
Abstract
Hematopoietic stem cell transplantation-associated thrombotic microangiopathy (HSCT-TMA) is a life-threatening syndrome that occurs in adult and pediatric patients after hematopoietic stem cell transplantation. Nonspecific symptoms, heterogeneity within study populations, and variability among current diagnostic criteria contribute to misdiagnosis and underdiagnosis of this syndrome. Hematopoietic stem cell transplantation and associated risk factors precipitate endothelial injury, leading to HSCT-TMA and other endothelial injury syndromes such as hepatic veno-occlusive disease/sinusoidal obstruction syndrome, idiopathic pneumonia syndrome, diffuse alveolar hemorrhage, capillary leak syndrome, and graft-versus-host disease. Endothelial injury can trigger activation of the complement system, promoting inflammation and the development of endothelial injury syndromes, ultimately leading to organ damage and failure. In particular, the lectin pathway of complement is activated by damage-associated molecular patterns (DAMPs) on the surface of injured endothelial cells. Pattern-recognition molecules such as mannose-binding lectin (MBL), collectins, and ficolins—collectively termed lectins—bind to DAMPs on injured host cells, forming activation complexes with MBL-associated serine proteases 1, 2, and 3 (MASP-1, MASP-2, and MASP-3). Activation of the lectin pathway may also trigger the coagulation cascade via MASP-2 cleavage of prothrombin to thrombin. Together, activation of complement and the coagulation cascade lead to a procoagulant state that may result in development of HSCT-TMA. Several complement inhibitors targeting various complement pathways are in clinical trials for the treatment of HSCT-TMA. In this article, we review the role of the complement system in HSCT-TMA pathogenesis, with a focus on the lectin pathway.
Similar content being viewed by others

Background
Over the last 50 years, hematopoietic stem cell transplantation (HSCT) has evolved into the standard of care for patients with otherwise fatal hematologic, metabolic, and neoplastic disorders [1, 2]. However, physical, chemical, and immunologic stressors during the transplantation process (conditioning regimens, radiotherapy, chemotherapy, immunosuppressive drugs, cytokines released during the engraftment process, and allogeneic reactions of donor-derived immune cells) perturb endothelial cells, precipitating endothelial injury [3]. Microvascular endothelial injury following HSCT places patients at risk for long-term organ damage and death [3].
Endothelial injury has been shown to be centrally involved in the pathophysiology of several HSCT-associated conditions [2, 4], collectively termed endothelial injury syndromes (EIS). EIS include hepatic veno-occlusive disease/sinusoidal obstruction syndrome, idiopathic pneumonia syndrome, diffuse alveolar hemorrhage, capillary leak syndrome, graft-versus-host disease (GVHD), and thrombotic microangiopathy (TMA) [2, 4]. These conditions are not discrete diseases, but different clinical manifestations stemming from endothelial injury. Thus, these syndromes often overlap in presentation and classification [2, 5]. In HSCT-associated TMA (HSCT-TMA), endothelial injury commonly affects the kidneys as well as the gastrointestinal tract, lungs, heart, central nervous system, and, rarely, the retina [3, 6]. These manifestations typically occur during the first 6 months after HSCT [7, 8] and reflect sites of underlying tissue damage [2].
Endothelial injury triggers activation of the complement system—significantly through the lectin pathway—via altered cell-surface patterns on injured endothelial cells, initiating an inflammatory response [7]. Activation of the lectin pathway further triggers the coagulation cascade [9]. Both complement activation and the coagulation cascade lead to a procoagulant state that reduces vascular integrity, promotes platelet adhesion, increases vasodilation, and promotes leukocyte infiltration [3]. Subsequent thrombus formation and tissue injury can lead to organ-specific damage, multiorgan dysfunction, or death [10].
Here we discuss evidence for the role of lectin pathway activation in endothelial injury-associated complications of HSCT and how targeting complement activity may provide therapeutic benefit for patients with HSCT-TMA.
Endothelial injury syndromes and clinical manifestations
Normal function of endothelial cells
Endothelial cells fulfill essential homeostatic functions within the circulatory system, as they form the contact interface between blood and the perfused tissues and organs [10]. Normally, a balance of procoagulant and anticoagulant pathways is poised to react to injury and to attenuate any imbalance before tissue damage [10]. Endothelial cells control these pathways by maintaining vascular tone and platelet activation, and by regulating prothrombotic and thrombolytic events [10]. In addition, endothelial cells mediate immune functions: healthy endothelium is responsible for diapedesis of leukocytes into injured and inflamed tissues and platelet-leukocyte interactions [10, 11].
Endothelial injury
In damaged endothelium, blood components such as C-reactive protein (CRP) enter the interstitial space and induce inflammation [10]. Impaired endothelial functions increase risk of disease by abrogating normal immune response, vascular tone, and transport of electrolytes and fluid [2]. Persistent activation of endothelium can lead to a procoagulant state, increasing the risk of stasis and endothelial injury [3, 12]. The resulting injury may cause organ damage and thrombosis, which can lead to arteriosclerosis, peripheral vascular disease, stroke, and hypertension [10]. Risk of endothelial injury is increased by diabetes, obesity, hypertension, and environmental factors such as smoking [10, 13, 14].
Endothelial injury is common in HSCT. Conditioning regimens (including chemotherapy and/or radiation) prior to transplantation, agents given for prevention of acute GVHD such as calcineurin or mammalian target of rapamycin (mTOR) inhibitors, infection, and the graft-versus-host reaction itself can all contribute to endothelial injury [3, 15]. The site and severity of endothelial injury associated with HSCT determine the presentation and classification of the syndrome (Table 1) [2].
Clinical manifestations of endothelial injury
We present EIS as different clinical manifestations stemming from the common underlying pathophysiology of endothelial injury, rather than as distinct diseases. Damage due to endothelial injury may occur throughout the body, with overlap in target sites due to similar characteristics across EIS [2]. The reported incidence of EIS in HSCT recipients varies widely due to differences among current diagnostic criteria and heterogeneity within study populations, especially in adults [12].
In some EIS, platelets adhere to endothelium and aggregate to form microthrombi [15]. These thrombi can cause thrombocytopenia and intravascular hemolysis due to red cell fragmentation, leading to tissue hypoxemia and organ damage and failure [15]. Hepatic veno-occlusive disease (VOD)/sinusoidal obstruction syndrome (SOS) occurs when sinusoidal endothelial injury from the conditioning regimen leads to hepatic central vein or sinusoidal thrombosis [16]. Extravasation of blood cells and release of cellular debris into the space of Disse further result in extraluminal compression of sinusoidal vessels, causing sinusoidal obstruction, portal hypertension, sodium-avid fluid retention, ascites, painful hepatomegaly from hepatic capsular distention, and jaundice [17]. Ultimately, multiorgan dysfunction and death can occur [16, 17].
Another of the organ-specific EIS, idiopathic pulmonary syndrome (IPS), is an umbrella term to describe any noninfectious disorder of the lungs characterized by multifocal acute lung injury, shortness of breath, cough, and hypoxemia that occurs within the first four months after HSCT [18]. The pathophysiology of IPS is not completely understood [12], and responses to high doses of corticosteroids are suboptimal [19]. Tumor necrosis factor-α (TNF-α) may play a role in the pathogenesis of IPS and the TNF inhibitor etanercept has been evaluated for management of IPS in combination with corticosteroids, although results were not definitively conclusive [20]. A subset of patients with IPS develop diffuse alveolar hemorrhage (DAH), a form of pulmonary injury that occurs at the time of engraftment, wherein diagnosis is established by the appearance of increasing bloody returns on serial bronchiolar lavage fluids [19]. DAH pathology is thought to be associated with damage to pulmonary arterioles, capillaries, venules, and the alveolar-capillary basement membrane [3].
EIS with systemic presentations include engraftment syndrome (ES), capillary leak syndrome (CLS), and fluid overload. ES occurs at the time of neutrophil engraftment after HSCT, and is believed to involve the unbridled release of proinflammatory cytokines, degranulation products, and activation of complement, leading to systemic endothelial injury [21]. ES usually manifests as fevers, shortness of breath from pulmonary vascular leak, and transient rash that generally respond well to a short course of corticosteroids [21]. CLS is characterized by the loss of intravascular fluids into interstitial spaces due to endothelial injury [22]. In fluid overload, fluid retention and weight gain may require ongoing diuretic therapy or be associated with organ dysfunction [5].
