]. Animals with digital dermatitis have higher bacterial diversity and increased prevalence of bacteria affiliated with Bacteroidetes, Proteobacteria, and Spirochaetes. In particular, Treponema spp. [81] are abundant in deep lesions and likely originate from the gut reservoir [72]. Sheep footrot is a similar infectious disease that results in lameness for entire sheep herds [82]. Dichelobacter nodosus likely initiates the disease, whereas Fusobacterium necrophorum plays a secondary role in infection [82]. Both digital dermatitis and sheep footrot are examples of polymicrobial diseases, where shifts in several skin microbiome taxa precede the onset of clinical symptoms. Dysbiosis also affects the fungal microbiota of vertebrate skin, with allergic dogs having lower diversity [83]. For dogs and cats with allergic skin disease, their mycobiota became very similar across different body sites [43]. Lastly, white nose syndrome devastates bat populations and is caused by the fungal pathogen Geomyces destructans [75].
Aquatic mammals
The skin of aquatic mammals has been studied to further marine conservation efforts. To date, cetaceans such as humpback whales, dolphins, and killer whales have been sampled. Significant differences were found between the microbial communities of bottledose dolphins and killer whales [84]. Skin biopsies and sloughed skin from 56 humpback whales (Megaptera novaeangliae) from the North Pacific, South Pacific, and North Atlantic oceans demonstrated Psychrobacter and Tenacibaculum as the core genera present on these free-swimming whales [85]. The abundance of these two genera varied significantly between humpback whales undergoing anabolic and catabolic metabolic states [85].
The cetacean skin microbiome varies geographically [86,87,88] (Fig. 1). Specifically, offshore bottlenose dolphins have higher skin microbial diversity than their coastal counterparts, who were more similar due to exposure from coastal runoff [87]. The skin microbiota of humpback whales was distinct from the surrounding seawater [86]. Likewise, skin samples of captive dolphins are also associated with distinct microbiota according to the environment where they are kept, being significantly influenced by food and air. Each environment maintained a distinct microbiota despite exposure incidents, implying that few exposures lead to permanent colonization [89]. Future studies aiming to provide evidence to improve the conservation status of wild animals affected by skin diseases should therefore include sampling of wild animals for the most accurate skin microbial community information.
Avians
Avian skin microbiota can be influenced by sex [90, 91], species [92], age, and habitat [90] (Fig. 1). European starlings (Sturnus vulgaris) and bluebirds (Sialia sialis) have distinct sex-dependent diversity associated with sampled plumage [90, 91]. These variations may be attributed to physiological variations between the sexes, such as pH [93]. In contrast, no differences were identified in the skin microbiota among male and female vultures [61]. The avian skin microbiota has also been linked to both nest location and age [90].
Birds are social animals whose sexual and social constructs aid in bacterial transmission [5]. For instance, the feathers from caged zebra finches (Taeniopygia guttata) infected with Bacillus licheniformis resulted in an oral-fecal-genital route of transmission. Preening led to autoinfection, which progressed to a sexual infection whose transmission rates varied by biological sex [5]. European starlings with larger brood sizes have more bacteria on their feathers [90]. Manipulating their brood size resulted in significantly different bacterial community composition on plumage, but not richness or feather degradation. Additionally, bluebirds sharing the same nest transmit plumage bacteria, based on results from culturing techniques [91]. Certain subsets of the microbiome are classified as “feather-degrading bacteria” and influence the condition of feathers and by extension avian health. These polyphyletic bacteria include OTUs affiliated with Bacillus spp. and hydrolyze ß-keratin [95], which is the predominant protein in feathers. The finding that nest sharing results in microbiome transmission has implications for the distribution of feather-degrading bacteria that are associated with body condition and feather coloration [91]. Recently, comparisons were made between three finch species [92]. Although these finches received the same diet and environmental exposures, each species had distinct overall skin communities, despite sharing conserved core OTUs. The authors hypothesized that the observed differences may contribute to odor production.
The eating habits of scavenger birds can alter the diversity of their skin microbiota. The skin microbiota of two species of New World vultures (Coragyps atratus and Cathartes aura) exceeds the diversity of their gut microbiota [94] (528 vs 72 OTUs, respectively). Frequent contact with carcasses may explain this increase in skin microbial diversity. Clostridia and Fusobacteria were dominant OTUs on vulture skin.
Very few virome studies involving the collection of nucleic acid sequences from the viral community in a habitat have been performed with animals. A single high-throughput sequencing study examined 15 healthy chickens (Gallus gallus domesticus) and determined that their skin was predominately inhabited by herpesvirus from the Mardivirus group [96]. The authors hypothesized that the viruses arose from vaccination or an asymptomatic infection. In addition, the skin virome of chickens differs from those of reptiles [97] and humans [42, 98]. Notably, chicken skin was absent of papillomaviruses and polyomaviruses that are typically detected on human skin [42, 98].
Reptiles
Despite links to numerous skin infections caused by viruses, bacteria, fungi, and parasites, very few studies have focused on the reptilian skin microbiome. A study on the oral and skin microbiome of komodo dragons (Varanus komodoensis) elucidated that captive dragons and their enclosure had similar microbial community composition and species richness [99] (Fig. 1). The Komodo dragon skin microbiome had higher diversity than either the oral or fecal microbiome. Dominant skin phyla included Bacteroidetes and Firmicutes, which were present in equal proportions [99].
Several studies have focused on microorganisms that are causative agents of reptile skin diseases. Reptiles are prone to infection by a variety of predominately Gram-negative commensal bacteria, including Aeromonas, Klebsiella, Proteus, Pseudomonas, and Salmonella [100]. Fungal dermatitis in the USA has affected numerous reptilian species, including dusky pigmy rattlesnakes (Sistrurus miliarius barbouri), garter snakes (Thamnophis sirtalis), and ribbon snakes (Thamnophis sauritis) [101]. The fungus Ophidiomyces ophiodiicola currently causes high mortality in snakes across Europe and North America [102]. A study of Eastern Massasauga snakes (Sistrurus catenatus) determined that infected snakes were more likely to have high populations of Serratia and Janthinobacterium. In contrast, Janthinobacterium has been associated beneficially with salamander populations to prevent Batrachochytrium dendrobatidis infections [103], whereas Serratia has been observed in the skin microbiome of immunocompromised human patients [104]. Moreover, a subset of OTUs such as Xylanimicrobium and Cellulosimicrobium were reduced in infected snakes, further indicating that snake fungal disease shifts the skin microbiome [102]. Another study determined that microbial communities did not differ significantly between snake populations of timber rattlesnakes (Crotalus horridus) and black racers (Coluber constrictor), indicating that snake fungal disease studies on model organisms may widely apply to multiple snake species [105]. Future studies will be able to leverage these findings to investigate whether a “protective microbiome” may help conservation efforts. For example, it may be possible to create a skin probiotic culture from microorganisms that have been experimentally determined to be protective against skin diseases. Developing a stable topical treatment may prove useful to shift the microbiome, just as this strategy has been moderately successful with human gut probiotics [106].
Additional studies have focused on the reptilian skin virome in relation to disease. A skin microbiome study of reptiles focused on the lizard virome [97]. Multiple viruses were associated with lethal skin lesions, including Ranavirus, Adenovirus, and Reovirus. Herpesvirus is currently infecting both wild and captive turtles and tortoises resulting in necrotizing lesions [100]. Examples of affected species include Argentine tortoises (Chelonoidis chilensis), Mediterranean tortoises (genus Testudo), Pacific pond turtles (Actinemys marmorata), and painted turtles (Chrysemys picta). Fibropapillomatosis affects wild populations of marine turtles, especially the green, loggerhead, and olive ridley sea turtles (Chelonia mydas, Caretta caretta, and Lepidochelys olivacea, respectively) [100]. This viral infection has spread globally and there is no current protocol to prevent transmission within wild populations.
