
Abstract
Metabolic reprogramming, fundamentally pivotal in carcinogenesis and progression of cancer, is considered as a promising therapeutic target against tumors. In chronic lymphocytic leukemia (CLL) cells, metabolic abnormalities mediate alternations in proliferation and survival compared with normal B cells. However, the role of metabolic reprogramming is still under investigation in CLL. In this review, the critical metabolic processes of CLL were summarized, particularly glycolysis, lipid metabolism and oxidative phosphorylation. The effects of T cells and stromal cells in the microenvironment on metabolism of CLL were also elucidated. Besides, the metabolic alternation is regulated by some oncogenes and tumor suppressor regulators, especially TP53, MYC and ATM. Thus, the agents targeting metabolic enzymes or signal pathways may impede the progression of CLL. Both the inhibitor of 3-hydroxy-3-methylglutaryl coenzyme A reductase (HMGCR) statins and the lipoprotein lipase inhibitor orlistat induce the apoptosis of CLL cells. In addition, a series of oxidative phosphorylation inhibitors play important roles in decreasing the proliferation of CLL cells. We epitomized recent advancements in metabolic reprogramming in CLL and discussed their clinical potentiality for innovative therapy options. Metabolic reprogramming plays a vital role in the initiation and progression of CLL. Therapeutic approaches targeting metabolism have their advantages in improving the survival of CLL patients. This review may shed novel light on the metabolism of CLL, leading to the development of targeted agents based on the reshaping metabolism of CLL cells.
Similar content being viewed by others
Introduction
Chronic lymphocytic leukemia (CLL) is characterized by malignant proliferation of mature monoclonal B lymphocytes in the blood, bone marrow and lymphoid tissues, with heterogeneous outcomes [1]. It is one of the most frequent types of leukemia in adults, which is caused by a complex balance between unrestrained cell proliferation and apoptotic death [2]. Although small targeted agents, for instance, Bruton’s tyrosine kinase inhibitors (BTKi), phosphatidylinositol 3-kinase inhibitors (PI3Ki) and BH3-only mimetics, improve the prognosis of CLL patients, it remains incurable and novel therapeutic options are urgently needed [3,4,5,6,7,8].
Rewiring of tumor cell metabolism, affected by various tumorigenic alterations, acts as a pivotal avenue to satisfy their needs of survival, malignant proliferation and division [9, 10]. They utilize mass of nutrients and augment biomass synthesis for satisfying energy demands [11, 12]. Metabolic reprogramming of CLL, changing with progression, includes the alterations of glucose metabolism, lipid metabolism and oxidative phosphorylation (OXPHOS) [13, 14]. Of note, recent data have suggested that metabolic reprogramming of CLL can be affected by extensive metabolic interactions with other nonmalignant cells in the microenvironment [15, 16]. Besides, CLL metabolic reprogramming is governed by the expression of aberrant oncogenes and tumor suppressor genes, including TP53, ATM and MYC [17].
To identify therapeutic opportunities specifically targeting metabolism, understanding the role of reprogrammed metabolism in CLL is essential [18, 19]. Herein we recapitulate evidence gathered in recent years regarding metabolic reprogramming in CLL cells, and discuss promising therapeutic strategies in metabolism.
Intracelluar metabolism
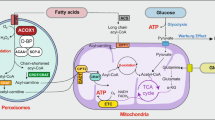
Abundant nutrients, including carbohydrates, fats and proteins, represent important drivers of metabolism [20]. Metabolic heterogeneities, induced by internal or external processes to cancer cells, have been found in human cancers, even in distinct regions of the same tumor [18]. Thus, we reviewed the metabolic reprogramming of CLL (shown in Fig. 1).

Metabolic reprogramming in chronic lymphocytic leukemia (CLL). In CLL cells, aerobic glycolysis, lipid synthesis, reductive carboxylation, beta-oxidation of fatty acids, and the consumption of glutamine are upregulated. These changes benefit CLL cells as they satisfy their demands of proliferation. CLL chronic lymphocytic leukemia, GLUT glucose transporter, G6P glucose 6-phosphate, TIGAR TP53-induced glycolysis and apoptosis regulator, TCA tricarboxylic acid, TG triglyceride, LPL lipoprotein lipase, LCFA-CoAs long-chain fatty acyl coenzyme A, HMG-CoA 3-hydroxy-3-methylglutaryl coenzyme A, HMGCR 3-hydroxy-3-methylglutaryl coenzyme A reductase, FFA free fatty acid, ApoA apolipoprotein A, CPT carnitine palmitoyl transferases, α-KG α-ketoglutarate, CAT-1 cationic amino acid transporter-1, STIM1 stromal interaction molecule 1, ROS reactive oxygen species
Glycolysis
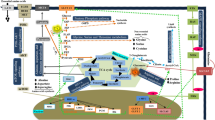
Tumor cells potentiate proliferation by enhancing cellular glucose metabolism [21, 22]. The metabolic intermediates of glycolysis provide cellular energy and play a pivotal role in various macromolecular biosynthesis [19]. Aerobic glycolysis is the principal glucose metabolic pathway in CLL cells. Different from normal cells depending on anaerobic glycolysis in the context of oxygen absence, cancer cells following the Warburg effect display a high proportion of glycolysis even in the presence of oxygen (aerobic glycolysis) [10, 23, 24]. Unlike other rapidly proliferating neoplasms, the energy supply of CLL depends on OXPHOS more than glycolysis (shown in Fig. 2). They increasingly rely on aerobic glycolysis for energy production only under a suitable stimulation, which is activated by the Notch-c-Myc axis partly [25,26,27]. Recent studies have shown that the primary function of aerobic glycolysis in CLL cells is to maintain high levels of glycolytic intermediates to sustain intracellular anabolic reactions. They participate in many biosynthesis processes, such as the pentose phosphate pathway (PPP) to generate NADPH, ribose-6-phosphate, amino acid, lipids and other cellular sources of energy [23, 28].

Aerobic glycolysis in CLL cells. A In normal cells, glucose is converted to pyruvate, which feeds the tricarboxylic acid (TCA) cycle for energy production under normoxia; B Pyruvate predominantly produces energy by lactic acid fermentation, even in the presence of oxygen (aerobic glycolysis) in cancer cells. The flux of pyruvate entering TCA cycle is decreased. C CLL cells do not follow the Warburg effect. They are not primarily dependent on glycolysis to produce energy, but increasing mitochondrial oxidative phosphorylation (OXPHOS). TCA tricarboxylic acid, OXPHOS oxidative phosphorylation
Besides, overexpression of glucose transporters (GLUT) facilitates glucose consumption in cancer cells [29, 30]. In glucose metabolism, p53 reduces glucose uptake by repressing the transcription of GLUT-1 and GLUT-4. And p53 suppresses glycolysis by negatively regulating PI3K/AKT signaling [31,32,33]. In addition, TP53-induced glycolysis and apoptosis regulator (TIGAR) suppresses glycolysis and subsequently prevents intracellular apoptosis reactive associated with oxygen species (ROS). Independently with wild-type p53, the overexpression of TIGAR is correlated with the reduction of spontaneous apoptosis in CLL cells and associated with poor clinical outcomes [34]. Potential therapeutic targets involved in the high level of glucose metabolism need to be further explored.
Lipid metabolism
Previous studies showed that fatty acid biosynthesis was significantly increased in cancer cells [35, 36]. CLL cells are like adipose cells and muscle cells [37], which can utilize more free fatty acids (FFA) to produce energy compared with normal B-cells or leukemia cells. Lipoprotein lipase (LPL), catalyzing the hydrolysis of triglyceride into FFA, was found aberrant expression in CLL cells. LPL is not expressed in normal lymphocytes, but its expression is significantly increased in IGHV unmutated subset CLL cells [38, 39]. LPL expression and proliferative phenotype in primary CLL B-cells could be induced in the leukemic clone, which promotes malignant B-cell growth [40]. Increased LPL, induced by the activation of signal transducer and activator of transcription-3 (STAT3) or downregulation of microRNA-125, mediates lipoprotein uptake and FFA utilization in CLL cells [41, 42]. LPL could contribute to cancer cell spreading, migration and be involved in CLL progression. The LPL inhibitor orlistat inhibits LPL induced by stimulation of B-cell receptor (BCR) in CLL cells. Treating primary CLL cells with orlistat results in the apoptosis of CLL cells, while no apoptosis is induced in the control group [43].
Lipidomics reveals that CLL cells have aberrant phospholipid levels. CLL membrane may have lower fluidity, resulting in chemotherapy resistance [44]. In addition, carnitine palmitoyl transferases (CPT) support the cell metabolism by transporting FFA into mitochondrial (Mt). CPT1 and CPT2 are upregulated in CLL cells. Perhexiline could inhibit CPT to reduce the cardiolipin, resulting in the damage of Mt membranes and clearing off leukemia cells [45]. Regulation of lipids is a potential therapeutic to improve treatment effects.
