
Abstract
The interaction between programmed cell death ligand 1 (PD-L1), which is expressed on the surface of tumor cells, and programmed cell death 1 (PD-1), which is expressed on T cells, impedes the effective activation of tumor antigen-specific T cells, resulting in the evasion of tumor cells from immune-mediated killing. Blocking the PD-1/PD-L1 signaling pathway has been shown to be effective in preventing tumor immune evasion. PD-1/PD-L1 blocking antibodies have garnered significant attention in recent years within the field of tumor treatments, given the aforementioned mechanism. Furthermore, clinical research has substantiated the efficacy and safety of this immunotherapy across various tumors, offering renewed optimism for patients. However, challenges persist in anti-PD-1/PD-L1 therapies, marked by limited indications and the emergence of drug resistance. Consequently, identifying additional regulatory pathways and molecules associated with PD-1/PD-L1 and implementing judicious combined treatments are imperative for addressing the intricacies of tumor immune mechanisms. This review briefly outlines the structure of the PD-1/PD-L1 molecule, emphasizing the posttranslational modification regulatory mechanisms and related targets. Additionally, a comprehensive overview on the clinical research landscape concerning PD-1/PD-L1 post-translational modifications combined with PD-1/PD-L1 blocking antibodies to enhance outcomes for a broader spectrum of patients is presented based on foundational research.
Similar content being viewed by others

Introduction
Programmed cell death 1 (PD-1) and its ligand PD-L1 have become pivotal in advancing tumor treatment by effectively modulating immune responses [1]. PD-L1 is expressed across various tumors, while PD-1 is primarily expressed on T cells within tumor tissues [2]. PD-L1 engages with PD-1, creating a molecular barrier that inhibits the cytotoxic actions of immune cells [3]. Overcoming this inhibition is possible through blocking antibodies or recombinant proteins that target signaling pathways, reactivating immune responses. Monoclonal antibodies against PD-1 and PD-L1 have demonstrated significant therapeutic success, indicating that immune checkpoint blockade therapy is a potent antitumor treatment. However, its current use mainly as a second-line treatment for advanced tumors and the emergence of drug resistance highlight ongoing challenges [4]. These factors underscore the necessity for continued research to potentially expand its use earlier in treatment protocols.
Exploring new biomarkers and developing combination drug therapies are essential for combating these challenges. Research has shown that PD-1 transcription can be increased by activating B-cell CLL/lymphoma 6 (BCL6), and various elements, such as cytokines, hypoxia, bromodomain-containing protein 4 (BRD4), and noncoding RNA, can elevate PD-L1 expression by influencing transcription [5, 6]. With advancements in detection technologies, numerous posttranslational modifications (PTMs) have been identified that play critical roles in human diseases and offer avenues for new treatments. Recent studies have focused on PTMs that impact PD-1/PD-L1 protein expression and their roles in immunosuppression. PD-1/PD-L1 is negatively regulated by mechanisms such as phosphorylation, ubiquitination, ubiquitin-like modification and methylation. Conversely, positive regulation occurs through processes such as deubiquitination, glycosylation, palmitoylation, adenosine diphosphate (ADP) ribosylation, and deacetylation [7]. A deeper understanding of these regulatory mechanisms and identification of novel targets for PD-1/PD-L1 modification are vital for advancing tumor immunotherapy toward precise treatments. Moreover, ongoing efforts are needed to discover and test safe, effective drug combinations to improve therapeutic outcomes.
Structures of PD-1/PD-L1 and their potential sites modulate PTMs
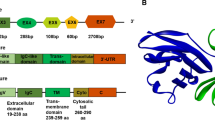
PD-1 (CD279), encoded by the PDCD1 gene on chromosome 2q37.3, is a type I transmembrane protein from the immunoglobulin superfamily, specifically the CD28 family. Unlike its family members, PD-1 uniquely exists as a monomer expressed on activated T cells, B cells, NK T cells, monocytes, and some dendritic cells (DCs) [8, 9]. It consists of 288 amino acids and features an ectodomain with a signal peptide, an N-loop, an IgV-like domain (with four N-linked glycosylation sites: N49, N58, N74, and N116, and a polyubiquitination site at K48), a transmembrane domain, and a cytoplasmic region with the key signaling motifs ITIM and ITSM and phosphorylation sites at Y223 and Y248 [10,11,12,13,14]. Recent research has identified additional O-glycosylation sites (T153, S157, S159, and T168) in the stalk region, indicating complex posttranslational modifications [15]. PD-1 interacts with two ligands, PD-L1 and PD-L2, from the B7 family, with PD-L2 binding to PD-1 with over three times the affinity of PD-L1 [16]. PD-L2 is distributed primarily on activated DCs and some macrophages, whereas PD-L1, which is more widely expressed on both immune and tumor cells, plays a crucial role in tumor immunity [17]. PD-L1 is a 40-kDa glycoprotein encoded by the CD274 gene on chromosome 9p24.1. Its structure includes a signal peptide, extracellular IgV and IgC domains, a transmembrane domain, and a cytoplasmic region. The extracellular regions contain four glycosylation sites (N35, N192, N200, and N219) [18], while the IgC domains have five phosphorylation sites (S176, T180, S184, S195, and T210) [19, 20]. The cytoplasmic tail includes an S-palmitoylation site at C272 [21], six methylation sites (K75, K89, K105, R113, K162, and R212) [22], and an acetylation site at K263 [23]. Moreover, recent studies have shown that the PD-L1 intracellular domain functions as an RNA binding protein [24] (Fig. 1).

Schematic of PD-1/PD-L1 proteins highlighting potential PTM sites on PD-1/PD-L1. a The full-length PD-1 protein is divided into three segments: the ectodomain, transmembrane (TM), and a cytoplasmic region (CR). The ectodomain comprises the signal peptide (SP), N-loop, IgV domain, and stalk region, with amino acid positions denoted by numbers. Potential N-glycosylation sites at N49, N58, N74, and N116 are marked with blue arrowheads. Yellow arrowheads indicate potential tyrosine phosphorylation sites at Y223 and Y248. The poly-ubiquitination site at K48 is denoted by pink arrowheads. O-glycosylation sites at T153, S157, S159, and T168 are indicated with red arrows. b Full-length PD-L1 is divided into an ectodomain, transmembrane, and cytoplasmic region. The ectodomain comprises the signal peptide, IgV, and IgC domains, with amino acid positions indicated by numbers. Potential N-glycosylation sites at N35, N192, N200, and N219 are marked with blue arrowheads. Serine/threonine phosphorylation sites, located at S176, T180, S184, S195, and T210, are denoted by orange arrowheads. The S-palmitoylation site at C272 is indicated with a purple arrowhead. The mono/multiubiquitination motif is situated in the IgV domain, while the polyubiquitination site is in the cytoplasmic region of PD-L1. The acetylation site K263 is highlighted with a red arrow. Methylation sites at K75, K89, K105, R113, K162, and R212 are indicated with yellow arrowheads
Preclinical study of PTMs inhibiting PD-L1 expression and function
PD-L1 phosphorylation inhibits PD-L1 protein expression by mediating ubiquitination
The process of protein phosphorylation involves the transfer of a phosphate group from ATP to amino acid residues of the target protein catalyzed by a series of protein kinases. This modification primarily occurs on two types of amino acids: serine (Ser or S) and threonine (Thr or T), as well as tyrosine (Tyr or Y). Protein phosphorylation plays a crucial role in regulating the activity of enzymes and other essential functional molecules, facilitating second messenger delivery and initiating enzymatic cascade reactions [25].
The nonglycosylated PD-L1 protein exhibits extreme instability, with a half-life of approximately 4 h. Serine/threonine phosphorylation of nonglycosylated PD-L1 mediates its ubiquitination and subsequent degradation. The interleukin 6 (IL-6)/Janus kinase 1 (JAK1) pathway phosphorylates PD-L1 at Y112, facilitating its binding to the N-glycosyltransferase STT3A, thus preventing PD-L1 ubiquitination and degradation [26]. Glycogen synthase kinase 3β (GSK3β), a serine/threonine protein kinase, initiates the phosphorylation of PD-L1 at sites S176, T180, and S184. This phosphorylation triggers the interaction with the E3 ligase β-TrCP, which targets proteins for proteasome degradation [27]. Additionally, D-mannose activates AMP-activated protein kinase (AMPK), leading to further phosphorylation of PD-L1 at S195 and proteasome degradation [28]. (Fig. 2).

Regulatory pathways governing PD-L1 via phosphorylation and ubiquitination. a In combination with JAK1, GSK3β, AMPK, EGF, STT3A, and IL-6 collaboratively facilitate serine/threonine phosphorylation of PD-L1, leading to subsequent ubiquitination. b β-TrCP, C-Cbl, Cbl-b, and DHA contribute to the increase in PD-L1 ubiquitination. Conversely, IFIT1 impedes PD-L1 ubiquitination. CDK4/6 enhances PD-L1 ubiquitination via SPOP. The E3 ubiquitin ligase HUWE1, in conjunction with PD-L1, facilitates PD-L1 ubiquitination. STUB1 stimulates PD-L1 ubiquitination, leading to subsequent degradation, while CMTM6/4 impedes the binding of PD-L1 to STUB1, thereby downregulating PD-L1 ubiquitination. Casp8 promotes PD-L1 ubiquitination by upregulating TNFAIP3. USP2 modulates the stability of VPRBP and facilitates PD-L1 ubiquitination. MIB2 catalyzes PD-L1 ubiquitination, mediating the trafficking of PD-L1 from the trans-Golgi network to the membrane through RAB8. Circular RNA-0000512 promotes PD-L1 ubiquitination by suppressing CMTM6 expression, while circular RNA-0067842 inhibits PD-L1 ubiquitination by upregulating CMTM6. The black arrows indicate positive regulation, and the red arrows indicate negative regulation
The expression of PD-L1 is negatively regulated by ubiquitination
Ubiquitination is the process of covalently attaching ubiquitin to a target protein under the catalysis of a series of enzymes. In monoubiquitination, a target protein binds to a single ubiquitin molecule. Multiubiquitination is the process by which a single ubiquitin molecule labels multiple lysine residues of a target protein. Polyubiquitination, on the other hand, occurs when multiple ubiquitin molecules label a single lysine residue of the target protein [29,30,31]. The E3 ligase plays a crucial and specific role in this process by regulating the activity of the ubiquitination system in these enzymatic cascades [29].