Other skin-associated infections, such as inclusion body disease (IBD), caused by a Retroviridae virus [107], are prevalent on multiple continents [100]. The distribution of IBD has primarily been reported on boid snakes, including Burmese pythons (Python bivittatus) and Boa constrictors (Boa constrictor) in Africa, Australia, Europe, and North America. Reptilian skin has been shown to harbor several viruses that lead to lesions and premature death [97]. Baseline high-throughput sequencing data of healthy and diseased skin states are required to implement conservation measures.
Amphibians
Many amphibian species have had their skin microbiota sampled to establish a microbial baseline (Additional file 1: Table S1), which is particularly important because of declining amphibian populations due to skin fungal infections [108]. Wild tiger salamanders (Ambystoma tigrinum), western chorus frogs (Pseudacris triseriata), and northern leopard frogs (Lithobates pipiens) harbor a similar level of diversity as human skin [109]. Of the 18 bacterial phyla observed on amphibian skin, Acidobacteria, Actinobacteria, Bacteroidetes, Cyanobacteria, Firmicutes, and Proteobacteria were most abundant [109]. Amphibian host species was the most important predictor of community composition in a study of five species that included toads, frogs, and newts [110]. This trend in amphibians is further supported by a study of Panamanian frog species, which determined that there were key differences among hosts at bacterial taxonomic levels below the phylum level [111]. Red-backed salamanders (Plethodon cinereus) had eight core OTUs, including Pseudomonas [112], present on > 90% of specimens. Italian steam frogs (Rana italica) were characterized by 16 distinct phyla [113] with a fifth of all detected OTUs present in all subjects [113]. A culture-based study of Cascade frogs (Rana cascadae) enumerated 20 higher order taxa and 31 genera [114].
Transmission of skin bacteria to four-toed salamander (Hemidactylium scutatum) embryos has been observed [115]. These salamanders can use communal nests with eggs from a minimum of two females, which leads to higher rates of offspring survival. These communal nests were more likely to contain skin bacteria that inhibit the fungus Mariannaea, which is lethal to four-toed salamanders. Only 27% of females have these beneficial skin bacteria and having multiple females in contact with a nest resulted in higher survivability rates than solitary nests with lower amounts of antifungal bacteria in their skin communities [115].
Amphibians have different skin microbial communities depending on the current stage in their life cycle. Tadpoles are associated with distinct skin microorganisms before they undergo metamorphosis [110]. The common coqui (Eleutherodactylus coqui) has skin microbial communities that differ between juvenile and adult forms [116]. Female four-toed salamander populations within a single nest location shared a greater proportion of common bacteria than those detected as being shared among all sampled individuals from all nests [117].
Differences exist among the body regions of fire-bellied toads (Bombina orientalis), such that the dorsal sides of wild toads associates with higher diversity and richness than ventral sides, whereas captive toads exhibit the opposite result [108]. Because some non-human vertebrate studies used a single swab to sample all body locations, future skin microbiome research should sample multiple body locations per animal to assess skin community heterogeneity. Lack of sex documentation is especially prevalent for amphibian studies (Additional file 1: Table S1), due to the difficulty of non-invasive sexing methods.
Skin bacterial communities in amphibians are influenced by diet, and their microbiota may also influence their behavior (Fig. 1). For instance, providing captive red-eyed tree frogs (Agalychnis callidryas) with a carotenoid-rich diet increased skin bacterial richness and abundance, including increases in Staphylococcus, Flavobacterium, Klebsiella, and Citrobacter [118]. Odor cues produced by bacteria are involved in influencing the behavioral activities of the red-eyed tree frogs, including mating, marking territory, and recognition [119]. Determining the distribution of microorganisms on vertebrate skin has the potential to answer several questions about animal behavior that were raised previously [120], such as how animals recognize individuals and kin and assess mate quality and social relationships.
Geographic location and seasonal variability have both been associated with shifts in amphibian skin populations (Fig. 1). A large study on five different amphibian species (i.e., Anaxyrus boreas, Pseudacris regilla, Taricha torosa, Lithobates catesbeianus, and Rana cascadae) in the USA determined that wetland site was the largest predictor of skin microbial community composition within each species [110]. Variations in microbial communities based on the geographic location the host inhabits can be partially explained by the microorganisms collected from local abiotic environments. A study of red-cheeked salamanders (Plethodon jordani) demonstrated that sampled salamanders shared their most abundant bacterial taxa with the moist forest floor debris [121]. In contrast, skin swab samples of redback salamanders, eastern newts (Notophthalamus viridescens), and larval bullfrogs (Rana catesbieana) were distinct from the water they inhabited [122]; amphibians cohabitating the same pond was not a significant factor influencing their community structure [109]. Seasonal variation has been observed in the lowland leopard frog (Lithobate yavapaiensis), which may be linked to disease incidence because frogs are at increased risk of B. dendrobatidis infection during winter [116]. It has been hypothesized that reduced immunity caused by exposure to lower temperatures [116] and humidity [111] contributed to the temporal bacterial community changes.
Amphibian skin microbial communities may also be affected by contaminants in the surrounding environment, potentially reducing skin defenses and immunity [28]. A study of the Perez’s frog (Pelophylax perezi) demonstrated that frogs living in a metal-rich environment had distinct skin microbiome profiles from frogs in uncontaminated environments [123]. All frog skin samples revealed bacteria predominately from the Actinobacteria and Alphaproteobacteria taxonomic groups, whereas those from contaminated sites had more OTUs associated with acid-metal contaminated water, such as Moraxella, Mycobacterium, and Hydrotalea. Testing the surrounding soil or water for both biotic and abiotic composition may therefore add more insight into factors that influence skin microbial community composition.
Similar to data previously shown for mammalians [58], wild amphibians have higher bacterial diversity levels on their skin than the same species in captivity (Fig. 1). Wild red-eyed tree frogs (Agalychnis callidryas) had over twice the number of bacterial OTUs on their skin as their captive counterparts, demonstrating that captive animals have a significant decrease in diversity [118]. The Panamanian golden frog (Atelopus zeteki) shares approximately 70% of bacterial OTUs on their skin between wild and captive specimens, although significant differences in richness, community structure, and phylogenies still existed [124]. Overall, wild fire-bellied toads had higher diversity than captive toads, which varied based on the presence of B. dendrobatidis infection [108].
Approximately 30% of all amphibian species are threatened with extinction [125]. Given their sensitivity to skin infection, amphibian skin has been relatively well studied among vertebrate classes in an effort to prevent infections within wild populations, such as those linked to Ranavirus, mycotic dermatitis, and chytridiomycosis [16, 112, 126]. A variety of fungi have been cultivated from the skin of injured hellbender salamanders (Cryptobranchus alleganiensis bishopi), including Acremonium, Cladosporium, Curvularia, Fusarium, Streptomycetes, and Penicillium [127]. In this same study, isolated opportunistic bacterial pathogens included Aerococcus viridans, Aeromonas hydrophila, Gordonai terrae, Granulicetella adiacens, Stenotrophomonas maltophilia, and Streptococcus pneumoniae. The cutaneous microbiota of two giant salamander subspecies (Cryptobranchus alleganiensis) were studied to better understand why the Ozark hellbender subspecies is affected by chronic wounds, whereas the eastern subspecies is not [128]. Salamanders with wounds had higher OTU abundances than those without wounds, potentially indicating that commensal environmental and skin-associated bacteria may constitute opportunistic colonizers. Greater understanding of the amphibian skin microbiome is important for creating effective conservation management programs for animals with declining populations due to skin diseases.