Mevalonate (MVA) metabolism not only produces cholesterol, but also contributes to cancer progression, including in the control of cell replication in CLL cells [46]. The statins inhibit 3-hydroxy-3-methylglutaryl-CoA reductase (HMGCR) in the MVA pathway. SREBP2 is the main transcription factor for MVA pathway-associated genes [47]. Tumor suppressor p53 leads to upregulation of the cholesterol-efflux transporter ABCA1, sequentially restricting SREBP2 and repressing the MVA pathway [48]. These results unveil that the MVA pathway has a key role in CLL. Statins are widely used for the prevention and management of cardiovascular disease [49]. In vitro, simvastatin decreases the survival of proliferation and induces apoptosis of CLL cells specifically [41, 50, 51]. It also can enhance the antitumor effect of venetoclax and ibrutinib [50]. Low-potency lipophilic statin could reduce the risk of CLL patients [49]. But statins negatively interfere with rituximab and other anti-CD20 antibodies. The inhibitors of squalene synthase and oxidosqualene cyclase inhibiting cholesterol biosynthesis regulate CD20 expression positively and enhance CLL chemoimmuno-sensitivity and apoptosis [52]. Preclinical studies have indicated that blocking cholesterol synthesis and trafficking blockade could hinder tumor formation and growth [53].
In CLL cells, low-density lipoprotein (LDL) can amplify signaling pathways, particularly cytokine-signaling [54]. Reducing LDL levels inhibits signaling and limits the proliferation of CLL cells. Besides, researchers have reported that the level of high-density lipoprotein (HDL) was decreased in patients with CLL [55]. Apolipoprotein A (ApoA) is one of the main components of HDL. A low level of ApoA is related to the advanced stage of CLL patients [56]. ApoA1 mimetics could inhibit the proliferation of CLL cells. ApoA5 has been found to play a significant function in triglyceride metabolism by stimulating LPL activity [57]. The role of ApoA in CLL requires additional investigation.
Amino acid metabolism
Amino acids are widely rewired to obtain the necessary energy to satisfy the increased need in cancer cells [58,59,60,61]. The level of isoleucine is decreased, while pyruvate and glutamate are increased in CLL patients [62, 63]. Among amino acids in vivo, glutamine (Gln) metabolism is important for cancer cell survival, which is the central position in carbon and nitrogen metabolism in tumor cells [64]. Previous research indicated the consumption of Gln was increased in CLL [65]. Elevated ammonia uptake and Gln synthetase expression are found in del11q CLL lymphocytes, favoring de novo Gln synthesis [66]. The expression of glutamate dehydrogenase (GDH) is decreased in del11q CLL cells, which benefits transaminase reactions using α-ketoglutarate for glutamate synthesis and reduces oxidative deamination of glutamate. Recent studies have suggested that the activity of membrane mechanistic target of rapamycin complex 1 (mTORC1) can be stimulated by extracellular Gln, which facilitates the transport of mTORC1-activating amino acids across the plasma. Apart from regulating mTOR, L-glutamate regulates translation to coordinate cell growth and proliferation [67]. However, ammonia, as a by-product of glutaminolysis, stimulates autophagy in a mTORC1-independent fashion [68]. Depending on these alterations, based on metabolism therapeutic options could be developed. Many novel mTOR inhibitors are being explored in clinical trials [69]. Affecting glutamine metabolism is one of the mechanisms of ibrutinib. Glutaminase inhibitor L-asparaginase catalyzes the conversion of Gln to glutamate in CLL cells, especially in those with del11q [70]. To conclude, the specific Gln metabolism characteristics in CLL are worth further study.
Besides Gln, arginine also plays an important role in CLL. Certain tumors have been verified to lose the ability to synthesize arginine independently, which is promising to be a therapeutic target [71, 72]. Argininosuccinate synthase (ASS) is not expressed in primary CLL cells, preventing arginine synthesis. As the only arginine importer expressed in CLL cells, cationic amino acid transporter-1 (CAT-1) is a novel target for CLL therapy [73]. The abnormal activation of amino acids maintains the metabolic balance in CLL.
Ion metabolism
One of the ion metabolic peculiarities of CLL is Ca2+ dysregulation [74]. The level of basal Ca2+ signaling is not uniformly increased in CLL cells. Compared with normal B cells, basal Ca2+ signaling in CLL cells is higher, especially in IGHV mutated CLL (M-CLL) [75]. Recent studies have shown a novel Ca2+ entry pathway, named constitutive Ca2+ entry (CE), which is constitutive and BCR-independent, is controlled by stromal interaction molecule 1 (STIM1) located at the plasma membrane [74]. Besides, the surface protein CD38 can enhance intracellular Ca2+ levels to promote RasGRP2/Rap1-mediated CLL cell adhesion and migration [76, 77]. Pathways associated with calcium ions, such as Rap1 signaling, may lead to potential therapeutic strategies targeting CLL treatment. Calcium concentration variation is directly through the BCR and chemokine receptors, or indirectly through co-stimulatory molecules. Transmit information depending on calcium signals is crucial to B-cell ontogeny, including specific signaling pathways essential for B cells development and activation [78, 79].
In addition, iron plays dual roles in the CLL cells. On the one hand, iron is a critical cofactor that is required for DNA synthesis [80]. The transferrin receptor (TfR) contributes to iron import and a higher TfR concentration has been found in CLL directly reflecting the large tumor burden [81]. Leukemia cells harbored TP53 mutation need a mass of iron to support rapid proliferation. P53 plays a role in iron homeostasis and mitochondrial iron homeostasis by modulating iron regulators [82]. Cells may go into canceration or demise when the homeostasis is disrupted [83]. Ferroptosis, a crucial component of p53-mediated tumor suppression, is an iron-dependent form of regulated cell death caused by unrestricted lipid peroxidation and subsequent plasma membrane rupture [84]. The activity of the selenoperoxidase Glutathione Peroxidase 4 (GPX4) is the cornerstone of the antiperoxidant defence [85]. The expression of GPX4 in ferroptosis depends on the presence of glutathione (GSH) [86]. Eprenetapopt, which could reactivate mutant forms of p53 and induce ferroptosis by GSH depletion, is currently being tested in the clinical trials involving patients with acute myeloid leukemia (NCT03931291) [87]. Cysteine availability is the main limiting factor in the synthesis of GSH. The depletion of extracellular cysteine leads to cell death in CLL. Cyst(e)inase, an engineered human enzyme, effectively degrades cysteine and induces ferroptosis in pancreatic ductal adenocarcinoma [86]. The strategy regulating extracellular cysteine levels opens up new therapeutic options for CLL patients with TP53 mutation. Nevertheless, the definitive mechanism that p53 modulates iron metabolism in CLL is unclear. Targeting altered iron metabolic pathway is specific to CLL patients with TP53 mutation.
Mitochondrial metabolism
Mt plays an important role in cellular metabolism, ATP synthesis, oxidative metabolism and the regulation of apoptosis [88, 89]. Compared with solid tumor cells, aerobic glycolysis is not increased in CLL cells, while the level of mitochondrial OXPHOS is elevated. Mt biogenesis, such as Mt mass, ATP production, Mt DNA, oxygen consumption and the production of ROS, has increased in CLL cells. The accumulation of ROS may contribute to the metabolic state of oxidative stress in CLL patients [90,91,92].
OXPHOS is upregulated in leukemias, including CLL. OXPHOS inhibitors could be used to improve treatment outcomes [93]. Recent studies have identified that PI3K/AKT signaling could be limited by the suppression of the expression and activity of the inhibitory phosphatase SH2-containing-inositol-5′-phosphatase-1 (SHIP1) in CLL cells. Increased Mt respiration and excessive accumulation of ROS lead to CLL cells death [94]. Besides, mtDNA mutations elevate the level of nitric oxide (NO), which can significantly influence Mt biogenesis. By inhibiting the expression of NO synthases (NOS), which can be induced by ROS stress, the NO-mediated Mt biogenesis in CLL cells can be changed [95]. Previous study verified the ability of 22 NOS inhibitors to induce CLL cell apoptosis, including L-NAME [96]. In addition, the Ser727-phosphorylated STAT3 molecule (pSTAT3Ser727) in Mt overactivity can enhance the antioxidant defense ability of CLL B cells that promotes their survival [97, 98].
Hence, recent studies have shown that combinations with mitochondrial targeting agents could be a promising cancer therapy. Of note, some unanticipated agents regulating metabolism also play roles in CLL. The OXPHOS inhibitor metformin (NCT01750567) alone or in combination with the GLUT4 inhibitor ritonavir (NCT02948283) was involved in the current clinical trials [70]. Metformin inhibits the OXPHOS of mitochondrial in CLL cells, while ritonavir induces CLL cells apoptosis [99]. The OXPHOS inhibitor IACS-010759 inhibits OXPHOS and diminishes intracellular ribonucleotide pools [100]. Berberine (BRB), used for metabolic disorders, induces impairment of OXPHOS and the associated increment of oxidative damage, with consequent inhibition of CLL cell activation and eventual cell death [101]. Besides, PK11195, the benzodiazepine derivate, blocks OXPHOS and induces apoptosis in CLL [102].
Role of the microenvironment in CLL metabolism
The specific microenvironment can promote the survival of CLL cells. In most human cancers, non-malignant cells in the microenvironment limit oxygen and nutrient transport to the cancer cells [36, 103]. Lipid metabolism in microenvironment plays the paradoxical role in anti-tumor and pro-tumor immune responses [104]. Therefore, cancer cells transfer their metabolic ability and adapt their microenvironment to support cancer cell growth and satisfy their biomass demands, even involving in cancer metastasis [105, 106].