The degradation of PD-L1 is intricately regulated by various ubiquitination-dependent proteasome pathways. Previous research has demonstrated that EGF can promote the expression of PD-L1 [32]. Horita et al. revealed that epidermal growth factor (EGF) induced PD-L1 monoubiquitination and polyubiquitination prior to EGF-mediated PD-L1 protein expression. Treatment of skin squamous carcinoma cells with gefitinib and SCH772984, chemical inhibitors of the EGF receptor (EGFR) pathway, inhibited the monoubiquitination and polyubiquitination of PD-L1 [33]. The chemical inhibitor PYR41 was also found to prevent the EGF-mediated increase in PD-L1 protein levels by inhibiting E1 ubiquitinase [33]. The E3 ubiquitinases Cbl-b and c-Cbl were shown to be involved in the downregulation of PD-L1 in EGFR wild-type non-small cell lung cancer (NSCLC) [34]. Cyclin-dependent kinase 4 (CDK4) and Cullin 3- speckle-type POZ protein (SPOP), an E3 ligase bound to Cullin3, can regulate the protein level of PD-L1 through the classical proteasome-mediated degradation pathway [35]. As mentioned in the PD-L1 phosphorylation section above, resveratrol can promote the GSK3β-β-TrCP-mediated degradation of polyubiquitinated PD-L1 [27]. Sun et al. proposed that leucine-rich repeat kinase 2 (LRRK2) inhibits ubiquitin‒proteasome degradation through the phosphorylation of PD-L1 [20]. Gao et al. reported that Fusobacterium nucleatum, which is enriched in colorectal cancer tissues, upregulates the expression of PD-L1 by reducing the ubiquitination-mediated degradation of PD-L1 through IFIT1 [36]. RIG-I can compete with SPOP for binding to PD-L1, leading to reduced polyubiquitination and proteasome degradation of PD-L1 [37]. Ring finger protein 125 (RNF125) promotes ubiquitin-mediated degradation of PD-L1 and downregulates PD-L1 in oral squamous cell carcinoma (OSCC) TSCCA cells [38]. Transmembrane and ubiquitin-like domain-containing protein 1 (TMUB1) inhibits PD-L1 K281 polyubiquitination in the endoplasmic reticulum (ER) by interacting with PD-L1 and competing with HECT, UBA, and WWE domain protein 1 (HUWE1), an E3 ubiquitin ligase [39]. Researchers have designed and synthesized PTPR, a peptide that competes with PD-L1 and weakens the regulatory role of TMUB1 at the cellular level [39]. As a substrate recognition subunit of the Cullin-4 (CUL4)-damage-specific DNA binding protein 1 (DDB1) ubiquitin E3 ligase complex, VPRBP directly induces ubiquitin-mediated PD-L1 degradation, and the stability of VPRBP is controlled by USP2 [40]. Docosahexaenoic acid (DHA) promotes PD-L1 degradation through the ubiquitin‒proteasome pathway, leading to decreased PD-L1 expression. This, in turn, reduces the PD-L1 and PD-1 interaction, reversing PD-L1-mediated immunosuppression and further promoting tumor growth inhibition [41]. Furthermore, the interaction between PD-L1 and Cullin-4 A facilitates the ubiquitination of PD-L1 [42]. Casp8 induces PD-L1 ubiquitination and promotes its degradation by upregulating TNFAIP3 (A20) expression in murine melanoma, suggesting that reduced Casp8 expression may serve as a potential biomarker for predicting sensitivity to anti-PD-L1 immunotherapies [43, 44]. (Fig. 2)
The ubiquitination of PD-L1 is not confined to the ER and Golgi apparatus but also involves the promotion of lysosomal degradation by endosomes. Mezzadra et al. demonstrated that CKLF-like MARVEL transmembrane domain-containing protein (CMTM)6 and CMTM4 bind to the ectodomain of PD-L1, preventing lysine residues in the cytoplasmic tail from interacting with the E3 ubiquitin ligase STUB1 [45]. This interference disrupts polyubiquitination, extending the half-life of the PD-L1 protein. Subsequently, CMTM6 enhances PD-L1 expression and inhibits the killing effect of tumor-specific T cells in mouse melanoma models [45]. Additionally, Burr et al. reported that CMTM6 can mediate PD-L1 ubiquitination-dependent proteolysis and lysosomal degradation simultaneously [46]. Wang et al.‘s research also supported the idea that CMTM6 and Huntingtin-interacting protein 1-related protein (HIP1R) participate in the lysosomal degradation of PD-L1. Overall, CMTM6 is a promising target for immunotherapy, as indicated by these findings [47]. (Fig. 2)
The ubiquitination of PD-L1 is also associated with its trafficking. Ding’s team found that sodium-glucose cotransporter 2 (SGLT2) colocalizes with PD-L1 in the cell membrane and circulating endosomes, preventing proteasome-mediated PD-L1 degradation [48]. Yu et al. discovered that MIB E3 ubiquitin protein ligase 2 (MIB2) catalyzes the nonproteolytic K63-linked polyubiquitination of PD-L1, facilitating its transport of PD-L1 from the trans-Golgi network (TGN) to the membrane through RAS-associated binding 8-mediated (RAB8-mediated) exocytosis [49]. (Fig. 2)
Noncoding RNAs significantly contribute to the ubiquitination of PD-L1. Knockdown of circ-0000512 enhances PD-L1 ubiquitination in triple-negative breast cancer (TNBC) cells by inhibiting CMTM6 [50]. The circular RNA hsa_circ_0067842 interacts with HuR, improving the stability of CMTM6 by influencing nuclear translocation. CMTM6, in turn, regulates the ubiquitination of PD-L1 and inhibits its degradation [51]. (Fig. 2)
Other PTMs inhibiting PD-L1 expression and function
Ectodomain shedding is a posttranslational modification involving the degradation of extracellular matrix components. Matrix metalloproteinases (MMPs) and disintegrin and metalloproteinases (ADAMs) convert transmembrane molecules into soluble forms in this process [52, 53]. The proteolytic cleavage of PD-L1 is attributed to the release of MMP-13 from fibroblasts. MMP-9 and MMP-13 have been identified as enzymes capable of cleaving the PD-1 binding domain of PD-L1, consequently inhibiting T-cell apoptosis [54]. Hira-Miyazawa et al. further confirmed that purified PD-L1 can undergo degradation by MMP-13 and MMP-7. A specific inhibitor of MMP-13 (CL82198) significantly restored the expression of PD-L1, providing additional evidence for the pivotal role of MMP-13 in the shedding/cleavage of PD-L1 [55]. Known as an effective inhibitor of MMPs, HE4 was investigated by Rowswell-Turner RB et al., who revealed its ability to inhibit MMP2, 9, and 13. This inhibition resulted in a significant increase in PD-L1 expression in both tumors and macrophages, and this effect was observed posttranscriptionally [56]. (Fig. 3a)

Negative regulatory pathways of PD-L1 mediated by other posttranslational modifications. a MMP-7, MMP-9, and MMP-13 cleave the PD-1 binding domain of PD-L1, inhibiting T-cell apoptosis. b PD-L1 proteins undergo methylation by SET7 and demethylation by LSD2. c UFL1 or UFM1 enhances the UFMylation of PD-L1. d ISG15 associates with glycosylated PD-L1, promoting its ISGylation and accelerating the glycosylation-mediated degradation of PD-L1. e Cullin3 promotes NEDDylation, which contributes to the degradation of the PD-L1 protein. f GSH upregulates SERCA activity, suppressing the NF-κB signaling cascade and consequently the transcription of PD-L1. g CK2 induces phosphorylation of ING4, leading to the activation of ING4 and subsequent inhibition of the proteolytic degradation of PD-L1. The black arrows indicate positive regulation, and the red arrows indicate negative regulation
Protein methylation is a prevalent modification that can occur on both histone and nonhistone proteins and typically affects arginine and lysine residues. Arginine methylation, a common posttranslational modification, involves the addition of methyl groups to arginine residues, altering the protein’s interactions with binding partners or regulating its activity [57]. Nonhistone methylation often participates in signal transduction, with many instances linked to cancer progression [58]. In a study by Huang et al., six monomethylation sites (K75, K89, K105, R113, K162, and R212) were identified on PD-L1 through mass spectrometry (MS) analysis. Interestingly, the K162R variant was the only variant demonstrated to enhance the engagement of PD-1/PD-L1. PD-L1 methylation at K162 restricted the interaction between PD-L1 and PD-1 [22]. SET domain containing lysine methyltransferase 7 (SETD7) catalyzes the methylation of PD-L1 at the K162 site, and this modification can be reversed by LSD2. Therefore, hypermethylation of PD-L1 has been identified as a key mechanism of resistance to PD-L1 therapy [22]. (Fig. 3b)
PD-L1 is also targeted by ubiquitin-fold modifier 1 (UFM1) modification (UFMylation) [59]. UFM1 is initially synthesized in its precursor form. Upon cleavage by UFSP1 or UFSP2, UFM1-G83 is activated. This activated form is processed by the specific E1-like activating enzyme UBA5 and then transferred to the E2-like binding enzyme UFC1. The final step involves the collaboration of UFC1 with the E3-like ligase UFL1 [60]. Silencing either UFL1 or UFM1 to suppress the UFMylation of PD-L1 can lead to its stabilization in various human and mouse cancer cells, which in turn disrupts anticancer immunity both in vitro and in mice [59]. (Fig. 3c)
Interferon-stimulating gene 15 (ISG15) modification (ISGylation) is a process similar to ubiquitination. During ISGylation, the target protein binds to ISG15, modifying the target protein. Subsequently, the modified target protein and ISG15 are separated by ISG15 depolymerase, and the separated ISG15 can be recycled [61]. ISG15 induces ISG modification and PD-L1 protein instability, thereby improving targeted immunotherapy targeting PD-L1 and inhibiting the growth of lung adenocarcinoma in vivo. Additionally, ISG15 enhances K48-linked ubiquitin modification of PD-L1, ultimately promoting the degradation of glycosylated PD-L1 through the proteasome pathway [62]. (Fig. 3d)
Neural precursor cell-expressed developmentally downregulated 8 (NEDD8) modification (NEDDylation), a process similar to ubiquitination, involves the coupling of the active ubiquitin-like protein NEDD8 with the scaffold Cullin protein by the E3 Cullin-RING ligase (CRL) [63]. Pevonedistat (MLN4924, TAK924) is a small molecule inhibitor of NEDD8. Pevonedistat blocks the degradation of the PD-L1 protein by inhibiting Cullin3 activity [64, 65], increasing the levels of PD-L1 mRNA and protein in a dose- and time-dependent manner [66]. (Fig. 3e)
S-glutathionylation is a common form of cysteine (Cys or C) modification that involves the reversible formation of mixed disulfide bonds with glutathione (GSH). According to Byun JK et al., inhibiting glutamine utilization increases PD-L1 levels in cancer cells, thereby inactivating cocultured T cells [67]. Restricting glutamine metabolism in cancer cells can impair sarcoplasmic/endoplasmic reticulum calcium ATPase (SERCA) activity by reducing S-glutathionylation due to low glutathione levels. This activates the calcium/NF-κB signaling cascade, ultimately leading to the transcriptional activation of PD-L1 [67]. (Fig. 3f)
Autophagy serves as the primary intracellular degradation system, ushering cytoplasmic substances into lysosomes for breakdown and generating new components and energy for cellular homeostasis [68]. Gou et al. demonstrated that growth inhibitory factor 4 (ING4) induces autophagic degradation of PD-L1, suppressing immune escape in NSCLC cells by enhancing T-cell activity. Additionally, casein kinase 2 (CK2) phosphorylates ING4 at S150, promoting its ubiquitination and degradation via the JFK ubiquitin ligase. Conversely, CK2 gene knockout strengthens ING4 protein stability and augments T-cell activity [69]. (Fig. 3g)
Preclinical study of PTMs promoting PD-L1 expression and function
Deubiquitination of PD-L1 upregulates its expression by enhancing protein stability
Deubiquitination is a process catalyzed by deubiquitination enzymes (DUBs), which reverse ubiquitination by removing ubiquitin molecules from ubiquitinated proteins [70, 71]. In contrast to ubiquitination, the deubiquitination of PD-L1 can enhance the stability of the protein. COP9 signaling body 5 (CSN5) plays a crucial role in the CSN complex, contributing to tumor immune escape by inducing the deubiquitination of PD-L1 [72]. Lim et al. reported that TNF-α secreted by macrophages in breast cancer (BC) impacts PD-L1 expression at the translational level. TNF-α induces the expression of CSN5 and CSN2 by activating p65 of NF-κB [73]. Subsequently, CSN5 binds to the C-terminus of PD-L1 and deubiquitinates it, thereby enhancing its stability. Although the MPN domain of CSN5 does not interact with PD-L1, disruption of the MPN domain affects the CSN5-mediated deubiquitination of PD-L1 and protein stability [73]. Protein disulfide isomerase A (PDIA)6 might upregulate the expression of CSN5 by regulating the formation of disulfide bonds in CSN5, increasing the stability of PD-L1 through deubiquitination in pancreatic cancer cells [74, 75]. (Fig. 4)

Regulatory pathways of PD-L1 via deubiquitination. The deubiquitination of PD-L1 involves a family of ubiquitin-specific proteases (USPs), namely, USP2, USP5, USP7, USP8, USP9X, USP20, USP22, USP28, and USP51, along with CSN5, UCHL1, OTUB1, and microRNAs (miR-199a-5p and miR-328-3p). Positive regulatory interactions are denoted by black arrows
USPs have been identified as novel deubiquitinases of PD-L1 in multiple cancers. USP22 specifically targets the C-terminus of PD-L1, leading to its deubiquitination and stabilization in liver cancer cells [76]. Additionally, USP22 enhances the stability of CSN5 both by deubiquitination and by directly regulating PD-L1 deubiquitination in NSCLC. This process enhances PD-L1 stability by removing the K6, K11, K27, K29, and K33 residues that bind to PD-L1 [77]. USP9X binds to PD-L1, inducing its deubiquitination and stabilizing protein expression in OSCC [78]. Thr288, Arg292, and Asp293 on USP2 regulate its binding to PD-L1, uncoupling the K48-linked residue on lysine 270 of PD-L1 to increase PD-L1 abundance. Deletion of USP2 leads to the degradation of ER-related PD-L1, which weakens the binding of PD-L1 to PD-1 and renders cancer cells susceptible to T-cell-mediated cytotoxicity [79]. USP51 enhances the stability of the PD-L1 protein by removing polyubiquitination, promoting chemotherapy resistance in NSCLC cells [80]. In pancreatic cancer, USP8 inhibits the ubiquitination-regulated proteasome degradation pathway by positively interacting with PD-L1 and upregulating its expression [81]. USP7 mediates the ubiquitination of PD-L1 and inhibits its degradation [82]. Additionally, UCHL1 promotes PD-L1 deubiquitination and upregulates its expression in NSCLC [83]. OTUB1 interacts with and removes K48-linked ubiquitin strands in the PD-L1 cytoplasmic domain via a process mediated by deubiquitinase activity, preventing PD-L1 degradation through the ER-associated degradation (ERAD) pathway [84]. (Fig. 4)
Noncoding RNAs are also known to be involved in regulating PD-L1 deubiquitination. The long noncoding RNA TINCR functions as a sponge of miR-199a-5p, enhancing the stability of USP20 mRNA through a competitive endogenous RNA mechanism. This causes PD-L1 to become ubiquitinated and increases its protein abundance [85]. . Zheng’s team found that the smoking-related lncRNA BCCE4 mutation rs62483508 G > A can disrupt the binding site of miR-328-3p, reducing the expression of USP18 and weakening the interaction between PD-L1/PD-1 to strengthen antitumor immune responsiveness in bladder tumors [86]. (Fig. 4)
Small ubiquitin-like modifier (SUMO) modification (SUMOylation) is correlated with deubiquitination [87]. Ma X et al. discovered that the E3 SUMO ligase tripartite motif-containing protein 28 (TRIM28) can stabilize the PD-L1 protein by inhibiting PD-L1 ubiquitination and promoting its SUMOylation in gastric cancer cells [88].
Glycosylation of PD-L1 promotes its protein expression and function
Glycosylation is a crucial modification that can significantly impact protein formation, function, and interactions with other proteins. The process of glycosylation involves the formation of glycoproteins with specific oligosaccharide chains in the ER, facilitated by various glycosyltransferases and glycosidases. Subsequently, glycoproteins move from the Cis surface to the Golgi body, where they undergo a series of ordered processing and modifications. N-linked glycosylation and O-linked glycosylation. N-linked glycosylation attaches a sugar chain to the -NH2 group of an asparagine residue, while O-linked glycosylation links a sugar chain to the oxygen of -OH groups in serine, threonine, or hydroxylysine residues of a polypeptide [89]. Hypoxia and abnormal glucose metabolism are known to alter protein glycosylation patterns in the tumor microenvironment. Notably, PD-L1 is highly glycosylated in most cells expressing it, while the unglycosylated form tends to have lower expression levels [18].
