
Abstract
Background
There are no head-to-head clinical studies comparing chimeric antigen receptor (CAR) T-cell therapies for the treatment of relapsed or refractory aggressive large B-cell lymphomas. Naive, indirect comparisons may be inappropriate, as the study designs and patient populations could differ substantially. Matching-adjusted indirect comparisons (MAIC) can reduce many biases associated with indirect comparisons between studies. To determine the comparative efficacy and safety of lisocabtagene maraleucel (liso-cel) to tisagenlecleucel, we describe an unanchored MAIC of the pivotal studies TRANSCEND NHL 001 (TRANSCEND; NCT02631044; liso-cel) and JULIET (NCT02445248; tisagenlecleucel).
Methods
Individual patient data (IPD) from TRANSCEND were available to the authors; for the JULIET pivotal study, summary-level data from the published study were used. To balance the populations between two studies, IPD from TRANSCEND were adjusted to match the marginal distribution (e.g., mean, variance) of clinical factors among patients from JULIET.
Results
Results from the primary MAIC showed liso-cel had statistically significant greater efficacy than tisagenlecleucel (objective response rate: odds ratio [OR] = 2.78, 95% confidence interval [CI]: 1.63‒4.74; complete response rate: OR = 2.01, 95% CI: 1.22‒3.30; progression-free survival: hazard ratio [HR] = 0.65, 95% CI: 0.47‒0.91; overall survival: HR = 0.67, 95% CI: 0.47‒0.95). MAIC of safety outcomes showed lower ORs for all-grade and grade ≥ 3 cytokine release syndrome, and grade ≥ 3 prolonged cytopenia for liso-cel when compared with tisagenlecleucel; there were no statistically significant differences detected for other safety outcomes.
Conclusions
Overall, this MAIC of two CAR T-cell therapies indicates liso-cel had favorable efficacy and a comparable or better safety profile relative to tisagenlecleucel.
Clinical trial registration: ClinicalTrials.gov identifiers: NCT02631044 and NCT02445248.
Similar content being viewed by others


Introduction
Non-Hodgkin lymphoma (NHL) is one of the most common types of cancer worldwide, with reported incidence rates of 6.7 per 100,000 in men and 4.7 per 100,000 in women in 2018 [1]. Diffuse large B-cell lymphoma (DLBCL) represents the most common NHL subtype, accounting for 30‒58% of NHL cases in Europe and 25% of cases in the United States [2, 3]. Between 2011 and 2012, the annual age-adjusted incidence rate of DLBCL was 3.8 per 100,000 persons in Europe and 6.9 per 100,000 persons in the United States [3, 4]. DLBCL can occur as de novo disease or arise as a transformation from other indolent forms of NHL. Treatment options for patients with relapsed or refractory (R/R) DLBCL are limited. These patients often receive salvage chemotherapies that confer poor survival outcomes; 4-year overall survival (OS) rate of 28% and median OS of 6 months in refractory patients [5].
Chimeric antigen receptor (CAR) T-cell therapies have shown clinical activity in patients with R/R large B-cell lymphoma, with objective response rates (ORR) and complete response (CR) rates ranging from 52 to 82% and from 40 to 54%, respectively [6,7,8]. Tisagenlecleucel, axicabtagene ciloleucel (axi-cel), and most recently, lisocabtagene maraleucel (liso-cel) have been approved in the United States for third-line or later treatment of large B-cell lymphoma (LBCL). While tisagenlecleucel and liso-cel utilize an anti-CD19 antigen-binding domain fused with the costimulatory 4-1BB and CD3ζ domains, the former has a CD8 hinge and transmembrane region, whereas the latter has an immunoglobulin G4 hinge region and CD28 transmembrane domain. Axi-cel utilizes an anti-CD19 antigen-binding domain fused to CD28 and CD3ζ costimulatory domains [9,10,11]. All three are single-dose products administered intravenously, though liso-cel has a defined composition of equal CD8+ and CD4+ cells with low variability. Dose and ratio of CD8+ and CD4+ CAR+ T cells may influence the incidence and severity of cytokine release syndrome (CRS) and neurological events (NE) [12,13,14]. It is unclear if the differences of these products affect clinical outcomes.
TRANSCEND NHL 001 (TRANSCEND; NCT02631044) was a phase 1, single-arm, multicenter, open-label study that sought to investigate the efficacy and safety of liso-cel as a treatment in patients with LBCL who have R/R disease after receiving at least two prior lines of therapy.[6] Patients with DLBCL not otherwise specified (de novo, transformed follicular lymphoma, and transformed indolent NHL), high-grade lymphoma with rearrangements in MYC and either BCL2, BCL6, or both, primary mediastinal B-cell lymphoma, and follicular lymphoma grade 3B were eligible if they had R/R positron emission tomography–positive disease after at least two lines of prior systemic therapy, including a CD20-targeted agent and anthracycline; had an Eastern Cooperative Oncology Group performance status (ECOG PS) of 0–2; and adequate organ function. Patients with secondary central nervous system (CNS) lymphoma or prior autologous or allogeneic hematopoietic stem cell transplantation (auto-HSCT or allo-HSCT, respectively) were permitted. However, patients with primary CNS lymphoma or allo-HSCT within 90 days of leukapheresis were excluded. Primary endpoints were adverse events (AE), dose-limiting toxicities, and ORR, as assessed by an independent review committee (IRC) per Lugano 2014 criteria [15]. Secondary endpoints included CR rate as assessed by IRC, duration of response, progression-free survival (PFS), and OS.