The microenvironment activates and protects CLL cells through several mechanisms [107]. CLL cells, primarily existing in peripheral blood and tissues and proliferate mainly in lymph nodes, interact directly with T cells, monocyte-derived cells (MDC) and stromal cells in proliferation centers. The signals, such as adhesion molecules, cell surface ligands, chemokines, cytokines and their respective receptors, mediate the interactions between CLL cells and the microenvironment. These signals promote an immunotolerant milieu in the CLL lymph node [108]. Thus, the microenvironment affects the metabolism of CLL cells significantly.
In CLL patients, T cells can activate mitochondrial metabolism, enhance chemo-resistance and promote cell proliferation. Follicular helper T cells (TFH) populations expand abnormally and produce a mass of cytokines and costimulatory molecules to help CLL cells proliferate [109, 110]. Besides, CLL cells express high levels of PD-L1 while T cells express PD-1 [111]. Blocking PD-1/PD-L1 might restore the glycolysis, phagocytosis and BTK signaling of monocytes/macrophages in CLL [112]. Direct contact with CLL cells induces T-cell dysfunction [113], such as inducing Rho GTPase signaling defects in T cells to evade immune recognition [114].
Stromal cells prolong the survival time of CLL cells by generating stromal cell-derived factor-1 (SDF-1) [115]. The survival signals delivered by stromal cells activate different pathways. Mesenchymal stromal cells (MSCs) in CLL patients signifcantly promote CLL cell proliferation compared with the control group [116]. In addition, MSC-derived extracellular vesicles (EVs) increase CLL cell migration and give CLL cell survival advantages [117]. Bone marrow stromal cells take in cystine and transform it into cysteine, which is transported to CLL cells for glutathione synthesis and enhances survival and drug resistance of CLL cells [118]. The selective inhibitor of the lipid kinase PI3Kδ idelalisib (NCT01539512) promotes apoptosis in primary CLL cells, and disrupts interactions between CLL cells and endothelial cells/bone marrow stromal cells [119, 120]. Bone marrow stromal cells induce the production of protein kinase C-β to promote CLL progression [121]. Protein kinase C-β lessens mitochondrial stress and facilitates glucose absorption. Disrupting this bidirectional communication between CLL cells and stromal cells can study novel treatment strategies.
Gene expression of metabolic reprogramming in CLL
Aberrant expression of oncogenes and tumor suppressors facilitates the metabolic reprogramming of cancer cells to enable increased nutrient acquisition and biosynthesis [15]. However, how gene expression affects metabolism of CLL cells is less understood. Therefore, we summarized the impact of gene expression on metabolic reprogramming of CLL cells (shown in Table 1).
The human tumor suppressor gene TP53, located on the short arm of chromosome 17, plays key roles in cell cycle arrest, apoptosis, DNA repair, autophagy and metabolism regulation in CLL [122, 123]. TP53 mutation and/or deletion of chromosome 17 is a poor prognostic biomarker and tailors the therapy in CLL patients [124,125,126]. Importantly, the protein p53, targeting various metabolism pathways, enables cells to maintain metabolic homeostasis and adapt to stress [127]. Researchers have indicated that mutant TP53 could conserve its tumor suppressor activity by decreasing reactive oxygen production and regulating energy metabolism [128]. In lipid metabolism, p53 inhibits fatty acid synthesis and even enhances fatty acid oxidation in cells [129]. Besides, p53 takes effect on OXPHOS, mitochondrial metabolism, serine metabolism, nucleotide metabolism and iron metabolism [129, 130]. In lymphomas, cellular metabolism and metabolic stress influence the activity of p53 conversely. Metabolic stress induced by glucose deprivation leads to cell-cycle arrest and apoptosis. The Gln metabolism and glycolysis also affect the transcription activity of the p53 protein [131]. Taken together, TP53 is a critical gene in CLL cellular metabolism and moderated by the metabolic status of the cells.
The MYC oncogene family, including c-MYC, N-MYC and L-MYC, encodes a group of nuclear phosphoproteins that take effect in cell proliferation, apoptosis and progression of cancers [132]. Endogenous and oncogenic MYC appears to share target genes involved in several facets of intermediary metabolism from glycolysis and glutaminolysis to nucleotide and lipid synthesis [133]. It was reported that the metabolic genes would be further amplified to support the bioenergetic needs of the growing cells when MYC is induced. In proliferative tumor cells, MYC increases the expression of glutamine transporters and glutaminase to promote mitochondrial glutamine utilization [134]. Through upregulation of enzymes in the PPP, MYC increases the shunting of glucose to the PPP in lymphocytes [135]. In addition, it regulates lymphocyte serine biosynthesis. Besides, MYC activates the expression of the enzymes ATP citrate lyase (ACLY), acetyl-CoA carboxylase alpha, fatty acid synthase (FASN) and stearoyl-CoA desaturase (SCD), involved in fatty acid synthesis from citrate [136, 137]. However, whether MYC takes similar effects in CLL needs further exploration. The expression of MYC in CLL is increased with disease progression, independent of other prognostic factors [70]. Richter syndrome (RS) is the transformation of CLL into an aggressive lymphoma and has a poor prognosis [138]. MYC aberrations are common in RS and enhanced glucose metabolism is detected in RS [139]. MYC may upregulate the glucose metabolism in CLL cells leading to disease progression.
The human tumor suppressor gene ATM, located on the long arm of chromosome 11, relates to DNA damage, cell cycle progression, p53 dysfunction and metabolism [66, 140]. In CLL, the ATM mutation is a poor independent prognostic biomarker for time to first treatment (TTFT) [141]. The underexpression of ATM is associated with the deletion of chromosome 11 (del11q), while del11q CLL lymphocytes reprogram glutamine metabolism and inhibit glucose metabolism [66]. Besides, increased expression of insulin receptors is found in del11q CLL [142]. Thus, del11q, associated with the low expression of ATM, inhibits glucose and glutamine metabolism pathways.
Besides the tumor suppressor genes and oncogenes above, other genes also take metabolic effects in CLL. Sucrase-isomaltase (SI) is a carbohydrate metabolism enzyme and SI gene mutations involve in the metabolism and development of cancer [143, 144]. In CLL, SI mutations result in metabolic reprogramming of glucose, heterocycle and cofactor metabolism. In this respect, SI could be an overlooked cancer gene. The oncogene AKT stimulates glucose metabolism and lactate production without increasing oxygen consumption in glioblastoma and hematopoietic cancer cell [145]. Active AKT signaling triggers CLL toward RS [146, 147]. While enhanced glucose metabolism is found in RS, the role of AKT plays in the glucose metabolic pathway of CLL may need further study. Due to the difference in cellular context, EZH2 acts as either an oncogene or a tumor suppressor gene [148, 149]. It alters metabolism of cancer cells involving glucose, lipid and amino acid metabolism [150]. In aggressive CLL, EZH2 upregulates the PI3K/AKT pathway through IGF1R and MYC, thus regulating glycolysis, glutaminolysis and mitochondrial biogenesis [151, 152]. Overexpression of EZH2 is associated with a poor prognosis of CLL [153]. Overall, some oncogenes and tumor suppressor genes could regulate metabolic reprogramming in CLL.
In the last decade, the biological basis of CLL pathogenesis studies has greatly expanded our knowledge of the progression of CLL remarkably, revealing a huge number of novel alterations that might drive the evolution of the disease. Therefore, we summarized the reprogrammed pathways or biomarkers related to metabolism in CLL (shown in Tables 2 and 3).
Other therapies targeting metabolic pathways in CLL
As mentioned, CLL cells alter the metabolic pathways to satisfy the need for proliferation and survival. In this context, metabolism is a novel target for CLL patients (detailed information is shown in Table 4).
Besides the treatments discussed above, the potential agents on the suite of CLL metabolism need more research to prove. For instance, a cardiac glycoside is a therapy for heart failure and arrhythmia. Ipecac alkaloids are used as anti-infective. They both repress hypoxia-inducible factor-1α (HIF-1α) and disturb intracellular redox homeostasis in CLL cells, as well as highly active against protected primary CLL cells [154, 155].
Ibrutinib, the BTK inhibitor, is the first choice for CLL [125]. In addition to targeting BCR signaling, ibrutinib participants in the control of lipid and mitochondrial metabolism [70]. Increased HDL level is found in CLL patients received ibrutinib therapy [156]. Ibrutinib affects Mt metabolism, and curtailing AMPK activity might sensitize ibrutinib-resistant clones to ibrutinib [157]. These results indicate that ibrutinib induces bioenergetic stress responses.
Metabolic reprogramming in other hematological malignancies
Metabolic heterogeneity is found in acute myeloid leukemia (AML) at different stages. Human leukemia stem cells (LSC) are dependent on OXPHOS for survival regulated by AMPK, as well as mTOR. Amino acid uptake, steady-state levels and catabolism are elevated in the LSC [158, 159]. Ecotropic Virus Integration site 1 protein homolog (EVI1) induces accelerated OXPHOS prior to activation of glycolysis in mixed lineage leukemia-rearrangement AML, with a higher dependency on Gln [160]. On the other hand, fatty acids oxidation (FAO) is a key metabolic pathway fostering the survival of chemoresistant LSC. Inhibiting very-long-chain acyl-CoA dehydrogenase, which supports FAO and OXPHOS in the mitochondrial metabolism in AML, is demonstrated preclinical activity [161]. In contrast to LSC, bulk AML blast cells rely on glycolysis to produce energy, and upon glucose deprivation have decreased viability in culture [59]. High expression of phosphomannonse isomerase (PMI), as a poor prognostic factor of AML, mobilizes mannose to glycolysis under glucose starvation in leukemia [162]. In addition, thioredoxin reductase (TrxR) directly regulates GAPDH leading to a disruption of glycolysis and an increase in flux through the PPP [163]. Combined inhibition of TrxR and PPP leads to leukemia growth inhibition.