The skin microbiome of amphibians may provide protective effects against skin pathogens [28]. Batrachochytrium dendrobatidis is a fungal pathogen that causes chytridiomycosis, which is responsible for amphibian population decline. Although B. dendrobatidis is associated with altered skin microbiome profiles [129], commensal skin bacteria are known to produce antifungal secondary metabolites that inhibit this pathogen [16]. Members of four bacterial genera (i.e., Bacillus, Chitinophaga, Janthinobacterium, and Pseudomonas) were isolated from red-backed salamanders and assayed to determine their ability to prevent B. dendrobatidis associated clinical signs, as measured by body mass and limb lifting. Whereas all of these bacteria acted synergistically to prevent infection, a co-culture of Bacillus and Chitinophaga was most effective at inhibiting the fungal pathogen, and this inhibition was linked to production of the metabolite tryptophol [16]. A reduced P. cinereus cutaneous community on redback salamanders has been linked to clinical signs of disease, namely weight loss and limb lifting [130]. Two closely related frog species (i.e., Rana sierra and Rana muscosa) were observed to have differential responses to B. dendrobatidis infections based on distinct skin microbiota. Most R. sierrae individuals had anti- B. dendrobatidis bacteria, such as Pseudomonas, and were able to persist with B. dendrobatidis for six years. In contrast, R. muscosa had lower proportions of this genus and went extinct within a year [131], indicating that Pseudomonas may protect frogs from B. dendrobatidis infections.
Over the past decade, a second distinct species of Batrachochytrium was identified as a causative agent for chytridiomycosis [132]. B. salamandrivorans (Bsal) causes lethal skin disease in salamanders and is responsible for declining populations in Europe [132]. For example, within the Netherlands, only 4% of the fire salamander population remains [133]. The remaining survivors currently do not have an increased resistance to Bsal, which has reservoirs in soil, water, and infected animals [134]. Currently, Bsal has not been detected in North American populations of salamanders [135]. The inability to initially diagnose chytridiomycosis infection in Europe resulted in crucial lost time to implement conservation strategies against the rapidly progressing disease. Researchers of other amphibian populations must therefore consider that chytridiomycosis has multiple known causative agents. Moreover, this finding is broadly applicable to researchers studying skin diseases of humans and other animals.
Fish
Analyzing the skin microbiota of numerous species of fish can provide insight into the microbial role in host health, which has economic implications for the fishing and aquaculture industries. Early culturing work on North Sea cod (Gadus morhua) showed that fish can undergo seasonal variations in skin bacterial abundances [136]. Predominant cultured isolates included Pseudomonas, Achromobacter, Corynebacterium, Flavobacterium, and Vibrio. The phyla Proteobacteria, Firmicutes, and Actinobacteria dominated several fish species [137]. The core OTU Aeribacillus was observed in all fish species, whereas other OTUs reflected species-specific distributions, such as Microbacterium in the northern red snapper (Lutjanus campechanus) and Neorickettsia in the flathead grey mullet (Mugil cephalus) [137]. Analysis of wild eel (Anguilla spp.) mucus has shown that mucosal pathogens associated with the Vibrio genus were highly abundant, implicating wild eels as a niche for their evolution and distribution [138]. A study of 102 fish from six species inhabiting the Gulf of Mexico (Mugil cephalus, Lutjanus campechanus, Cynoscion nebulosus, Cynoscion arenarius, Micropogonias undulatus, and Lagodon rhomboides) determined that each species had a distinct skin community [137].
Seasonal variations of fish skin microbial communities, which at times are coupled with geographic location, have been supported by analyzing lemon sole (Microstomus kitt) and skate fish (Rajidae spp.) [139] (Fig. 1). These variations may be due to the timing of plankton blooms and changes in the microbial community from the surrounding water. Other factors that may influence the aquatic skin microbiome include pH, dissolved oxygen concentration, and temperature [88, 140]. Fish located in warmer waters have higher proportions of mesophiles [136], whereas those near coast lines possess higher proportions of halotolerant bacteria [140]. Salmon also have varying bacterial loads based on whether they are in marine or freshwater environments [141]. However, a recent study on farmed salmon determined that there was little correlation between the microbial community on the fish and their surrounding water [142]. Manipulating salinity resulted in a reproducible shift in the microbial community that is significantly different from that of surrounding water in the enclosure [143]. As with other vertebrates, geographical location also influenced the bacterial community of six fish species significantly [137].
Fish skin microbiota can also change based on the metabolic status and mutualism of the host (Fig. 1). Salmon (Salmo salar) that are deprived of food have significant differences in both bacterial and fungal community composition and density, which was hypothesized to be a result of a decrease in the number of mucosal cells [49]. A study of 44 species of coral reef fish determined that both host diet and phylogeny influenced skin microbial communities [144]. The authors presented two hypotheses to explain this observation. First, various diets may result in shifts in the fish gut microbiome, which would indirectly transfer to the skin in an aquatic environment. Alternately, variations in diet are known to result in changes in the metabolites within surface mucus composition, thereby shifting microbial communities. An additional influence on the fish microbiome is mutualism [145]. Clownfish (Amphiprioninae spp.) that associate with anemone undergo a significant shift in their skin microbiota. This reversible shift likely occurs when microorganisms are transferred directly between the animals; however, changes in mucus thickness or chemical substrates may also contribute.
The composition of the fish skin microbiota and related skin pathogens have been studied to prevent large economic losses affecting the fishing and aquaculture industries. For example, colonization of rainbow trout (Oncorhynchus mykiss) skin by the pathogen Vibrio anguillarum is an important step for disease spread to other body regions [146]. Fish have also been shown to possess beneficial skin bacteria that help prevent infections. For example, rainbow trout have commensal lactic acid bacteria on their skin that prevent Lactococcus garvieae from colonizing by producing inhibitory compounds and outcompeting for nutrients [147]. Moreover, the gut health of yellowtail kingfish (Seriola lalandii) helps define both skin and gill microbial communities [148]. Fish with chronic lymphocytic enteritis exhibited lower overall diversity and increases in members of the Proteobacteria and Actinobacteria phyla [148]. Therefore, intestinal diseases have the potential to influence the skin microbiome.
Of methodological concern for fish microbiome studies, fish replicates sampled prior to handling had more variability [142], implying that the process of netting and handling fish alters the sampled skin microbiome. Lack of sex documentation is especially prevalent in fish studies (Additional file 1: Table S1), due to the difficulty of non-invasive sexing methods. Cultivation studies have estimated a range of 102–107 culturable microorganisms/cm2 skin [140]. This wide range has been attributed to variations among capture techniques. Trawling leads to larger microbial loads than capture with a baited line, due to sediment contamination from contact with the seabed and release of fish gastrointestinal contents [149]. Additionally, salmon have higher microbial loads in their spawning grounds than their marine habitat due to varying numbers of bacteria in the water [141]. The true skin microbial density of fish is likely several orders of magnitude higher because culturing techniques only enumerate a subset of the total microbial community. These findings should be considered while designing future aquatic microbiome studies.
Phylosymbiosis and the vertebrate skin microbiome
Phylosymbiosis describes the eco-evolutionary pattern that occurs when the host phylogenetic trees parallel the similarities observed among the corresponding host-associated microbial communities [150]. This pattern can occur via several mechanisms, including maternal transfer, co-speciation, or selection from the external environment via similar diets or behavior [151]. In other words, co-evolution of hosts and host-associated microbial communities is not the only mechanism underlying phylosymbiosis. Several studies of the gut microbiome demonstrate phylosymbiosis across a range of hosts including, hominids [152], mammals [151], birds [153], fish [154], and insects [150]. A key invertebrate gut microbiome study conducted with a controlled laboratory environment demonstrated topological congruence between insect host phylogenies and microbiota dendrograms [150]. However, this type of analysis has more confounding factors for skin microbiome studies, because the skin is constantly exposed to environmental influences. Despite external influences, congruence between host and microbial phylogentic trees and skin microbiota dendrograms, respectively, was recently reported for the Artiodactyla and Perissodactyla mammalian orders [37]. This study provided the first evidence that phylosymbiosis can be detected for vertebrates and their corresponding skin microbial communities.