JULIET (NCT02445248) was a phase 2, single-arm, multicenter, open-label, registrational study of the efficacy and safety of tisagenlecleucel in patients with R/R LBCL [8]. Eligible patients had DLBCL, high-grade lymphoma with MYC rearrangement plus rearrangement of BCL2, BCL6, or both, or transformed follicular lymphoma; received at least two prior lines of therapy, including rituximab and an anthracycline; and were ineligible for or had disease progression after auto-HSCT. Patients were excluded if they had primary mediastinal B-cell lymphoma, had previously received allo-HSCT, or had secondary CNS lymphoma. The primary endpoint was best ORR, as assessed by IRC per Lugano 2014 criteria [15], and key secondary endpoints included duration of response, OS, and safety. CRS was originally graded according to the University of Pennsylvania criteria, but a secondary analysis aligned to the Lee 2014 criteria, which was used for this comparative analysis [16]. There are no head-to-head clinical studies comparing the CAR T-cell therapies to inform treatment decisions, policy decision-making, and other health care–related issues. Naive, indirect comparisons may be inappropriate, as the study designs and patient populations could differ substantially. Comparing interventions using matching-adjusted indirect comparison (MAIC) analyses can reduce many biases associated with indirect comparisons between studies by adjusting for differences in patient and study characteristics [17]. MAICs are increasingly being included in submissions to regulators and/or health technology assessment agencies. To determine the comparative efficacy and safety of liso-cel versus tisagenlecleucel, we describe a MAIC analysis of the pivotal studies TRANSCEND (liso-cel) and JULIET (tisagenlecleucel).
Methods
Data sources and study characteristics
MAIC methodology was used to estimate population-adjusted relative treatment effects associated with liso-cel compared with tisagenlecleucel. Table 1 summarizes the data sets used, and Table 2 lists study design characteristics and eligibility criteria for TRANSCEND and JULIET.
Patient characteristics
Of the 17 baseline patient characteristics reported in both studies, definitions or minimum/maximum thresholds differed between the studies for nine patient characteristics. Definitions or categorizations of these patient characteristics as used in TRANSCEND were aligned to JULIET either by recategorizing or recalculating the corresponding variables from the TRANSCEND individual patient data (IPD; details presented in Table 2), thereby allowing their inclusion in analyses and reducing bias owing to differences between studies.
Outcomes
All analyses conducted for the patient populations included those patients who were enrolled and received ≥ 1 dose of CAR T cells (ie, were infused). Outcomes of interest included efficacy (ORR, CR rate, PFS, and OS) and safety (CRS per Lee 2014 criteria, NEs per study-specified definitions [including aphasia and encephalopathy], infections, hypogammaglobulinemia, and prolonged cytopenia [defined as grade ≥ 3 cytopenias not resolved by day 29 after infusion]).
Statistical analysis
Relevant clinical factors for matching and adjusting were identified via literature search, which was reviewed by a panel of external clinical experts. A ranked list of clinical prognostic factors and treatment-effect modifiers was derived per outcome by evaluating the strength of association between each clinical factor to each efficacy outcome endpoint (i.e., data-driven rank) using classification-based random forest models for binary outcomes (CR rate and ORR) and survival-based random forest models for time-to-event outcomes (OS and PFS) [18,19,20]. Data-driven ranks were then reviewed by the panel of experts and a final evidence-informed ranked list of factors was determined for each outcome by consolidating expert clinical opinion (Additional file 1: Table S1).
For a given set of ranked clinical prognostic factors and treatment-effect modifiers, separate MAICs were conducted sequentially, adjusting for one additional variable at a time, in order of ranked importance. After fitting each model, the performance and suitability of each MAIC model was assessed based on the following criteria: effective sample size (ESS; a proxy for sample size when patients are weighted, which is required to achieve a given level of precision), distribution of patient weights (wherein the goal is to avoid extreme patient weights), summary statistics (assessment of balance between study populations), and assumption of proportional hazards for OS and PFS. Balance was assessed using the absolute value of the standardized mean difference (SMD) for each covariate, a standard diagnostic for propensity score-based methods that enables comparability across factors and analyses [21]. Primary analyses were selected to strike a balance between these criteria (e.g., by retaining ESS and mitigating extreme patient weights, while adjusting for the most important factors), whereas sensitivity analyses prioritized adjustment of more factors over ESS.
After completing the matching phase of the MAIC, the remaining patients from TRANSCEND were weighted using a method-of-moments propensity score algorithm. Method-of-moments was chosen because only summary level data were available from JULIET and this method would guarantee an exact balancing of clinical factors of interest [22]. Generalized linear models for binary outcomes (i.e., ORR, CR rate, and safety outcomes) were used to estimate odds ratios (OR) and Cox proportional hazards models for time-to-event outcomes (i.e., OS and PFS) were used to estimate hazard ratios (HR).
All analyses were conducted using R Project for Statistical Computing, version 3.6.1 (R Core Team, Vienna, Austria; https://www.r-project.org/).
Results
Clinical factors before and after matching and adjusting
For each efficacy outcome, comparisons of clinical factors at baseline were conducted for TRANSCEND versus JULIET naively, without matching or adjusting infused patients from TRANSCEND. This exercise showed that few factors were similar (i.e., SMD < 0.1) between TRANSCEND and JULIET (Table 3). Notable differences (i.e., SMD ≥ 0.1) were observed for age, ECOG PS score, active secondary CNS lymphoma, disease histology, cell of origin, double or triple hit, prior allo-HSCT and auto-HSCT, bridging therapy, number of prior lines of therapy, R/R to last therapy, pre-lymphodepletion creatinine clearance, left ventricular ejection fraction at screening, and pre-leukapheresis absolute lymphocyte count. The matching phase of the MAIC involved removing TRANSCEND patients with primary mediastinal B-cell lymphoma or follicular lymphoma grade 3B disease histology (efficacy outcomes only), an ECOG PS of 2 at screening, secondary CNS lymphoma, or prior allo-HSCT. Bridging therapy was not matched in this analysis, as both trial protocols permitted the use of bridging therapy per investigator’s discretion. In both primary and sensitivity analyses, matching and adjusting patients from the TRANSCEND to the JULIET population produced substantial improvements in the balance of clinical factors between studies. For example, in the primary analysis of OS, the proportion of ranked clinical factors achieving SMD < 0.1 increased from the naive rate of 17.6% to 41.2%, which further improved in the sensitivity analysis to 88.2%. Factors with SMD < 0.1 after matching and adjusting in the primary analysis included International Prognostic Index score, ECOG PS, active secondary CNS lymphoma, disease histology, prior allo- and auto-HSCT, and R/R to last therapy. Similar improvements in balance were observed in the primary analyses conducted for ORR, CR rate, and PFS (Additional file 1: Tables S2–S4).