Similar to lymphomas and AML, aberrant glycolysis and OXPHOS are the main altered metabolic processes in multiple myeloma (MM). Lactate dehydrogenase isoform A (LDHA), a key enzyme in glycolysis, is highly expressed in MM [164]. In MM, the PPARγ coactivator-1β (PGC1β) promotes the transcription of LDHA, thus modulates glycolysis and mitochondrial function. Otherwise, MM cells with overexpression of PLR-3 have higher aerobic glycolytic rate, OXPHOS and ATP production by promoting glucose uptake and lactate excretion, enhancing the levels of proteins regulating glycolysis and enzymes in the serine/glycine synthesis pathway [165]. Besides, lipid metabolism and microenvironment affect the cell proliferation in MM. In bone marrow, MM cells induce lipolysis of adipocytes. Subsequently, released FFA is taken up by myeloma cells through FFA transporter proteins, leading to growth or lipotoxicity [166].
One of the common metabolic characteristics among some B-cell derived lymphomas is the increased OXPHOS. According to consensus cluster classification, diffuse large B-cell lymphoma (DLBCL) is separated into three clusters with distinct metabolic fringerpringts: OXPHOS-DLBCL, BCR-DLBCL and host response-DLBCL [167]. OXPHOS-DLBCL displays a prominent mitochondrial component with elevated OXPHOS. Quantitative evidence validates marked increased mitochondrial FAO and palmitate is a predominant respiratory fuel in OXPHOS-DLBCL [168]. In contrast, non-OXPHOS-DLBCL is metabolically rewired to aerobic glycolysis. In addition, overexpression of transporters of lactate in DLBCL cells, such as monocarboxylate transporter 1 (MCT1) and TOMM20, promotes the TCA cycle of malignant cells in the process of reverse Warburg effect [169]. Similarly, Hodgkin and Reed Sternber cells with high expression of MCT1 and TOMM20 are of increased mitochondrial metabolism, while tumor-associated macrophages with high expression of MCT4 present elevated glycolysis in the microenvironment [170]. mTOR signaling plays a pivotal role in the aberrant OXHPHOS and glycolysis in B-cell derived lymphoma, including DLBCL, follicular lymphoma (FL), mantle cell lymphoma (MCL) [171, 172]. However, rapalogs, the inhibitor of mTORC1, fails to improve the prognosis of refractory/relapsed DLBCL, FL and MCL in the clinical studies [171]. mTOR signaling pathway is a potential therapeutic target for the B-cell derived lymphomas.
To some extent, the metabolism rewiring of CLL is in accordance with partly B-cell derived lymphomas, in which the survival and proliferation of cells depend on elevated OXPHOS or aerobic glycolysis. Although metabolic alternations of other hematological malignancies involve in lipid metabolism, the overexpression of LPL is not reported in the hematological malignancies except for CLL. Similar to adipocytes, lipid-like vesicles uptaked by LPL are detected in CLL cells [42]. Several proteins that drive steps in glycolysis and FFA biosynthesis are overexpressed, while proteins involved in the citric acid cycle are at low levels in CLL cells. This suggests CLL cells increase both endogenous lipid synthesis and exogenous lipid uptake [44]. Compared to other hematological malignancies, CLL cells are more inclined to rely on lipid metabolic pathways to support their survival.
Conclusions
Extensive research over the last decade has provided compelling evidence for metabolic reprogramming in CLL. Unlike normal cells which rely on aerobic glucose metabolism, CLL cells depend on the aberrant lipid metabolism and mitochondrial OXPHOS to support survival and growth. Altered metabolism of amino acids and ions contribute to the intracellular signal transduction in CLL cells. In addition, the microenvironment, activating the metabolism through several mechanisms, plays a vital role in the survival of CLL cells. Besides, the process of metabolic rewiring in CLL is driven by some oncogenes and tumor suppressor genes, especially TP53, MYC and ATM. Exploring novel agents with high selectivity and specificity by regulating the metabolic activity provides an opportunity to benefit CLL patients. Furthermore, the synergistic effects of metabolism-regulated drugs with targeted therapy need to be tested comprehensively in the clinic to facilitate development of novel treatment strategies for CLL patients. With improved understanding of CLL metabolic reprogramming, the rise of innovative therapeutic interventions that target metabolic pathways is anticipated.
Availability of data and materials
Not applicable.
Abbreviations
- CLL:
-
Chronic lymphocytic leukemia
- BTKi:
-
Bruton’s tyrosine kinase inhibitor
- FFAs:
-
Free fatty acids
- GLUT:
-
Glucose transporter
- PPP:
-
Pentose phosphate pathway
- OXPHOS:
-
Oxidative phosphorylation
- LPL:
-
Lipoprotein lipase
- STAT3:
-
Signal transducer and activator of transcription-3
- HDL:
-
High-density lipoprotein
- MVA:
-
Mevalonate
- HMGCR:
-
3-Hydroxy-3-methylglutaryl-CoA reductase
- ApoA:
-
Apolipoprotein A
- Gln:
-
Glutamine
- GDH:
-
Glutamate dehydrogenase
- ASS:
-
Argininosuccinate synthase
- CAT-1:
-
Cationic amino acid transporter-1
- ROS:
-
Reactive oxygen species
- STIM1:
-
Stromal interaction molecule 1
- Mt:
-
Mitochondrial
- CPT:
-
Carnitine palmitoyl transferases
- SHIP1:
-
SH2-containing-inositol-5′-phosphatase-1
- BRB:
-
Berberine
- mTORC1:
-
Membrane mechanistic target of rapamycin complex 1
- NO:
-
Nitric oxide
- NOS:
-
Nitric oxide synthase
- CE:
-
Constitutive Ca2+ entry
- TfR:
-
Transferrin receptor
- GPX4:
-
Glutathione peroxidase 4
- GSH:
-
Glutathione
- pSTAT3Ser727:
-
Ser727-phosphorylated STAT3 molecule
- MDC:
-
Monocyte-derived cells
- TFH :
-
Follicular helper T cells
- ACLY:
-
ATP citrate lyase
- FASN:
-
Fatty acid synthase
- SCD:
-
Stearoyl-CoA desaturase
- SDF-1:
-
Stromal cell-derived factor-1
- HIF-1α:
-
Hypoxia-inducible factor-1α
- MSCs:
-
Mesenchymal stromal cells
- EVs:
-
Extracellular vesicles
- TIGAR:
-
TP53-induced glycolysis and apoptosis regulator
- TTFT:
-
Time to first treatment
- RS:
-
Richter syndrome
- SI:
-
Sucrase-isomaltase
- SYK:
-
Spleen tyrosine kinase
- DLBCL:
-
Diffuse large B-cell lymphoma
- FAO:
-
Fatty acids oxidation
- FL:
-
Follicular lymphoma
- MCL:
-
Mantle cell lymphoma
- AML:
-
Acute myeloid leukemia
- LSC:
-
Leukemia stem cell
- EVI1:
-
Ecotropic Virus Integration site 1 protein homolog
- PMI:
-
Phosphomannonse isomerase
- TrxR:
-
Thioredoxin reductase
- MM:
-
Multiple myeloma
- MCT:
-
Monocarboxylate transporter
- LDHA:
-
Lactate dehydrogenase isoform A
- PGC1β:
-
PPARγ coactivator-1β
- HMG-CoA:
-
3-Hydroxy-3-methylglutaryl coenzyme A
- BCR:
-
B-cell receptor
References
Scarfo L, Ferreri AJ, Ghia P. Chronic lymphocytic leukaemia. Crit Rev Oncol Hematol. 2016;104:169–82.
DeBerardinis RJ, Thompson CB. Cellular metabolism and disease: what do metabolic outliers teach us? Cell. 2012;148(6):1132–44.
Zhang Y, Zhou X, Li Y, Xu Y, Lu K, Li P, et al. Inhibition of maternal embryonic leucine zipper kinase with OTSSP167 displays potent anti-leukemic effects in chronic lymphocytic leukemia. Oncogene. 2018;37(41):5520–33.
Lu K, Wang X. Therapeutic advancement of chronic lymphocytic leukemia. J Hematol Oncol. 2012;5:55.
Klener P, Sovilj D, Renesova N, Andera L. BH3 mimetics in hematologic malignancies. Int J Mol Sci. 2021;22(18):10157.
Billard C. Design of novel BH3 mimetics for the treatment of chronic lymphocytic leukemia. Leukemia. 2012;26(9):2032–8.
Tian Z, Liu M, Fang X, Zhou X, Li P, Li Y, et al. Distinct age-related clinical features and risk assessment in Chinese with chronic lymphocytic leukemia. Front Oncol. 2022. https://doi.org/10.3389/fonc.2022.885150.
Zhang J, Lu X, Li J, Miao Y. Combining BTK inhibitors with BCL2 inhibitors for treating chronic lymphocytic leukemia and mantle cell lymphoma. Biomark Res. 2022;10(1):17.
Wang S, Fu JL, Hao HF, Jiao YN, Li PP, Han SY. Metabolic reprogramming by traditional Chinese medicine and its role in effective cancer therapy. Pharmacol Res. 2021;170:105728.