GVHD is the most commonly expected complication of allogeneic HSCT [23]. Acute GVHD is mediated by donor T lymphocytes targeting host tissue, causing unspecified vascular inflammation and endothelial injury early after HSCT, while chronic GVHD tends to occur later and is mediated by a complex interplay between donor effector and regulatory T cells, B cells, and tissue macrophages [2, 23]. Both acute and chronic GVHD can establish conditions that increase the risk of HSCT-TMA, while the presence of HSCT-TMA increases the risk of GVHD [2, 3, 24]. Endothelial injury may represent a common link between GVHD and HSCT-TMA [25]: it is considered the common denominator for both conditions, but excessive complement activation distinguishes HSCT-TMA from GVHD [26].
HSCT-TMA, also known as transplant-associated thrombotic microangiopathy (TA-TMA), occurs when endothelial injury and microthrombus formation cause microangiopathic hemolytic anemia, thrombocytopenia, and organ damage [3]. Reported incidence rates for HSCT-TMA range from 3 to 39% in children [24, 27,28,29], and from 4 to 68% in adults [8, 30,31,32,33,34,35,36]. Differing awareness of HSCT-TMA and screening practices across institutions may reflect differences in identifying the condition, rather than variable incidence in the population. In addition, these reported rates may underestimate the incidence of HSCT-TMA given the nonspecific symptoms, variability among current diagnostic criteria, and heterogeneity within study populations, which have contributed to misdiagnosis and underdiagnosis of the disorder [37,38,39,40,41].
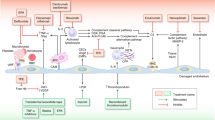
The pathophysiology of HSCT-TMA is believed to occur in three phases (Fig. 1). The initiation phase of HSCT-TMA pathogenesis results from endothelial injury caused by immunosuppressive agents such as calcineurin or mTOR inhibitors, acute GVHD, infection, or conditioning with cytotoxic agents and/or total body irradiation [15]. During the progression phase, complement activation including the lectin pathway, which is activated by altered carbohydrate and acetylated ligand patterns on injured endothelial cells, causes additional damage, particularly in the microvasculature [7, 15]. In the thrombosis phase, platelets aggregate and form microthrombi, causing consumptive thrombocytopenia, hemolysis from red cell fragmentation, and organ damage [15].

Role of the complement system, including the lectin pathway, in pathophysiology of HSCT-TMA [7, 15]. In Phase 1 (Initiation), factors associated with hematopoietic stem cell transplantation such as calcineurin and mTOR inhibitors, acute graft-versus-host disease, infection, or total body irradiation lead to endothelial injury. In Phase 2 (Progression), the lectin pathway of complement is activated and complement proteins cause further endothelial injury, leading to platelet aggregation and microthrombi formation. In Phase 3 (Thrombosis), further microthrombi formation and mechanical damage lead to HSCT-TMA, organ damage, and organ failure. HSCT-TMA hematopoietic stem cell transplantation-associated thrombotic microangiopathy, mTOR mammalian target of rapamycin
Endothelial injury activates the complement system
A cascade of cellular distress signals can trigger complement activation, leading to a plethora of biologic events: activation of the cellular immune system, chemotactic direction of immune cells to sites of injury, proinflammatory stimulation of leukocytes, promotion of cell‒cell interactions, and the generation of lytic or sublytic membrane attack complexes (MACs) that lyse or mark targeted cells [42, 43]. In a balanced system, the three complement activation pathways—the classical, lectin, and alternative pathways (Fig. 2)—eliminate or clear infection or damaged host cells [42, 43]. When the complement system is dysregulated, excessive complement activation results in damage [44].

Complement activation pathways [42, 48, 52]. The three complement activation pathways—the classical, lectin, and alternative pathways—eliminate or clear infection or damaged host cells. The classical pathway initiates complement activation through antibody binding to immune complexes. The lectin pathway is initiated when pattern-recognition molecules bind to certain molecular patterns presented on damaged, malignant or distressed self-tissue or on microbes. The alternative pathway acts as an amplification loop of the classical or lectin pathways. All three pathways converge to mediate cleavage of C3, leading to initiation of the terminal pathway and assembly of the MAC. The coagulation cascade can be activated via MASP-2 cleavage of prothrombin to thrombin and cleavage of Factor XII to XIIa. MAC membrane attack complex, MASP mannan-binding lectin-associated serine proteases, MBL mannose-binding lectin
Complement pathways
The classical pathway initiates complement activation through antibody binding to immune complexes: the globular heads of C1q bind to the Fc portion of IgM or to clusters of IgG fixed to antigens [43]. C1q may also identify foreign surfaces containing proteoglycan patterns (e.g., chondroitin sulfate [serglycin]) [43]. When C1q binds a target, the C1q-associated protease C1r is activated through a conformational change to cleave its only substrate C1s, which in turn cleaves and activates complement proteins C4 and C2 [43]. The resultant complex C4b2b (formerly C4b2a) is known as C3 convertase [45]. The classical pathway was thought to be the predominant pathway activated by endothelial injury [46, 47], but subsequent studies have elucidated that endothelial injury primarily activates the lectin pathway of complement [48,49,50,51].
Activation of the lectin pathway, a scavenger system, is initiated when pattern-recognition molecules (PRMs) bind to certain molecular patterns presented on damaged, malignant or distressed self-tissue, or ligands on microbes [43, 52]. Specific molecular patterns exposed on the surface of necrotic, apoptotic, distressed or otherwise injured host cells are termed damage-associated molecular patterns (DAMPs) and those on microbes are termed pathogen-associated molecular patterns [43, 52]. PRMs may also bind fragments and debris of viruses. In HSCT, highly cytotoxic treatment leaves behind many injured endothelial cells that trigger lectin pathway activation.
The lectin pathway-activating carbohydrate pattern-binding class of PRMs known as lectins include mannose-binding lectin (MBL), collectins (CL-10 and CL-11), and ficolins (ficolin-1, ficolin-2, and ficolin-3), which bind to specific ligands on bacteria, viruses, and injured cells [53]. Lectin pathway-specific proteases known as MBL-associated serine proteases 1, 2, and 3 (MASP-1, MASP-2, and MASP-3) form activation complexes with the collectins and ficolins [53]. Juxtaposition of discrete PRM/MASP activation complexes initiates lectin pathway activation [54]; binding of these activation complexes to DAMPs in close proximity to each other facilitates the conversion of MASPs from their zymogen form into their enzymatically active form [53, 55,56,57]. Cleavage of C4 and C2 via MASP-2 results in formation of C3 convertase [53, 55,56,57]. Notably, MASP-2 is the only MASP that can cleave both C2 and C4; hence, in the absence of MASP-2, the lectin pathway cannot generate C3 convertase [58,59,60,61].
The alternative pathway balances a low-grade steady state of activation with the ability to respond to damage or infection [43, 52]. Factor D cleaves Factor B associated with C3b or C3(H2O), generating a C3 convertase [C3bBb or C3(H2O)Bb] [62, 63]. MASP-3 was recently shown to be essential for alternative pathway functional activity, as it is required to convert pro-Factor D into its enzymatically active form [63,64,65,66]. Hence, in the absence of MASP-3 functional activity, the alternative pathway is deficient [63, 65, 66]. The alternative pathway primarily acts as an amplification loop of the classical or lectin pathways, triggered by formation of C3b [59, 64,65,66]. Spontaneous hydrolysis of C3 to C3(H2O) allows for continuous turnover of C3 and generation of C3 convertase to initiate the alternative pathway [43]. Crosstalk between different pathways of complement supports a rapid response to triggers, and tight regulation prevents collateral damage [60, 67]. A single study observed that properdin may act as a PRM and bind DAMPs on injured host cells, initiating complement activation via the alternative pathway [68]. However, no follow-up studies have been published that corroborate a functional role of properdin as a PRM.
All three pathways converge to mediate the cleavage of C3 into C3a and C3b. C3a, along with C5a, is a potent anaphylatoxin with proinflammatory, prothrombotic, and chemotactic functions that trigger leukocyte recruitment and cytokine production [42, 53]. Accumulation of C3b on the cell surface leads to opsonization of debris and bacteria for clearance [42]. Binding of C3b to C3 convertase forms C5 convertase (C4b2b3b from the classical/lectin pathway, or C3bBb3b from the alternative pathway), which cleaves C5 into C5a and C5b, initiating the terminal pathway [42]. C5b recruits C6, C7, C8, and multiple C9s, resulting in formation of the MAC [42]. The MAC promotes further endothelial damage and may result in apoptosis [69].