N-glycosylation of PD-L1 positively regulates its protein stability and interaction with PD-1
Glycosylation of PD-L1 plays a crucial role in promoting its protein stability. Specifically, the N192, N200, and N219 sites on the PD-L1 protein hinder the interaction between GSK3β and PD-L1 [90]. The inhibition of GSK3β facilitates the glycosylation of PD-L1 in breast cancer, preventing its degradation by the 26 S proteasome [91]. Sigma1 has been implicated in regulating the glycosylation of newly synthesized PD-L1 in the ER and Golgi compartments to promote the expression of PD-L1 [91]. FK506 binding protein 51 s (FKBP51 s), which are specifically expressed in glioblastoma, promote the glycosylation of PD-L1 in the ER and upregulate its expression on cell membranes [92]. Glycosyltransferase 1 domain 1 (GLT1D1) enhances the stability of PD-L1 through N-glycosylation, promoting immunosuppression and tumor growth [93]. The GDP-fucose transporter (GFT) is a critical molecule involved in fucosylation of PD-L1. Knockout of the GFT gene SLC35C1 significantly decreases PD-L1 fucosylation, leading to increased ubiquitination of PD-L1 [94]. Beta-1,4-galactosyltransferase 1 (B4GALT1) directly mediates the N-glycosylation of PD-L1, preventing its degradation. Inhibition of B4GALT1 increases the abundance and activity of CD8+ T cells, enhancing antitumor immunity against PD-1 therapy in vivo [95]. In breast cancer tumor stem cells, the enrichment of PD-L1 is considered crucial for tumor stem cell immune escape. The mechanism involves β-catenin inducing the transcription of the N-glycosyltransferase STT3 to promote the oligoglycosylation of PD-L1 in the ER and upregulate PD-L1 expression [96]. PD-L1 enhances its stability by activating the N-glycosyltransferases STT3A and STT3B through PAR2 [97]. Additionally, TMUB1 enhances the N-glycosylation and stability of PD-L1 by recruiting STT3A, which promotes PD-L1 maturation and facilitates tumor immune escape [39]. TGF-β1 activates the c-Jun/STT3A signaling pathway, promoting the N-glycosylation of PD-L1 [98]. FAT atypical cadherin-4 (FAT4) overexpression not only reduces PD-L1 mRNA expression but also inhibits STT3A by promoting β-catenin degradation. This triggers aberrant glycosylation of PD-L1, causing its accumulation in the ER and degradation by ubiquitin-dependent pathways [99]. The gene SEC61G, located adjacent to the EGFR chromosome, promotes the translocation of immune checkpoint ligands (PD-L1, PVR, and PD-L2) to the ER, facilitating their glycosylation, stability, and membrane presentation [100]. Monocarboxylate transporter 4 (MCT4) has been found to promote the glycosylation of PD-L1 through the classical WNT pathway, stabilizing PD-L1. The high coexpression of MCT4 and PD-L1 suggests a more effective target for treating TNBC, potentially improving the immune checkpoint treatment of TNBC [101]. (Fig. 5)

Regulatory pathways of PD-L1 through glycosylation. a Sigma1 promotes N-glycosylation in both the ER and Golgi, and FKBP51 s also augment N-glycosylation of PD-L1 in the ER. EGF upregulates B3GNT3, facilitating the glycosylation of PD-L1 in the Golgi apparatus. Conversely, GSK3β inhibits PD-L1 glycosylation. GLT1D1, GFT, and B4GALT1 promote the N-glycosylation of PD-L1. β-catenin induces N-glycosyltransferase STT3 transcription, stabilizing the oligosaccharide chains of PD-L1 in the ER and upregulating PD-L1. PAR2 activates the N-glycosyltransferases STT3A and STT3B, enhancing the glycosylation of PD-L1. TMUB1 enhances the N-glycosylation and stability of PD-L1 by recruiting STT3A. TGF-β1 activates the c-Jun/STT3A signaling pathway, promoting N-glycosylation of PD-L1. At the transcriptional level, FAT4 reduces PD-L1 mRNA expression and downregulates STT3A through β-catenin, resulting in abnormal glycosylation of PD-L1. SEC61G induces PD-L1 translation and N-glycosylation. MCT4 stabilizes PD-L1 by promoting its glycosylation through the classical WNT pathway. Notably, secreted PD-L1 splicing variants exist, with only those possessing N-linked glycosylation sites exhibiting stable secretion. b GALNT2/14 and GFAT1 potentially increase the O-glycosylation of PD-L1. The black arrows denote positive regulation, while the red arrows indicate negative regulation
PD-L1 glycosylation is essential for the interaction of PD-L1 with PD-1. While the signaling of costimulatory molecules can function effectively without glycosylation, the signaling of coinhibitory molecules, including PD-L1, requires glycosylation, particularly N-linked glycosylation [90]. Furthermore, activation of the EGF/EGFR signaling pathway has been shown to upregulate beta-1,3-N-acetylglucosaminyltransferase 3 (B3GNT3), promoting the glycosylation of poly-N-acetyllactosamine at the N192 and N200 sites of PD-L1 in the Golgi apparatus. This enhanced glycosylation, mediated by B3GNT3, increases the affinity of PD-L1 for binding to PD-1 [90]. Molecular dynamics simulations of the PD-L1/PD-1 interaction with N-glycans suggest that N-glycosylation of the PD-L1 N219 site may influence the interaction with PD-1 [18]. (Fig. 5)
The glycosylation of PD-L1 appears to be involved in the promotion of tumor metastasis. Erlichman and his team reported that PD-L1 activates STAT1 and STAT3 to promote breast cancer cell metastasis both in vitro and in vivo and that PD-L1 is required for N-glycosylation at the N219 site [102]. In addition, the glycosylation sites N192 and N200 (depending on cell type) contribute to the autonomous cell migration function of PD-L1 in vitro [102]. (Fig. 5)
The glycosylation of secreted PD-L1 variants has been implicated in drug resistance to PD-L1 antibodies. Gong et al. identified five secreted PD-L1 splicing variants in patients resistant to anti-PD-L1 antibodies: PD-L1 v174, PD-L1 v178, PD-L1 v229, PD-L1 v242, and PD-L1 v265. Among these variants, PD-L1 v242 and PD-L1 v229 contain three N-glycosylation sites (N192, N200, and N219), which contribute to the stabilization of PD-L1, allowing it to be stably secreted and induce resistance to anti-PD-L1 antibodies [103]. Conversely, PD-L1 v178 lacks N-glycosylation sites, making it unstable and poorly secreted. As a splicing variant of PD-L1, PD-L1-vInt4 functions as bait in anti-PD-L1 antibody therapy, further contributing to drug resistance [104]. This finding sheds light on a novel mechanism of drug resistance against anti-PD-L1 antibodies. (Fig. 5)
O-linked glycosylation of PD-L1 may be related to its expression
GALNT2/14, which are polypeptide N-acetyl glucosaminyl transferase 2/14, play a role in initiating mucin O-glycosylation in the Golgi apparatus. Research has demonstrated a positive correlation between the expression of GALNT2/14 and that of PD-L1 [105]. However, conflicting studies have suggested that the stability of the PD-L1 protein might not be dependent on O-linked glycosylation [106]. Chen et al. reported that although inhibiting L-glutamine: D-fructose-6-phosphate aminotransferase 1 (GFAT1) reduces overall protein O-GlcNAcylation, it does not seem to affect the stability of PD-L1. The increase in PD-L1 protein degradation is attributed to the decrease in N-linked glycosylation, even though other mechanisms cannot be ruled out [106]. (Fig. 5)
Other PTMs inhibiting PD-L1 expression and function
Secreted and membrane proteins often contain numerous disulfide bonds formed by the oxidation of two Cys residues, which are crucial for their structural stability and function. Incorrect disulfide bond formation can cause protein misfolding in the ER, triggering the unfolded protein response (UPR) to manage protein folding [107]. ERO1-α, an ER oxidase often overexpressed in tumors, works with protein disulfide isomerase (PDI) to form disulfide bonds. Studies by Tanaka et al. have shown that ERO1-α enhances PD-L1 expression by facilitating the folding of oxidized proteins in PD-L1 [108]. Chen et al. reported that silencing PDIA5 in human glioma cells upregulates PD-L1 expression, suggesting that PDIA5, by modifying disulfide bonds and activating the UPR, may influence PD-L1 expression, although the exact mechanisms involved are unexplored [109]. (Fig. 6a)

Positive regulatory pathways of PD-L1 mediated by other posttranslational modifications. a PDIA5 appears to exert a negative regulatory effect on PD-L1, while ERO1-a enhances PD-L1 expression by facilitating the proper formation of disulfide bonds in PD-L1. ERO1-α additionally upregulates HIF-1a protein, resulting in increased PD-L1 mRNA and protein levels. b S-palmitoylation occurs within the Golgi apparatus. ZDHHC9, DHHC3, DHHC5, and FASN have been identified as promoters of PD-L1 palmitoylation and thereby contribute to the stabilization of the PD-L1 protein. Conversely, DHA inhibits FASN, thereby suppressing the palmitoylation of PD-L1. c STAT5, which promotes glycolysis, leads to lactic acid accumulation, subsequently facilitating E3BP nuclear translocation and histone lactylation, culminating in the induction of PD-L1 transcription. d HDAC2 facilitates nuclear translocation through PD-L1 deacetylation, whereas p300 promotes acetylation, enhancing its interaction with TRAPPC4 and facilitating PD-L1 recycling to the membrane. e PDGF/ARF6/AMAP1 enhances the recycling of PD-L1 to the membrane by augmenting the ADP-ribosylation of PD-L1. The black arrows denote positive regulation, while the red arrows signify negative regulation
Palmitoylation, a lipid modification, is essential for regulating membrane proteins and includes S-palmitoylation, N-palmitoylation, and O-palmitoylation [110]. S-palmitoylation involves attaching a 16-carbon fatty acid palmitate to Cys residues via an unstable covalent bond, which is typically catalyzed by DHHC palmitoyl transferase [110, 111]. This is a pivotal modification in several cancer-related proteins, including PD-L1, where C272 palmitoylation helps stabilize the protein, thus protecting tumor cells from being eliminated by T cells. In breast cancer, ZDHHC9 enhances PD-L1 stability through palmitoylation [112, 113], and ZDHHC9 deficiency in lung cancer prevents PD-L1 degradation, enhancing the effectiveness of anti-PD-L1 immunotherapy [115]. Similarly, ZDHHC3 increases PD-L1 palmitoylation at C272 in colorectal and pancreatic cancer models, reducing its degradation [116]. Shahid et al. reported that fatty acid synthase (FASN) in cisplatin-resistant bladder cancer cells enhances PD-L1 expression by regulating palmitate formation [117]. Moreover, DHA downregulates FASN, inhibits DHHC5, and promotes PD-L1 degradation [41]. Addressing PD-L1 palmitoylation may be an effective way to counteract tumor immune evasion strategies. (Fig. 6b)
Succinylation correlates with increased PD-L1 expression in prostate cancer, suggesting its significant role in regulating PD-L1 levels [118,119,120,121]. Additionally, lactic acid, a precursor of histone lysine modifications, is linked to glycolytic gene activation by STAT5 in AML, leading to increased PD-L1 transcription via enhanced histone lactylation and nuclear translocation of E3BP [122, 123]. These modifications reveal intricate connections between metabolic processes and immune regulation in cancer. (Fig. 6c)
Protein lysine acetylation, which is reversible via lysine acetylases (KATs), influences protein stability and localization [124, 125]. Recent findings have shown that nuclear PD-L1, which is acetylated at K263 by p300 and deacetylated by HDAC2, acts as a transcription factor that alters gene expression related to antigen presentation and inflammatory pathways, affecting cytotoxic T lymphocyte activity and tumor immune evasion [126]. Nuclear PD-L1 also upregulates other immune checkpoint genes and angiogenesis markers in breast cancer. EGF enhances PD-L1 acetylation [33], while VPA increases PD-L1 recycling to the membrane, highlighting complex regulatory mechanisms [127]. (Fig. 6d)
ADP-ribosylation is a dynamic, reversible posttranslational modification that involves the attachment of an ADP-ribose group to proteins, affecting their degradation and vesicle transport between organelles [128,129,130,131,132]. This modification is initiated by NAD+ cleavage, leading to either mono- or multi-ADP-ribosylation. Hashimoto et al. reported that PDGF binding to its receptor, PDGFRβ, activates ADP-ribosylation factors such as ARF6 and AMAP1, promoting PD-L1 recycling to the cell membrane; silencing these factors reduces PD-L1 surface expression, illustrating the role of ADP-ribosylation in vesicle transport [130, 131]. (Fig. 6e)
Preclinical study of PTMs regulating PD-1 expression and function
Phosphorylation of PD-1 affects its immunosuppressive effect
Tyrosine phosphorylation within the PD-1 ITSM domain is a pivotal step in the activation of downstream immunosuppressive pathways. Upon interaction between PD-1 and PD-L1, phosphorylation occurs at the PD-1 ITIM (Y223) and ITSM (Y248). The phosphorylation of ITSM results in the recruitment of protein tyrosine phosphatase 2 (SHP2), which subsequently dephosphorylates the ζ chains and ζ chain-related tyrosine kinase 70 (ZAP70) within CD28 and the T-cell receptor (TCR)/CD3 complex. This inhibition affects the downstream PLCγ1, PI3K/AKT, and ERK1/2 signaling pathways, leading to reduced IL-2 secretion and glucose metabolism. Consequently, T-cell function is further inactivated, playing a negative role in immune regulation [27]. Hui et al. reported that CD28 and PD-1 cluster briefly and concentrically near the TCR when PD-1 on T cells binds to PD-L1. The TCR phosphorylation kinase Lck effectively phosphorylates PD-1, while SHP2 dephosphorylates PD-1, rendering PD-1 unstable. In the absence of SHP2, SHP1 can assume its role [133]. Similarly, upon binding of PD-L1 to PD-1 on B cells, tyrosine in the PD-1 ITSM domain undergoes phosphorylation [134]. Furthermore, ERK can phosphorylate the T234 site of PD-1, subsequently promoting the interaction between PD-1 and USP5, which results in deubiquitination and enhanced stability of PD-1 [135]. (Fig. 7)

Posttranslational modification of PD-1. As a transmembrane protein, PD-1 undergoes intricate posttranslational modifications. The primary site of focus for PD-1 is within the Golgi apparatus. Fut8 plays a pivotal role in promoting the core structure of the PD-1 protein, thereby contributing to the stabilization of PD-1. Upon binding to PD-L1, the intracellular domain of PD-1 undergoes phosphorylation, recruiting SHP2-2 and subsequently initiating immunosuppressive signaling. Lck enhances the phosphorylation of PD-1, intensifying its downstream effects. IL-2 promotes the transcription of FBXO38, which, in turn, binds to the cytoplasmic region of PD-1, facilitating polyubiquitination and subsequent proteasome-mediated degradation. MARCH5, c-Cbl, and FBW7 are also implicated in promoting PD-1 ubiquitination. MDM2 facilitates the interaction between NGLY1 and PD-1, leading to the deglycosylation of PD-1. Furthermore, DHHC9 promoted the palmitoylation of PD-1 to enhance its interaction with Rab11. Inhibition of palmitoylation diminishes the transport of PD-1 to the recycling endosome, promoting its degradation in the lysosome. This process is also associated with a notable enhancement in the interaction between PD-1 and mTOR signal effector proteins (S6K and eIF4E). The black arrows indicate positive regulatory pathways, while the red arrows indicate negative regulatory pathways
Ubiquitination of PD-1 mediates its degradation and regulates the antitumor immunity of T cells
Factors within the tumor microenvironment can induce high expression of the inhibitory receptor PD-1 on functional T cells. However, there is limited understanding of the degradation mechanism of PD-1. FBXO38 is recognized as the E3 ligase responsible for PD-1, directly targeting the PD-1 cytoplasmic domain and mediating its K48-linked polyubiquitination, followed by proteasome degradation [13]. IL-2 treatment significantly enhances the transcription of F-box protein 38 (Fbxo38), reducing PD-1 levels and boosting anticancer activity in mice [13]. Lv et al. elucidated that cytokine-inducible SH2 domain-containing protein (CISH) promotes PD-1 expression by inhibiting FBXO38 expression, suggesting novel strategies to enhance CAR-T-cell therapeutic efficacy by inhibiting CISH [136]. The C-terminus of c-Cbl interacts with the cytoplasmic tail of PD-1 and destabilizes PD-1 through ubiquitination-proteasome degradation in mouse colorectal cancer [137]. Additionally, F-box and wd repeat domain containing 7 (FBW7) has been shown to promote the ubiquitination of PD-1 and subsequent proteasome hydrolysis [12]. Recently, Wu et al. demonstrated that ubiquitination and breakdown of PD-1 require elimination of N-linked glycosylation and identified MDM2 as an E3 ligase for deglycosylation of PD-1. These enzymes facilitate the interaction between glycosylated PD-1 and N-glycanase 1 (NGLY1), leading to further deglycosylation of PD-1 catalyzed by NGLY1 [138]. These preclinical studies suggest that the ubiquitination of PD-1 is expected to become a new focus in the development of anticancer medications [139]. (Fig. 7)
N-glycosylation of PD-1 impacts its protein expression and interaction with PD-L1
The attachment of PD-1 to its ligands is dependent on the N49, N58, N74, and N116 glycosylation sites located in the PD-1 IgV domain [11]. Core fucosylation at N49 and N74 regulates PD-1 expression. The inhibition of core fucosylation through the use of 2-fluoro-L-fucose (2 F-Fuc), which targets the fucosyltransferase Fut8, results in decreased PD-1 expression and T-cell activation [140]. (Fig. 7)
Palmitoylation of PD-1 upregulates its expression and interaction with mTOR signaling effectors
Palmitoylation of PD-1 plays a crucial role in inhibiting lysosomal degradation, thereby stabilizing the protein. Yao et al. reported that DHHC9 promotes the palmitoylation of PD-1, leading to interaction with Rab11, which is a pivotal molecule facilitating the transport of cargo proteins to recycled endosomes [141]. Blocking palmitoylation reduces PD-1 transport to recycled endosomes and enhances lysosomal degradation. Intriguingly, PD-1 palmitoylation significantly enhances the interaction between PD-1 and mTOR signaling effectors (S6K and eIF4E), activating mTOR signaling and promoting tumor growth [141]. (Fig. 7)
Therapeutic prospects and clinical transformation of PD-1/PD-L1 PTMs
Building on foundational research into PTMs of PD-1/PD-L1 that regulate their expression and function, researchers have developed targeted therapies tested in cell and mouse models (Table 1; Fig. 8). Currently, these promising results are moving toward clinical applications, with multiple treatment regimens involving PTM-targeting drugs and immune checkpoint inhibitors actively progressing through clinical trials. These efforts aim to validate and expand the use of these innovative therapies in clinical settings (Table 2).