Li Z, Geng M, Ye X, Ji Y, Li Y, Zhang X, et al. IRF7 inhibits the Warburg effect via transcriptional suppression of PKM2 in osteosarcoma. Int J Biol Sci. 2022;18(1):30–42.
Sneeggen M, Guadagno NA, Progida C. Intracellular transport in cancer metabolic reprogramming. Front Cell Dev Biol. 2020;8:597608.
Sun L, Suo C, Li ST, Zhang H, Gao P. Metabolic reprogramming for cancer cells and their microenvironment: beyond the Warburg effect. Biochim Biophys Acta Rev Cancer. 2018;1870(1):51–66.
La Vecchia S, Sebastián C. Metabolic pathways regulating colorectal cancer initiation and progression. Semin Cell Dev Biol. 2020;98:63–70.
Luo X, Cheng C, Tan Z, Li N, Tang M, Yang L, et al. Emerging roles of lipid metabolism in cancer metastasis. Mol Cancer. 2017;16(1):76.
Pavlova NN, Thompson CB. The emerging hallmarks of cancer metabolism. Cell Metab. 2016;23(1):27–47.
Xia L, Oyang L, Lin J, Tan S, Han Y, Wu N, et al. The cancer metabolic reprogramming and immune response. Mol Cancer. 2021;20(1):28.
Landau DA, Tausch E, Taylor-Weiner AN, Stewart C, Reiter JG, Bahlo J, et al. Mutations driving CLL and their evolution in progression and relapse. Nature. 2015;526(7574):525–30.
Faubert B, Solmonson A, DeBerardinis RJ. Metabolic reprogramming and cancer progression, vol. 368. New York: Science; 2020.
Ganapathy-Kanniappan S, Geschwind JF. Tumor glycolysis as a target for cancer therapy: progress and prospects. Mol Cancer. 2013;12:152.
Bonora M, Patergnani S, Rimessi A, De Marchi E, Suski JM, Bononi A, et al. ATP synthesis and storage. Purinergic Signal. 2012;8(3):343–57.
Han X, Raun SH, Carlsson M, Sjoberg KA, Henriquez-Olguin C, Ali M, et al. Cancer causes metabolic perturbations associated with reduced insulin-stimulated glucose uptake in peripheral tissues and impaired muscle microvascular perfusion. Metabolism. 2020;105:154169.
Li X, Yu C, Luo Y, Lin J, Wang F, Sun X, et al. Aldolase A enhances intrahepatic cholangiocarcinoma proliferation and invasion through promoting glycolysis. Int J Biol Sci. 2021;17(7):1782–94.
Kalyanaraman B. Teaching the basics of cancer metabolism: developing antitumor strategies by exploiting the differences between normal and cancer cell metabolism. Redox Biol. 2017;12:833–42.
Pascale RM, Calvisi DF, Simile MM, Feo CF, Feo F. The Warburg effect 97 years after its discovery. Cancers. 2020;12(10):2819.
Lu J, Bottcher M, Walther T, Mougiakakos D, Zenz T, Huber W. Energy metabolism is co-determined by genetic variants in chronic lymphocytic leukemia and influences drug sensitivity. Haematologica. 2019;104(9):1830–40.
Jitschin R, Braun M, Qorraj M, Saul D, Le Blanc K, Zenz T, et al. Stromal cell-mediated glycolytic switch in CLL cells involves Notch-c-Myc signaling. Blood. 2015;125(22):3432–6.
Christiansen JR, Kanellopoulos A, Lund MB, Massey R, Dalen H, Kiserud CE, et al. Impaired exercise capacity and left ventricular function in long-term adult survivors of childhood acute lymphoblastic leukemia. Pediatr Blood Cancer. 2015;62(8):1437–43.
Lunt SY, Vander Heiden MG. Aerobic glycolysis: meeting the metabolic requirements of cell proliferation. Annu Rev Cell Dev Biol. 2011;27:441–64.
Macheda ML, Rogers S, Best JD. Molecular and cellular regulation of glucose transporter (GLUT) proteins in cancer. J Cell Physiol. 2005;202(3):654–62.
Barron CC, Bilan PJ, Tsakiridis T, Tsiani E. Facilitative glucose transporters: implications for cancer detection, prognosis and treatment. Metabolism. 2016;65(2):124–39.
Schwartzenberg-Bar-Yoseph F, Armoni M, Karnieli E. The tumor suppressor p53 down-regulates glucose transporters GLUT1 and GLUT4 gene expression. Cancer Res. 2004;64(7):2627–33.
Liu J, Zhang C, Lin M, Zhu W, Liang Y, Hong X, et al. Glutaminase 2 negatively regulates the PI3K/AKT signaling and shows tumor suppression activity in human hepatocellular carcinoma. Oncotarget. 2014;5(9):2635–47.
Gomes AS, Ramos H, Soares J, Saraiva L. p53 and glucose metabolism: an orchestra to be directed in cancer therapy. Pharmacol Res. 2018;131:75–86.
Hong M, Xia Y, Zhu Y, Zhao HH, Zhu H, Xie Y, et al. TP53-induced glycolysis and apoptosis regulator protects from spontaneous apoptosis and predicts poor prognosis in chronic lymphocytic leukemia. Leuk Res. 2016;50:72–7.
Guo W, Wang X, Li Y, Bai O. Function and regulation of lipid signaling in lymphomagenesis: a novel target in cancer research and therapy. Crit Rev Oncol Hematol. 2020;154:103071.
Liu Y, Zhou X, Wang X. Targeting the tumor microenvironment in B-cell lymphoma: challenges and opportunities. J Hematol Oncol. 2021;14(1):125.
Bilban M, Heintel D, Scharl T, Woelfel T, Auer MM, Porpaczy E, et al. Deregulated expression of fat and muscle genes in B-cell chronic lymphocytic leukemia with high lipoprotein lipase expression. Leukemia. 2006;20(6):1080–8.
Heintel D, Kienle D, Shehata M, Krober A, Kroemer E, Schwarzinger I, et al. High expression of lipoprotein lipase in poor risk B-cell chronic lymphocytic leukemia. Leukemia. 2005;19(7):1216–23.
Friedman DR. Lipids and their effects in chronic lymphocytic leukemia. EBioMedicine. 2017;15:2–3.
Abreu C, Moreno P, Palacios F, Borge M, Morande P, Landoni AI, et al. Methylation status regulates lipoprotein lipase expression in chronic lymphocytic leukemia. Leuk Lymphoma. 2013;54(8):1844–8.
Rozovski U, Hazan-Halevy I, Barzilai M, Keating MJ, Estrov Z. Metabolism pathways in chronic lymphocytic leukemia. Leuk Lymphoma. 2016;57(4):758–65.
Rozovski U, Grgurevic S, Bueso-Ramos C, Harris DM, Li P, Liu Z, et al. Aberrant LPL expression, driven by STAT3, mediates free fatty acid metabolism in CLL cells. Mol Cancer Res. 2015;13(5):944–53.
Pallasch CP, Schwamb J, Königs S, Schulz A, Debey S, Kofler D, et al. Targeting lipid metabolism by the lipoprotein lipase inhibitor orlistat results in apoptosis of B-cell chronic lymphocytic leukemia cells. Leukemia. 2008;22(3):585–92.
Thurgood LA, Dwyer ES, Lower KM, Chataway TK, Kuss BJ. Altered expression of metabolic pathways in CLL detected by unlabelled quantitative mass spectrometry analysis. Br J Haematol. 2019;185(1):65–78.
Liu PP, Liu J, Jiang WQ, Carew JS, Ogasawara MA, Pelicano H, et al. Elimination of chronic lymphocytic leukemia cells in stromal microenvironment by targeting CPT with an antiangina drug perhexiline. Oncogene. 2016;35(43):5663–73.
Larson RA, Yachnin S. Mevalonic acid induces DNA synthesis in chronic lymphocytic leukemia cells. Blood. 1984;64(1):257–62.
Mullen PJ, Yu R, Longo J, Archer MC, Penn LZ. The interplay between cell signalling and the mevalonate pathway in cancer. Nat Rev Cancer. 2016;16(11):718–31.
Huang B, Song BL, Xu C. Cholesterol metabolism in cancer: mechanisms and therapeutic opportunities. Nat Metab. 2020;2(2):132–41.
Righolt CH, Zhang G, Ye X, Banerji V, Johnston JB, Gibson S, et al. Statin use and chronic lymphocytic leukemia incidence: a nested case-control study in manitoba, Canada. Cancer Epidemiol Biomark Prevent. 2019;28(9):1495–501.
Gimenez N, Tripathi R, Giró A, Rosich L, López-Guerra M, López-Oreja I, et al. Systems biology drug screening identifies statins as enhancers of current therapies in chronic lymphocytic leukemia. Sci Rep. 2020;10(1):22153.
Podhorecka M, Halicka D, Klimek P, Kowal M, Chocholska S, Dmoszynska A. Simvastatin and purine analogs have a synergic effect on apoptosis of chronic lymphocytic leukemia cells. Ann Hematol. 2010;89(11):1115–24.
Benakanakere I, Johnson T, Sleightholm R, Villeda V, Arya M, Bobba R, et al. Targeting cholesterol synthesis increases chemoimmuno-sensitivity in chronic lymphocytic leukemia cells. Exp Hematol Oncol. 2014;3:24.