Induction of a procoagulant state can be initiated not only by C3a and C5a but also via MASP-2 cleavage of prothrombin to thrombin; MASP-2 can activate the coagulation cascade via activation of prothrombin [70] and by the cleavage of Factor XII to XIIa [71], and activation of the cascade drives fibrin deposition and clot formation [9, 72]. The coagulation cascade [70]—in concert with endothelial damage arising from complement activation—leads to thrombosis, which can result in stroke, hypertension, and peripheral vascular disease [10].
Endothelial injury triggers lectin pathway activation in related diseases
As the lectin pathway of complement is activated in response to endothelial injury, evidence for complement activation has been demonstrated in ischemia of various organs, including the kidney and heart [48, 73, 74]. Direct activation of the lectin pathway was identified in animal models of ischemia/reperfusion associated with skeletal muscle, intestinal, myocardial, and kidney injuries [51]. In certain instances of kidney ischemia, the alternative pathway amplifies cleavage of C3 after initiation through the lectin pathway [51, 75]. Activation of the lectin pathway is triggered by local CL-11 upregulation in post-ischemic kidney tissue [76], and CL-11–driven lectin pathway activation may play a central role in tubulointerstitial injury broadly associated with proteinuric renal diseases [77, 78]. Lectin pathway activity also occurs during progression of ischemic brain damage [79,80,81,82,83], with the lectin pathway recognition molecules MBL [84, 85] and ficolin-3 [82] identified as independent predictors of ischemic stroke outcome. Furthermore, MASP-2 was found to play a crucial role in ischemia/reperfusion of a murine model: MASP-2‒deficient mice were protected against myocardial and gastrointestinal injury arising from ischemia/reperfusion [58]. In a later study, use of an anti–MASP-2 antibody in a murine model conferred cardioprotection against myocardial infarction [86].
Rapidly developing research in severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) has demonstrated the role of endothelial injury in COVID-19. Endothelial injury in patients with COVID-19 activates the complement system, leading to thrombosis and acute respiratory distress syndrome (ARDS) [87]. Microvascular deposits of C5b-9, C4d, and MASP-2 in patients with COVID-19 suggest the role of the lectin pathway in proinflammatory sequelae [87]. Lectin pathway PRMs such as MBL, ficolin-2, and CL-11 bind to spike and nucleocapsid (N) proteins of SARS-CoV-2, contributing to subsequent lectin pathway-mediated deposition of C3b and C4b [88]. Moreover, N proteins within the coronavirus family, including SARS-CoV, MERS-CoV, and SARS-CoV-2, are bound to MASP-2 through an evolutionarily conserved motif [88, 89]. This interaction results in hyperactivation of the complement system and suggests that the lectin pathway is a promising target for coronavirus-induced pneumonia [89]. In small case series, anti-complement therapies have been associated with COVID-19 improvements [89,90,91,92,93]. The C5 inhibitor eculizumab resulted in recovery and reduced mean CRP levels in four patients with COVID-19 [92], although the global Phase 3 clinical trial evaluating the longer-acting C5 inhibitor ravulizumab was recently discontinued for lack of efficacy [94]. The C5a inhibitor BDB-001 has shown promising results in patients with severe COVID-19 [95]. The C3 inhibitor AMY-101 resulted in successful treatment of a patient with severe ARDS due to COVID-19 pneumonia [91] and is being investigated for the management of ARDS caused by COVID-19 in a Phase 2 placebo-controlled trial [96], although a Phase 1/2 trial of the C3 inhibitor APL-9, a pegylated form of AMY-101, for the treatment of severe COVID-19 was recently discontinued due to lack of efficacy [97]. The MASP-2 inhibitor narsoplimab was associated with rapid and sustained reduction of circulating endothelial cell counts and serum IL-6, IL-8, CRP, and lactate dehydrogenase (LDH) in six patients with COVID-19 on mechanical ventilation, correlating with clinical improvement [93].
Collectively, these data suggest a key role for complement, and particularly the lectin pathway, in the pathogenesis of diseases associated with endothelial injury.
Complement activation underlies the pathology and diagnosis of HSCT-TMA
Complement activation plays an essential role in the pathology of EIS, including HSCT-TMA. The “Three-Hit Hypothesis” (Table 2) outlines sequential risks that facilitate development and progression of HSCT-TMA [7, 15, 98]. The first “hit” toward HSCT-TMA comprises inherent or nonmodifiable risk factors, such as underlying predisposition to complement activation via genetic risk factors or prior endothelial injury [15]. The second “hit” involves transplant-associated risk factors, such as cytotoxic conditioning regimens during HSCT that cause endothelial injury [7, 15]. The third “hit” toward development of HSCT-TMA includes post-transplant risk factors (medications, GVHD, infection, and/or circulating antibodies) that may initiate complement activation [7, 15].
Evidence for the role of complement in HSCT-TMA can be found in both pediatric and adult populations. Elevated serum levels of the terminal complement complex C5b-9 are observed in both children and adults with HSCT-TMA [24, 29, 99,100,101,102]. In addition, a higher level of complement activation is detected in sera or plasma from patients with HSCT-TMA compared with patients without TMA after HSCT [26, 103].
Genetic evidence indicates a role for complement in HSCT-TMA
Genetic abnormalities in complement system proteins and regulatory components are associated with increased complement activity and risk of TMA in HSCT recipients [102, 104]. Recent genetic analysis demonstrated that adult patients with HSCT-TMA possessed significantly more pathogenic, rare variants in regulatory and coding regions of ADAMTS13, C3, CFB, CFH, CFI, CD46, CFHR3, CD55, and THBD than non-TMA HSCT control recipients [102]. These variants were detected in approximately three fifths of patients who did not respond to conventional treatment and approximately two thirds of patients who died from transplant-associated complications [102].
Genetic variations have also been detected in pediatric HSCT-TMA populations. In an analysis of 17 candidate genes known to participate in complement activation, variations were identified in 65% of patients with HSCT-TMA versus 9% of non-TMA HSCT controls [104]. Incidence of HSCT-TMA and number of gene variants were both higher in nonwhite versus white HSCT recipients [104]. In a different study, transcriptome analyses collected before HSCT, at onset of HSCT-TMA, and after resolution of HSCT-TMA in children showed upregulation of all three complement pathways during active HSCT-TMA that then returned to normal levels after treatment with eculizumab [105].
Diagnostic and prognostic markers in HSCT-TMA
Pediatric diagnostic criteria for HSCT-TMA are relatively well established [29]; however, diagnostic criteria in adults are less clear due to the lack of robust natural history studies [37,38,39,40,41]. Variability across current guidelines for HSCT-TMA diagnosis is shown in Table 3, demonstrating the need for universally accepted diagnostic criteria for HSCT-TMA in adults. Moreover, uniform standards for diagnosis and prognosis of HSCT-TMA may expand understanding of markers for disease onset and progression [29] and would be important for clinical management [3].
Tissue histology remains the gold standard in HSCT-TMA diagnosis, but the inherent risk involved with biopsy for HSCT recipients has led clinicians to seek other less-invasive diagnostic markers [106]. Based on results of pediatric studies, the earliest indications of endothelial injury can occur 10 to 14 days before HSCT-TMA diagnosis [29]. In pediatric populations, hypertension, proteinuria, and elevated serum LDH levels have emerged as early signals of HSCT-TMA [29, 30]. Grade II to IV acute GVHD is also an independent risk factor for pediatric HSCT-TMA [30].
Increased markers of endothelial and complement activation correlate with TMA and other EIS after HSCT. Immunohistochemistry of complement has been used to diagnose and characterize HSCT-TMA [26, 106,107,108]. Deposition of C5b-9 and C4d have been observed in blood vessels and organs of patients with HSCT-TMA [106, 108], and these patients have higher levels of soluble C5b-9 and endothelial activation markers (e.g., thrombomodulin) compared with patients without HSCT-related complications [26, 29]. Another method to identify patients with increased complement activity is the modified Ham test, which has been used to show a higher level of complement activation in patients with HSCT-TMA versus control recipients of HSCT [26, 103, 109]. Finally, significantly higher levels of MASP-2 have been reported in patients with TMAs after allogeneic HSCT [110].