Regulatory networks and corresponding therapeutic interventions targeting PD-1/PD-L1 posttranslational modifications. The figure illustrates various therapies targeting PD-L1 and PD-1 post-translational modifications. The colored regions—purple, red, blue, green, brown, pink, gray, orange, and yellow—correspond sequentially to deubiquitination, glycosylation, ectodomain shedding, acetylation, UFMylation, phosphorylation, ubiquitination, autophagy degradation, and S-palmitoylation modifications. Adjacent to each colored region, the outer grids display the related molecules and potential therapeutic drugs targeting these specific post-translational modifications
PD-L1 phosphorylation-related therapeutic prospects and clinical transformation
Therapeutic promotion and clinical transformation of drugs that inhibit the EGFR pathway
Blocking the EGFR pathway is linked to an increase in PD-L1 levels in tumor cells, leading to improved outcomes from PD-1/PD-L1 blockade therapy in cancers such as breast cancer and NSCLC [27, 142]. Clinical trials are currently investigating combinations of EGFR inhibitors with PD-1/PD-L1 blockade therapies and EGF tumor vaccines (Table 2). However, the efficacy of these combinations is under scrutiny due to serious treatment-related toxicity, such as a 22% incidence of interstitial lung disease in the TATTON study [143] and a 71.4% rate of severe hepatotoxicity in another study involving pembrolizumab and gefitinib [144]. Despite these challenges, studies such as KEYNOTE-021 reported manageable toxicity and a 41.7% objective response rate (ORR) for the combination of pembrolizumab and erlotinib [144]. Additionally, cetuximab is being evaluated in trials for its potential to enhance immune checkpoint therapy in head and neck squamous cell carcinoma (HNSCC), as it has shown promising progression-free survival (PFS) rates and a 45% ORR [145, 146]. Final results on the safety and efficacy of these combination therapies are highly anticipated [147].
Therapeutic prospects and clinical transformation of PARP inhibitors
Jiao et al. reported that the drug olaparib, a PARP1 inhibitor, increases PD-L1 levels in cancer cells by deactivating GSK3β [148]. When olaparib was combined with anti-PD-L1 therapy, it was more effective in treating cancer in live models than when each drug was used alone [148]. As a result, many clinical trials are now testing combinations of PARP1 inhibitors and anti-PD-1/PD-L1 therapies (Table 2). Some of these trials have shown that these combinations are superior to standard treatments. For example, in a study involving patients with advanced kidney, bile duct, and liver cancers, one combination therapy resulted in a 23% ORR, which increased to 30% with a higher dose [149]. Common side effects included mild to moderate fatigue, diarrhea, and nausea [149]. Another trial showed that combining specific drugs for advanced liver cancer achieved a 30% response rate, which was better than the 13.3% response rate of the standard treatment [150]. This trial also revealed high response rates in patients with certain genetic markers, such as β-catenin, and even in those who did not express PD-L1, a target of the treatment [150]. Other trials exploring different combinations for breast and ovarian cancer have shown promising results with good tolerability [151,152,153,154,155,156,157].
Therapeutic promise and clinical transformation of metformin
Metformin activates AMPK, which phosphorylates PD-L1, disrupting its normal assembly and leading to its degradation. This interaction suggests that combining metformin with immune therapies such as CTLA4 blockers could improve cancer treatment outcomes [158]. However, metformin shows limited effectiveness against cancer under certain conditions where PD-L1 cannot be phosphorylated [158]. Current clinical trials are testing the effectiveness of combining metformin with anti-PD-1 therapy [159] (Table 2).
Therapeutic promise of LRRK2 inhibitors
LRRK2 is an enzyme that modifies PD-L1 by adding a phosphate group to it, which prevents PD-L1 from being broken down in cells. Inhibiting LRRK2 enhances the effects of PD-L1-targeted treatments in mice, increasing the therapeutic response. Adenosine cobalamin, a form of vitamin B12, effectively blocks LRRK2 and improves the response to PD-L1 immunotherapy in mice with pancreatic cancer. This approach, in which PD-L1 blockade is combined with LRRK2 inhibition, appears promising as a new treatment strategy for pancreatic cancer [20].
PD-1/PD-L1 ubiquitination treatment prospects and clinical transformation
PROTACs targeting PD-1/PD-L1
The use of proteolysis-targeting chimeras (PROTACs), which target and degrade difficult-to-drug proteins, is a new method for cancer treatment [160, 161]. Wang et al. developed a PROTAC called 21a that breaks down the PD-L1 protein in various cancers [162]. Another PROTAC, P22, specifically disrupts the PD-1/PD-L1 interaction, enhancing therapeutic efficacy [163]. Cotton et al. proposed the use of antibody-based PROTACs (AbTACs), which use the E3 ligase RNF43 to target PD-L1 for lysosomal destruction [164]. Su et al. introduced carbon-based PROTACs (CDTACs), which also target PD-L1 but for proteasome degradation, showing promise in preclinical studies by inhibiting tumor growth and boosting the immune response [165]. Sun et al. developed ROTACs, a type of PROTAC that targets and degrades specific signaling molecules, using a chimera called R2PD1 to efficiently degrade PD-L1 in melanoma cells, outperforming existing treatments in activating immune responses and inhibiting tumor growth [166]. These developments suggest that PROTACs could significantly improve PD-1/PD-L1-targeted therapies for cancer [167].
Prospects and clinical translation of CDK4/CDK6 inhibitors
Research has revealed that CDK4/CDK6 inhibitors, by increasing CDH1 levels, promote the degradation of the SPOP protein, which in turn increases PD-L1 expression through a pathway involving cyclin D-CDK4. Using the CDK4/6 inhibitor palbociclib and PD-1 immunotherapy in a mouse colon cancer model resulted in significant tumor shrinkage and extended survival, highlighting a new regulatory mechanism involving cyclin kinase and ubiquitin ligase for PD-L1 [35]. In another discovery, Ding et al. reported that the diabetes drug canagliflozin disrupts the interaction between SGLT2 and PD-L1, allowing PD-L1 recognition and degradation by the Cullin 3-spopoe3 ligase and enhancing T-cell attack on tumor cells [48]. This finding illustrates a potential strategy for using existing drugs to decrease PD-L1 and boost immune responses against cancer. Additionally, Lin’s team identified PIK-93, a compound that increases the binding of PD-L1 to Cullin-4 A, thus improving the effectiveness of anti-PD-L1 immunotherapy [42]. These findings have propelled multiple clinical trials testing combinations of CDK4/6 inhibitors with PD-1/PD-L1 therapies [168, 169]. A phase I trial on advanced non-small cell lung cancer reported that 53% of patients showed clinical improvement and tolerated the treatment well, indicating a promising avenue for enhancing cancer immunotherapy [168] (Table 2).
Prospects and clinical translation of targeting the deubiquitination of PD-1/PD-L1
Zhang et al. discovered that the USP22 inhibitors Rottlerin and Morusin promote the breakdown of PD-L1 and Sirt1 proteins, suggesting a new method for cancer therapy [170]. In related research, combining a USP2 inhibitor with an anti-PD1 antibody led to complete tumor regression in models with functional p53, emphasizing the therapeutic potential of targeting protein stability [40]. Li et al. reported that the flavonoid dihydromyricetin (DHM) acts as a USP51 inhibitor, enhancing lung cancer cell sensitivity to chemotherapy by promoting PD-L1 degradation [80]. Similarly, a study on a USP8 inhibitor demonstrated its effectiveness in suppressing pancreatic tumor growth by activating killer T cells, especially when combined with anti-PD-L1 therapy [81]. Additionally, A11, an inhibitor of USP7, showed promising antitumor effects by blocking PD-L1’s ability to help tumors evade immune detection, and when combined with PD-1 antibody therapy, it showed enhanced antitumor activity [82]. Additionally, the application of the CSN5 inhibitor curcumin inhibited the ubiquitination of PD-L1, reduced PD-L1 expression, and increased the sensitivity of tumor cells to CTLA4 immunotherapy [73].
Treatment prospects and clinical transformation associated with PD-1/PD-L1 glycosylation
PD-1 glycosylation enhances the binding of PD-1 to antibodies and reduces immune escape
The glycosylation of PD-1, particularly at the N58 site, significantly influences its interaction with certain antibodies. Glycosylation enhances the effectiveness of camrelizumab by improving its binding to PD-1 [171], whereas the interaction between cemiplimab and PD-1 mirrors that of camrelizumab [172,173,174]. Other antibodies, such as nivolumab and toripalimab, do not depend on glycosylation for their function [173, 175]. To address the challenges posed by the large size of typical IgG antibodies, researchers have developed smaller proteins, JYQ12 and JYQ12-2, from the extracellular domains of PD-1. These proteins, which are only 14–17 kDa and contain a single N-linked glycan chain, not only bind effectively to PD-L1 and PD-L2 but also enhance the proliferation of human T cells, showing promising potential for therapeutic and diagnostic applications in cancer immunotherapy [176].
Targeting the N-glycosylation site of PD-L1 blocks its interaction with PD-1
The glycosylation of PD-L1 strengthens its interaction with PD-1, suppressing immune responses and aiding tumor escape. To counter this, new drugs have been developed to target glycosylation sites. For example, the antibody STM108 targets glycosylated PD-L1 at specific sites (N35, N192, and N200), effectively blocking the PD-L1/PD-1 interaction. This finding demonstrates the potential of using glycosylation-specific antibodies in cancer therapy to prevent tumors from escaping the immune system [90].
Glycosylation of PD-L1 affects clinical immunohistochemistry
The glycosylation of PD-L1 can interfere with its detection by immunohistochemical antibodies, potentially causing false-negative results in tests that assess PD-L1 expression in cancer patients. This issue arises when glycosyl structures on the PD-L1 protein prevent antibody binding [90]. To address this issue, researchers have developed a method of removing these sugars—called deglycosylation—before testing. This technique significantly improves the accuracy of PD-L1 detection and correlates better with patients’ responses to anti-PD-1/PD-L1 therapies [177], and it has been patented (UTSC.P1325US. P1) due to its substantial clinical value.
Antitumor activity of the PD-L1 glycosylation inhibitor
Glycosylation inhibitors of PD-L1 are promising antitumor agents. In a phase I trial, the fucosylation inhibitor SGN-2FF combined with pembrolizumab yielded promising results in patients with advanced solid tumors, including a complete response in an HNSCC patient and significant tumor reduction in a TNBC patient (Table 2). However, the trial was stopped due to thromboembolism risks [178]. Newer inhibitors, such as A2F1P and B2FF1P, have shown greater effectiveness than SGN-2FF due to improved cellular retention and efficiency [179, 180]. Other developments include IPAG and SAFit, which inhibit PD-L1 glycosylation and degrade PD-L1 in cells, enhancing the potential for cancer therapy [84, 85]. Additionally, drugs such as BAY-876 inhibit glycolysis in TNBC, reducing PD-L1 glycosylation and enhancing the efficacy of anti-PD-L1 therapies [181]. These advancements demonstrate significant potential for developing drugs that target the PD-1/PD-L1 pathway, although safety and impact on normal tissues remain critical considerations [182].
Etoposide can inhibit the enzyme STT3, which is involved in N-glycosylation, through its anti-EMT effects, reducing PD-L1 levels and increasing the effectiveness of anti-Tim3 therapy [96]. Clinical trials of etoposide combined with anti-PD-1/PD-L1 immunotherapy are currently underway [183,184,185,186,187,188,189] (Table 2). In a phase III trial for extensive small cell lung cancer, compared with placebo, pembrolizumab combined with etoposide and platinum significantly improved 12-month PFS (13.6% vs. 3.1%, P = 0.0023), enhancing patient quality of life [183, 184]. Another study revealed that atezolizumab combined with carboplatin and etoposide increased overall survival (OS) to 12.3 months from 10.3 months with chemotherapy alone (P = 0.0154) and was well tolerated [185, 186]. Similarly, tislelizumab or serplulimab with the same regimen in different trials extended OS and PFS [188, 189].
Targeting the PD-L1 dimer inhibits PD-L1 function
PD-L1 can form homodimers and tetramers, and its complex glycosylation is linked to the homodimeric structure of its intracellular domain [190]. Natural compounds such as capsaicin, 6-gingerol, and curcumin may block the PD-1/PD-L1 interaction by targeting PD-L1 dimerization, enhancing anticancer immunity [191]. The small molecule BMS-202, with modified carbonyl to hydroxyl groups, produces two enantiomers, MS and MR, both of which disrupt PD-L1 function by targeting its dimerization [192]. Furthermore, compounds such as α-mangostin and ethanol extracts can inhibit PD-L1 glycosylation and promote its degradation by binding within the pocket of the PD-L1 dimer [193]. These findings from preclinical studies highlight the potential of designing inhibitors that target PD-L1 dimers to enhance immunotherapy efficacy.