Xu H, Zhou S, Tang Q, Xia H, Bi F. Cholesterol metabolism: new functions and therapeutic approaches in cancer. Biochim Biophys Acta Rev Cancer. 2020;1874(1):188394.
McCaw L, Shi Y, Wang G, Li YJ, Spaner DE. Low density lipoproteins amplify cytokine-signaling in chronic lymphocytic leukemia cells. EBioMedicine. 2017;15:24–35.
Yavasoglu I, Sargin G, Yilmaz F, Altındag S, Akgun G, Tombak A, et al. Cholesterol levels in patients with chronic lymphocytic leukemia. J Natl Med Assoc. 2017;109(1):23–7.
Yun X, Sun X, Hu X, Zhang H, Yin Z, Zhang X, et al. Prognostic and therapeutic value of apolipoprotein a and a new risk scoring system based on apolipoprotein a and adenosine deaminase in chronic lymphocytic leukemia. Front Oncol. 2021;11:698572.
Hubacek JA. Apolipoprotein A5 fifteen years anniversary: lessons from genetic epidemiology. Gene. 2016;592(1):193–9.
Li Z, Zhang H. Reprogramming of glucose, fatty acid and amino acid metabolism for cancer progression. Cell Mol Life Sci. 2015;73(2):377–92.
Jiang H, Zhang N, Tang T, Feng F, Sun H, Qu W. Target the human alanine/serine/cysteine transporter 2(ASCT2): achievement and future for novel cancer therapy. Pharmacol Res. 2020;158:104844.
Wei Z, Liu X, Cheng C, Yu W, Yi P. Metabolism of amino acids in cancer. Front Cell Dev Biol. 2020;8:603837.
Ding M, Bu X, Li Z, Xu H, Feng L, Hu J, et al. NDRG2 ablation reprograms metastatic cancer cells towards glutamine dependence via the induction of ASCT2. Int J Biol Sci. 2020;16(16):3100–15.
MacIntyre DA, Jimenez B, Lewintre EJ, Martin CR, Schafer H, Ballesteros CG, et al. Serum metabolome analysis by 1H-NMR reveals differences between chronic lymphocytic leukaemia molecular subgroups. Leukemia. 2010;24(4):788–97.
Shen Y, Zhang Y, Li W, Chen K, Xiang M, Ma H. Glutamine metabolism: from proliferating cells to cardiomyocytes. Metabolism. 2021;121:154778.
Shanware NP, Mullen AR, DeBerardinis RJ, Abraham RT. Glutamine: pleiotropic roles in tumor growth and stress resistance. J Mol Med. 2011;89(3):229–36.
Mayer RL, Schwarzmeier JD, Gerner MC, Bileck A, Mader JC, Meier-Menches SM, et al. Proteomics and metabolomics identify molecular mechanisms of aging potentially predisposing for chronic lymphocytic leukemia. Mol Cell Proteom. 2018;17(2):290–303.
Galicia-Vazquez G, Smith S, Aloyz R. Del11q-positive CLL lymphocytes exhibit altered glutamine metabolism and differential response to GLS1 and glucose metabolism inhibition. Blood Cancer J. 2018;8(1):13.
Nicklin P, Bergman P, Zhang B, Triantafellow E, Wang H, Nyfeler B, et al. Bidirectional transport of amino acids regulates mTOR and autophagy. Cell. 2009;136(3):521–34.
Eng CH, Yu K, Lucas J, White E, Abraham RT. Ammonia derived from glutaminolysis is a diffusible regulator of autophagy. Sci Signal. 2010;3(119):ra31.
Hua H, Kong Q, Zhang H, Wang J, Luo T, Jiang Y. Targeting mTOR for cancer therapy. J Hematol Oncol. 2019;12(1):71.
Galicia-Vázquez G, Aloyz R. Metabolic rewiring beyond Warburg in chronic lymphocytic leukemia: how much do we actually know? Crit Rev Oncol Hematol. 2019;134:65–70.
Zou S, Wang X, Liu P, Ke C, Xu S. Arginine metabolism and deprivation in cancer therapy. Biomed Pharmacother. 2019;118:109210.
Zhu D, Zhang Y, Wang S. Histone citrullination: a new target for tumors. Mol Cancer. 2021;20(1):90.
Werner A, Pieh D, Echchannaoui H, Rupp J, Rajalingam K, Theobald M, et al. Cationic amino acid transporter-1-mediated arginine uptake is essential for chronic lymphocytic leukemia cell proliferation and viability. Front Oncol. 2019;9:1268.
Debant M, Burgos M, Hemon P, Buscaglia P, Fali T, Melayah S, et al. STIM1 at the plasma membrane as a new target in progressive chronic lymphocytic leukemia. J Immunother Cancer. 2019;7(1):111.
Muggen AF, Pillai SY, Kil LP, van Zelm MC, van Dongen JJ, Hendriks RW, et al. Basal Ca(2+) signaling is particularly increased in mutated chronic lymphocytic leukemia. Leukemia. 2015;29(2):321–8.
van de Donk NW, Janmaat ML, Mutis T, van LammertsBueren JJ, Ahmadi T, Sasser AK, et al. Monoclonal antibodies targeting CD38 in hematological malignancies and beyond. Immunol Rev. 2016;270(1):95–112.
Mele S, Devereux S, Pepper AG, Infante E, Ridley AJ. Calcium-RasGRP2-Rap1 signaling mediates CD38-induced migration of chronic lymphocytic leukemia cells. Blood Adv. 2018;2(13):1551–61.
Debant M, Hemon P, Brigaudeau C, Renaudineau Y, Mignen O. Calcium signaling and cell fate: how can Ca2+ signals contribute to wrong decisions for chronic lymphocytic leukemic B lymphocyte outcome? Int J Dev Biol. 2015;59(7–9):379–89.
Hemon P, Renaudineau Y, Debant M, Le Goux N, Mukherjee S, Brooks W, et al. Calcium signaling: from normal B cell development to tolerance breakdown and autoimmunity. Clin Rev Allergy Immunol. 2017;53(2):141–65.
Laubach K, Zhang J, Chen X. The p53 family: a role in lipid and iron metabolism. Front Cell Dev Biol. 2021;9:715974.
Metzgeroth G, Schultheis B, Kuhn C, Dorn-Beineke A, LaRosée P, Hehlmann R, et al. The soluble transferrin receptor reflects tumor load in chronic lymphocytic leukemia. Clin Chem Lab Med. 2007;45(10):1313–8.
Zhang J, Chen X. p53 tumor suppressor and iron homeostasis. FEBS J. 2019;286(4):620–9.
Wang Y, Yu L, Ding J, Chen Y. Iron metabolism in cancer. Int J Mol Sci. 2018;20(1):95.
Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. 2012;149(5):1060–72.
Ursini F, Maiorino M. Lipid peroxidation and ferroptosis: the role of GSH and GPx4. Free Radic Biol Med. 2020;152:175–85.
Chen X, Kang R, Kroemer G, Tang D. Broadening horizons: the role of ferroptosis in cancer. Nat Rev Clin Oncol. 2021;18(5):280–96.
Cosialls E, El Hage R, Dos Santos L, Gong C, Mehrpour M, Hamaï A. Ferroptosis: cancer stem cells rely on iron until “to die for” it. Cells. 2021;10(11):2981.
Barbato A, Scandura G, Puglisi F, Cambria D, La Spina E, Palumbo GA, et al. Mitochondrial bioenergetics at the onset of drug resistance in hematological malignancies: an overview. Front Oncol. 2020;10:604143.
Auger C, Vinaik R, Appanna VD, Jeschke MG. Beyond mitochondria: alternative energy-producing pathways from all strata of life. Metabolism. 2021;118:154733.
Jitschin R, Hofmann AD, Bruns H, Giessl A, Bricks J, Berger J, et al. Mitochondrial metabolism contributes to oxidative stress and reveals therapeutic targets in chronic lymphocytic leukemia. Blood. 2014;123(17):2663–72.
Roy Chowdhury S, Banerji V. Targeting mitochondrial bioenergetics as a therapeutic strategy for chronic lymphocytic leukemia. Oxid Med Cell Longev. 2018;2018:2426712.
Benfeitas R, Uhlen M, Nielsen J, Mardinoglu A. New challenges to study heterogeneity in cancer redox metabolism. Front Cell Dev Biol. 2017;5:65.
Ashton TM, McKenna WG, Kunz-Schughart LA, Higgins GS. Oxidative phosphorylation as an emerging target in cancer therapy. Clin Cancer Res. 2018;24(11):2482–90.
Ecker V, Stumpf M, Brandmeier L, Neumayer T, Pfeuffer L, Engleitner T, et al. Targeted PI3K/AKT-hyperactivation induces cell death in chronic lymphocytic leukemia. Nat Commun. 2021;12(1):3526.
Carew JS, Nawrocki ST, Xu RH, Dunner K, McConkey DJ, Wierda WG, et al. Increased mitochondrial biogenesis in primary leukemia cells: the role of endogenous nitric oxide and impact on sensitivity to fludarabine. Leukemia. 2004;18(12):1934–40.
Levesque MC, Ghosh DK, Beasley BE, Chen Y, Volkheimer AD, O’Loughlin CW, et al. CLL cell apoptosis induced by nitric oxide synthase inhibitors: correlation with lipid solubility and NOS1 dissociation constant. Leuk Res. 2008;32(7):1061–70.