Suppressor of tumorigenicity 2 (ST2) is another molecular marker under consideration for diagnosis of HSCT-TMA [25]. When measured 14 days after HSCT, elevated ST2 is associated with an increased risk of HSCT-TMA in pediatric and young-adult recipients of HSCT [25]. ST2 is also associated with treatment resistance and death in patients with GVHD [111, 112]. In adults, preliminary evidence from the Mount Sinai Acute GVHD International Consortium (MAGIC) suggests that a combined test for ST2 and regenerating islet-derived 3α (REG3α) may predict development of HSCT-TMA [113]. When measurement of these two biomarkers from blood samples taken one week after allogeneic HSCT was used to determine risk category (high vs low), HSCT-TMA developed in seven of 18 high-risk patients and 13 of 88 low-risk patients (p = 0.041) [113]. Other diagnostic tests in development include measuring levels of thrombomodulin, calpain, and haptoglobin degradation product [7].
A few prognostic markers have been identified in pediatric populations, but further characterization is necessary in adult recipients of HSCT. Initial approaches to characterize early symptom patterns in both adults and children during HSCT-TMA may be associated with better treatment outcomes [108]. In pediatric patients predicted to have a moderate risk of HSCT-TMA, elevated soluble C5b-9 levels were associated with higher risk of mortality than was nephrotic range proteinuria [24]. Expression of ST2, a possible diagnostic marker of HSCT-TMA as previously discussed, before HSCT was also found to be a prognostic marker for one-year nonrelapse mortality and severe GVHD [112].
The Endothelial Activation and Stress Index (EASIX)—a composite measure of LDH, creatinine, and platelet levels—has demonstrated prognostic value for risk of death in patients with GVHD [114,115,116]. In adult patients with GVHD and HSCT-TMA, soluble C5b-9 levels were strongly associated with EASIX score 100 days after transplant and at last follow-up [115]. When EASIX was calculated before conditioning, the score was a significant prognostic factor for HSCT-TMA and was predictive of overall survival after HSCT, independent of other assessments [114]. EASIX score before conditioning also correlated with biomarkers of endothelial homeostasis (e.g., CXCL8/IL-18 and free IL-18) [114].
Established risk factors for mortality associated with HSCT-TMA reflect the multifactorial nature of this condition [15, 33]. In pediatric recipients of HSCT, proteinuria and plasma levels of soluble C5b-9 are negatively associated with survival: 1-year survival rates are significantly lower in patients with elevated plasma C5b-9 levels or proteinuria 30 mg/dL or higher [29]. Furthermore, the antecedent conditions of HSCT-TMA affect survival rates: patients with idiopathic or drug-related HSCT-TMA have longer survival after diagnosis than patients with other precipitating events [33]. Additional risk factors for mortality may include anemia (hemoglobin below 9 g/dL), liver dysfunction, and gastrointestinal bleeding [30]. Intestinal TMA following HSCT emerges as a distinct condition from HSCT-TMA that results in higher mortality rates [117] and is an unfavorable predictor of overall survival by multivariate analysis (p = 0.048) [118].
Treatment of HSCT-TMA
Standard of care
The standard of care for HSCT-TMA aims to resolve the physiologic stress that leads to complement activation and endothelial injury. For patients who do not meet the high-risk criteria for HSCT-TMA, recommended management strategies include reducing or withdrawing calcineurin inhibitor treatment (following risk–benefit assessment) and providing supportive care [7, 108]. For patients with high-risk HSCT-TMA, these measures do not markedly improve survival: one study reported similar rates of hematologic resolution in patients withdrawn from calcineurin inhibitor treatment versus those who continued calcineurin inhibitor treatment (28% vs 29%, respectively) [33]. Hazard ratios for death were not appreciably different between the two groups even after adjusting for covariates [33].
Therapeutic plasma exchange (TPE) historically has been used as an urgent treatment for TMAs, including HSCT-TMA [119, 120]. In complement-mediated TMAs, TPE is thought to remove activated complement components and replenish complement regulators [120]. Early initiation of TPE in ten patients with HSCT-TMA unresponsive to conventional care was associated with laboratory resolution of microangiopathy in nine patients and improved kidney function and HSCT-TMA survival in five patients, suggesting that early TPE may be beneficial in selected patients [119]. However, many patients with HSCT-TMA do not respond to TPE, and the long-term results of TPE in complement-mediated TMAs are poor [7, 121].
Defibrotide is approved by the FDA and EMA for treatment of VOD/SOS [122, 123] and due to its endothelial protective properties, several retrospective studies and case series have investigated defibrotide for the treatment of HSCT-TMA. Outcomes have been varied: an early retrospective study reported death of all five pediatric HSCT-TMA patients treated with defibrotide [124], while another study reported response to defibrotide in four of five children and two of seven adults with HSCT-TMA [125]. In a large retrospective study of 539 HSCT recipients, 64 of whom developed HSCT-TMA, defibrotide treatment was associated with a favorable outcome based on univariate analysis (p = 0.02) [126]. More recent retrospective surveys have found response rates to defibrotide ranging from 65 to 77% in adult and pediatric patients with HSCT-TMA [127, 128]. In a case series, three adults with HSCT-TMA responded to treatment with low-dose defibrotide (< 10 mg/kg/day), although one later died of sepsis [129].
Targeting the complement system
Reducing or inhibiting complement activity has shown promise in selected patients with HSCT-TMA during clinical trials (Table 4). Several therapeutics under clinical investigation target the terminal pathway by inhibiting C5, while one targets the lectin pathway via inhibition of MASP-2. These potential treatments are discussed in further detail below. Additional complement inhibitors that have been approved or under development for other complement-mediated diseases [130] may also be relevant in HSCT-TMA, but their clinical utility in this syndrome is currently not known.
The monoclonal antibody eculizumab binds C5 to prevent cleavage into C5a and C5b, inhibiting generation of C5a (one of two primary complement anaphylatoxins) and terminal complement activity [131]. Eculizumab has been studied in small trials and case series with mixed outcomes for adult and pediatric patients [24, 101, 132,133,134,135,136]. Data supporting treatment of adult patients with HSCT-TMA are limited; in two case reports, adult patients with HSCT-TMA had hematologic and/or renal responses to eculizumab treatment [133, 134]. A retrospective analysis of nine adults and three children treated with eculizumab for TMA showed a hematologic response in six patients, including four adults [135]. In a different case series, initial hematologic responses to eculizumab were observed, but long-term overall survival was poor in adult patients [136]. Despite limitations of availability and early initiation, complement inhibition seems to offer improved survival compared to best available treatment so far. Better patient selection might help to identify patients who are in need of a complement inhibitor or patients who might be resistant to eculizumab [102, 137].
Among pediatric populations, the largest eculizumab trial to date established a diagnostic protocol to identify patients with high-risk HSCT-TMA, which has the poorest prognosis [24, 138]. Across pediatric patients (N = 64), 64% had measurable responses, 56% achieved complete remission, and there were no HSCT-TMA relapses during the study [24]. Patients with high-risk HSCT-TMA and elevated complement activation showed poorer outcomes after eculizumab treatment (odds ratio = 0.15, p = 0.0014) and received more doses of eculizumab (r = 0.43, p = 0.0004) [24]. Taken together, these results indicate that early initiation of eculizumab treatment and adjustment of dosing seems to be beneficial in this pediatric subpopulation. Since patients with HSCT-TMA suffer from multiple comorbidities, long-term effects of eculizumab require further investigation.
Ravulizumab (ALXN1210) is a C5 inhibitor engineered from eculizumab that possesses a longer terminal half-life, allowing for extended dosing intervals [139]. Like eculizumab, ravulizumab binds C5 with high affinity [140]. In a Phase 3 trial for atypical hemolytic uremic syndrome, ravulizumab treatment resulted in complete TMA response in 54% of patients with no unexpected adverse events [139]. Phase 3 trials for ravulizumab in both adult and pediatric populations with HSCT-TMA are currently ongoing [141, 142].
Nomacopan (formerly Coversin) is a bifunctional inhibitor of C5 and leukotriene B4 that blocks terminal complement activity [143]. In a case report of pediatric HSCT-TMA resistant to eculizumab (due to a C5 variant), nomacopan showed promising results. Daily treatment with nomacopan improved LDH levels and reticulocyte count and decreased classical pathway hemolytic assay (CH50) below the lower limit of normal [144]. A two-part Phase 3 trial evaluating dosing and efficacy of nomacopan for pediatric HSCT-TMA is ongoing [145].