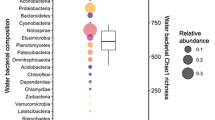
Subsequent studies have further demonstrated a strong host influence on skin microbiota (Fig. 1). A study of salamanders and frogs in Guatemala and Mexico observed phylosymbiosis at higher host taxonomic levels, such as order, which was not observed within genera and species [155]. Indeed, at lower taxonomic resolution, the habitat geography became the most important influence [155]. In contrast, a study of amphibians from Madagascar observed a greater influence from the ecology (arboreal vs aquatic vs terrestrial) of the frogs compared to phylosymbiosis [156]. Within tropical coral fish, a weak phylosymbiosis pattern was observed [144]. The authors hypothesized that several confounding factors may have contributed to the weak pattern such as the plasticity of the fish immune system, the age of the fish, and health status. Additionally, this study included 138 fish from 44 species. The possibility exists that including more samples per species would strengthen the observed phylosymbiosis patterns. Moreover, conducting phylosymbiosis studies in controlled laboratory environments may be helpful because many environmental confounding factors can be removed. Indeed, a study of three finch species observed that each bird species had distinct skin microbial communities [92], despite identical diets and environmental exposures. Together, such studies contribute to the hypothesis that vertebrates share an evolutionary pattern with their skin microbiome, which is a first step to identifying the mechanisms responsible for phylosymbiosis.
From contigs to conservation
Skin microbiome research is currently at an exciting crossroads. Researchers now have high-throughput sequencing technologies and standardized protocols to sample and sequence the skin microbiome from a plethora of animals. The next step is to translate such large-scale survey datasets into testable hypotheses with meaningful outcomes that improve the lives of studied animals. One option to advance the field is to follow the processes used in gut microbiome studies, which have progressed from basic surveys of the microorganisms present to studies that manipulate the intestinal environment to establish clinical therapies.
A translatable application for skin microbiome studies involves developing skin probiotics that could protect animals from skin infections. Initial research in skin probiotics determined that certain strains of the human skin commensal bacterium Propionibacterium acnes can ferment glycerol to mitigate methicillin-resistant Staphylococcus aureus (MRSA) [157]. Currently, endangered amphibian populations have the greatest need for such a product to protect against chytridiomycosis because conservationists do not have the required tools to mitigate population collapses. Several OTUs of interest isolated from amphibian skin are being examined for their ability to prevent Bd infections. Janthinobacterium, Rhodococcus, and Pseudomonas spp. all have the potential to inhibit Bd [158]. A mixed culture with diverse skin commensals may prove critical for conservation efforts. Moreover, exploring the mechanisms behind why Bd and Bsal infect different amphibian hosts may elucidate valuable information on ways to prevent their transmission to susceptible animals. In addition to improving conservation efforts, development of such products would also be beneficial to other animals and could be applicable to the aquaculture industry, animal husbandry, and the pet industry. For example, effectively preventing Lactococcus garvieae or Vibrio anguillarum infections in aquaculture would be beneficial from an economic perspective as well as providing healthier food for human consumption.
Conclusions and recommendations for future studies
Although the animal subphylum Vertebrata possesses a highly diverse range of animal species with varying skin physiology, social constructs, and skin conditions, several common trends are apparent. The habitat and geographic location of an animal, maternal effects, and disease status are factors that affect vertebrate skin microbiota. Additionally, biological sex, age, species, and disease state affect a wide range of vertebrates. Recent evidence suggests that phylosymbiosis occurrs between vertebrates and their skin community and is observable within higher host taxonomic classifications such as the order. Studies that sample hosts from evolutionarily distant branches of the vertebrate tree are therefore positioned to analyze phylosymbiosis and the complex interplay of factors that likely contribute to skin microbial community assemblages.
It is crucial to sample skin microbiomes from a wide range of animals to have meaningful baseline data for conservation management programs. Numerous skin diseases have been linked to population declines and threaten the extinction of a variety of animals. More information is needed on the disease transmission mechanisms of both wild and captive animals. Deeper sequencing should therefore be completed on a wider variety of species and include bacteria, archaea, fungi, and viruses. As stated earlier, there are currently very few reptilian skin microbiome studies, despite their numerous skin diseases. Using standardized methodology and rigorous metadata collection, further characterization of skin microbiota for a wide range of vertebrates in both healthy and diseased states will provide crucial baseline data for conservation efforts, with applications extending to animal care in farming and pet industries. Additionally, each of these classes of animals does not exist in a vacuum. Observations and future potential probiotics that work for one species’ skin microbiome may be translatable for conservation of other animals. Researchers are encouraged to delve further than “skin deep” into studies from other animals that may translate to their animal population of interest.
References
Colston TJ, Jackson CR. Microbiome evolution along divergent branches of the vertebrate tree of life: what is known and unknown. Mol Ecol. 2016;25:3776–800.
Gomez D, Sunyer JO, Salinas I. The mucosal immune system of fish: the evolution of tolerating commensals while fighting pathogens. Fish Shellfish Immunol. 2013;35:1729–39.
Woodhams DC, Brandt H, Baumgartner S, Kielgast J, Küpfer E, Tobler U, et al. Interacting symbionts and immunity in the amphibian skin mucosome predict disease risk and probiotic effectiveness. PLoS ONE. 2014;9:e96375.
Mańkowska-Wierzbicka D, Karczewski J, Dobrowolska-Zachwieja A, Adamski Z. The microbiome and dermatological diseases. Postep Hig Med Dosw. 2015;69:978–85.
Kulkarni S, Heeb P. Social and sexual behaviours aid transmission of bacteria in birds. Behav Processes. 2007;74:88–92.
Pessier AP. An overview of amphibian skin disease. Semin Avian Exot Pet Med. 2002;11:162–74.
Divya S, Sriharsha M, Narotham RK, Krupa SN, Siva TRK. Role of diet in dermatological conditions. J Nutr Food Sci. 2015;5:5.
Leyden JJ, McGiley KJ, Mills OH, Kligman AM. Age-related changes in the resident bacterial flora of the human face. J Invest Dermatol. 1975;65:379–81.
Capone KA, Dowd SE, Stamatas GN, Nikolovski J. Diversity of the human skin microbiome early in life. J Invest Dermatol. 2011;131:2026–32.
Bouslimani A, Porto C, Rath CM, Wang M, Guo Y, Gonzalez A, et al. Molecular cartography of the human skin surface in 3D. Proc Natl Acad Sci U S A. 2015;112:E2120–9.
Costello EK, Lauber CL, Hamady M, Fierer N, Gordon JI, Knight R. Bacterial community variation in human body habitats across space and time. Science. 2009;326:1694–7.
Grice EA, Segre JA. The skin microbiome. Nat Rev Microbiol. 2011;9:244–53.
Cundell AM. Microbial ecology of the human skin. Microb Ecol. 2016:1–8.
Byrd AL, Belkaid Y, Segre JA. The human skin microbiome. Nat Rev Microbiol. 2018;16:143–55.
Bang C, Schmitz RA. Archaea associated with human surfaces: not to be underestimated. FEMS Microbiol Rev. 2015;39:631–48.