There were six clinical prognostic factors and treatment-effect modifiers used in the primary efficacy analyses. While the adjustment factors differed for each efficacy outcome, the matching criteria of disease histology, ECOG PS, secondary CNS lymphoma, and prior allo-HSCT were consistently used across the primary efficacy analyses. All available clinical factors except bridging therapy were adjusted for in the sensitivity analyses (Additional file 1: Table S1). There were four clinical factors adjusted for in the primary safety analysis; three clinical factors (secondary CNS lymphoma, ECOG PS, and prior allo-HSCT) were related to trial eligibility criteria and were used to match the TRANSCEND and JULIET populations. One additional clinical factor (number of prior therapies) was then adjusted to minimize differences between studies in the remaining patients according to the final rank-order for all safety outcomes (Additional file 1: Table S5).
Efficacy analyses
Overall, the results of the MAIC showed a statistically significant greater odds of response for liso-cel than for tisagenlecleucel. Naive ORRs were higher for liso-cel (72.7% [n = 256]) than for tisagenlecleucel (51.6% [n = 93]) (Table 4). This corresponded to significantly greater odds of overall response for liso-cel than for tisagenlecleucel (OR = 2.49, 95% confidence interval [CI]: 1.52‒4.07; P < 0.001). In the primary analysis that matched and adjusted for six factors, liso-cel had an ORR of 74.7% (ESS = 164). The odds of overall response were significantly greater for liso-cel than for tisagenlecleucel (OR = 2.78, 95% CI: 1.63‒4.74; P < 0.001). Similarly, in the sensitivity analysis that matched and adjusted for all available clinical factors except for bridging therapy, liso-cel had an ORR of 80.8% (ESS = 37.3). The odds of overall response were, again, significantly greater for liso-cel than for tisagenlecleucel (OR = 3.95; 95% CI: 1.64‒9.51; P = 0.002).
Naive CR rates were higher for liso-cel (53.1% [n = 256]) than for tisagenlecleucel (39.8% [n = 93]; Table 4). This corresponded to significantly greater odds of CR for liso-cel than for tisagenlecleucel (OR = 1.71, 95% CI: 1.06‒2.78; P = 0.029). In the primary analysis (six factors), liso-cel was associated with a CR rate of 57.0% (ESS = 200.1). The odds of CR were significantly greater for liso-cel than for tisagenlecleucel (OR = 2.01, 95% CI: 1.22‒3.30; P = 0.006). In the sensitivity analysis (all available clinical factors except for bridging therapy), liso-cel had a CR rate of 60.6% (ESS = 37.3). The odds of CR were also significantly greater for liso-cel than for tisagenlecleucel (OR = 2.33; 95% CI: 1.06‒5.10; P = 0.034).
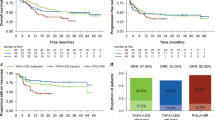
In naive comparisons, liso-cel had a longer median PFS (6.8 months; 95% CI: 3.5‒17.7; N = 256) than tisagenlecleucel (2.8 months; 95% CI: 2.3‒4.2; N = 111). This corresponded to a significantly lower rate of disease progression for liso-cel than for tisagenlecleucel (HR = 0.67, 95% CI: 0.49‒0.91; P = 0.009; Table 5). In the primary analysis (six factors), liso-cel had a median PFS of 6.7 months (95% CI: 3.5‒not reached [NR]; ESS = 149.3). The rate of disease progression was significantly lower for liso-cel than for tisagenlecleucel (HR = 0.65, 95% CI: 0.47‒0.91; P = 0.012; Fig. 1a). In the sensitivity analysis (all available clinical factors except for bridging therapy), the median PFS for liso-cel was 5.9 months (95% CI: 3.1‒NR; ESS = 24.8). Similar to the primary analysis, the rate of disease progression was significantly lower for liso-cel than for tisagenlecleucel (HR = 0.55; 95% CI: 0.32‒0.96; P = 0.035; Table 5).

Kaplan–Meier curves for PFS (a) and OS (b) in infused patients, matched-adjusted comparison (primary analysis). CI confidence interval, ESS effective sample size, liso-cel lisocabtagene maraleucel, NR not reached, OS overall survival, PFS progression-free survival
In naive comparisons, liso-cel had a longer median OS (21.1 months; 95% CI: 13.3‒NR; N = 256) than tisagenlecleucel (11.7 months; 95% CI: 7.2‒NR; N = 111) but the mortality rate was not significantly different between treatments (HR = 0.73, 95% CI: 0.52‒1.02; P = 0.062; Table 5). In the primary analysis (six factors), liso-cel had a median OS of 22.0 months (95% CI: 16.8‒NR; ESS = 180.0). For this comparison, the mortality rate was significantly lower for liso-cel than for tisagenlecleucel (HR = 0.67, 95% CI: 0.47‒0.95; P = 0.026; Fig. 1b). In the sensitivity analysis (all available clinical factors except for bridging therapy), median OS for liso-cel was 19.9 months (95% CI: 9.2‒NR; ESS = 51.0). For this comparison, the mortality rate was not significantly different between liso-cel and tisagenlecleucel (HR = 0.68, 95% CI: 0.42‒1.10; P = 0.115; Table 5).