Narsoplimab (OMS721) is a fully human immunoglobulin gamma 4 (IgG4) monoclonal antibody that binds MASP-2, the effector enzyme of the lectin pathway, and thereby blocks lectin-mediated complement activation [110]. Targeting the lectin pathway without affecting the function of the classical pathway maintains the body’s ability to adopt adaptive immune defense mechanisms for protection against encapsulated organisms, such as Neisseria meningitis [146]. Narsoplimab was studied in adult patients with severe HSCT-TMA (N = 28) in a single-arm, open-label trial [147]. Overall, 61% of patients (17/28) who received at least one dose of narsoplimab achieved a response based on improvement in laboratory markers and organ function, and 74% of patients (17/23) who received per-protocol narsoplimab (at least four doses) responded to treatment [148]. One-hundred–day survival post-HSCT-TMA diagnosis was 68% among all patients, 83% among the per-protocol population, and 94% among responders [148]. The most commonly reported adverse events were fever, diarrhea, vomiting, nausea, neutropenia, fatigue, and hypokalemia [148]. These results suggest efficacy and safety of narsoplimab as treatment for HSCT-TMA.
In patients with HSCT-TMA, there is an inherent risk of infection due to intensive conditioning regimens and prolonged immunosuppression associated with the transplantation process. HSCT recipients should be given vaccinations according to published guidelines [149], although there is limited evidence regarding the efficacy of meningococcal vaccinations in HSCT patients receiving terminal complement inhibitors [150, 151]. Antibiotic prophylaxis should be considered for patients receiving C5 inhibitors despite vaccination status [151]. Following HSCT, and in those receiving complement inhibitors for HSCT-TMA, it is critical that patients are closely monitored for infection.
Conclusions
The characterization of endothelial injury and complement activity in human diseases has improved our understanding of HSCT-TMA and other EIS. Genetic, histologic, and clinical evidence supports the “Three-Hit Hypothesis” for HSCT-TMA, demonstrating that pre-existing physiologic conditions as well as peri-transplant events and immunologic agents add to the risk of endothelial injury in patients undergoing HSCT.
Currently, there is limited knowledge of the natural history of HSCT-TMA in adult patients. A better understanding of the clinical course of HSCT-TMA in both adult and pediatric patients is needed to provide appropriate treatment. Diagnostic and prognostic markers will be important for distinguishing between patients who may benefit from supportive care versus anti-complement therapy.
Understanding the pathophysiology of complement activation in EIS, including activation of the lectin pathway, has provided an opportunity for evidence-based and mechanism-based, targeted therapy for HSCT-TMA. Preliminary single-arm clinical trials evaluating complement inhibitors for treatment of severe HSCT-TMA have provided promising results for this life-threatening condition. Lectin pathway inhibitors hold potential for treatment of HSCT-TMA.
Availability of data and materials
Not applicable.
Abbreviations
- ARDS:
-
Acute respiratory distress syndrome
- CH50:
-
Classical pathway hemolytic assay
- CL:
-
Collectin
- CLS:
-
Capillary leak syndrome
- CRP:
-
C-reactive protein
- DAH:
-
Diffuse alveolar hemorrhage
- DAMP:
-
Damage-associated molecular pattern
- EASIX:
-
Endothelial Activation and Stress Index
- EIS:
-
Endothelial injury syndromes
- ES:
-
Engraftment syndrome
- GVHD:
-
Graft-versus-host disease
- HSCT:
-
Hematopoietic stem cell transplantation
- HSCT-TMA:
-
Hematopoietic stem cell transplantation-associated thrombotic microangiopathy
- IgG4:
-
Immunoglobulin gamma 4
- IPS:
-
Idiopathic pulmonary syndrome
- LDH:
-
Lactate dehydrogenase
- MAC:
-
Membrane attack complex
- MAGIC:
-
Mount Sinai Acute GVHD International Consortium
- MASP:
-
Mannan-binding lectin-associated serine protease
- MBL:
-
Mannose-binding lectin
- mTOR:
-
Mammalian target of rapamycin
- PRM:
-
Pattern-recognition molecule
- REG3α:
-
Regenerating islet-derived 3α
- SARS-CoV-2:
-
Severe acute respiratory syndrome coronavirus 2
- SOS:
-
Sinusoidal obstruction syndrome
- ST2:
-
Suppressor of tumorigenicity 2
- TA-TMA:
-
Transplant-associated thrombotic microangiopathy
- TMA:
-
Thrombotic microangiopathy
- TNF-α:
-
Tumor necrosis factor-α
- TPE:
-
Therapeutic plasma exchange
- VOD:
-
Veno-occlusive disease
References
Storb R. HSCT: Historical Perspective. In: Carreras E, Dufour C, Mohty M, Kröger N, editors. The EBMT handbook: hematopoietic stem cell transplantation and cellular therapies. Cham (CH): Springer International Publishing; 2019. p. 3–9.
Carreras E, Diaz-Ricart M. The role of the endothelium in the short-term complications of hematopoietic SCT. Bone Marrow Transplant. 2011;46(12):1495–502.
Pagliuca S, Michonneau D, de Fontbrune FS, del Galy AS, Xhaard A, Robin M, et al. Allogeneic reactivity-mediated endothelial cell complications after HSCT: a plea for consensual definitions. Blood Adv. 2019;3(15):2424–35.
Carreras E, Palomo M, Ricart MD, Martínez-Sánchez J, on behalf of the Barcelona Endothelium Team. Vascular endothelial syndromes after HCT: 2020 update. Bone Marrow Transplant. 2020;55(10):1885–7.
Rondon G, Saliba RM, Chen J, Ledesma C, Alousi AM, Oran B, et al. Impact of fluid overload as new toxicity category on hematopoietic stem cell transplantation outcomes. Biol Blood Marrow Transplant. 2017;23(12):2166–71.
Castillo P, Voigt K, Crockett C, Stout JT, Krance R, Naik S. Purtscher-like retinopathy: a rare presentation of hematopoietic stem cell transplant-associated thrombotic microangiopathy. Pediatr Transplant. 2019;23(2):e13344.
Dvorak CC, Higham C, Shimano KA. Transplant-associated thrombotic microangiopathy in pediatric hematopoietic cell transplant recipients: a practical approach to diagnosis and management. Front Pediatr. 2019;7:133.
Postalcioglu M, Kim HT, Obut F, Yilmam OA, Yang J, Byun BC, et al. Impact of thrombotic microangiopathy on renal outcomes and survival after hematopoietic stem cell transplantation. Biol Blood Marrow Transplant. 2018;24(11):2344–53.
Gulla KC, Gupta K, Krarup A, Gal P, Schwaeble WJ, Sim RB, et al. Activation of mannan-binding lectin-associated serine proteases leads to generation of a fibrin clot. Immunology. 2010;129(4):482–95.
Rajendran P, Rengarajan T, Thangavel J, Nishigaki Y, Sakthisekaran D, Sethi G, et al. The vascular endothelium and human diseases. Int J Biol Sci. 2013;9(10):1057–69.
Mai J, Virtue A, Shen J, Wang H, Yang X-F. An evolving new paradigm: endothelial cells—conditional innate immune cells. J Hematol Oncol. 2013;6(1):61.
Hildebrandt GC, Chao N. Endothelial cell function and endothelial-related disorders following haematopoietic cell transplantation. Br J Haematol. 2020;190(4):508–19.
Engin A. Endothelial dysfunction in obesity. In: Engin AB, Engin A, editors. Obesity and lipotoxicity. Cham: Springer International Publishing; 2017. p. 345–79.
Saeid G, Lars E, Ismail L. Smoking and endothelial dysfunction. Curr Vasc Pharmacol. 2020;18(1):1–11.
Khosla J, Yeh AC, Spitzer TR, Dey BR. Hematopoietic stem cell transplant-associated thrombotic microangiopathy: current paradigm and novel therapies. Bone Marrow Transplant. 2018;53(2):129–37.
Corbacioglu S, Carreras E, Ansari M, Balduzzi A, Cesaro S, Dalle JH, et al. Diagnosis and severity criteria for sinusoidal obstruction syndrome/veno-occlusive disease in pediatric patients: a new classification from the European Society for Blood and Marrow Transplantation. Bone Marrow Transplant. 2018;53(2):138–45.