Loudon AH, Holland JA, Umile TP, Burzynski EA, Minbiole KPC, Harris RN, et al. Interactions between amphibians’ symbiotic bacteria cause the production of emergent anti-fungal metabolites. Front Microbiol. 2014;5:1–8.
Fredricks DN. Microbial ecology of human skin in health and disease. J Investig Dermatol Symp Proc. 2001;6:167–9.
Percival SL, Emanuel C, Cutting KF, Williams DW. Microbiology of the skin and the role of biofilms in infection. Int Wound J. 2012;9:14–32.
Kong HH, Segre JA. Skin microbiome: looking back to move forward. J Invest Dermatol. 2012;132:933–9.
Grice EA, Kong HH, Renaud G, Young AC, NISC CSP, Bouffard GG, et al. A diversity profile of the human skin microbiota. Genome Res. 2008;18:1043–50.
Ogai K, Nagase S, Mukai K, Iuchi T, Mori Y, Collado MC. A comparison of techniques for collecting skin microbiome samples: swabbing versus tape-stripping. Front Microbiol. 2018;9:2362.
Folk GE, Semken HA. The evolution of sweat glands. Int J Biometeorol. 1991;35:180–6.
Couteaudier M, Denesvre C. Marek’s disease virus and skin interactions. Vet Res. 2014;45:1–12.
Sawyer RH, Knapp LW. Avian skin development and the evolutionary origin of feathers. J Exp Zool. 2003;298:57–72.
Dhouailly D. A new scenario for the evolutionary origin of hair, feather, and avian scales. J Anat. 2009;214:587–606.
Alibardi L. Adaptation to the land: The skin of reptiles in comparison to that of amphibians and endotherm amniotes. J Exp Zool. 2003;298:12–41.
Heatwole HE, Barthalmus GT. The integument. In: Amphibian biology. Chipping Norton: Surrey Beatty & Sons; 1994.
Varga JFA, Bui-Marinos MP, Katzenback BA. Frog skin innate immune defences: sensing and surviving pathogens. Front Immunol. 2019;9:3128.
Scheele BC, Pasmans F, Skerratt LF, Berger L, Martel A, Beukema W, et al. Amphibian fungal panzootic causes catastrophic and ongoing loss of biodiversity. Science. 2019;363:1459–63.
Sharpe PT. Fish scale development: hair today, teeth and scales yesterday? Curr Biol. 2001;11:751–2.
Kanno T, Nakai T, Muroga K. Scanning electron microscopy on the skin surface of ayu Plecoglossus altivelis infected with Vibrio anguillarum. Dis Aquat Organ. 1990;8:73–5.
Merrifield DL, Rodiles A. The fish microbiome and its interactions with mucosal tissues. In: Beck BH, Peatman E, editors. Mucosal health in aquaculture: Academic Press; 2015.
Shewan JM, Hobbs G. The bacteriology of fish spoilage and preservation. Prog. Ind. Microbiol. London: Iliffe Books Ltd; 1967.
Weese JS. The canine and feline skin microbiome in health and disease. Vet Dermatol. 2013;24:137–46.
Kloos WE, Zimmerman RJ, Smith RF. Preliminary studies on the characterization and distribution of Staphylococcus and Micrococcus species on animal skin. Appl Environ Microbiol. 1976;31:53–9.
Nagase N, Sasaki A, Yamashita K, Shimizu A, Wakita Y, Kitai S, et al. Isolation and species distribution of staphylococci from animal and human skin. J Vetrinary Med Sci. 2001;64:245–50.
Ross AA, Müller KM, Weese JS, Neufeld JD. Comprehensive skin microbiome analysis reveals the uniqueness of human skin and evidence for phylosymbiosis within the class Mammalia. Proc Natl Acad Sci U S A. 2018;115:E5786–95.
Council SE, Savage AM, Urban JM, Ehlers ME, JHP S, Platt ML, et al. Diversity and evolution of the primate skin microbiome. Proc R Soc B Biol Sci. 2016;283:2586.
Hoffmann AR, Patterson AP, Diesel A, Lawhon SD, Ly HJ, Stephenson CE, et al. The skin microbiome in healthy and allergic dogs. PLoS ONE. 2014;9:e83197.
Older CE, Diesel A, Patterson AP, Meason-Smith C, Johnson TJ, Mansell J, et al. The feline skin microbiota: The bacteria inhabiting the skin of healthy and allergic cats. PLoS ONE. 2017;12:e0178555.
Kamus LJ, Theoret C, Costa MC. Use of next generation sequencing to investigate the microbiota of experimentally induced wounds and the effect of bandaging in horses. PLoS ONE. 2018;13:e0206989.
Oh J, Byrd AL, Deming C, Conlan S, Barnabas B, Blakesley R, et al. Biogeography and individuality shape function in the human skin metagenome. Nature. 2014;514:59–64.
Meason-Smith C, Diesel A, Patterson AP, Older CE, Johnson TJ, Mansell JM, et al. Characterization of the cutaneous mycobiota in healthy and allergic cats using next generation sequencing. Vet Dermatol. 2016;28:71–e17.
Older CE, Diesel AB, Lawhon SD, Queiroz CRR, Henker LC, Hoffmann AR. The feline cutaneous and oral bacterial and fungal microbiota is influenced by breed and environment: Submitted.
Findley K, Oh J, Yang J, Conlan S, Deming C, Meyer JA, et al. Topographic diversity of fungal and bacterial communities in human skin. Nature. 2013;498:367–70.
Avena CV, Parfrey LW, Leff JW, Archer H, Frick WF, Langwig K, et al. Deconstructing the bat skin microbiome: influences of the host and the environment. Front Microbiol. 2016;7:1753.
Fierer N, Hamady M, Lauber CL, Knight R. The influence of sex, handedness, and washing on the diversity of hand surface bacteria. Proc Natl Acad Sci U S A. 2008;105:17994–9.
Oh J, Conlan S, Polley EC, Segre JA, Kong HH. Shifts in human skin and nares microbiota of healthy children and adults. Genome Med. 2012;4:77.
Landeira-Dabarca A, Sieiro C, Álvarez M. Change in food ingestion induces rapid shifts in the diversity of microbiota associated with cutaneous mucus of Atlantic salmon Salmo salar. J Fish Biol. 2013;82:893–906.
Kang D, Shi B, Erfe MC, Craft N, Li H. Vitamin B12 modulates the transcriptome of the skin microbiota in acne pathogenesis. Sci Transl Med. 2015;7:293ra103.
HMP Consortium. Structure, function and diversity of the healthy human microbiome. Nature. 2012;486:207–14.
Ross AA, Doxey AC, Neufeld JD. The skin microbiome of cohabiting couples. mSystems. 2017;2:e00043–17.
Clemente JC, Pehrsson EC, Blaser MJ, Sandhu K, Gao Z, Wang B, et al. The microbiome of uncontacted Amerindians. Sci Adv. 2015;1:e1500183.
Klepeis NE, Nelson WC, Ott WR, Robinson JP, Tsang AM, Switzer P, et al. The national human activity pattern survey (NHAPS): a resource for assessing exposure to environmental pollutants. J Expo Anal Environ Epidemiol. 2001;11:231–52.
Lavrinienko A, Tukalenko E, Mappes T, Watts PC. Skin and gut microbiomes of a wild mammal respond to different environmental cues. Microbiome. 2018;6:209.
Ley RE, Hamady M, Lozupone C, Turnbaugh PJ, Ramey RR, Bircher JS, et al. Evolution of mammals and their gut microbes. Science. 2008;320:1647–51.
Meason-Smith C, Older CE, Ocana R, Dominguez B, Lawhon SD, Wu J, et al. Novel association of Psychrobacter and Pseudomonas with malodour in bloodhound dogs, and the effects of a topical product composed of essential oils and plant-derived essential fatty acids in a randomized, blinded, placebo-controlled study. Vet Dermatol. 2018;29:465–e158.