Capron C, Jondeau K, Casetti L, Jalbert V, Costa C, Verhoeyen E, et al. Viability and stress protection of chronic lymphoid leukemia cells involves overactivation of mitochondrial phosphoSTAT3Ser727. Cell Death Dis. 2014;5:e1451.
Zou S, Tong Q, Liu B, Huang W, Tian Y, Fu X. Targeting STAT3 in cancer immunotherapy. Mol Cancer. 2020;19(1):145.
Adekola KU, Dalva Aydemir S, Ma S, Zhou Z, Rosen ST, Shanmugam M. Investigating and targeting chronic lymphocytic leukemia metabolism with the human immunodeficiency virus protease inhibitor ritonavir and metformin. Leuk Lymphoma. 2015;56(2):450–9.
Vangapandu HV, Alston B, Morse J, Ayres ML, Wierda WG, Keating MJ, et al. Biological and metabolic effects of IACS-010759, an OxPhos inhibitor, on chronic lymphocytic leukemia cells. Oncotarget. 2018;9(38):24980–91.
Ravera S, Ghiotto F, Tenca C, Gugiatti E, Santamaria S, Ledda B, et al. Berberine affects mitochondrial activity and cell growth of leukemic cells from chronic lymphocytic leukemia patients. Sci Rep. 2020;10(1):16519.
Santidrián AF, Cosialls AM, Coll-Mulet L, Iglesias-Serret D, de Frias M, González-Gironès DM, et al. The potential anticancer agent PK11195 induces apoptosis irrespective of p53 and ATM status in chronic lymphocytic leukemia cells. Haematologica. 2007;92(12):1631–8.
Zhang X, Xiang J. Remodeling the microenvironment before occurrence and metastasis of cancer. Int J Biol Sci. 2019;15(1):105–13.
Yu W, Lei Q, Yang L, Qin G, Liu S, Wang D, et al. Contradictory roles of lipid metabolism in immune response within the tumor microenvironment. J Hematol Oncol. 2021;14(1):187.
Gouirand V, Guillaumond F, Vasseur S. Influence of the tumor microenvironment on cancer cells metabolic reprogramming. Front Oncol. 2018;8:117.
Wang C, Luo D. The metabolic adaptation mechanism of metastatic organotropism. Exp Hematol Oncol. 2021;10(1):30.
Jimenez I, Tazon-Vega B, Abrisqueta P, Nieto JC, Bobillo S, Palacio-Garcia C, et al. Immunological and genetic kinetics from diagnosis to clinical progression in chronic lymphocytic leukemia. Biomark Res. 2021;9(1):37.
van Attekum MH, Eldering E, Kater AP. Chronic lymphocytic leukemia cells are active participants in microenvironmental cross-talk. Haematologica. 2017;102(9):1469–76.
Wu X, Fajardo-Despaigne JE, Zhang C, Neppalli V, Banerji V, Johnston JB, et al. Altered T follicular helper cell subsets and function in chronic lymphocytic leukemia. Front Oncol. 2021;11:674492.
Wang YA, Li XL, Mo YZ, Fan CM, Tang L, Xiong F, et al. Effects of tumor metabolic microenvironment on regulatory T cells. Mol Cancer. 2018;17(1):168.
Brusa D, Serra S, Coscia M, Rossi D, D’Arena G, Laurenti L, et al. The PD-1/PD-L1 axis contributes to T-cell dysfunction in chronic lymphocytic leukemia. Haematologica. 2013;98(6):953–63.
Qorraj M, Bruns H, Böttcher M, Weigand L, Saul D, Mackensen A, et al. The PD-1/PD-L1 axis contributes to immune metabolic dysfunctions of monocytes in chronic lymphocytic leukemia. Leukemia. 2017;31(2):470–8.
Ramsay AG, Clear AJ, Fatah R, Gribben JG. Multiple inhibitory ligands induce impaired T-cell immunologic synapse function in chronic lymphocytic leukemia that can be blocked with lenalidomide: establishing a reversible immune evasion mechanism in human cancer. Blood. 2012;120(7):1412–21.
Ramsay AG, Evans R, Kiaii S, Svensson L, Hogg N, Gribben JG. Chronic lymphocytic leukemia cells induce defective LFA-1-directed T-cell motility by altering Rho GTPase signaling that is reversible with lenalidomide. Blood. 2013;121(14):2704–14.
Han TT, Fan L, Li JY, Xu W. Role of chemokines and their receptors in chronic lymphocytic leukemia: function in microenvironment and targeted therapy. Cancer Biol Ther. 2014;15(1):3–9.
Pontikoglou C, Kastrinaki MC, Klaus M, Kalpadakis C, Katonis P, Alpantaki K, et al. Study of the quantitative, functional, cytogenetic, and immunoregulatory properties of bone marrow mesenchymal stem cells in patients with B-cell chronic lymphocytic leukemia. Stem Cells Dev. 2013;22(9):1329–41.
Dubois N, Crompot E, Meuleman N, Bron D, Lagneaux L, Stamatopoulos B. Importance of crosstalk between chronic lymphocytic leukemia cells and the stromal microenvironment: direct contact, soluble factors, and extracellular vesicles. Front Oncol. 2020;10:1422.
Zhang W, Trachootham D, Liu J, Chen G, Pelicano H, Garcia-Prieto C, et al. Stromal control of cystine metabolism promotes cancer cell survival in chronic lymphocytic leukaemia. Nat Cell Biol. 2012;14(3):276–86.
Robak P, Robak T. A targeted therapy for protein and lipid kinases in chronic lymphocytic leukemia. Curr med Chem. 2012;19(31):5294–318.
Brown JR, Byrd JC, Coutre SE, Benson DM, Flinn IW, Wagner-Johnston ND, et al. Idelalisib, an inhibitor of phosphatidylinositol 3-kinase p110δ, for relapsed/refractory chronic lymphocytic leukemia. Blood. 2014;123(22):3390–7.
von Heydebrand F, Fuchs M, Kunz M, Voelkl S, Kremer AN, Oostendorp RAJ, et al. Protein kinase C-beta-dependent changes in the glucose metabolism of bone marrow stromal cells of chronic lymphocytic leukemia. Stem Cells. 2021;39(6):819–30.
Monti P, Menichini P, Speciale A, Cutrona G, Fais F, Taiana E, et al. Heterogeneity of TP53 mutations and P53 protein residual function in cancer: does it matter? Front Oncol. 2020;10:593383.
Moia R, Boggione P, Mahmoud AM, Kodipad AA, Adhinaveni R, Sagiraju S, et al. Targeting p53 in chronic lymphocytic leukemia. Expert Opin Ther Targets. 2020;24(12):1239–50.
Yun X, Zhang Y, Wang X. Recent progress of prognostic biomarkers and risk scoring systems in chronic lymphocytic leukemia. Biomark Res. 2020;8:40.
Wierda WG, Byrd JC, Abramson JS, Bilgrami SF, Bociek G, Brander D, et al. Chronic lymphocytic leukemia/small lymphocytic lymphoma, version 4.2020, NCCN clinical practice guidelines in oncology. J Natl Compr Cancer Netw. 2020;18(2):185–217.
Hallek M, Cheson BD, Catovsky D, Caligaris-Cappio F, Dighiero G, Döhner H, et al. iwCLL guidelines for diagnosis, indications for treatment, response assessment, and supportive management of CLL. Blood. 2018;131(25):2745–60.
Liu J, Zhang C, Hu W, Feng Z. Tumor suppressor p53 and its mutants in cancer metabolism. Cancer Lett. 2015;356(2 Pt A):197–203.
Li T, Kon N, Jiang L, Tan M, Ludwig T, Zhao Y, et al. Tumor suppression in the absence of p53-mediated cell-cycle arrest, apoptosis, and senescence. Cell. 2012;149(6):1269–83.
Liu J, Zhang C, Hu W, Feng Z. Tumor suppressor p53 and metabolism. J Mol Cell Biol. 2019;11(4):284–92.
Yang L, Li A, Lei Q, Zhang Y. Tumor-intrinsic signaling pathways: key roles in the regulation of the immunosuppressive tumor microenvironment. J Hematol Oncol. 2019;12(1):125.
Calvo-Vidal MN, Cerchietti L. The metabolism of lymphomas. Curr Opin Hematol. 2013;20(4):345–54.
Zajac-Kaye M. Myc oncogene: a key component in cell cycle regulation and its implication for lung cancer. Lung Cancer. 2001;34(Suppl 2):S43–6.
Stine ZE, Walton ZE, Altman BJ, Hsieh AL, Dang CV. MYC, metabolism, and cancer. Cancer Discov. 2015;5(10):1024–39.
Wise DR, Thompson CB. Glutamine addiction: a new therapeutic target in cancer. Trends Biochem Sci. 2010;35(8):427–33.
Wang R, Dillon CP, Shi LZ, Milasta S, Carter R, Finkelstein D, et al. The transcription factor Myc controls metabolic reprogramming upon T lymphocyte activation. Immunity. 2011;35(6):871–82.
Morrish F, Noonan J, Perez-Olsen C, Gafken PR, Fitzgibbon M, Kelleher J, et al. Myc-dependent mitochondrial generation of acetyl-CoA contributes to fatty acid biosynthesis and histone acetylation during cell cycle entry. J Biol Chem. 2010;285(47):36267–74.