Treatment prospects and clinical transformation of PD-L1/PD-1 palmitoylation
Palmitoylation of PD-L1 stabilizes the protein, and targeting this modification enhances PD-L1 immunotherapy efficacy. Porcupine, a membrane-bound o-acyltransferase, is targeted by inhibitors such as LGK974, ETC-1,922,159, CGX1321, and RXC004 and is now in phase I trials [194]. These inhibitors have also been tested in combination with anti-PD-1/PD-L1 antibodies in clinical trials (Table 2). Research shows that chloroquine derivatives improve anti-PD-1 therapy in melanoma by targeting palmitoyl protein thioesterase 1 (PPT1) [195]. Combining PPT1 inhibitors with anti-PD-1 antibodies activates T cells, enhancing tumor immunity [196]. Innovative therapies include HHAT and APT1/2 inhibitors and 2-bromopalmitate (2-BP) in polymer-lipid hybrid nanoparticles (2-BP/CPT-PLNs) that replace anti-PD-L1 antibodies in immune checkpoint blockade, showing potent antitumor effects and improved survival in melanoma models [197,198,199]. Additionally, a novel peptide (CPP-S1) that inhibits PD-L1 palmitoylation and promotes its degradation offers another strategy to enhance immunotherapy efficacy [21].
Dai et al. demonstrated that targeting PD-L1 palmitoylation was more effective than direct targeting [200]. Additionally, Shi et al. created a PROTAC (SP-PROTAC) using an anastomotic peptide targeting the palmitoyl transferase ZDHHC3, which significantly reduced PD-L1 expression in a human cervical cancer cell line [201].
ZDHHC9 palmitoylates cGAS at Cys 404/405, enhancing its activation, while depalmitoylation byLYPLAL1 impairs cGAS function. TargetingLYPLAL1-mediated cGAS depalmitoylation could boost cGAS activation and improve antitumor immunotherapy efficacy [202]. As ZDHHC9 also affects PD-L1 palmitoylation, inhibitingLYPLAL1 might enhance overall immunotherapy outcomes.
Therapeutic promise of other PD-L1/PD-1 posttranslational modifications
Several preclinical treatments targeting PD-1/PD-L1 posttranslational modifications are being developed. HDAC2 inhibitors combined with PD-1 antibodies have been shown to significantly delay tumor growth and improve survival in syngeneic MC38 mouse models [22]. JQ-1, which reduces PD-L1 expression through acetylation, shows potential for treating pancreatic cancer [127]. Pevonedistat, a NEDDylation inhibitor, is undergoing clinical trials for various cancers and may upregulate PD-L1 expression, although its effectiveness is still under study [203, 204]. Zhou et al. discovered a UFSP2 inhibitor that enhances UFMyation, decreases PD-L1 expression, and supports PD-1 blockade [59]. CK2 inhibitors trigger PD-L1 autophagic degradation and enhance antitumor immunotherapy when combined with PD-1 antibodies [61]. Additionally, paclitaxel, which increases MMP-13 in certain cancer cells, shows promise for head and neck cancer treatment when used with anti-PD-1 therapy [65]. These methods represent promising strategies for cancer immunotherapy.
Summary and prospective
In this review, we summarize the PTMs of PD-1/PD-L1 and their regulatory mechanisms and propose new targets for biomarkers and combination therapies to enhance PD-1/PD-L1 blockade in immunotherapy. Despite these advances, many aspects of PD-1/PD-L1 PTMs remain elusive. For diagnosis, PD-L1 glycosylation can obscure antibody binding sites, causing false negatives [177]. Additionally, the degradation of the glycan region of the PD-L1 epitope may lead to a loss of staining on immunohistochemistry [205]. The absolute and effective glycosylation levels may also vary significantly [206]. In treatment contexts, PD-L1 PTMs can contribute to tumor progression. In addition to PD-1/PD-L1 blockade, PTMs are vital for antigen presentation, CAR-T-cell therapy, and vaccine development [207]. Innovations such as multifluorescence resonance energy transfer (multi-FRET) are enhancing PTM research, offering new avenues for advancing tumor immunotherapy [208].
Data availability
No datasets were generated or analysed during the current study.
Abbreviations
- 2F‒Fuc:
-
2-fluoro-L-fucose
- ADCC:
-
Antibody-dependent cytotoxicity
- ADPR:
-
ADP-ribose
- ARF9:
-
ADP-ribosylation factor-6
- BC:
-
Breast cancer
- BCL:
-
B-cell lymphoma
- BL:
-
Burkitt lymphoma
- CISH:
-
Cytokine-inducible SH2 domain-containing protein
- CPT:
-
Camptothecin
- CR:
-
Complete response
- CRC:
-
Colorectal cancer
- CSCC:
-
Cutaneous squamous cell carcinoma
- CTLs:
-
Cytotoxic T lymphocytes
- CxCa:
-
Cervical cancer
- Cys or C:
-
Cysteine
- DHHC:
-
Aspartic acid-histidine-histidine-cysteine
- DOR:
-
Duration of response
- ESCC:
-
Esophageal squamous cell carcinoma
- FASN:
-
Fatty acid synthase
- GBM:
-
Glioblastoma
- GC:
-
Gastric cancer
- Gly:
-
Glycine
- HCC:
-
Hepatocellular carcinoma
- HNSCC:
-
Head and neck squamous cell carcinoma
- HRD:
-
Hyperprogressive disease
- ICB:
-
Immune checkpoint blockade
- ICD:
-
Immunogenic cell death
- KATs:
-
Lysine acetylases
- LUAD:
-
Lung adenocarcinoma
- MD:
-
Molecular dynamics
- MHC:
-
Major histocompatibility complex
- MULTI-FRET:
-
Multifluorescence resonance energy transfer
- NPC:
-
Nasopharyngeal carcinoma
- NSCLC:
-
Non-small cell lung cancer
- NSQ:
-
Nonsquamous
- OC:
-
Ovarian cancer
- ORR:
-
Objective response rate
- OS:
-
Overall survival
- OSCC:
-
Oral squamous cell carcinoma
- PC:
-
Prostate cancer
- PCA:
-
pancreatic cancer
- PD-1:
-
Programmed cell death protein 1
- PD-L1:
-
Programmed death-ligand 1
- PDAC:
-
Pancreatic ductal adenocarcinoma
- PFS:
-
Progression-free survival
- PROTACs:
-
PROteolysis-Targeting Chimeras
- PTM:
-
Posttranslational modification
- SCLC:
-
Small cell lung cancer
- SD:
-
Stable disease
- Ser or S:
-
Seronine
- SQ:
-
Squamous
- TCR:
-
T-cell antigen receptor
- Thr or T:
-
Threonine
- TNBC:
-
Triple-negative breast cancer
- TRAE:
-
Treatment-related adverse events
- Tyr or Y:
-
Tyrosine
- ZAP70:
-
ζchain-related tyrosine kinase 70
References
Weber EW, Maus MV, Mackall CL. The Emerging Landscape of Immune Cell therapies. Cell. 2020;181(1):46–62.
Sun C, Mezzadra R, Schumacher TN. Regulation and function of the PD-L1 checkpoint. Immunity. 2018;48(3):434–52.
Feng C, Zhang L, Chang X, Qin D, Zhang T. Regulation of post-translational modification of PD-L1 and advances in tumor immunotherapy. Front Immunol. 2023;14:1230135.
Yang Y, Zhou T, Chen X, Li J, Pan J, He X, et al. Efficacy, safety, and biomarker analysis of Camrelizumab in previously treated recurrent or metastatic nasopharyngeal carcinoma (CAPTAIN study). J Immunother Cancer. 2021;9(12):e003790.
Zhu H, Bengsch F, Svoronos N, Rutkowski MR, Bitler BG, Allegrezza MJ, et al. BET bromodomain inhibition promotes anti-tumor immunity by suppressing PD-L1 expression. Cell Rep. 2016;16(11):2829–37.
Wang J, Ge J, Wang Y, Xiong F, Guo J, Jiang X, et al. EBV miRNAs BART11 and BART17-3p promote immune escape through the enhancer-mediated transcription of PD-L1. Nat Commun. 2022;13(1):866.
Hsu JM, Li CW, Lai YJ, Hung MC. Posttranslational modifications of PD-L1 and their applications in Cancer Therapy. Cancer Res. 2018;78(22):6349–53.
Ikemizu S, Gilbert RJC, Fennelly JA, Collins AV, Harlos K, Jones EY, et al. Structure and dimerization of a soluble form of B7-1. Immunity. 2000;12(1):51–60.
Zhang X, Schwartz JC, Almo SC, Nathenson SG. Crystal structure of the receptor-binding domain of human B7-2: insights into organization and signaling. Proc Natl Acad Sci U S A. 2003;100(5):2586–91.
Zhang X, Schwartz JC, Guo X, Bhatia S, Cao E, Chen L, et al. Structural and functional analysis of the costimulatory receptor programmed death-1. Immunity. 2004;20(3):337–47.
Sun L, Li CW, Chung EM, Yang R, Kim YS, Park AH, et al. Targeting glycosylated PD-1 induces potent Antitumor immunity. Cancer Res. 2020;80(11):2298–310.
Liu J, Wei L, Hu N, Wang D, Ni J, Zhang S, et al. FBW7-mediated ubiquitination and destruction of PD-1 protein primes sensitivity to anti-PD-1 immunotherapy in non-small cell lung cancer. J Immunother Cancer. 2022;10(9):e005116.
Meng X, Liu X, Guo X, Jiang S, Chen T, Hu Z, et al. FBXO38 mediates PD-1 ubiquitination and regulates anti-tumour immunity of T cells. Nature. 2018;564(7734):130–35.
Patsoukis N, Wang Q, Strauss L, Boussiotis VA. Revisiting the PD-1 pathway. Sci Adv. 2020;6(38):eabd2712.
Tit-oon P, Wonglangka A, Boonkanta K, Ruchirawat M, Fuangthong M, Khongmanee A, et al. Intact mass analysis reveals the novel O-linked glycosylation on the stalk region of PD-1 protein. Sci Rep. 2023;13(1):9631.
Freeman GJ, Long AJ, Iwai Y, Bourque K, Chernova T, Nishimura H, et al. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J Exp Med. 2000;192(7):1027–34.
Cheng X, Veverka V, Radhakrishnan A, Waters LC, Muskett FW, Morgan SH, et al. Structure and interactions of the human programmed cell death 1 receptor. J Biol Chem. 2013;288(17):11771–85.
Benicky J, Sanda M, Brnakova Kennedy Z, Grant OC, Woods RJ, Zwart A, et al. PD-L1 glycosylation and its impact on binding to clinical antibodies. J Proteome Res. 2021;20(1):485–97.
Cha JH, Chan LC, Li CW, Hsu JL, Hung MC. Mechanisms Controlling PD-L1 expression in Cancer. Mol Cell. 2019;76(3):359–70.
Sun K, Zhang X, Lao M, He L, Wang S, Yang H, et al. Targeting leucine-rich repeat serine/threonine-protein kinase 2 sensitizes pancreatic ductal adenocarcinoma to anti-PD-L1 immunotherapy. Mol Ther. 2023;31(10):2929–47.
Yao H, Lan J, Li C, Shi H, Brosseau JP, Wang H, et al. Inhibiting PD-L1 palmitoylation enhances T-cell immune responses against tumours. Nat Biomed Eng. 2019;3(4):306–17.
Gao Y, Nihira NT, Bu X, Chu C, Zhang J, Kolodziejczyk A, et al. Acetylation-dependent regulation of PD-L1 nuclear translocation dictates the efficacy of anti-PD-1 immunotherapy. Nat Cell Biol. 2020;22(9):1064–75.
Huang C, Ren S, Chen Y, Liu A, Wu Q, Jiang T, et al. PD-L1 methylation restricts PD-L1/PD-1 interactions to control cancer immune surveillance. Sci Adv. 2023;9(21):eade4186.
Tu X, Qin B, Zhang Y, Zhang C, Kahila M, Nowsheen S, et al. PD-L1 (B7-H1) competes with the RNA exosome to regulate the DNA damage response and can be targeted to sensitize to Radiation or Chemotherapy. Mol Cell. 2019;74(6):1215–e264.
Singh V, Ram M, Kumar R, Prasad R, Roy BK, Singh KK. Phosphorylation: implications in Cancer. Protein J. 2017;36(1):1–6.
Chan LC, Li CW, Xia W, Hsu JM, Lee HH, Cha JH, et al. IL-6/JAK1 pathway drives PD-L1 Y112 phosphorylation to promote cancer immune evasion. J Clin Invest. 2019;129(8):3324–38.
Li CW, Lim SO, Xia W, Lee HH, Chan LC, Kuo CW, et al. Glycosylation and stabilization of programmed death ligand-1 suppresses T-cell activity. Nat Commun. 2016;7:12632.
Zhang R, Yang Y, Dong W, Lin M, He J, Zhang X, et al. D-mannose facilitates immunotherapy and radiotherapy of triple-negative breast cancer via degradation of PD-L1. Proc Natl Acad Sci U S A. 2022;119(8):e2114851119.
Park HB, Baek KH. E3 ligases and deubiquitinating enzymes regulating the MAPK signaling pathway in cancers. Biochim Biophys Acta Rev Cancer. 2022;1877(3):188736.
Rape M. Ubiquitylation at the crossroads of development and disease. Nat Rev Mol Cell Biol. 2018;19(1):59–70.
Popovic D, Vucic D, Dikic I. Ubiquitination in disease pathogenesis and treatment. Nat Med. 2014;20(11):1242–53.
Chen N, Fang W, Zhan J, Hong S, Tang Y, Kang S, et al. Upregulation of PD-L1 by EGFR activation mediates the Immune escape in EGFR-Driven NSCLC: implication for Optional Immune targeted therapy for NSCLC patients with EGFR Mutation. J Thorac Oncol. 2015;10(6):910–23.
Horita H, Law A, Hong S, Middleton K. Identifying Regulatory Posttranslational modifications of PD-L1: a focus on Monoubiquitinaton. Neoplasia. 2017;19(4):346–53.
Wang S, Xu L, Che X, Li C, Xu L, Hou K, et al. E3 ubiquitin ligases Cbl-b and c-Cbl downregulate PD-L1 in EGFR wild-type non-small cell lung cancer. FEBS Lett. 2018;592(4):621–30.
Zhang J, Bu X, Wang H, Zhu Y, Geng Y, Nihira NT, et al. Cyclin D-CDK4 kinase destabilizes PD-L1 via cullin 3-SPOP to control cancer immune surveillance. Nature. 2018;553(7686):91–5.