Mohty M, Malard F, Abecassis M, Aerts E, Alaskar AS, Aljurf M, et al. Revised diagnosis and severity criteria for sinusoidal obstruction syndrome/veno-occlusive disease in adult patients: a new classification from the European Society for Blood and Marrow Transplantation. Bone Marrow Transplant. 2016;51(7):906–12.
Afessa B, Litzow MR, Tefferi A. Bronchiolitis obliterans and other late onset non-infectious pulmonary complications in hematopoietic stem cell transplantation. Bone Marrow Transplant. 2001;28(5):425–34.
Afessa B, Tefferi A, Litzow MR, Krowka MJ, Wylam ME, Peters SG. Diffuse alveolar hemorrhage in hematopoietic stem cell transplant recipients. Am J Respir Crit Care Med. 2002;166(5):641–5.
Yanik GA, Horowitz MM, Weisdorf DJ, Logan BR, Ho VT, Soiffer RJ, et al. Randomized, double-blind, placebo-controlled trial of soluble tumor necrosis factor receptor: enbrel (etanercept) for the treatment of idiopathic pneumonia syndrome after allogeneic stem cell transplantation: Blood and Marrow Transplant Clinical Trials Network protocol. Biol Blood Marrow Transplant. 2014;20(6):858–64.
Spitzer TR. Engraftment syndrome: double-edged sword of hematopoietic cell transplants. Bone Marrow Transplant. 2015;50(4):469–75.
Lucchini G, Willasch AM, Daniel J, Soerensen J, Jarisch A, Bakhtiar S, et al. Epidemiology, risk factors, and prognosis of capillary leak syndrome in pediatric recipients of stem cell transplants: a retrospective single-center cohort study. Pediatr Transplant. 2016;20(8):1132–6.
Ghimire S, Weber D, Mavin E, Wang XN, Dickinson AM, Holler E. Pathophysiology of GvHD and other HSCT-related major complications. Front Immunol. 2017;8:79.
Jodele S, Dandoy CE, Lane A, Laskin BL, Teusink-Cross A, Myers KC, et al. Complement blockade for TA-TMA: lessons learned from a large pediatric cohort treated with eculizumab. Blood. 2020;135(13):1049–57.
Rotz SJ, Dandoy CE, Davies SM. ST2 and endothelial injury as a link between GVHD and microangiopathy. N Engl J Med. 2017;376(12):1189–90.
Gavriilaki E, Chrysanthopoulou A, Sakellari I, Batsis I, Mallouri D, Touloumenidou T, et al. Linking complement activation, coagulation, and neutrophils in transplant-associated thrombotic microangiopathy. Thromb Haemost. 2019;119(9):1433–40.
Schoettler M, Lehmann LE, Margossian S, Lee M, Kean LS, Kao PC, et al. Risk factors for transplant-associated thrombotic microangiopathy and mortality in a pediatric cohort. Blood Adv. 2020;4(11):2536–47.
Dandoy CE, Rotz SR, Alonso PB, Lane A, Higham C, Dvorak CC, et al. Incidence and outcomes of patients with thrombotic microangiopathy after transplant: results of prospective screening through a multi-institutional collaborative. Biol Blood Marrow Transplant. 2020;26(3 Supplement):S92.
Jodele S, Davies SM, Lane A, Khoury J, Dandoy C, Goebel J, et al. Diagnostic and risk criteria for HSCT-associated thrombotic microangiopathy: a study in children and young adults. Blood. 2014;124(4):645–53.
Ye Y, Zheng W, Wang J, Hu Y, Luo Y, Tan Y, et al. Risk and prognostic factors of transplantation-associated thrombotic microangiopathy in allogeneic haematopoietic stem cell transplantation: a nested case control study. Hematol Oncol. 2017;35(4):821–7.
Zeisbrich M, Becker N, Benner A, Radujkovic A, Schmitt K, Beimler J, et al. Transplant-associated thrombotic microangiopathy is an endothelial complication associated with refractoriness of acute GvHD. Bone Marrow Transplant. 2017;52(10):1399–405.
Sridharan M, Go RS, Shah MV, Litzow MR, Hogan WJ, Alkhateeb HB. Incidence and mortality outcomes in allogeneic transplant-associated thrombotic microangiopathy. Biol Blood Marrow Transplant. 2018;24(3):S329–30.
Li A, Wu Q, Davis C, Kirtane KS, Pham PD, Sorror ML, et al. Transplant-associated thrombotic microangiopathy is a multifactorial disease unresponsive to immunosuppressant withdrawal. Biol Blood Marrow Transplant. 2019;25(3):570–6.
Kraft S, Bollinger N, Bodenmann B, Heim D, Bucher C, Lengerke C, et al. High mortality in hematopoietic stem cell transplant-associated thrombotic microangiopathy with and without concomitant acute graft-versus-host disease. Bone Marrow Transplant. 2019;54(4):540–8.
Sakellari I, Gavriilaki E, Boussiou Z, Batsis I, Mallouri D, Constantinou V, et al. Transplant-associated thrombotic microangiopathy: an unresolved complication of unrelated allogeneic transplant for hematologic diseases. Hematol Oncol. 2017;35(4):932–4.
Wall SA, Zhao Q, Yearsley M, Blower L, Agyeman A, Ranganathan P, et al. Complement-mediated thrombotic microangiopathy as a link between endothelial damage and steroid-refractory GVHD. Blood Adv. 2018;2(20):2619–28.
Ho VT, Cutler C, Carter S, Martin P, Adams R, Horowitz M, et al. Blood and Marrow Transplant Clinical Trials Network toxicity committee consensus summary: thrombotic microangiopathy after hematopoietic stem cell transplantation. Biol Blood Marrow Transplant. 2005;11(8):571–5.
Ruutu T, Barosi G, Benjamin RJ, Clark RE, George JN, Gratwohl A, et al. Diagnostic criteria for hematopoietic stem cell transplant-associated microangiopathy: results of a consensus process by an International Working Group. Haematologica. 2007;92(1):95–100.
Cho BS, Yahng SA, Lee SE, Eom KS, Kim YJ, Kim HJ, et al. Validation of recently proposed consensus criteria for thrombotic microangiopathy after allogeneic hematopoietic stem-cell transplantation. Transplantation. 2010;90(8):918–26.
Shayani S, Palmer J, Stiller T, Liu X, Thomas SH, Khuu T, et al. Thrombotic microangiopathy associated with sirolimus level after allogeneic hematopoietic cell transplantation with tacrolimus/sirolimus-based graft-versus-host disease prophylaxis. Biol Blood Marrow Transplant. 2013;19(2):298–304.
Uderzo CC, Jodele S, Missiry ME, Ciceri F, Busca A, Bacigalupo A, et al. Transplant-associated thrombotic microangiopathy (TA-TMA) and consensus based diagnostic and therapeutic recommendations: which TA-TMA patients to treat and when? J Bone Marrow Res. 2014;2:152.
Merle NS, Church SE, Fremeaux-Bacchi V, Roumenina LT. Complement system part I—molecular mechanisms of activation and regulation. Front Immunol. 2015;6:262.
Eriksson O, Mohlin C, Nilsson B, Ekdahl KN. The human platelet as an innate immune cell: interactions between activated platelets and the complement system. Front Immunol. 2019;10:1590.
Ricklin D, Reis ES, Lambris JD. Complement in disease: a defence system turning offensive. Nat Rev Nephrol. 2016;12(7):383–401.
Bohlson SS, Garred P, Kemper C, Tenner AJ. Complement nomenclature-deconvoluted. Front Immunol. 2019;10:1308.
Buerke M, Prüfer D, Dahm M, Oelert H, Meyer J, Darius H. Blocking of classical complement pathway inhibits endothelial adhesion molecule expression and preserves ischemic myocardium from reperfusion injury. J Pharmacol Exp Ther. 1998;286(1):429–38.
Weiser MR, Williams JP, Moore FD Jr, Kobzik L, Ma M, Hechtman HB, et al. Reperfusion injury of ischemic skeletal muscle is mediated by natural antibody and complement. J Exp Med. 1996;183(5):2343–8.
Collard CD, Vakeva A, Morrissey MA, Agah A, Rollins SA, Reenstra WR, et al. Complement activation after oxidative stress: role of the lectin complement pathway. Am J Pathol. 2000;156(5):1549–56.