Cheng Y, Fox S, Pemberton D, Hogg C, Papenfuss AT, Belov K. The Tasmanian devil microbiome—implications for conservation and management. Microbiome. 2015;3:76.
Winter AS, Hathaway JJM, Kimble JC, Buecher DC, Valdez EW, Porras-alfaro A, et al. Skin and fur bacterial diversity and community structure on American southwestern bats: effects of habitat, geography and bat traits. PeerJ. 2017;5:e3944.
Zeng B, Zhao J, Guo W, Zhang S, Hua Y, Tang J, et al. High-altitude living shapes the skin microbiome in humans and pigs. Front Microbiol. 2017;8:1929.
Torres S, Clayton JB, Danzeisen JL, Ward T, Huang H, Knights D, et al. Diverse bacterial communities exist on canine skin and are impacted by cohabitation and time. PeerJ. 2017;5:e3075.
Misic AM, Davis MF, Tyldsley AS, Hodkinson BP, Tolomeo P, Hu B, et al. The shared microbiota of humans and companion animals as evaluated from Staphylococcus carriage sites. Microbiome. 2015;3:2.
Song SJ, Lauber C, Costello EK, Lozupone CA, Humphrey G, Berg-Lyons D, et al. Cohabiting family members share microbiota with one another and with their dogs. eLife. 2013;2:e00458.
Lax S, Smith DP, Hampton-Marcell J, Owens SM, Handley KM, Scott NM, et al. Longitudinal analysis of microbial interaction between humans and the indoor environment. Sci Rep. 2012;345:1048–52.
Hoffmann AR. The cutaneous ecosystem: the roles of the skin microbiome in health and its association with inflammatory skin conditions in humans and animals. Vet Dermatol. 2017;28:60–e15.
Rohmer L, Hocquet D, Miller SI. Are pathogenic bacteria just looking for food? Metabolism and microbial pathogenesis. Trends Microbiol. 2011;19:341–8.
Elias PM, Schmuth M, Elias PM. Abnormal skin barrier in the etiopathogenesis of atopic dermatitis. Curr Allergy Asthma Rep. 2009;9:265–72.
Nemoto-hasebe I, Akiyama M, Nomura T, Sandilands A, Mclean WHI. Clinical severity correlates with impaired barrier in filaggrin-related eczema. J Invest Dermatol. 2009;129:682–9.
Agrawal R, Woodfold JA. Skin barrier defects in atopic dermatitis. Curr Allergy Asthma Rep. 2015;14:433.
Bradley CW, Morris DO, Rankin SC, Cain CL, Misic AM, Houser T, et al. Longitudinal evaluation of the skin microbiome and association with microenvironment and treatment in canine atopic dermatitis. J Invest Dermatol. 2016;136:1182–90.
Steiner M, Pierezan F, Olivry T, Paps JS, Lawhon SD, Wu J, et al. The skin microbiome in allergen-induced canine atopic dermatitis. Vet Dermatol. 2016;27:332–e82.
Zinicola M, Lima F, Lima S, Machado V, Gomez M, Döpfer D, et al. Altered microbiomes in bovine digital dermatitis lesions, and the gut as a pathogen reservoir. PLoS ONE. 2015;10:e0120504.
Shrestha D, Thapa B, Rawal G, Dhakal S, Sharma B. Prevalence of demodectic mange in canines of Kathmandu Valley having skin disorder and its associated risk factors. Int J Appl Sci Biotechnol. 2015;3:459–63.
Blehert DS, Hicks AC, Behr M, Meteyer CU, Berlowski-zier BM, Buckles EL, et al. Bat white-nose syndrome: an emerging fungal pathogen? Science. 2009;323:227.
Gargas A, Trest MT, Christensen M, Volk TJ, Blehert DS. Geomyces destructans sp. nov. associated with bat white-nose syndrome. Mycotaxon. 2009;108:147–54.
Ayalew Y, Assefa A, Mekonen N, Belete S, Ayisheshim A. A review on camel dermatophilosis. Adv Biol Rev. 2015;9:363–72.
Huang S, Seston S, Xing J, Hickey R, Ferna ANA. How stable is stable? Function versus community composition. Appl Environ Microbiol. 1999;65:3697–704.
Oh J, Byrd AL, Park M, Kong HH, Segre JA, Oh J, et al. Temporal stability of the human skin microbiome. Cell. 2016;165:854–66.
Kong HH, Oh J, Deming C, Conlan S, Grice EA, Beatson MA, et al. Temporal shifts in the skin microbiome associated with disease flare and treatment in children with atopic dermatitis. Genome Res. 2015;22:850–9.
Wilson-Welder JH, Alt DP, Nally JE. Digital dermatitis in cattle: current bacterial and immunological findings. Animals. 2015;5:1114–35.
Klitgaard K, Boye M, Capion N, Jensen TK. Evidence of multiple Treponema phylotypes involved in bovine digital dermatitis as shown by 16S rRNA gene analysis and fluorescence in situ hybridization. J Clin Microbiol. 2008;46:3012–20.
Witcomb LA, Green LE, Kaler J, Ul-Hassan A, Calvo-Bado LA, Medley GF, et al. A longitudinal study of the role of Dichelobacter nodosus and Fusobacterium necrophorum load in initiation and severity of footrot in sheep. Prev Vet Med. 2014;115:48–55.
Meason-Smith C, Diesel A, Patterson AP, Older CE, Mansell JM, Suchodolski JS, et al. What is living on your dog’s skin? Characterization of the canine cutaneous mycobiota and fungal dysbiosis in canine allergic dermatitis. FEMS Microbiol Ecol. 2015;91:fiv139.
Chiarello M, Villé S, Bo C, Auguet JC, Bouvier T. Captive bottlenose dolphins and killer whales harbor a species- specific skin microbiota that varies among individuals. Sci Rep. 2017;7:15269.
Apprill A, Robbins J, Eren AM, Pack AA, Reveillaud J, Mattila D, et al. Humpback whale populations share a core skin bacterial community: towards a health index for marine mammals? PLoS ONE. 2014;9:e90785.
Apprill A, Mooney TA, Lyman E, Stimpert AK, Rappé MS. Humpback whales harbour a combination of specific and variable skin bacteria. Environ Microbiol Rep. 2011;3:223–32.
Russo CD, Weller DW, Nelson KE, Chivers SJ, Torralba M, Grimes DJ. Bacterial species identified on the skin of bottlenose dolphins off southern California via next generation sequencing techniques. Microb Ecol. 2018;75:303–9.
Bierlich KC, Miller C, Deforce E, Friedlaender AS, Johnston DW, Apprill A. Temporal and regional variability in the skin microbiome of humpback whales along the western Antarctic peninsula. Appl Environ Microbiol. 2017;84:1–15.
Cardona C, Lax S, Larson P, Stephens B, Hampton-Marcell J, Edwardson C, et al. Environmental sources of bacteria differentially influence host-associated microbial dynamics. mSystems. 2018;3:e00052–18.
Lucas FS, Moureau B, Jourdie V, Heeb P. Brood size modifications affect plumage bacterial assemblages of European starlings. Mol Ecol. 2005;14:639–46.
Gunderson AR, Forsyth MH, Swaddle JP. Evidence that plumage bacteria influence feather coloration and body condition of eastern bluebirds Sialia sialis. J Avian Biol. 2009;40:440–7.
Engel K, Sauer J, Jünemann S, Winkler A, Wibberg D, Kalinowski J, et al. Individual- and species-specific skin microbiomes in three different estrildid finch species revealed by 16S amplicon sequencing. Microb Ecol. 2018;76:518–29.