Gao Y, Zou T, Xu P, Wang Y, Jiang Y, Chen YX, et al. Fusobacterium nucleatum stimulates cell proliferation and promotes PD-L1 expression via IFIT1-related signal in colorectal cancer. Neoplasia. 2023;35:100850.
Zhang Y, Zeng L, Wang M, Yang Z, Zhang H, Gao L, et al. RIG-I promotes immune evasion of colon cancer by modulating PD-L1 ubiquitination. J Immunother Cancer. 2023;11(9):e007313.
Jiang C, He L, Xiao S, Wu W, Zhao Q, Liu F. E3 ubiquitin ligase RNF125 suppresses Immune escape in Head and Neck squamous cell carcinoma by regulating PD-L1 expression. Mol Biotechnol. 2023;65(6):891–903.
Shi C, Wang Y, Wu M, Chen Y, Liu F, Shen Z, et al. Promoting anti-tumor immunity by targeting TMUB1 to modulate PD-L1 polyubiquitination and glycosylation. Nat Commun. 2022;13(1):6951.
Yi J, Tavana O, Li H, Wang D, Baer RJ, Gu W. Targeting USP2 regulation of VPRBP-mediated degradation of p53 and PD-L1 for cancer therapy. Nat Commun. 2023;14(1):1941.
Zhang H, Chen H, Yin S, Fan L, Jin C, Zhao C, et al. Docosahexaenoic acid reverses PD-L1-mediated immune suppression by accelerating its ubiquitin-proteasome degradation. J Nutr Biochem. 2023;112:109186.
Lin CY, Huang KY, Kao SH, Lin MS, Lin CC, Yang SC, et al. Small-molecule PIK-93 modulates the tumor microenvironment to improve immune checkpoint blockade response. Sci Adv. 2023;9(14):eade9944.
Zou J, Xia H, Zhang C, Xu H, Tang Q, Zhu G, et al. Casp8 acts through A20 to inhibit PD-L1 expression: the mechanism and its implication in immunotherapy. Cancer Sci. 2021;112(7):2664–78.
Guo W, Ma J, Guo S, Wang H, Wang S, Shi Q, et al. A20 regulates the therapeutic effect of anti-PD-1 immunotherapy in melanoma. J Immunother Cancer. 2020;8(2):e001866.
Mezzadra R, Sun C, Jae LT, Gomez-Eerland R, de Vries E, Wu W, et al. Identification of CMTM6 and CMTM4 as PD-L1 protein regulators. Nature. 2017;549(7670):106–10.
Burr ML, Sparbier CE, Chan Y-C, Williamson JC, Woods K, Beavis PA, et al. CMTM6 maintains the expression of PD-L1 and regulates anti-tumour immunity. Nature. 2017;549(7670):101–05.
Wang H, Yao H, Li C, Shi H, Lan J, Li Z, et al. HIP1R targets PD-L1 to lysosomal degradation to alter T cell–mediated cytotoxicity. Nat Chem Biol. 2019;15:42–50.
Ding L, Chen X, Zhang W, Dai X, Guo H, Pan X, et al. Canagliflozin primes antitumor immunity by triggering PD-L1 degradation in endocytic recycling. J Clin Invest. 2023;133(1):e154754.
Yu X, Li W, Liu H, Wang X, Coarfa C, Cheng C, et al. PD-L1 translocation to the plasma membrane enables tumor immune evasion through MIB2 ubiquitination. J Clin Invest. 2023;133(3):e160456.
Dong LF, Chen FF, Fan YF, Zhang K, Chen HH. circ-0000512 inhibits PD-L1 ubiquitination through sponging miR-622/CMTM6 axis to promote triple-negative breast cancer and immune escape. J Immunother Cancer. 2023;11(6):e005461.
Li J, Dong X, Kong X, Wang Y, Li Y, Tong Y, et al. Circular RNA hsa_circ_0067842 facilitates tumor metastasis and immune escape in breast cancer through HuR/CMTM6/PD-L1 axis. Biol Direct. 2023;18(1):48.
Le NT, Xue M, Castelnoble LA, Jackson CJ. The dual personalities of matrix metalloproteinases in inflammation. Front Biosci. 2007;12:1475–87.
Egeblad M, Werb Z. New functions for the matrix metalloproteinases in cancer progression. Nat Rev Cancer. 2002;2(3):161–74.
Dezutter-Dambuyant C, Durand I, Alberti L, Bendriss-Vermare N, Valladeau-Guilemond J, Duc A, et al. A novel regulation of PD-1 ligands on mesenchymal stromal cells through MMP-mediated proteolytic cleavage. Oncoimmunology. 2015;5(3):e1091146.
Hira-Miyazawa M, Nakamura H, Hirai M, Kobayashi Y, Kitahara H, Bou-Gharios G, et al. Regulation of programmed-death ligand in the human head and neck squamous cell carcinoma microenvironment is mediated through matrix metalloproteinase-mediated proteolytic cleavage. Int J Oncol. 2018;52(2):379–88.
Rowswell-Turner RB, Singh RK, Urh A, Yano N, Kim KK, Khazan N, et al. HE4 overexpression by Ovarian Cancer promotes a suppressive Tumor Immune Microenvironment and enhanced tumor and macrophage PD-L1 expression. J Immunol. 2021;206(10):2478–88.
Raposo AE, Piller SC. Protein arginine methylation: an emerging regulator of the cell cycle. Cell Div. 2018;13:3.
Dai X, Ren T, Zhang Y, Nan N. Methylation multiplicity and its clinical values in cancer. Expert Rev Mol Med. 2021;23:e2.
Zhou J, Ma X, He X, Chen B, Yuan J, Jin Z, et al. Dysregulation of PD-L1 by UFMylation imparts tumor immune evasion and identified as a potential therapeutic target. Proc Natl Acad Sci U S A. 2023;120(11):e2215732120.
Tatsumi K, Sou Y, Tada N, Nakamura E, Iemura S, Natsume T, et al. A novel type of E3 ligase for the Ufm1 conjugation system. J Biol Chem. 2010;285(8):5417–27.
Malakhov MP, Kim KI, Malakhova OA, Jacobs BS, Borden EC, Zhang DE. High-throughput immunoblotting. Ubiquitiin-like protein ISG15 modifies key regulators of signal transduction. J Biol Chem. 2003;278(19):16608–13.
Qu T, Zhang W, Yan C, Ren D, Wang Y, Guo Y, et al. ISG15 targets glycosylated PD-L1 and promotes its degradation to enhance antitumor immune effects in lung adenocarcinoma. J Transl Med. 2023;21(1):341.
Enchev RI, Schulman BA, Peter M. Protein neddylation: beyond cullin-RING ligases. Nat Rev Mol Cell Biol. 2015;16(1):30–44.
Wang X, Chen C, Vuong D, Rodriguez-Rodriguez S, Lam V, Roleder C, et al. Pharmacologic targeting of Nedd8-activating enzyme reinvigorates T-cell responses in lymphoid neoplasia. Leukemia. 2023;37(6):1324–35.
Zhou S, Zhao X, Yang Z, Yang R, Chen C, Zhao K, et al. Neddylation inhibition upregulates PD-L1 expression and enhances the efficacy of immune checkpoint blockade in glioblastoma. Int J Cancer. 2019;145(3):763–74.
Zhang S, You X, Xu T, Chen Q, Li H, Dou L, et al. PD-L1 induction via the MEK-JNK-AP1 axis by a neddylation inhibitor promotes cancer-associated immunosuppression. Cell Death Dis. 2022;13(10):844.
Byun JK, Park M, Lee S, Yun JW, Lee J, Kim JS, et al. Inhibition of glutamine utilization synergizes with Immune checkpoint inhibitor to promote Antitumor Immunity. Mol Cell. 2020;80(4):592–e6068.
Mizushima N, Komatsu M. Autophagy: renovation of cells and tissues. Cell. 2011;147(4):728–41.
Gou Q, Chen H, Chen M, Shi J, Jin J, Liu Q et al. Inhibition of CK2/ING4 pathway facilitates Non-small Cell Lung Cancer Immunotherapy. Adv Sci (Weinh). 2023;e2304068.
Antao AM, Tyagi A, Kim KS, Ramakrishna S. Advances in deubiquitinating enzyme inhibition and applications in Cancer therapeutics. Cancers (Basel). 2020;12(6):1579.
Mennerich D, Kubaichuk K, Kietzmann T. DUBs, Hypoxia, and Cancer. Trends Cancer. 2019;5(10):632–53.
Jumpertz S, Hennes T, Asare Y, Schütz AK, Bernhagen J. CSN5/JAB1 suppresses the WNT inhibitor DKK1 in colorectal cancer cells. Cell Signal. 2017;34:38–46.
Lim SO, Li CW, Xia W, Cha JH, Chan LC, Wu Y, et al. Deubiquitination and stabilization of PD-L1 by CSN5. Cancer Cell. 2016;30(6):925–39.
Wang Y, Lu H, Fang C, Xu J. Palmitoylation as a Signal for Delivery. Adv Exp Med Biol. 2020;1248:399–424.
Ma Y, Xia P, Wang Z, Xu J, Zhang L, Jiang Y. PDIA6 promotes pancreatic cancer progression and immune escape through CSN5-mediated deubiquitination of β-catenin and PD-L1. Neoplasia. 2021;23(9):912–28.
Huang X, Zhang Q, Lou Y, Wang J, Zhao X, Wang L, et al. USP22 Deubiquitinates CD274 to suppress anticancer immunity. Cancer Immunol Res. 2019;7(10):1580–90.
Wang Y, Sun Q, Mu N, Sun X, Wang Y, Fan S, et al. The deubiquitinase USP22 regulates PD-L1 degradation in human cancer cells. Cell Commun Signal. 2020;18(1):112.
Wu J, Guo W, Wen D, Hou G, Zhou A, Wu W. Deubiquitination and stabilization of programmed cell death ligand 1 by ubiquitin-specific peptidase 9, X-linked in oral squamous cell carcinoma. Cancer Med. 2018;7(8):4004–11.
Kuang Z, Liu X, Zhang N, Dong J, Sun C, Yin, et al. USP2 promotes tumor immune evasion via deubiquitination and stabilization of PD-L1. Cell Death Differ. 2023;30(10):2249–64.
Li J, Xiao X, Ou Y, Cao L, Guo M, Qi C, et al. USP51/PD-L1/ITGB1-deployed juxtacrine interaction plays a cell-intrinsic role in promoting chemoresistant phenotypes in non-small cell lung cancer. Cancer Commun (Lond). 2023;43(7):765–87.
Yang H, Zhang X, Lao M, Sun K, He L, Xu J, et al. Targeting ubiquitin-specific protease 8 sensitizes anti-programmed death-ligand 1 immunotherapy of pancreatic cancer. Cell Death Differ. 2023;30(2):560–75.
Yu ZZ, Liu YY, Zhu W, Xiao D, Huang W, Lu SS, et al. ANXA1-derived peptide for targeting PD-L1 degradation inhibits tumor immune evasion in multiple cancers. J Immunother Cancer. 2023;11(3):e006345.
Mao R, Tan X, Xiao Y, Wang X, Wei Z, Wang J, et al. Ubiquitin C-terminal hydrolase L1 promotes expression of programmed cell death-ligand 1 in non-small-cell lung cancer cells. Cancer Sci. 2020;111(9):3174–83.
Zhu D, Xu R, Huang X, Tang Z, Tian Y, Zhang J, et al. Deubiquitinating enzyme OTUB1 promotes cancer cell immunosuppression via preventing ER-associated degradation of immune checkpoint protein PD-L1. Cell Death Differ. 2021;28(6):1773–89.
Wang Q, Li G, Ma X, Liu L, Liu J, Yin Y, et al. LncRNA TINCR impairs the efficacy of immunotherapy against breast cancer by recruiting DNMT1 and downregulating MiR-199a-5p via the STAT1-TINCR-USP20-PD-L1 axis. Cell Death Dis. 2023;14(2):76.
Zheng R, Gao F, Mao Z, Xiao Y, Yuan L, Huang Z, et al. LncRNA BCCE4 genetically enhances the PD-L1/PD-1 Interaction in Smoking-related bladder Cancer by modulating mir-328-3p-USP18 signaling. Adv Sci (Weinh). 2023;10(30):e2303473.
Sahin U, de Thé H, Lallemand-Breitenbach V. Sumoylation in Physiology, Pathology and Therapy. Cells. 2022;11(5):814.
Ma X, Jia S, Wang G, Liang M, Guo T, Du H, et al. TRIM28 promotes the escape of gastric cancer cells from immune surveillance by increasing PD-L1 abundance. Signal Transduct Target Ther. 2023;8(1):246.
Wang J, Wang Y, Jiang X, Xu M, Wang M, Wang R, et al. Unleashing the power of immune checkpoints: post-translational modification of novel molecules and clinical applications. Cancer Lett Published Online Febr. 2024;22. https://doi.org/10.1016/j.canlet.2024.216758.
Li CW, Lim SO, Chung EM, Kim YS, Park AH, Yao J, et al. Eradication of Triple-negative breast Cancer cells by targeting glycosylated PD-L1. Cancer Cell. 2018;33(2):187–e20110.
Maher CM, Thomas JD, Haas DA, Longen CG, Oyer HM, Tong JY, et al. Small-molecule Sigma1 modulator induces autophagic degradation of PD-L1. Mol Cancer Res. 2018;16(2):243–55.
D’Arrigo P, Russo M, Rea A, Tufano M, Guadagno E, De Del Basso ML, et al. A regulatory role for the co-chaperone FKBP51s in PD-L1 expression in glioma. Oncotarget. 2017;8(40):68291–304.
Liu X, Zhang Y, Han Y, Lu W, Yang J, Tian J, et al. Overexpression of GLT1D1 induces immunosuppression through glycosylation of PD-L1 and predicts poor prognosis in B-cell lymphoma. Mol Oncol. 2020;14(5):1028–44.
Zhang Y, Zhang N, Song W, Yousuf S, Li W. Ablation of the GDP-fucose transporter suppresses lung cancer cell proliferation and migration by reducing expression of PD-L1. J Cancer. 2023;14(17):3295–308.
Cui Y, Li J, Zhang P, Yin D, Wang Z, Dai J, et al. B4GALT1 promotes immune escape by regulating the expression of PD-L1 at multiple levels in lung adenocarcinoma. J Exp Clin Cancer Res. 2023;42(1):146.
Hsu JM, Xia W, Hsu YH, Chan LC, Yu WH, Cha JH, et al. STT3-dependent PD-L1 accumulation on cancer stem cells promotes immune evasion. Nat Commun. 2018;9(1):1908. https://doi.org/10.1038/s41467-018-04313-6. Published 2018 May 15.