Edmunds LR, Sharma L, Kang A, Lu J, Vockley J, Basu S, et al. c-Myc programs fatty acid metabolism and dictates acetyl-CoA abundance and fate. J Biol Chem. 2015;290(33):20100.
Parikh SA, Kay NE, Shanafelt TD. How we treat Richter syndrome. Blood. 2014;123(11):1647–57.
Bruzzi JF, Macapinlac H, Tsimberidou AM, Truong MT, Keating MJ, Marom EM, et al. Detection of Richter’s transformation of chronic lymphocytic leukemia by PET/CT. J Nuclear Med. 2006;47(8):1267–73.
Weston VJ, Oldreive CE, Skowronska A, Oscier DG, Pratt G, Dyer MJ, et al. The PARP inhibitor olaparib induces significant killing of ATM-deficient lymphoid tumor cells in vitro and in vivo. Blood. 2010;116(22):4578–87.
Nadeu F, Delgado J, Royo C, Baumann T, Stankovic T, Pinyol M, et al. Clinical impact of clonal and subclonal TP53, SF3B1, BIRC3, NOTCH1, and ATM mutations in chronic lymphocytic leukemia. Blood. 2016;127(17):2122–30.
Brown JR. Insulin receptor activation in deletion 11q chronic lymphocytic leukemia. Clin Cancer Res. 2011;17(9):2605–7.
Rodríguez D, Ramsay AJ, Quesada V, Garabaya C, Campo E, Freije JM, et al. Functional analysis of sucrase-isomaltase mutations from chronic lymphocytic leukemia patients. Hum Mol Genet. 2013;22(11):2273–82.
Rodríguez D, Bretones G, Arango JR, Valdespino V, Campo E, Quesada V, et al. Molecular pathogenesis of CLL and its evolution. Int J Hematol. 2015;101(3):219–28.
Abdel-Wahab AF, Mahmoud W, Al-Harizy RM. Targeting glucose metabolism to suppress cancer progression: prospective of anti-glycolytic cancer therapy. Pharmacol Res. 2019;150:104511.
Kohlhaas V, Blakemore SJ, Al-Maarri M, Nickel N, Pal M, Roth A, et al. Active Akt signaling triggers CLL toward Richter transformation via overactivation of Notch1. Blood. 2021;137(5):646–60.
Brown JR. AKT: a key to RT? Blood. 2021;137(5):582–4.
Rinke J, Chase A, Cross NCP, Hochhaus A, Ernst T. EZH2 in myeloid malignancies. Cells. 2020;9(7):1639.
Mirzaei S, Gholami MH, Hushmandi K, Hashemi F, Zabolian A, Canadas I, et al. The long and short non-coding RNAs modulating EZH2 signaling in cancer. J Hematol Oncol. 2022;15(1):18.
Zhang T, Gong Y, Meng H, Li C, Xue L. Symphony of epigenetic and metabolic regulation-interaction between the histone methyltransferase EZH2 and metabolism of tumor. Clin Epigenet. 2020;12(1):72.
Kosalai ST, Morsy MHA, Papakonstantinou N, Mansouri L, Stavroyianni N, Kanduri C, et al. EZH2 upregulates the PI3K/AKT pathway through IGF1R and MYC in clinically aggressive chronic lymphocytic leukaemia. Epigenetics. 2019;14(11):1125–40.
Jellusova J, Rickert RC. The PI3K pathway in B cell metabolism. Crit Rev Biochem Mol Biol. 2016;51(5):359–78.
Rabello Ddo A, Lucena-Araujo AR, Alves-Silva JC, da Eira VB, de Vasconcellos MC, de Oliveira FM, et al. Overexpression of EZH2 associates with a poor prognosis in chronic lymphocytic leukemia. Blood Cells Mol Dis. 2015;54(1):97–102.
Geng X, Wang F, Tian D, Huang L, Streator E, Zhu J, et al. Cardiac glycosides inhibit cancer through Na/K-ATPase-dependent cell death induction. Biochem Pharmacol. 2020;182:114226.
Yosifov DY, Idler I, Bhattacharya N, Reichenzeller M, Close V, Ezerina D, et al. Oxidative stress as candidate therapeutic target to overcome microenvironmental protection of CLL. Leukemia. 2020;34(1):115–27.
Molica S, Lentini M, Zappalà D, Levato L. Effects of ibrutinib on glucose-lipid metabolism in patients with chronic lymphocytic leukemia (CLL). Leuk Lymphoma. 2020;61(11):2778–80.
Sharif-Askari B, Doyon D, Paliouras M, Aloyz R. Bruton’s tyrosine kinase is at the crossroads of metabolic adaptation in primary malignant human lymphocytes. Sci Rep. 2019;9(1):11069.
Jones CL, Stevens BM, D’Alessandro A, Reisz JA, Culp-Hill R, Nemkov T, et al. Inhibition of amino acid metabolism selectively targets human leukemia stem cells. Cancer Cell. 2018;34(5):724-740.e4.
Stubbins RJ, Maksakova IA, Sanford DS, Rouhi A, Kuchenbauer F. Mitochondrial metabolism: powering new directions in acute myeloid leukemia. Leuk Lymphoma. 2021;62(10):2331–41.
Saito Y, Sawa D, Kinoshita M, Yamada A, Kamimura S, Suekane A, et al. EVI1 triggers metabolic reprogramming associated with leukemogenesis and increases sensitivity to l-asparaginase. Haematologica. 2020;105(8):2118–29.
Tabe Y, Konopleva M. Break the lifeline of AML cells. Blood. 2021;137(25):3465–7.
Saito Y, Kinoshita M, Yamada A, Kawano S, Liu HS, Kamimura S, et al. Mannose and phosphomannose isomerase regulate energy metabolism under glucose starvation in leukemia. Cancer Sci. 2021;112(12):4944–56.
Karunanithi S, Liu R, Hou Y, Gonzalez G, Oldford N, Roe AJ, et al. Thioredoxin reductase is a major regulator of metabolism in leukemia cells. Oncogene. 2021;40(33):5236–46.
Zhang H, Li L, Chen Q, Li M, Feng J, Sun Y, et al. PGC1β regulates multiple myeloma tumor growth through LDHA-mediated glycolytic metabolism. Mol Oncol. 2018;12(9):1579–95.
Abdollahi P, Vandsemb EN, Elsaadi S, Røst LM, Yang R, Hjort MA, et al. Phosphatase of regenerating liver-3 regulates cancer cell metabolism in multiple myeloma. FASEB J. 2021;35(3):e21344.
Panaroni C, Fulzele K, Mori T, Siu KT, Onyewadume C, Maebius A, et al. Multiple myeloma cells induce lipolysis in adipocytes and uptake fatty acids through fatty acid transporter proteins. Blood. 2022;139(6):876–88.
Böttcher M, Baur R, Stoll A, Mackensen A, Mougiakakos D. Linking immunoevasion and metabolic reprogramming in B-cell-derived lymphomas. Front Oncol. 2020;10:594782.
Caro P, Kishan AU, Norberg E, Stanley IA, Chapuy B, Ficarro SB, et al. Metabolic signatures uncover distinct targets in molecular subsets of diffuse large B cell lymphoma. Cancer Cell. 2012;22(4):547–60.
Gooptu M, Whitaker-Menezes D, Sprandio J, Domingo-Vidal M, Lin Z, Uppal G, et al. Mitochondrial and glycolytic metabolic compartmentalization in diffuse large B-cell lymphoma. Semin Oncol. 2017;44(3):204–17.
Mikkilineni L, Whitaker-Menezes D, Domingo-Vidal M, Sprandio J, Avena P, Cotzia P, et al. Hodgkin lymphoma: a complex metabolic ecosystem with glycolytic reprogramming of the tumor microenvironment. Semin Oncol. 2017;44(3):218–25.
Ricci JE, Chiche J. Metabolic reprogramming of non-Hodgkin’s B-cell lymphomas and potential therapeutic strategies. Front Oncol. 2018;8:556.
Zhang L, Yao Y, Zhang S, Liu Y, Guo H, Ahmed M, et al. Metabolic reprogramming toward oxidative phosphorylation identifies a therapeutic target for mantle cell lymphoma. Sci Transl Med. 2019;11(491):eaau1167.
Acknowledgements
Not applicable.
Funding
This study was funded by National Natural Science Foundation (Nos. 82000195, 82070203 and 81770210); Key Research and Development Program of Shandong Province (No. 2018CXGC1213); Translational Research Grant of NCRCH (Nos. 2021WWB02, 2020ZKMB01); Taishan Scholars Program of Shandong Province; Shandong Provincial Natural Science Foundation (No. ZR2020QH094); Shandong Provincial Engineering Research Center of Lymphoma; Technology Development Project of Jinan City (No. 202019182); Academic Promotion Programme of Shandong First Medical University (Nos. 2019QL018, 2020RC007); Shandong Provincial Hospital Youth Talent Plan.
Author information
Authors and Affiliations
Contributions
YN and XY collected related literature and drafted this manuscript. XW and YZ conceived, designed and revised the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
All authors declare no relevant conflicts of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Nie, Y., Yun, X., Zhang, Y. et al. Targeting metabolic reprogramming in chronic lymphocytic leukemia. Exp Hematol Oncol 11, 39 (2022). https://doi.org/10.1186/s40164-022-00292-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40164-022-00292-z