Dao H, Kazin RA. Gender differences in skin: A review of the literature. Gend Med. 2007;4:308–28.
Roggenbuck M, Schnell IB, Blom N, Bælum J, Bertelsen MF, Sicheritz-Pontén T, et al. The microbiome of new world vultures. Nat Commun. 2014;5:5498.
Burtt EH, Ichida JM. Occurrene of feather-degrading bacilli in the plumage of birds. Auk. 1999;116:364–72.
Denesvre C, Dumarest M, Rémy S, Gourichon D, Eloit M. Chicken skin virome analyzed by high-throughput sequencing shows a composition highly different from human skin. Virus Genes. 2015;51:209–16.
Stöhr AC, Blahak S, Heckers KO, Wiechert J, Behncke H, Mathes K, et al. Ranavirus infections associated with skin lesions in lizards. Vet Res. 2013;44:84.
Foulongne V, Sauvage V, Hebert C, Dereure O, Cheval J, Gouilh MA, et al. Human skin microbiota: high diversity of DNA viruses identified on the human skin by high throughput sequencing. PLoS ONE. 2012;7:e38499.
Hyde ER, Navas-Molina JA, Song SJ, Kueneman JG, Ackermann G, Cardona C, et al. The oral and skin microbiomes of captive Komodo dragons are significantly shared with their habitat. mSystems. 2016;1:e00046–16.
Schumacher J. Selected infectious diseases of wild reptiles and amphibians. J Exot Pet Med. 2006;15:18–24.
Cheatwood JL, Jacobson ER, May PG, Farrell TM, Homer BL, Samuelson DA, et al. An outbreak of fungal dermatitis and stomatitis in a free-ranging population of pigmy rattlesnakes (Sistrurus miliarius barbouri) in Florida. J Wildl Dis. 2003;39:329–37.
Allender MC, Baker S, Britton M, Kent AD. Snake fungal disease alters skin bacterial and fungal diversity in an endangered rattlesnake. Sci Rep. 2018;8:12147.
Brucker RM, Harris RN, Schwantes CR, Gallaher TN, Flaherty DC, Lam BA, et al. Amphibian chemical defense: antifungal metabolites of the microsymbiont Janthinobacterium lividum on the salamander Plethodon cinereus. J Chem Ecol. 2008;34:1422–9.
Oh J, Freeman AF, Park M, Sokolic R, Candotti F, Holland SM, et al. The altered landscape of the human skin microbiome in patients with primary immunodeficiencies. Genome Res. 2013;23:2103–14.
Hill AJ, Leys JE, Bryan D, Erdman FM, Malone KS, Russell GN, et al. Common cutaneous bacteria isolated from snakes inhibit growth of Ophidiomyces ophiodiicola. Ecohealth. 2018;15:109–20.
Gareau MG, Sherman PM, Walker WA. Probiotics and the gut microbiota in intestinal health and disease. Nat Rev Gastro Hep. 2010;7:503–14.
Schumacher J, Jacobson ER, Homer BL, Gaskin JM. Inclusion body disease in boid snakes. J Zoo Wild Med. 1994;25:511–24.
Bataille A, Lee-Cruz L, Tripathi B, Kim H, Waldman B. Microbiome variation across amphibian skin regions: implications for chytridiomycosis mitigation efforts. Microb Ecol. 2016;71:221–32.
McKenzie VJ, Bowers RM, Fierer N, Knight R, Lauber CL. Co-habiting amphibian species harbor unique skin bacterial communities in wild populations. ISME J. 2012;6:588–96.
Kueneman JG, Parfrey LW, Woodhams DC, Archer HM, Knight R, McKenzie VJ. The amphibian skin-associated microbiome across species, space and life history stages. Mol Ecol. 2014;23:1238–50.
Belden LK, Hughey MC, Rebollar EA, Umile TP, Loftus SC, Burzynski EA, et al. Panamanian frog species host unique skin bacterial communities. Front Microbiol. 2015;6:1–21.
Loudon AH, Woodhams DC, Parfrey LW, Archer H, Knight R, McKenzie V, et al. Microbial community dynamics and effect of environmental microbial reservoirs on red-backed salamanders (Plethodon cinereus). ISME J. 2014;8:830–40.
Federici E, Rossi R, Fidati L, Paracucchi R, Scargetta S, Montalbani E, et al. Characterization of the skin microbiota in Italian stream frogs (Rana italica) infected and uninfected by a cutaneous parasitic disease. Microbes Environ. 2015;30:262–9.
Roth T, Foley J, Worth J, Piovia-Scott J, Pope K, Lawler S. Bacterial flora on Cascades frogs in the Klamath mountains of California. Comp Immunol Microbiol Infect Dis. 2013;36:591–8.
Banning JL, Weddle AL, Wahl GW, Simon MA, Lauer A, Walters RL, et al. Antifungal skin bacteria, embryonic survival, and communal nesting in four-toed salamanders, Hemidactylium scutatum. Oecologia. 2008;156:423–9.
Longo AV, Savage AE, Hewson I, Zamudio KR. Seasonal and ontogenetic variation of skin microbial communities and relationships to natural disease dynamics in declining amphibians. R Soc Open Sci. 2015;2:140377.
Lauer A, Simon MA, Banning JL, Lam BA, Harris RN. Diversity of cutaneous bacteria with antifungal activity isolated from female four-toed salamanders. ISME J. 2008;2110:145–57.
Antwis RE, Haworth RL, Engelmoer DJP, Ogilvy V, Fidgett AL, Preziosi RF. Ex situ diet influences the bacterial community associated with the skin of red-eyed tree frogs (Agalychnis callidryas). PLoS ONE. 2014;9:1–8.
Kuhn F, Natsch A. Body odour of monozygotic human twins: a common pattern of odorant carboxylic acids released by a bacterial aminoacylase from axilla secretions contributing to an inherited body odour type. J R Soc Interface. 2009;6:377–92.
Archie EA, Theis KR. Animal behaviour meets microbial ecology. Anim Behav. 2011;82:425–36.
Fitzpatrick BM, Allison AL. Similarity and differentiation between bacteria associated with skin of salamanders (Plethodon jordani) and free-living assemblages. FEMS Microbiol Ecol. 2014;88:482–94.
Culp CE, Falkinham JO, Belden LK. Identification of the natural bacterial microflora on the skin of eastern newts, bullfrog tadpoles and redback salamanders. Herpetologica. 2007;63:66–71.
Costa S, Lopes I, Proença DN, Ribeiro R, Morais PV. Diversity of cutaneous bacterial community of Pelophylax perezi populations inhabiting different environments. Sci Total Environ. 2016;572:995–1004.
Becker MH, Richards-Zawacki CL, Gratwicke B, Belden LK. The effect of captivity on the cutaneous bacterial community of the critically endangered Panamanian golden frog (Atelopus zeteki). Biol Conserv. 2014;176:199–206.
Stuart SN, Chanson JS, Cox NA, Young BE, Rodrigues ASL, Fischman DL, et al. Status and trends of amphibian declines and extinctions. Science. 2004;306:1783–5.
Taylor SK, Williams ES, Thorne ET, Mills KW, Withers DI, Pier AC. Causes of mortality of the Wyoming toad. J Wildl Dis. 1999;35:49–57.
Nickerson CA, Ott CM, Castro SL, Garcia VM, Molina TC, Briggler JT, et al. Evaluation of microorganisms cultured from injured and repressed tissue regeneration sites in endangered giant aquatic Ozark Hellbender salamanders. PLoS ONE. 2011;6:e28906.
Hernández-Gómez O, Kimble SJA, Briggler JT, Williams RN. Characterization of the cutaneous bacterial communities of two giant salamander subspecies. Microb Ecol. 2016;73:445–54.