Paul S, Das K, Ghosh A, Chatterjee A, Bhoumick A, Basu A et al. Coagulation factor VIIa enhances programmed death-ligand 1 expression and its stability in breast cancer cells to promote breast cancer immune evasion. J Thromb Haemost. 2023;S1538-7836(23)00632-3.
Ma XM, Luo YF, Zeng FF, Su C, Liu X, Li XP, et al. TGF-β1-Mediated PD-L1 glycosylation contributes to Immune escape via c-Jun/STT3A pathway in nasopharyngeal carcinoma. Front Oncol. 2022;12:815437.
Wang D, Wu S, He J, Sun L, Zhu H, Zhang Y, et al. FAT4 overexpression promotes antitumor immunity by regulating the β-catenin/STT3/PD-L1 axis in cervical cancer. J Exp Clin Cancer Res. 2023;42(1):222.
Zeng K, Zeng Y, Zhan H, Zhan Z, Wang L, Xie Y, et al. SEC61G assists EGFR-amplified glioblastoma to evade immune elimination. Proc Natl Acad Sci U S A. 2023;120(32):e2303400120.
Duan X, Xie Y, Yu J, Hu X, Liu Z, Li N, et al. MCT4/Lactate promotes PD-L1 glycosylation in Triple-negative breast Cancer cells. J Oncol. 2022;2022:3659714.
Erlichman N, Meshel T, Baram T, Abu Raiya A, Horvitz T, Ben-Yaakov H, et al. The Cell-Autonomous Pro-metastatic activities of PD-L1 in breast Cancer are regulated by N-Linked glycosylation-dependent activation of STAT3 and STAT1. Cells. 2023;12(19):2338.
Gong B, Kiyotani K, Sakata S, Nagano S, Kumehara S, Baba S, et al. Secreted PD-L1 variants mediate resistance to PD-L1 blockade therapy in non-small cell lung cancer. J Exp Med. 2019;216(4):982–1000.
Sagawa R, Sakata S, Gong B, Seto Y, Takemoto A, Takagi S, et al. Soluble PD-L1 works as a decoy in lung cancer immunotherapy via alternative polyadenylation. JCI Insight. 2022;7(1):e153323.
Yu Y, Wang Z, Zheng Q, Li J. GALNT2/14 overexpression correlate with prognosis and methylation: potential therapeutic targets for lung adenocarcinoma. Gene. 2021;790:145689.
Chen W, Saxton B, Tessema M, Belinsky SA. Inhibition of GFAT1 in lung cancer cells destabilizes PD-L1 protein. Carcinogenesis. 2021;42(9):1171–78.
Hetz C, Zhang K, Kaufman RJ. Mechanisms, regulation and functions of the unfolded protein response. Nat Rev Mol Cell Biol. 2020;21(8):421–38.
Tanaka T, Kutomi G, Kajiwara T, Kukita K, Kochin V, Kanaseki T, et al. Cancer-associated oxidoreductase ERO1-α promotes immune escape through up-regulation of PD-L1 in human breast cancer. Oncotarget. 2017;8(15):24706–18.
Chen Y, He J, Chen R, Wang Z, Dai Z, Liang X, et al. Pan-cancer Analysis of the immunological role of PDIA5: a potential target for Immunotherapy. Front Immunol. 2022;13:881722.
Ko PJ, Dixon SJ. Protein palmitoylation and cancer. EMBO Rep. 2018;19(10):e46666.
Das T, Yount JS, Hang HC. Protein S-palmitoylation in immunity. Open Biol. 2021;11(3):200411.
Runkle KB, Kharbanda A, Stypulkowski E, Cao XJ, Wang W, Garcia BA et al. Inhibition of DHHC20-Mediated EGFR Palmitoylation creates a dependence on EGFR Signaling. Mol Cell 2016,62(3):385–96.
Yang Y, Hsu JM, Sun L, Chan LC, Li CW, Hsu JL, et al. Palmitoylation stabilizes PD-L1 to promote breast tumor growth. Cell Res. 2019;29(1):83–6.
Li Z, Jiang D, Liu F, Li Y. Involvement of ZDHHC9 in lung adenocarcinoma: regulation of PD-L1 stability via palmitoylation. Vitro Cell Dev Biol Anim. 2023;59(3):193–203.
Lin Z, Huang K, Guo H, Jia M, Sun Q, Chen X, et al. Targeting ZDHHC9 potentiates anti-programmed death-ligand 1 immunotherapy of pancreatic cancer by modifying the tumor microenvironment. Biomed Pharmacother. 2023;161:114567.
Lin Z, Lv Z, Liu X, Huang K. Palmitoyl transferases act as novel drug targets for pancreatic cancer. J Transl Med. 2023;21(1):249.
Shahid M, Kim M, Jin P, Zhou B, Wang Y, Yang W, et al. S-Palmitoylation as a Functional Regulator of Proteins Associated with Cisplatin Resistance in bladder Cancer. Int J Biol Sci. 2020;16(14):2490–505.
Zhang Z, Chen Y, Fang L, Zhao J, Deng S. The involvement of high succinylation modification in the development of prostate cancer. Front Oncol. 2022;12:1034605.
Yang Y, Gibson GE. Succinylation Links metabolism to protein functions. Neurochem Res. 2019;44(10):2346–59.
Mu R, Ma Z, Lu C, Wang H, Cheng X, Tuo B, et al. Role of succinylation modification in thyroid cancer and breast cancer. Am J Cancer Res. 2021;11(10):4683–99.
Dai X, Zhou Y, Han F, Li J. Succinylation and redox status in cancer cells. Front Oncol. 2022;12:1081712.
Yang H, Zou X, Yang S, Zhang A, Li N, Ma Z. Identification of lactylation related model to predict prognostic, tumor infiltrating immunocytes and response of immunotherapy in gastric cancer. Front Immunol. 2023;14:1149989.
Huang ZW, Zhang XN, Zhang L, Liu LL, Zhang JW, Sun YX, et al. STAT5 promotes PD-L1 expression by facilitating histone lactylation to drive immunosuppression in acute myeloid leukemia. Signal Transduct Target Ther. 2023;8(1):391.
Shvedunova M, Akhtar A. Modulation of cellular processes by histone and non-histone protein acetylation. Nat Rev Mol Cell Biol. 2022;23(5):329–49.
Narita T, Weinert BT, Choudhary C. Functions and mechanisms of non-histone protein acetylation. Nat Rev Mol Cell Biol. 2019;20(3):156–74.
Yu J, Zhuang A, Gu X, Hua Y, Yang L, Ge S, et al. Nuclear PD-L1 promotes EGR1-mediated angiogenesis and accelerates tumorigenesis. Cell Discov. 2023;9(1):33.
Romeo MA, Gilardini Montani MS, Santarelli R, Benedetti R, Arena A, Cirone M. Acetylation increases expression, interaction with TRAPPC4 and surface localization of PD-L1. Discov Oncol. 2023;14(1):152.
Cohen MS, Chang P. Insights into the biogenesis, function, and regulation of ADP-ribosylation. Nat Chem Biol. 2018;14(3):236–43.
Hashimoto S, Furukawa S, Hashimoto A, Tsutaho A, Fukao A, Sakamura Y, et al. ARF6 and AMAP1 are major targets of KRAS and TP53 mutations to promote invasion, PD-L1 dynamics, and immune evasion of pancreatic cancer. Proc Natl Acad Sci U S A. 2019;116(35):17450–59.
Di Girolamo M, Fabrizio G, Scarpa ES, Di Paola S. NAD+-dependent enzymes at the endoplasmic reticulum. Curr Top Med Chem. 2013;13(23):3001–10.
Nickel W, Huber LA, Kahn RA, Kipper N, Barthel A, Fasshauer D, et al. ADP ribosylation factor and a 14-kD polypeptide are associated with heparan sulfate-carrying post-trans-golgi network secretory vesicles in rat hepatocytes. J Cell Biol. 1994;125(4):721–32.
D’Souza-Schorey C, van Donselaar E, Hsu VW, Yang C, Stahl PD, Peters PJ. ARF6 targets recycling vesicles to the plasma membrane: insights from an ultrastructural investigation. J Cell Biol. 1998;140(3):603–16.
Hui E, Cheung J, Zhu J, Su X, Taylor MJ, Wallweber HA, et al. T cell costimulatory receptor CD28 is a primary target for PD-1-mediated inhibition. Science. 2017;355(6332):1428–33.
Celis-Gutierrez J, Blattmann P, Zhai Y, Jarmuzynski N, Ruminski K, Grégoire C, et al. Quantitative interactomics in primary T cells provides a rationale for concomitant PD-1 and BTLA Coinhibitor Blockade in Cancer Immunotherapy. Cell Rep. 2019;27(11):3315–e307.
Xiao X, Shi J, He C, Bu X, Sun Y, Gao M, et al. ERK and USP5 govern PD-1 homeostasis via deubiquitination to modulate tumor immunotherapy. Nat Commun. 2023;14(1):2859.
Lv J, Qin L, Zhao R, Wu D, Wu Z, Zheng D, et al. Disruption of CISH promotes the antitumor activity of human T cells and decreases PD-1 expression levels. Mol Ther Oncolytics. 2022;28:46–58.
Lyle C, Richards S, Yasuda K, Napoleon MA, Walker J, Arinze N, et al. c-Cbl targets PD-1 in immune cells for proteasomal degradation and modulates colorectal tumor growth. Sci Rep. 2019;9(1):20257.
Wu Z, Cao Z, Yao H, Yan X, Xu W, Zhang M, et al. Coupled deglycosylation-ubiquitination cascade in regulating PD-1 degradation by MDM2. Cell Rep. 2023;42(7):112693.
Lee TA, Tsai EY, Liu SH, Hsu Hung SD, Chang SJ, et al. Post-translational modification of PD-1: potential targets for Cancer Immunotherapy. Cancer Res. 2024;84(6):800–7.
Okada M, Chikuma S, Kondo T, Hibino S, Machiyama H, Yokosuka T, et al. Blockage of Core Fucosylation reduces cell-surface expression of PD-1 and promotes Anti-tumor Immune responses of T cells. Cell Rep. 2017;20(5):1017–28.
Yao H, Li C, He F, Song T, Brosseau JP, Wang H, et al. A peptidic inhibitor for PD-1 palmitoylation targets its expression and functions. RSC Chem Biol. 2020;2(1):192–205.
Sugiyama E, Togashi Y, Takeuchi Y, Shinya S, Tada Y, Kataoka K, et al. Blockade of EGFR improves responsiveness to PD-1 blockade in EGFR-mutated non-small cell lung cancer. Sci Immunol. 2020;5(43):eaav3937.
Oxnard GR, Yang JC-H, Yu H, Kim S-W, Saka H, Horn L, et al. TATTON: a multi-arm, phase Ib trial of osimertinib combined with selumetinib, savolitinib, or durvalumab in EGFR-mutant lung cancer. Ann Oncol. 2020;31(4):507–16.
Yang JC, Gadgeel SM, Sequist LV, Wu CL, Papadimitrakopoulou VA, Su WC, et al. Pembrolizumab in Combination with Erlotinib or Gefitinib as First-Line Therapy for Advanced NSCLC with sensitizing EGFR Mutation. J Thorac Oncol. 2019;14(3):553–59.
Sacco AG, Chen R, Worden FP, Wong DJL, Adkins D, Swiecicki P, et al. Pembrolizumab plus Cetuximab in patients with recurrent or metastatic head and neck squamous cell carcinoma: an open-label, multi-arm, non-randomised, multicentre, phase 2 trial. Lancet Oncol. 2021;22(6):883–92.
Bonomo P, Desideri I, Loi M, Mangoni M, Sottili M, Marrazzo L, et al. Anti PD-L1 DUrvalumab combined with Cetuximab and RadiOtherapy in locally advanced squamous cell carcinoma of the head and neck: a phase I/II study (DUCRO). Clin Transl Radiat Oncol. 2018;9:42–7.
Gillison ML, Ferris RL, Harris J, Colevas AD, Mell LK, Kong C, et al. Safety of Nivolumab added to Chemoradiation Therapy platforms for Intermediate and High-Risk Locoregionally Advanced Head and Neck Squamous Cell Carcinoma: RTOG Foundation 3504. Int J Radiat Oncol Biol Phys. 2023;115(4):847–60.
Jiao S, Xia W, Yamaguchi H, Wei Y, Chen MK, Hsu JM, et al. PARP inhibitor upregulates PD-L1 expression and enhances Cancer-Associated Immunosuppression. Clin Cancer Res. 2017;23(14):3711–20.
Yarchoan M, Powderly JD, Bastos BR, Karasic TB, Crysler OV, Munster PN, et al. A phase 1 study of TPST-1120 as a single agent and in combination with nivolumab in subjects with advanced solid tumors. J Clin Oncol. 2022;40(16suppl):3005–3005.
Tempest Therapeutics. TPST-1120 Randomized Data in First-Line HCC. Tempest Therapeutics. Published online October 11. 2023. https://ir.tempesttx.com/static-files/c2c921e9-1435-4f8a-b853-793da79f6765.
Yap TA, Bardia A, Dvorkin M, Galsky MD, Beck JT, Wise DR, et al. Avelumab Plus Talazoparib in patients with Advanced Solid tumors: the JAVELIN PARP Medley Nonrandomized Controlled Trial. JAMA Oncol. 2023;9(1):40–50.
Konstantinopoulos PA, Waggoner S, Vidal GA, Mita M, Moroney JW, Holloway R, et al. Single-arm phases 1 and 2 trial of Niraparib in Combination with Pembrolizumab in patients with recurrent platinum-resistant ovarian carcinoma. JAMA Oncol. 2019;5(8):1141–49.
Post CCB, Westermann AM, Boere IA, Witteveen PO, Ottevanger PB, Sonke GS, et al. Efficacy and safety of durvalumab with olaparib in metastatic or recurrent endometrial cancer (phase II DOMEC trial). Gynecol Oncol. 2022;165(2):223–29.
Domchek SM, Postel-Vinay S, Im S-A, Park YH, Delord J-P, Italiano A, et al. Olaparib and durvalumab in patients with germline BRCA-mutated metastatic breast cancer (MEDIOLA): an open-label, multicentre, phase 1/2, basket study. Lancet Oncol. 2020;21(9):1155–64.
Drew Y, Kim JW, Penson RT, O’Malley DM, Parkinson C, Roxburgh P, et al. Olaparib plus Durvalumab, with or without Bevacizumab, as treatment in PARP Inhibitor-Naïve platinum-sensitive relapsed ovarian Cancer: a phase II Multi-cohort Study. Clin Cancer Res. 2024;30(1):50–62.
Friedlander M, Meniawy T, Markman B, Mileshkin L, Harnett P, Millward M, et al. Pamiparib in combination with tislelizumab in patients with advanced solid tumours: results from the dose-escalation stage of a multicentre, open-label, phase 1a/b trial. Lancet Oncol. 2019;20(9):1306–15.