Hart ML, Ceonzo KA, Shaffer LA, Takahashi K, Rother RP, Reenstra WR, et al. Gastrointestinal ischemia-reperfusion injury is lectin complement pathway dependent without involving C1q. J Immunol. 2005;174(10):6373–80.
Gorsuch WB, Chrysanthou E, Schwaeble WJ, Stahl GL. The complement system in ischemia-reperfusion injuries. Immunobiology. 2012;217(11):1026–33.
Farrar CA, Asgari E, Schwaeble WJ, Sacks SH. Which pathways trigger the role of complement in ischaemia/reperfusion injury? Front Immunol. 2012;3:341.
Ricklin D, Mastellos DC, Reis ES, Lambris JD. The renaissance of complement therapeutics. Nat Rev Nephrol. 2018;14(1):26–47.
Garred P, Genster N, Pilely K, Bayarri-Olmos R, Rosbjerg A, Ma YJ, et al. A journey through the lectin pathway of complement-MBL and beyond. Immunol Rev. 2016;274(1):74–97.
Degn SE, Kjaer TR, Kidmose RT, Jensen L, Hansen AG, Tekin M, et al. Complement activation by ligand-driven juxtaposition of discrete pattern recognition complexes. Proc Natl Acad Sci. 2014;111(37):13445–50.
Matsushita M, Fujita T. Activation of the classical complement pathway by mannose-binding protein in association with a novel C1s-like serine protease. J Exp Med. 1992;176(6):1497–502.
Thiel S, Vorup-Jensen T, Stover CM, Schwaeble W, Laursen SB, Poulsen K, et al. A second serine protease associated with mannan-binding lectin that activates complement. Nature. 1997;386(6624):506–10.
Dahl MR, Thiel S, Matsushita M, Fujita T, Willis AC, Christensen T, et al. MASP-3 and its association with distinct complexes of the mannan-binding lectin complement activation pathway. Immunity. 2001;15(1):127–35.
Schwaeble WJ, Lynch NJ, Clark JE, Marber M, Samani NJ, Ali YM, et al. Targeting of mannan-binding lectin-associated serine protease-2 confers protection from myocardial and gastrointestinal ischemia/reperfusion injury. Proc Natl Acad Sci. 2011;108(18):7523–8.
Dobo J, Schroeder V, Jenny L, Cervenak L, Zavodszky P, Gal P. Multiple roles of complement MASP-1 at the interface of innate immune response and coagulation. Mol Immunol. 2014;61(2):69–78.
Dobo J, Pal G, Cervenak L, Gal P. The emerging roles of mannose-binding lectin-associated serine proteases (MASPs) in the lectin pathway of complement and beyond. Immunol Rev. 2016;274(1):98–111.
Matsushita M, Thiel S, Jensenius JC, Terai I, Fujita T. Proteolytic activities of two types of mannose-binding lectin-associated serine protease. J Immunol. 2000;165(5):2637–42.
Oroszlan G, Kortvely E, Szakacs D, Kocsis A, Dammeier S, Zeck A, et al. MASP-1 and MASP-2 do not activate pro-Factor D in resting human blood, whereas MASP-3 is a potential activator: kinetic analysis involving specific MASP-1 and MASP-2 inhibitors. J Immunol. 2016;196(2):857–65.
Dobó J, Szakács D, Oroszlán G, Kortvely E, Kiss B, Boros E, et al. MASP-3 is the exclusive pro-factor D activator in resting blood: the lectin and the alternative complement pathways are fundamentally linked. Sci Rep. 2016;6:31877.
Iwaki D, Kanno K, Takahashi M, Endo Y, Matsushita M, Fujita T. The role of mannose-binding lectin-associated serine protease-3 in activation of the alternative complement pathway. J Immunol. 2011;187(7):3751–8.
Pihl R, Jensen L, Hansen AG, Thogersen IB, Andres S, Dagnaes-Hansen F, et al. Analysis of factor D isoforms in Malpuech-Michels-Mingarelli-Carnevale patients highlights the role of MASP-3 as a maturase in the alternative pathway of complement. J Immunol. 2017;199:2158–70.
Hayashi M, Machida T, Ishida Y, Ogata Y, Omori T, Takasumi M, et al. Cutting edge: role of MASP-3 in the physiological activation of Factor D of the alternative complement pathway. J Immunol. 2019;203(6):1411–6.
Dobo J, Kocsis A, Gal P. Be on target: strategies of targeting alternative and lectin pathway components in complement-mediated diseases. Front Immunol. 2018;9:1851.
Spitzer D, Mitchell LM, Atkinson JP, Hourcade DE. Properdin can initiate complement activation by binding specific target surfaces and providing a platform for de novo convertase assembly. J Immunol. 2007;179(4):2600.
Ma YJ, Lee BL, Garred P. An overview of the synergy and crosstalk between pentraxins and collectins/ficolins: their functional relevance in complement activation. Exp Mol Med. 2017;49(4):e320.
Krarup A, Wallis R, Presanis JS, Gal P, Sim RB. Simultaneous activation of complement and coagulation by MBL-associated serine protease 2. PLoS ONE. 2007;2(7):e623.
Demopulos GA, Dudler T, Nilsson B, inventors; Omeros Corporation, assignee. Compositions and methods of inhibiting MASP-2 for the treatment of various thrombotic diseases and disorders. Patent WO 2019/246367 A1. 2019.
Kozarcanin H, Lood C, Munthe-Fog L, Sandholm K, Hamad OA, Bengtsson AA, et al. The lectin complement pathway serine proteases (MASPs) represent a possible crossroad between the coagulation and complement systems in thromboinflammation. J Thromb Haemost. 2016;14(3):531–45.
Monsinjon T, Richard V, Fontaine M. Complement and its implications in cardiac ischemia/reperfusion: strategies to inhibit complement. Fundam Clin Pharmacol. 2001;15(5):293–306.
Castellano G, Melchiorre R, Loverre A, Ditonno P, Montinaro V, Rossini M, et al. Therapeutic targeting of classical and lectin pathways of complement protects from ischemia-reperfusion-induced renal damage. Am J Pathol. 2010;176(4):1648–59.
Zhou W, Farrar CA, Abe K, Pratt JR, Marsh JE, Wang Y, et al. Predominant role for C5b–9 in renal ischemia/reperfusion injury. J Clin Invest. 2000;105(10):1363–71.
Farrar CA, Tran D, Li K, Wu W, Peng Q, Schwaeble W, et al. Collectin-11 detects stress-induced L-fucose pattern to trigger renal epithelial injury. J Clin Invest. 2016;126(5):1911–25.
Alghadban S, Kenawy HI, Dudler T, Schwaeble WJ, Brunskill NJ. Absence of the lectin activation pathway of complement ameliorates proteinuria-induced renal injury. Front Immunol. 2019;10(2238):2238.
Nauser CL, Howard MC, Fanelli G, Farrar CA, Sacks S. Collectin-11 (CL-11) is a major sentinel at epithelial surfaces and key pattern recognition molecule in complement-mediated ischaemic injury. Front Immunol. 2023;2018(9):2023.
Orsini F, Chrysanthou E, Dudler T, Cummings WJ, Takahashi M, Fujita T, et al. Mannan binding lectin-associated serine protease-2 (MASP-2) critically contributes to post-ischemic brain injury independent of MASP-1. J Neuroinflammation. 2016;13(1):213.
Cervera A, Planas AM, Justicia C, Urra X, Jensenius JC, Torres F, et al. Genetically-defined deficiency of mannose-binding lectin is associated with protection after experimental stroke in mice and outcome in human stroke. PLoS ONE. 2010;5(2):e8433.
Osthoff M, Walder B, Delhumeau C, Trendelenburg M, Turck N. Association of lectin pathway protein levels and genetic variants early after injury with outcomes after severe traumatic brain injury: a prospective cohort study. J Neurotrauma. 2017;34(17):2560–6.
Fust G, Munthe-Fog L, Illes Z, Szeplaki G, Molnar T, Pusch G, et al. Low ficolin-3 levels in early follow-up serum samples are associated with the severity and unfavorable outcome of acute ischemic stroke. J Neuroinflamm. 2011;8:185.