Safety analyses
Safety analyses were conducted for the infused patient populations (TRANSCEND, N = 269; JULIET, N = 111). After matching and adjusting for four factors, the ORs for most safety endpoints were similar for both treatments or were lower for liso-cel (ESS = 122.9) than for tisagenlecleucel. Specifically, liso-cel had statistically significant lower odds after MAIC of all-grade and grade ≥ 3 CRS and grade ≥ 3 prolonged cytopenia (Table 6).
The naive rate of CRS per Lee 2014 criteria was lower for liso-cel than for tisagenlecleucel (Table 6) and corresponded to significantly lower odds of all-grade (OR = 0.55, 95% CI: 0.35‒0.86; P = 0.009) and grade ≥ 3 (OR = 0.11, 95% CI: 0.04‒0.29; P < 0.001) CRS events for liso-cel. After matching and adjusting (four factors), there were statistically significant lower odds of all-grade (OR = 0.53, 95% CI: 0.32‒0.89; P = 0.016) and grade ≥ 3 (OR = 0.10, 95% CI: 0.03‒0.31; P < 0.001) CRS events for liso-cel than for tisagenlecleucel.
There were no statistically significant differences in study-specific NE rates for liso-cel compared with tisagenlecleucel. The naive rate of NEs was higher for liso-cel than for tisagenlecleucel (Table 6) and corresponded to numerically greater odds of all-grade NEs for liso-cel (OR = 1.59, 95% CI: 0.94‒2.70; P = 0.085). The inverse was true for grade ≥ 3 NEs, for which naive rates were lower for liso-cel than for tisagenlecleucel and corresponded to numerically lower odds of grade 3 events (OR = 0.82, 95% CI: 0.41‒1.65; P = 0.576). After matching and adjusting, the odds of all-grade NEs were numerically greater for liso-cel (OR = 1.36, 95% CI: 0.76‒2.44; P = 0.306) but grade ≥ 3 NEs were numerically lower for liso-cel (OR = 0.79, 95% CI: 0.36‒1.73; P = 0.551).
The naive rates of study-specific NEs of all-grade encephalopathy events were similar for liso-cel and tisagenlecleucel (OR = 1.06, 95% CI: 0.42‒2.67; P = 0.907; Table 6). After matching and adjusting, there were no statistically significant differences in all-grade encephalopathy events. The naive rates of study-specific NEs of all-grade aphasia were numerically higher for liso-cel (OR = 2.88, 95% CI: 0.89‒9.33; P = 0.078). After matching and adjusting, there were no statistically significant differences in all-grade aphasia events.
Rates of laboratory-confirmed grade ≥ 3 prolonged cytopenia were significantly lower for liso-cel for both the naive (OR = 0.53, 95% CI: 0.34‒0.83; P = 0.006) and matching-adjusted (OR = 0.44, 95% CI: 0.26‒0.73; P = 0.002) data sets. There were no statistically significant differences in infection or hypogammaglobulinemia rates between liso-cel and tisagenlecleucel.
Discussion
Liso-cel had favorable efficacy and a comparable or better safety profile relative to tisagenlecleucel after matching and adjusting for important clinical prognostic factors and treatment-effect modifiers in this MAIC. The MAIC approach is a form of population adjustment designed to mitigate between-study differences in eligibility criteria, adjust for between-study differences in baseline characteristics, reconcile differences in varying definitions, and reduce sensitivity to effect measures. An assessment identified 17 clinical factors reported in both TRANSCEND and JULIET that were available for adjustment. The primary efficacy analysis that matched on and adjusted for six clinical factors showed that the odds of response were significantly greater, while the odds of disease progression and mortality were significantly lower for liso-cel than tisagenlecleucel. To assess the robustness of the primary efficacy analysis, sensitivity analyses were conducted by matching and adjusting for all available clinical factors except for bridging therapy, at the expense of ESS. Sensitivity analyses supported the primary findings, except for OS, for which there was no longer a statistically significant greater OS for liso-cel. However, because the sensitivity analyses adjusted for more factors, the corresponding estimates were based on a lower ESS, which produced greater uncertainty in statistical estimates (i.e., wider CIs). Furthermore, a large drop in the sensitivity analysis Kaplan–Meier curve for OS was estimated at around 20 months because of loss to follow-up (i.e., censoring), accentuating a large patient weight in the remaining risk set.
After matching and adjusting for four clinical factors, the ORs for most safety endpoints were similar for both treatments or were lower for liso-cel than for tisagenlecleucel. Importantly, liso-cel had statistically significant lower odds after MAIC of all-grade and grade ≥ 3 CRS and grade ≥ 3 prolonged cytopenia.
Two MAIC analyses assessing the relative efficacy and safety between axi-cel (ZUMA-1) and tisagenlecleucel (JULIET) have been performed. A recently published MAIC by Oluwole et al. matched and adjusted the ZUMA-1 population to JULIET [23]. The authors found that, after adjusting for differences in patient characteristics between studies, axi-cel was associated with a higher ORR and CR rate than tisagenlecleucel among patients who underwent infusion, and OS comparisons favored axi-cel. They also found a higher rate of grades 1–2 CRS in ZUMA-1 compared with JULIET, though similar rates of grade ≥ 3 CRS and study-specific NEs. However, they noted significant limitations that could have led to bias since definitions of relapsed disease differed and they could not account for the impact bridging chemotherapy had on relative outcomes. In contrast, Zhang et al. matched and adjusted the JULIET population to ZUMA-1 [24]. The authors concluded that differences between the JULIET and ZUMA-1 patient populations were substantial, rendering estimates of relative treatment effects (via MAIC or other adjusted indirect treatment comparison methods) unreliable, due to small ESSs, after aligning patient population data sets. For example, matching on bridging therapy alone (0% in ZUMA-1 and > 90% in JULIET) would have resulted in < 10% of patients remaining in IPD from JULIET. Furthermore, the authors discussed sources of bias that could not be accounted for in statistical analyses, such as manufacturing times in the enrollment process, which could undermine the accuracy of indirect treatment comparison estimates.