Liu J, Gaillard S, Hendrickson AW, Moroney J, Yeku O, Diver E, et al. An open-label phase II study of dostarlimab (TSR-042), bevacizumab (bev), and niraparib combination in patients (pts) with platinum-resistant ovarian cancer (PROC): cohort a of the OPAL trial. Gynecol Oncol. 2021;162(S1):S17–8.
Cha JH, Yang WH, Xia W, Wei Y, Chan LC, Lim SO, et al. Metformin promotes Antitumor Immunity via Endoplasmic-Reticulum-Associated degradation of PD-L1. Mol Cell. 2018;71(4):606–. – 20.e7.
Zandberg DP, Menk AV, Velez M, Normolle D, DePeaux K, Liu A, et al. Tumor hypoxia is associated with resistance to PD-1 blockade in squamous cell carcinoma of the head and neck. J Immunother Cancer. 2021;9(5):e002088.
Békés M, Langley DR, Crews CM. PROTAC targeted protein degraders: the past is prologue. Nat Rev Drug Discov. 2022;21(3):181–200.
Paiva SL, Crews CM. Targeted protein degradation: elements of PROTAC design. Curr Opin Chem Biol. 2019;50:111–19.
Wang Y, Zhou Y, Cao S, Sun Y, Dong Z, Li C, et al. In vitro and in vivo degradation of programmed cell death ligand 1 (PD-L1) by a proteolysis targeting chimera (PROTAC). Bioorg Chem. 2021;111:104833.
Cheng B, Ren Y, Cao H, Chen J. Discovery of novel resorcinol diphenyl ether-based PROTAC-like molecules as dual inhibitors and degraders of PD-L1. Eur J Med Chem. 2020;199:112377.
Cotton AD, Nguyen DP, Gramespacher JA, Seiple IB, Wells JA. Development of antibody-based PROTACs for the degradation of the cell-surface Immune checkpoint protein PD-L1. J Am Chem Soc. 2021;143(2):593–98.
Su W, Tan M, Wang Z, Zhang J, Huang W, Song H, et al. Targeted degradation of PD-L1 and activation of the STING pathway by Carbon-dot-based PROTACs for Cancer Immunotherapy. Angew Chem Int Ed Engl. 2023;62(11):e202218128.
Sun R, Meng Z, Lee H, Offringa R, Niehrs C. ROTACs leverage signaling-incompetent R-spondin for targeted protein degradation. Cell Chem Biol. 2023;30(7):739–e528.
Li S, Chen T, Liu J, Zhang H, Li J, Wang Z, et al. PROTACs: novel tools for improving immunotherapy in cancer. Cancer Lett. 2023;560:216128.
Solomon B, Callejo A, Bar J, Berchem G, Bazhenova L, Saintigny P, et al. A WIN Consortium phase I study exploring avelumab, palbociclib, and axitinib in advanced non-small cell lung cancer. Cancer Med. 2022;11(14):2790–800.
Mayer EL, Ren Y, Wagle N, Mahtani R, Ma C, DeMichele A, et al. PACE: a randomized phase II study of Fulvestrant, Palbociclib, and Avelumab after Progression on cyclin-dependent kinase 4/6 inhibitor and aromatase inhibitor for hormone Receptor-Positive/Human epidermal growth factor receptor-negative metastatic breast Cancer. J Clin Oncol Published Online March. 2024;21. https://doi.org/10.1200/JCO.23.01940.
Zhang Y, Song J, Zhou Y, Jia H, Zhou T, Sun Y, et al. Discovery of selective and potent USP22 inhibitors via structure-based virtual screening and bioassays exerting anti-tumor activity. Bioorg Chem. 2023;141:106842.
Liu K, Tan S, Jin W, Guan J, Wang Q, Sun H, et al. N-glycosylation of PD-1 promotes binding of camrelizumab. EMBO Rep. 2020;21(12):e51444.
Jiang M, Liu M, Liu G, Ma J, Zhang L, Wang S. Advances in the structural characterization of complexes of therapeutic antibodies with PD-1 or PD-L1. MAbs. 2023;15(1):2236740.
Chen D, Tan S, Zhang H, Wang H, He W, Shi R, et al. The FG Loop of PD-1 serves as a Hotspot for therapeutic monoclonal antibodies in Tumor Immune Checkpoint Therapy. iScience. 2019;14:113–24.
Lu D, Xu Z, Zhang D, Jiang M, Liu K, He J, et al. PD-1 N58-Glycosylation-dependent binding of monoclonal antibody cemiplimab for Immune Checkpoint Therapy. Front Immunol. 2022;13:826045.
Tan S, Zhang H, Chai Y, Song H, Tong Z, Wang Q, et al. An unexpected N-terminal loop in PD-1 dominates binding by Nivolumab. Nat Commun. 2017;8:14369.
Ha JY, Chun K-J, Ko S, Lee HW, Hwang OK, Lim CS, et al. Glycan-controlled human PD-1 variants displaying broad-spectrum high binding to PD-1 Ligands Potentiate T cell. Mol Pharm. 2023;20(4):2170–80.
Lee HH, Wang YN, Xia W, Chen CH, Rau KM, Ye L, et al. Removal of N-Linked glycosylation enhances PD-L1 detection and predicts Anti-PD-1/PD-L1 therapeutic efficacy. Cancer Cell. 2019;36(2):168–e784.
Do KT, Chow LQM, Reckamp K, Sanborn RE, Burris H, Robert F, et al. First-In-Human, First-In-Class, Phase I trial of the Fucosylation inhibitor SGN-2FF in patients with Advanced Solid tumors. Oncologist. 2021;26(11):925–e1918.
Pijnenborg JFA, Visser EA, Noga M, et al. Cellular Fucosylation inhibitors based on Fluorinated Fucose-1-phosphates*. Chemistry. 2021;27(12):4022–27.
Pijnenborg JFA, Visser EA, Noga M, Rossing E, Veizaj R, Lefeber DJ, et al. Fluorinated rhamnosides inhibit cellular fucosylation. Nat Commun. 2021;12(1):7024.
Ren X, Cheng Z, He J, Yao X, Liu Y, Cai K, et al. Inhibition of glycolysis-driven immunosuppression with a nano-assembly enhances response to immune checkpoint blockade therapy in triple negative breast cancer. Nat Commun. 2023;14(1):7021.
Mao C, Li J, Feng L, Gao W. Beyond antibody fucosylation: α-(1,6)-fucosyltransferase (Fut8) as a potential new therapeutic target for cancer immunotherapy. Antib Ther. 2023;6(2):87–96.
Rudin CM, Awad MM, Navarro A, Gottfried M, Peters S, Csőszi T, et al. Pembrolizumab or Placebo Plus Etoposide and Platinum as First-Line therapy for extensive-stage small-cell Lung Cancer: Randomized, Double-Blind, phase III KEYNOTE-604 study. J Clin Oncol. 2020;38(21):2369–79.
Kim HR, Awad MM, Navarro A, Gottfried M, Peters S, Csőszi T, et al. Patient-reported Health-Related Quality of Life in KEYNOTE-604: Pembrolizumab or Placebo added to Etoposide and Platinum as First-Line therapy for extensive-stage SCLC. JTO Clin Res Rep. 2023;4(11):100572.
Horn L, Mansfield AS, Szczęsna A, Havel L, Krzakowski M, Hochmair MJ, et al. First-line atezolizumab plus chemotherapy in extensive-stage small-cell Lung Cancer. N Engl J Med. 2018;379(23):2220–29.
Liu SV, Reck M, Mansfield AS, Mok T, Scherpereel A, Reinmuth N, et al. Updated overall survival and PD-L1 subgroup analysis of patients with extensive-stage small-cell lung Cancer treated with atezolizumab, carboplatin, and Etoposide (IMpower133). J Clin Oncol. 2021;39(6):619–30.
Bria E, Morgillo F, Garassino MC, Ciardiello F, Ardizzoni A, Stefani A, et al. Atezolizumab Plus Carboplatin and Etoposide in patients with untreated extensive-stage small-cell Lung Cancer: interim results of the MAURIS phase IIIb trial. Oncologist. Published Online Febr. 2024;20. https://doi.org/10.1093/oncolo/oyad342.
Wang Z, Zhao J, Ma Z, Cui J, Shu Y, Liu Z, et al. A phase 2 study of Tislelizumab in Combination with Platinum-based chemotherapy as first-line treatment for Advanced Lung Cancer in Chinese patients. Lung Cancer. 2020;147:259–68.
Cheng Y, Han L, Wu L, Chen J, Sun H, Wen G, et al. Effect of First-Line Serplulimab vs Placebo added to Chemotherapy on Survival in patients with extensive-stage small cell Lung Cancer: the ASTRUM-005 Randomized Clinical Trial. JAMA. 2022;328(12):1223–32.
Zhou L, Chai F, He Y, Zhou Z, Guo S, Li P, et al. Homodimerized cytoplasmic domain of PD-L1 regulates its complex glycosylation in living cells. Commun Biol. 2022;5(1):887.
Wu X, Wang N, Liang J, Wang B, Jin Y, Liu B, et al. Is the triggering of PD-L1 dimerization a potential mechanism for Food-Derived Small molecules in Cancer Immunotherapy? A study by Molecular Dynamics. Int J Mol Sci. 2023;24(2):1413.
Liang J, Wang B, Yang Y, Liu B, Jin Y. Approaching the dimerization mechanism of small molecule inhibitors targeting PD-L1 with Molecular Simulation. Int J Mol Sci. 2023;24(2):1280.
Naing S, Sandech N, Maiuthed A, Chongruchiroj S, Pratuangdejkul J, Lomarat P. Garcinia mangostana L. Pericarp Extract and its active compound α-Mangostin as potential inhibitors of Immune Checkpoint programmed death Ligand-1. Molecules. 2023;28(19):6991.
Shah K, Panchal S, Patel B. Porcupine inhibitors: Novel and emerging anti-cancer therapeutics targeting the wnt signaling pathway. Pharmacol Res. 2021;167:105532.
Sharma G, Ojha R, Noguera-Ortega E, Rebecca VW, Attanasio J, Liu S, et al. PPT1 inhibition enhances the antitumor activity of anti-PD-1 antibody in melanoma. JCI Insight. 2020;5(17):e133225.
Weng J, Liu S, Zhou Q, Xu W, Xu M, Gao D, et al. Intratumoral PPT1-positive macrophages determine immunosuppressive contexture and immunotherapy response in hepatocellular carcinoma. J Immunother Cancer. 2023;11(6):e006655.
Lin DT, Conibear E. ABHD17 proteins are novel protein depalmitoylases that regulate N-Ras palmitate turnover and subcellular localization. Elife. 2015;4:e11306.
Cortes JE, Gutzmer R, Kieran MW, Solomon JA. Hedgehog signaling inhibitors in solid and hematological cancers. Cancer Treat Rev. 2019;76:41–50.
Tan X, Wang C, Zhou H, Zhang S, Liu X, Yang X, et al. Bioactive fatty acid analog-derived hybrid nanoparticles confer antibody-independent chemo-immunotherapy against carcinoma. J Nanobiotechnol. 2023;21(1):183.
Dai MY, Shi YY, Wang AJ, Liu XL, Liu M, Cai HB. High-potency PD-1/PD-L1 degradation induced by Peptide-PROTAC in human cancer cells. Cell Death Dis. 2022;13(11):924.
Shi YY, Dong DR, Fan G, Dai MY, Liu M. A cyclic peptide-based PROTAC induces intracellular degradation of palmitoyltransferase and potently decreases PD-L1 expression in human cervical cancer cells. Front Immunol. 2023;14:1237964.
Fan Y, Gao Y, Nie L, Ma J, Wei W, Li L, et al. Targeting LYPLAL1-mediated cGAS depalmitoylation enhances the response to anti-tumor immunotherapy. Mol Cell. 2023;83(19):3520–e35327.
Lockhart AC, Bauer TM, Aggarwal C, Lee CB, Harvey RD, Cohen RB, et al. Phase ib study of pevonedistat, a NEDD8-activating enzyme inhibitor, in combination with docetaxel, carboplatin and paclitaxel, or gemcitabine, in patients with advanced solid tumors. Invest New Drugs. 2019;37(1):87–97.
Swords RT, Coutre S, Maris MB, Zeidner JF, Foran JM, Cruz J, et al. Pevonedistat, a first-in-class NEDD8-activating enzyme inhibitor, combined with azacitidine in patients with AML. Blood. 2018;131(13):1415–24.
Fernandez AI, Gaule P, Rimm DL. Tissue age affects antigenicity and scoring for the 22C3 immunohistochemistry Companion Diagnostic Test. Mod Pathol. 2023;36(7):100159.
Chongsaritsinsuk J, Steigmeyer AD, Mahoney KE, Rosenfeld MA, Lucas TM, Ince D, et al. Glycoproteomic landscape and structural dynamics of TIM family immune checkpoints enabled by mucinase SmE. Nat Commun. 2023;14(1):6169.
Long J, Wang Y, Jiang X, Ge J, Chen M, Zheng B, et al. Nanomaterials boost CAR-T therapy for solid tumors. Adv Healthc Mater Published Online March. 2024;14. https://doi.org/10.1002/adhm.202304615.
Fu Y, Qian H, Yang Y, Li J, Xie G. Enhanced imaging of protein-specific palmitoylation with HCR-based cis-membrane multi-FRET. Talanta. 2024;266(Pt 1):124972.
Acknowledgements
Figures were created by PowerPoint (Microsoft Office 2016) and some materials of the figures were provided by the Servier Medical Art (https://smart.servier.com/) under the CC BY 4.0 license.
Funding
This work was supported partially by grants from the National Natural Science Foundation of China (82303069 [J.W.], 52102276 [J.L.], 82303126 [Y.W.]), the Natural Science Foundation of Fujian Province (2023J05038 [J.W.]), the Fujian Provincial Project of Education and Science for Young and Middle-aged Teachers (JAT220076 [J.W.]), the Startup Foundation of Fujian Medical University (2022QH1003 [J.W.], 2022QH1783 [M.C.]), and the Natural Science Foundation of Hunan Province (2024JJ6325 [Y.W.]).
Author information
Authors and Affiliations
Contributions
R.W. and S.H. collected the related paper and finished the manuscript and figures, R.W. and S.H. contributed equally to this work. J.W. and J.L. gave constructive guidance and made critical revisions. Y.W., X.J., M.C. participated in the design of this review. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethical approval
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Wang, R., He, S., Long, J. et al. Emerging therapeutic frontiers in cancer: insights into posttranslational modifications of PD-1/PD-L1 and regulatory pathways. Exp Hematol Oncol 13, 46 (2024). https://doi.org/10.1186/s40164-024-00515-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40164-024-00515-5