Jani AJ, Briggs CJ. The pathogen Batrachochytrium dendrobatidis disturbs the frog skin microbiome during a natural epidemic and experimental infection. Proc Natl Acad Sci U S A. 2014;111:E5049–58.
Becker MH, Harris RN. Cutaneous bacteria of the redback salamander prevent morbidity associated with a lethal disease. PLoS ONE. 2010;5:1–6.
Lam BA, Walke JB, Vredenburg VT, Harris RN. Proportion of individuals with anti-Batrachochytrium dendrobatidis skin bacteria is associated with population persistence in the frog Rana muscosa. Biol Conserv. 2010;143:529–31.
Martel A, der Sluijs AS, Blooi M, Bert W, Ducatelle R, Fisher MC. Batrachochytrium salamandrivorans sp. nov. causes lethal chytridiomycosis in amphibians. Proc Natl Acad Sci U S A. 2013;110:15325–9.
der Sluijs AS, Spikmans F, Bosman W, de Zeeuw M. Rapid enigmatic decline drives the fire salamander (Salamandra salamandra) to the edge of extinction in the Netherlands. Amphibia Reptilia. 2013;34:233–9.
Stegen G, Pasmans F, Schmidt BR, Rouffaer LO, van Praet S, Schaub M, et al. Drivers of salamander extirpation mediated by Batrachochytrium salamandrivorans. Nature. 2017;544:353–6.
Bales EK, Hyman OJ, Loudon AH, Harris RN, Lipps G, Chapman E, et al. Pathogenic chytrid fungus Batrachochytrium dendrobatidis, but not B . salamandrivorans, detected on Eastern Hellbenders. PLoS ONE. 2015;10:e0116405.
Georgala DL. The bacterial flora of the skin of north sea cod. J Gen Microbiol. 1958;18:84–91.
Larsen A, Tao Z, Bullard SA, Arias CR. Diversity of the skin microbiota of fishes: evidence for host species specificity. FEMS Microbiol Ecol. 2013;85:483–94.
Carda-Diéguez M, Ghai R, Rodríguez-valera F, Amaro C. Wild eel microbiome reveals that skin mucus of fish could be a natural niche for aquatic mucosal pathogen evolution. Microbiome. 2017;6:162.
Liston J. Quantitative variations in the bacterial flora of flatfish. J Gen Microbiol. 1956;15:305–14.
Horsley RW. A review of the bacterial flora of teleosts and elasmobranchs, including methods for its analysis. J Fish Biol. 1977;10:529–53.
Horsley RW. The bacterial flora of the Atlantic salmon (Salmo salar L.) in relation to its environment. J Appl Bacteriol. 1973;36:377–86.
Minniti G, Hagen LH, Porcellato D, Jørgensen SM, Pope PB, Vaaje-kolstad G. The skin-mucus microbial community of farmed atlantic salmon (Salmo salar). Front Microbiol. 2017;8:2043.
Schmidt VT, Smith KF, Melvin DW, Amaral-Zettler LA. Community assembly of a euryhaline fish microbiome during salinity acclimation. Mol Ecol. 2015;24:2537–50.
Chiarello M, Auguet J, Bettarel Y, Bouvier C, Claverie T, Graham NAJ, et al. Skin microbiome of coral reef fish is highly variable and driven by host phylogeny and diet. Microbiome. 2018;6:147.
Pratte ZA, Pattin NV, McWhirt ME, Caughman AM, Parris DJ, Stewart FJ. Association with a sea anemone alters the skin microbiome of clownfish. Coral Reefs. 2018;37:1119–25.
Weber B, Chen C, Milton DL. Colonization of fish skin is vital for Vibrio anguillarum to cause disease. Environ Microbiol Rep. 2010;2:133–9.
Pérez-Sánchez T, Balcázar JL, Garcia Y, Halaihel N, Vendrell D, de Blas I, et al. Identification and characterization of lactic acid bacteria isolated from rainbow trout, Oncorhynchus mykiss (Walbaum), with inhibitory activity against Lactococcus garvieae. J Fish Dis. 2011;34:499–507.
Legrand TPRA, Catalano SR, Wos-oxley ML, Stephens F, Landos M, Bansemer MS, et al. The inner workings of the outer surface: skin and gill microbiota as indicators of changing gut health in yellowtail kingfish. Front Microbiol. 2018;8:2664.
Shewan JM. Some bacteriological aspects of handling, processing and distribution of fish. J R Sanit Inst. 1949;69:394–421.
Brooks AW, Kohl KD, Brucker RM, van Opstal EJ, Bordenstein SR. Phylosymbiosis: relationships and functional effects of microbial communities across host evolutionary history. PLoS Biol. 2016;14:e2000225.
Groussin M, Mazel F, Sanders JG, Smillie CS, Thuiller W, Alm EJ. Unraveling the processes shaping mammalian gut microbiomes over evolutionary time. Nat Clim Chang. 2017;8:14319.
Ochman H, Worobey M, Kuo CH, Ndjango JN, Peeters M, Hahn BH, et al. Evolutionary relationships of wild hominids recapitulated by gut microbial communities. PLoS Biol. 2010;8:e1000546.
Kubov J, Dagmar C, Kreisinger J. Codiversification of gastrointestinal microbiota and phylogeny in passerines is not explained by ecological divergence. Mol Ecol. 2017;26:5292–304.
Miyake S, Ngugi DK, Stingl U. Phylogenetic diversity, distribution, and cophylogeny of giant bacteria (Epulopiscium) with their surgeonfish hosts in the red sea. Front Microbiol. 2016;7:285.
Ellison S, Rovito S, Vredenburg VT. The influence of habitat and phylogeny on the skin microbiome of amphibians in Guatemala and Mexico. Microb Ecol. 2018:1–11.
Bletz MC, Archer H, Harris RN, Mckenzie VJ, Rabemananjara FCE, Rakotoarison A, et al. Host ecology rather than host phylogeny drives amphibian skin microbial community structure in the biodiversity hotspot of madagascar. Front Microbiol. 2017;8:1530.
Shu M, Wang Y, Yu J, Kuo S, Coda A, Jiang Y, et al. Fermentation of Propionibacterium acnes, a commensal bacterium in the human skin microbiome, as skin Probiotics against methicillin-resistant Staphylococcus aureus. PLoS ONE. 2013;8:e55380.
Woodhams DC, Bletz M, Kueneman J, Mckenzie V. Managing amphibian disease with skin microbiota. Trends Microbiol. 2016;24:161–4.
Acknowledgements
Not applicable
Funding
AAR was funded by a Canada Graduate Scholarship from the Canadian Institutes for Health Research (CIHR) and an Ontario Graduate Scholarship (OGS). JDN acknowledges funding from a Discovery Grant from the Natural Sciences and Engineering Research Council of Canada (NSERC).
Availability of data and materials
Data sharing is not applicable to this article as no datasets were generated or analyzed during the current study.
Author information
Authors and Affiliations
Contributions
AAR prepared the first draft and AAR, AHR, and JDN edited and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
All human and non-human animal studies cited in this review received ethics approval at their respective institutions. Please refer to the original publications for the ethics committee names and reference numbers.
Consent for publication
Not applicable
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional file
Additional file 1:
Table S1. Molecular studies investigating the non-human vertebrate skin microbiome. Only studies that used culture-independent methods were included. Studies within a vertebrate clade are listed in alphabetical order according to first author. (DOCX 80 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Ross, A.A., Rodrigues Hoffmann, A. & Neufeld, J.D. The skin microbiome of vertebrates. Microbiome 7, 79 (2019). https://doi.org/10.1186/s40168-019-0694-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40168-019-0694-6