A recent MAIC analysis comparing efficacy-evaluable patients in JULIET (N = 115; data cutoff February 2020) to TRANSCEND (N = 256; data cutoff August 2019) was conducted to evaluate the comparative efficacy of tisagenlecleucel versus liso-cel, and found no evidence of differences in ORR, CR rate, OS, and PFS between the two CAR T-cell therapies [25, 26]. Several of the following analytical approaches employed by the authors are worth noting: (1) 8 patients who did not receive lymphodepleting chemotherapy and 1 patient with DLBCL misclassification were first removed from the JULIET dataset before analysis (n = 106); (2) TRANSCEND enrolled a broader patient population (e.g., primary mediastinal B-cell lymphoma and follicular lymphoma grade 3B subtypes, ECOG PS of 2, secondary CNS lymphoma, prior allo-HSCT, impaired renal function, no prespecified threshold for blood counts) that could not be emulated using patients enrolled in JULIET; (3) proportion of patients who did not receive bridging therapy in JULIET (n = 11 of 106) was up-weighted from 10.4% to 42.4% to match that from TRANSCEND (n = 106 of 256). MAIC resulted in an ESS of 29 compared with an initial sample of 106 patients in the JULIET study. The low ESS may be because of the initial small sample size of JULIET and/or matching to the proportion of patients receiving bridging therapy in TRANSCEND. A small ESS is an indication that the patient weights are highly variable owing to a lack of population overlap, and that the estimate may be unstable. The distribution of weights themselves should also be examined directly alongside pre- and post-MAIC balance in baseline characteristics (e.g., via SMDs) to diagnose population overlap and to highlight any overly influential individuals. Furthermore, there is an additional challenge to directly compare patients who received bridging therapy in the two studies, since the manufacturing times differed for the two CAR T-cell therapies (median time from enrollment to infusion in JULIET: 54 days [90% of patients received infusions between 30 and 92 days after enrollment] vs median time from leukapheresis to infusion in TRANSCEND: 37 days [range, 27‒224 days]). As bridging therapy was administered at the discretion of the investigator, the shorter time to CAR T-cell availability for liso-cel may have resulted in bridging therapy being administered preferentially to patients with more aggressive or rapidly progressing disease, whereas the longer time to CAR T-cell availability for tisagenlecleucel may have resulted in administering bridging therapy to a broader group of patients, as 90% of patients received bridging therapy in JULIET versus 59% of patients in TRANSCEND. Taken together, this suggests that the author's main findings are unlikely applicable to the intended target population represented by TRANSCEND. Given these limitations, the study design was likely insufficient for detecting clinically relevant effect sizes for the intended comparison. This highlights the importance of compatibility assessment between trials as a first step for an MAIC. As we have shown in our analysis, the broader patient population of TRANSCEND is better suited for matching and adjusting to the JULIET patient population, resulting in a higher degree of alignment for comparisons.
Our MAIC analysis has several notable strengths. Multiple JULIET data sources were evaluated to identify the most compatible cohorts to those available in the TRANSCEND pivotal trial for each outcome. The analysis employed a rigorous, multifaceted process to identify and rank-order clinically relevant factors. Clinical experts rank-ordered factors from most to least important to include in our models, which, when paired with data-driven rankings, resulted in the final list of evidence-informed ranking of factors. Accounting for these challenges to matching and adjustment, the ESS remained robust enough to allow for clinically relevant conclusions about the comparison of these two CAR T-cell therapies. However, there were several limitations we should note. Absence of a common comparator in TRANSCEND and JULIET meant that only an unanchored MAIC could be performed. Given the degree of imbalance between the two studies, it was not feasible to match and adjust on all identified factors without losing substantial ESS. Though rank-ordering the factors helped ensure the most important factors were prioritized for inclusion in the model, only a subset of those identified could be included in the primary analyses. Enrollment and manufacturing times differed between studies; the impact of differences in manufacturing process and time could not be fully accounted for in the analysis, which has been posited as a potentially significant bias factor [24]. The inability to control for the factors of bulky disease and tumor burden may have impacted the overall results. Finally, though sensitivity analyses involving additional clinical factors offered alternative estimated relative treatment effects, they often relied on reduced ESS. This manifested in less-reliable Kaplan–Meier curve estimates at longer follow-up times, where the number-at-risk set is small and estimation was predominantly based upon a few patients. Despite these limitations, it is encouraging that the primary and sensitivity analyses were similar, indicating a statistically significant efficacy advantage for liso-cel compared with tisagenlecleucel, with the exception of the sensitivity analysis for OS.
While the liso-cel and tisagenlecleucel CAR constructs both contain a 4-1BB costimulatory domain, there are differences in the CAR T-cell manufacturing process and composition of the two products. The liso-cel manufacturing process purifies T cells from the leukapheresis to minimize tumor cell residuals and includes T-cell–specific activation for a consistent reduction of non–T-cell impurities. CD8+ and CD4+ cells are positively selected from fresh leukapheresis, and each population is separately activated, transduced, and expanded. Liso-cel is a defined composition product administered as a sequential infusion of separate CD8+ and CD4+ components at equal target doses. Preclinical studies have show that CD4+ cells affect CD8+ effector T-cell expansion, memory formation, trafficking, and cytolytic effector T-cell function [27,28,29], indicating that at least some CD8+ function may be optimized by controlling the dose of CD8+ and CD4+ cell components [30]. In animal models, a 1:1 ratio of CD8+:CD4+ CAR T cells showed improved expansion and activity over treatment with either T-cell component alone [31]. The tisagenlecleucel manufacturing process begins with a frozen leukapheresis sample, after which the vector is introduced into T cells selected from thawed peripheral blood mononuclear cells using CD3/CD28-coated magnetic beads. Unlike the liso-cel manufacturing process, tisagenlecleucel manufacturing does not select for the T-cell subpopulations [32], leading to heterogeneity of the CD8+:CD4+ ratio in the final product. The cellular composition and final cell number vary between individual patient batches. This heterogeneity may contribute to differences in efficacy and safety profiles of the different CAR T-cell products.
Conclusions
In summary, an unanchored MAIC leveraging IPD from TRANSCEND and summary level data from JULIET was used to derive indirect comparisons while accounting for between-study differences in eligibility criteria and baseline characteristics. Overall, after matching and adjusting for important clinical prognostic factors and treatment-effect modifiers, liso-cel had favorable efficacy and a comparable or better safety profile relative to tisagenlecleucel. This analysis, which was bound by the context and limitations of the single-arm studies, does not replace a head-to-head, randomized controlled study, and these results should be further validated in a real-world clinical setting.
Availiability of data and materials
Bristol Myers Squibb policy on data sharing may be found at https://www.bms.com/researchers-and-partners/independent-research/data-sharing-request-process.html.
Abbreviations
- ABC:
-
Activated B cell
- AE:
-
Adverse event
- ALC:
-
Absolute lymphocyte count
- allo-HSCT:
-
Allogeneic hematopoietic stem cell transplantation
- auto-HSCT:
-
Autologous hematopoietic stem cell transplantation
- axi-cel:
-
Axicabtagene ciloleucel
- CAR:
-
Chimeric antigen receptor
- CI:
-
Confidence interval
- CNS:
-
Central nervous system
- CR:
-
Complete response
- CrCl:
-
Creatinine clearance
- CRS:
-
Cytokine release syndrome
- CT:
-
Computed tomography
- DLBCL:
-
Diffuse large B-cell lymphoma
- ECOG PS:
-
Eastern Cooperative Oncology Group performance status
- EMA:
-
European Medicines Agency
- EOS:
-
End of study
- ESS:
-
Effective sample size
- FDA:
-
Food and Drug Administration
- FL3B:
-
Follicular lymphoma grade 3B
- GCB:
-
Germinal center B cell
- HGBCL:
-
High-grade B-cell lymphoma
- HR:
-
Hazard ratio
- IPD:
-
Individual patient data
- IPI:
-
International Prognostic Index
- IRC:
-
Independent review committee
- IV:
-
Intravenous
- LBCL:
-
Large B-cell lymphoma
- liso-cel:
-
Lisocabtagene maraleucel
- LVEF:
-
Left ventricular ejection fraction
- MAIC:
-
Matching-adjusted indirect comparison
- NCI CTCAE:
-
National Cancer Institute Common Terminology Criteria for Adverse Events
- NE:
-
Neurological event
- NHL:
-
Non-Hodgkin lymphoma
- NOS:
-
Not otherwise specified
- NR:
-
Not reached
- OR:
-
Odds ratio
- ORR:
-
Objective response rate
- OS:
-
Overall survival
- PET:
-
Positron emission tomography
- PFS:
-
Progression-free survival
- PMBCL:
-
Primary mediastinal B-cell lymphoma
- R/R:
-
Relapsed or refractory
- SD:
-
Standard deviation
- SMD:
-
Standardized mean difference
- SOC:
-
System Organ Class
- SPD:
-
Sum of the product of perpendicular diameters
- TEAE:
-
Treatment-emergent adverse event
- tFL:
-
Transformed follicular lymphoma
- tiNHL:
-
Transformed from indolent non-Hodgkin lymphoma
- ULN:
-
Upper limit of normal
References
Ferlay J, Colombet M, Soerjomataram I, Mathers C, Parkin DM, Pineros M, et al. Estimating the global cancer incidence and mortality in 2018: GLOBOCAN sources and methods. Int J Cancer. 2019;144:1941–53.
Chihara D, Ito H, Matsuda T, Shibata A, Katsumi A, Nakamura S, et al. Differences in incidence and trends of haematological malignancies in Japan and the United States. Br J Haematol. 2014;164:536–45.
Tilly H, Gomes da Silva M, Vitolo U, Jack A, Meignan M, Lopez-Guillermo A, et al. Diffuse large B-cell lymphoma (DLBCL): ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2015;26(Suppl 5):v116–25.
Teras LR, DeSantis CE, Cerhan JR, Morton LM, Jemal A, Flowers CR. 2016 US lymphoid malignancy statistics by World Health Organization subtypes. CA Cancer J Clin. 2016;66:443–59.
Zelenetz AD, Gordon LI, Abramson JS, Advani RH, Bartlett NL, Caimi PF, et al. NCCN Guidelines Insights: B-Cell Lymphomas, Version 3.2019. J Natl Compr Canc Netw. 2019;17:650–61.
Abramson JS, Palomba ML, Gordon LI, Lunning MA, Wang M, Arnason J, et al. Lisocabtagene maraleucel for patients with relapsed or refractory large B-cell lymphomas (TRANSCEND NHL 001): a multicentre seamless design study. Lancet. 2020;396:839–52.
Locke FL, Ghobadi A, Jacobson CA, Miklos DB, Lekakis LJ, Oluwole OO, et al. Long-term safety and activity of axicabtagene ciloleucel in refractory large B-cell lymphoma (ZUMA-1): a single-arm, multicentre, phase 1–2 trial. Lancet Oncol. 2019;20:31–42.
Schuster SJ, Bishop MR, Tam CS, Waller EK, Borchmann P, McGuirk JP, et al. Tisagenlecleucel in adult relapsed or refractory diffuse large B-cell lymphoma. N Engl J Med. 2019;380:45–56.
KYMRIAH (tisagenlecleucel). [Package insert]. East Hanover, NJ: Novartis Pharmaceuticals Corporation; 2018.
YESCARTA (axicabtagene ciloleucel). [Package insert]. Santa Monica, CA: Kite Pharma, Inc.; 2020.
BREYANZI (lisocabtagene maraleucel). [Prescribing information]. Princeton, NJ: Bristol Myers Squibb; 2021.
DeAngelo DJ, Ghobadi A, Park JH, Dinner SN, Mannis GN, Lunning MA, et al. Clinical outcomes for the phase 2, single-arm, multicenter trial of JCAR015 in adult B-ALL (ROCKET Study). J Immunother Cancer. 2017;5:305–6.
Neelapu SS, Locke FL, Bartlett NL, Lekakis LJ, Miklos DB, Jacobson CA, et al. Axicabtagene ciloleucel CAR T-cell therapy in refractory large B-cell lymphoma. N Engl J Med. 2017;377:2531–44.
Turtle CJ, Hanafi LA, Berger C, Hudecek M, Pender B, Robinson E, et al. Immunotherapy of non-Hodgkin’s lymphoma with a defined ratio of CD8+ and CD4+ CD19-specific chimeric antigen receptor-modified T cells. Sci Transl Med. 2016;8:355ra116.
Cheson BD, Fisher RI, Barrington SF, Cavalli F, Schwartz LH, Zucca E, et al. Recommendations for initial evaluation, staging, and response assessment of Hodgkin and non-Hodgkin lymphoma: the Lugano classification. J Clin Oncol. 2014;32:3059–68.
Schuster SJ, Maziarz RT, Rusch ES, Li J, Signorovitch JE, Romanov VV, et al. Grading and management of cytokine release syndrome in patients treated with tisagenlecleucel in the JULIET trial. Blood Adv. 2020;4:1432–9.
Signorovitch JE, Sikirica V, Erder MH, Xie J, Lu M, Hodgkins PS, et al. Matching-adjusted indirect comparisons: a new tool for timely comparative effectiveness research. Value Health. 2012;15:940–7.
Altmann A, Tolosi L, Sander O, Lengauer T. Permutation importance: a corrected feature importance measure. Bioinformatics. 2010;26:1340–7.
Breiman L. Random forests. Mach Learn. 2001;45:5–32.
Ishwaran H, Kogalur U, Blackstone E, Lauer M. Random survival forests. Ann Appl Stat. 2008;2:841–60.
Austin PC, Stuart EA. Moving towards best practice when using inverse probability of treatment weighting (IPTW) using the propensity score to estimate causal treatment effects in observational studies. Stat Med. 2015;34:3661–79.
Phillippo DM, Ades AE, Dias S, Palmer S, Abrams KR, Welton NJ. NICE DSU Technical Support Document 18: Methods for population-adjusted indirect comparisons in submission to NICE. 2016. Accessed December 13, 2020. https://research-information.bris.ac.uk/ws/portalfiles/portal/94868463/Population_adjustment_TSD_FINAL.pdf. 2016.
Oluwole OO, Jansen JP, Lin VW, Chan K, Keeping S, Navale L, et al. Comparing efficacy, safety, and preinfusion period of axicabtagene ciloleucel versus tisagenlecleucel in relapsed/refractory large B cell lymphoma. Biol Blood Marrow Transplant. 2020;26:1581–8.
Zhang J, Li J, Ma Q, Yang H, Signorovitch J, Wu E. A review of two regulatory approved anti-CD19 CAR T-cell therapies in diffuse large B-cell lymphoma: why are indirect treatment comparisons not feasible? Adv Ther. 2020;37:3040–58.
Schuster SJ, Zhang J, Yang H, Tang W, Martinez-Prieto M, Bollu V, et al. Comparative efficacy of tisagenlecleucel (tisa-cel) and lisocabtagene maraleucel (liso-cel) in patients with relapsed/refractory diffuse large B-cell lymphoma (r/r DLBCL). J Clin Oncol. 2021;39(Suppl 15):7535.
Kersten MJ, Zhang J, Yang H, Agarwal A, Tang W, Martinez-Prieto M, et al. Comparative efficacy of tisagenlecleucel (tisa-cel) and lisocabtagene maraleucel (liso-cel) in patients with relapsed/refractory diffuse large B-cell lymphoma (r/r DLBCL). To be presented at The European Haematology Association 2021 Congress. June 11: Abstract EP510. Available from: https://library.ehaweb.org/eha/2021/eha2021-virtual-congress/325270/marie.jose.kersten.comparative.efficacy.of.tisagenlecleucel.28tisa-cel29.and.html?f=listing%3D1%2Abrowseby%3D8%2Asortby%3D2%2Amedia%3D3%2Ace_id%3D2035%2Alabel%3D21989%2Aot_id%3D25552%2Asearch%3Dtisagenlecleucel%2Amarker%3D1286.
Adusumilli PS, Cherkassky L, Villena-Vargas J, Colovos C, Servais E, Plotkin J, et al. Regional delivery of mesothelin-targeted CAR T cell therapy generates potent and long-lasting CD4-dependent tumor immunity. Sci Transl Med. 2014;6:261ra151.
Bos R, Sherman LA. CD4+ T-cell help in the tumor milieu is required for recruitment and cytolytic function of CD8+ T lymphocytes. Cancer Res. 2010;70:8368–77.
Toes RE, Ossendorp F, Offringa R, Melief CJ. CD4 T cells and their role in antitumor immune responses. J Exp Med. 1999;189:753–6.
Riddell SR, Sommermeyer D, Berger C, Liu LS, Balakrishnan A, Salter A, et al. Adoptive therapy with chimeric antigen receptor-modified T cells of defined subset composition. Cancer J. 2014;20:141–4.
Sommermeyer D, Hudecek M, Kosasih PL, Gogishvili T, Maloney DG, Turtle CJ, et al. Chimeric antigen receptor-modified T cells derived from defined CD8+ and CD4+ subsets confer superior antitumor reactivity in vivo. Leukemia. 2016;30:492–500.
Vairy S, Garcia JL, Teira P, Bittencourt H. CTL019 (tisagenlecleucel): CAR-T therapy for relapsed and refractory B-cell acute lymphoblastic leukemia. Drug Des Devel Ther. 2018;12:3885–98.
European Medicines Agency. Assessment report. Procedure No. EMEA/H/C/004090/0000; 2018. Accessed 13 Dec 2020. https://www.ema.europa.eu/en/documents/assessment-report/kymriah-epar-public-assessment-report_en.pdf.
Kymriah. Summary of product characteristics. Novartis Pharmaceuticals UK Ltd.; 2018. Accessed 13 Dec 2020. https://www.ema.europa.eu/en/documents/assessment-report/kymriah-epar-public-assessment-report_en.pdf.
Kymriah. Summary basis for regulatory action. Novartis Pharmaceuticals UK Ltd.; 2018. Accessed 13 Dec 2020. https://www.fda.gov/media/113215/download.
Lee DW, Gardner R, Porter DL, Louis CU, Ahmed N, Jensen M, et al. Current concepts in the diagnosis and management of cytokine release syndrome. Blood. 2014;124:188–95.
Abramson JS, Palomba ML, Gordon LI, Lunning MA, Wang MA, Arnason JE, et al. Pivotal safety and efficacy results from Transcend NHL 001, a multicenter phase 1 study of lisocabtagene maraleucel (liso-cel) in relapsed/refractory (R/R) large B cell lymphomas. Blood. 2019;134(Suppl 1):241.
Acknowledgements
This study was funded by Bristol Myers Squibb. All authors contributed to and approved the manuscript; writing and editorial assistance were provided by Jeremy Henriques, PhD, of The Lockwood Group (Stamford, CT, USA), funded by Bristol Myers Squibb. Presented at the 64th American Society of Hematology Annual Meeting & Exposition.
Funding
The analysis was funded by Bristol Myers Squibb.
Author information
Authors and Affiliations
Contributions
DGM and GC designed research, analyzed and interpreted data, and wrote and reviewed the manuscript. CPF, JH, JK, and DL analyzed and interpreted data and contributed to manuscript writing. AB and YZ performed research, contributed to collection, analysis, and interpretation of data, performed statistical analysis, and wrote/reviewed the manuscript. FFL designed and performed research, analyzed and interpreted data, and wrote and reviewed the manuscript. AK collected, analyzed, and interpreted data, performed statistical analysis, and wrote/reviewed the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Both TRANSCEND and JULIET were conducted in accordance with the Declaration of Helsinki, International Conference on Harmonization Good Clinical Practice guidelines, institutional review boards at participating institutions approved the study protocols and amendments, and all patients provided written informed consent.
Consent for publication
All the authors have signed the form of consent to publication.
Competing interests
Guillaume Cartron has received consultancy fees from Celgene, a Bristol-Myers Squibb Company, and F. Hoffmann-La Roche; and honoraria from AbbVie, Celgene, a Bristol-Myers Squibb Company, F. Hoffmann-La Roche, Gilead Sciences, Janssen, and Sanofi. Christopher P. Fox has received honoraria from AbbVie, Adienne, AstraZeneca, Atara Biotherapeutics, Celgene, a Bristol-Myers Squibb Company, Genmab, Gilead Sciences, Incyte, Roche, Sunesis Pharmaceuticals, and Takeda; and grants for research from AbbVie, Adienne, Gilead Sciences, Roche, and Takeda. Fei Fei Liu, Ana Kostic, and Daniel Li are employees of Bristol Myers Squibb and hold stock in Bristol Myers Squibb. Jens Hasskarl was an employee of Celgene, a Bristol-Myers Squibb Company, at the time of this analysis and may hold stock in Bristol Myers Squibb. Ashley Bonner and Yixie Zhang are employees of EVERSANA, which received funding from Bristol Myers Squibb to conduct the analyses. David G. Maloney reports scientific advisory board membership for A2 Biotherapeutics for which he receives consultancy fees; equity holdings in A2 Biotherapeutics for which he has stock options; honoraria from Amgen, BioLineRx, Bristol Myers Squibb, Celgene, a Bristol-Myers Squibb Company, Genentech, Gilead Sciences, Janssen, Juno Therapeutics, a Bristol-Myers Squibb Company, Kite Pharma, a Gilead Company, Legend Biotech, MorphoSys, Novartis, and Pharmacyclics; intellectual property patents with Juno, a Bristol-Myers Squibb Company (not licensed, no royalties); and research funding paid directly to his institution from Celgene, a Bristol-Myers Squibb Company, Juno Therapeutics, a Bristol-Myers Squibb Company, and Kite Pharma, a Gilead Company. John Kuruvilla reports consultancy from AbbVie, Bristol Myers Squibb, Gilead, Karyopharm, Merck, Roche, and Seattle Genetics; honoraria from Amgen, Antengene, AstraZeneca, Celgene, a Bristol-Myers Squibb Company, Gilead, Janssen, Karyopharm, Merck, Novartis, Pfizer, Roche, Seattle Genetics, and TG Therapeutics; and research funding from AstraZeneca, Janssen, and Roche.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Table S1
. Clinical factors included for primary and sensitivity analyses comparing liso-cel with tisagenlecleucel. Table S2. Comparison of clinical factors before and after MAIC for primary and sensitivity analyses of PFS in TRANSCEND and JULIET. Table S3. Comparison of clinical factors before and after MAIC for primary and sensitivity analyses of CR in TRANSCEND and JULIET. Table S4. Comparison of clinical factors before and after MAIC for primary and sensitivity analyses of ORR in TRANSCEND and JULIET. Table S5. Comparison of clinical factors before and after MAIC for safety analysis in TRANSCEND and JULIET.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Cartron, G., Fox, C.P., Liu, F.F. et al. Matching-adjusted indirect treatment comparison of chimeric antigen receptor T-cell therapies for third-line or later treatment of relapsed or refractory large B-cell lymphoma: lisocabtagene maraleucel versus tisagenlecleucel. Exp Hematol Oncol 11, 17 (2022). https://doi.org/10.1186/s40164-022-00268-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40164-022-00268-z