Zanier ER, Zangari R, Munthe-Fog L, Hein E, Zoerle T, Conte V, et al. Ficolin-3-mediated lectin complement pathway activation in patients with subarachnoid hemorrhage. Neurology. 2014;82(2):126–34.
Song FY, Wu MH, Zhu LH, Zhang ZQ, Qi QD, Lou CL. Elevated serum mannose-binding lectin levels are associated with poor outcome after acute ischemic stroke in patients with type 2 diabetes. Mol Neurobiol. 2015;52(3):1330–40.
Zhang ZG, Wang C, Wang J, Zhang Z, Yang YL, Gao L, et al. Prognostic value of mannose-binding lectin: 90-day outcome in patients with acute ischemic stroke. Mol Neurobiol. 2015;51(1):230–9.
Clark JE, Dudler T, Marber MS, Schwaeble W. Cardioprotection by an anti-MASP-2 antibody in a murine model of myocardial infarction. Open Heart. 2018;5(1):e000652.
Magro C, Mulvey JJ, Berlin D, Nuovo G, Salvatore S, Harp J, et al. Complement associated microvascular injury and thrombosis in the pathogenesis of severe COVID-19 infection: a report of five cases. Transl Res. 2020;220:1–13.
Ali YM, Ferrari M, Lynch NJ, Yaseen S, Dudler T, Gragerov S, et al. Lectin pathway mediates complement activation by SARS-CoV-2 proteins. Front Immunol. 2021. https://doi.org/10.3389/fimmu.2021.714511.
Gao T, Hu M, Zhang X, Li H, Zhu L, Liu H, et al. Highly pathogenic coronavirus N protein aggravates lung injury by MASP-2-mediated complement over-activation. medRxiv. 2020. https://doi.org/10.1101/2020.03.29.20041962.
Gavriilaki E, Brodsky RA. Severe COVID-19 infection and thrombotic microangiopathy: success does not come easily. Br J Haematol. 2020;189(6):e227–30.
Mastaglio S, Ruggeri A, Risitano AM, Angelillo P, Yancopoulou D, Mastellos DC, et al. The first case of COVID-19 treated with the complement C3 inhibitor AMY-101. Clin Immunol. 2020;215:108450.
Diurno F, Numis FG, Porta G, Cirillo F, Maddaluno S, Ragozzino A, et al. Eculizumab treatment in patients with COVID-19: preliminary results from real life ASL Napoli 2 Nord experience. Eur Rev Med Pharmacol Sci. 2020;24(7):4040–7.
Rambaldi A, Gritti G, Mico MC, Frigeni M, Borleri G, Salvi A, et al. Endothelial injury and thrombotic microangiopathy in COVID-19: treatment with the lectin-pathway inhibitor narsoplimab. Immunobiology. 2020;225(6):152001.
Alexion provides update on phase 3 study of ULTOMIRIS® (ravulizumab-cwvz) in hospitalized patients with severe COVID-19. https://ir.alexion.com/news-releases/news-release-details/alexion-provides-update-phase-3-study-ultomirisr-ravulizumab. Accessed 11 Feb 2021.
Staidson links with Pivotal to test BDB-001 in severe COVID-19 patients. https://www.thepharmaletter.com/article/staidson-links-with-pivotal-to-test-bdb-001-in-severe-covid-19-patients. Accessed 29 Jan 2021.
A Study of the C3 Inhibitor AMY-101 in Patients With ARDS Due to COVID-19 (SAVE). https://clinicaltrials.gov/ct2/show/NCT04395456. Accessed 12 March 2021.
Apellis Provides Update on APL-9 for Severe COVID-19. https://investors.apellis.com/news-releases/news-release-details/apellis-provides-update-apl-9-severe-covid-19. Accessed 2 Apr 2021.
Gavriilaki E, Sakellari I, Anagnostopoulos A, Brodsky RA. Transplant-associated thrombotic microangiopathy: opening Pandora’s box. Bone Marrow Transplant. 2017;52(10):1355–60.
Horvath O, Kallay K, Csuka D, Mezo B, Sinkovits G, Kassa C, et al. Early increase in complement terminal pathway activation marker sC5b-9 is predictive for the development of thrombotic microangiopathy after stem cell transplantation. Biol Blood Marrow Transplant. 2018;24(5):989–96.
Qi J, Wang J, Chen J, Su J, Tang Y, Wu X, et al. Plasma levels of complement activation fragments C3b and sC5b-9 significantly increased in patients with thrombotic microangiopathy after allogeneic stem cell transplantation. Ann Hematol. 2017;96(11):1849–55.
Jodele S, Fukuda T, Mizuno K, Vinks AA, Laskin BL, Goebel J, et al. Variable eculizumab clearance requires pharmacodynamic monitoring to optimize therapy for thrombotic microangiopathy after hematopoietic stem cell transplantation. Biol Blood Marrow Transplant. 2016;22(2):307–15.
Gavriilaki E, Touloumenidou T, Sakellari I, Batsis I, Mallouri D, Psomopoulos F, et al. Pretransplant genetic susceptibility: clinical relevance in transplant-associated thrombotic microangiopathy. Thromb Haemost. 2020;120(4):638–46.
Rotz SJ, Luebbering N, Dixon BP, Gavriilaki E, Brodsky RA, Dandoy CE, et al. In vitro evidence of complement activation in transplantation-associated thrombotic microangiopathy. Blood Adv. 2017;1(20):1632–4.
Jodele S, Zhang K, Zou F, Laskin B, Dandoy CE, Myers KC, et al. The genetic fingerprint of susceptibility for transplant-associated thrombotic microangiopathy. Blood. 2016;127(8):989–96.
Jodele S, Medvedovic M, Luebbering N, Chen J, Dandoy CE, Laskin BL, et al. Interferon-complement loop in transplant-associated thrombotic microangiopathy. Blood Adv. 2020;4(6):1166–77.
Jodele S, Dandoy CE, Myers KC, El-Bietar J, Nelson A, Wallace G, et al. New approaches in the diagnosis, pathophysiology, and treatment of pediatric hematopoietic stem cell transplantation-associated thrombotic microangiopathy. Transfus Apher Sci. 2016;54(2):181–90.
Rossoff JE, Schneiderman J, Chaudhury S, Arva NC. Diagnostic utility of complement immunohistochemical studies in post-stem cell transplant intestinal thrombotic microangiopathy: case report. J Pediatr Hematol Oncol. 2017;39(4):282–6.
Jodele S. Complement in pathophysiology and treatment of transplant-associated thrombotic microangiopathies. Semin Hematol. 2018;55(3):159–66.
Gavriilaki E, Yuan X, Ye Z, Ambinder AJ, Shanbhag SP, Streiff MB, et al. Modified Ham test for atypical hemolytic uremic syndrome. Blood. 2015;125(23):3637–46.
Elhadad S, Chapin J, Copertino D, Van Besien K, Ahamed J, Laurence J. MASP2 levels are elevated in thrombotic microangiopathies: association with microvascular endothelial cell injury and suppression by anti-MASP2 antibody narsoplimab. Clin Exp Immunol. 2020;203(1):96–104.
Vander Lugt MT, Braun TM, Hanash S, Ritz J, Ho VT, Antin JH, et al. ST2 as a marker for risk of therapy-resistant graft-versus-host disease and death. N Engl J Med. 2013;369(6):529–39.
Rowan CM, Pike F, Cooke KR, Krance R, Carpenter PA, Duncan C, et al. Assessment of ST2 for risk of death following graft-versus-host disease in pediatric and adult age groups. Blood. 2020;135(17):1428–37.
Sampson ME, Lane A, Lake KE, Luebbering N, Dandoy CE, Davies SM. Ann Arbor Score predicts thrombotic microangiopathy but not graft-versus-host disease in pediatric bone marrow transplant recipients. Biol Blood Marrow Transplant. 2019;25:S230–1.
Luft T, Benner A, Terzer T, Jodele S, Dandoy CE, Storb R, et al. EASIX and mortality after allogeneic stem cell transplantation. Bone Marrow Transplant. 2020;55(3):553–61.
Gavriilaki E, Sakellari I, Chatziconstantinou T, Mallouri D, Batsis I, Vardi A, et al. EASIX is strongly associated with complement activation and overall survival in adult allogeneic hematopoietic cell transplantation recipients. Blood. 2019;134(Supplement 1):4520.
Gavriilaki E, Sakellari I, Chatzikonstantinou T, Mallouri D, Batsis I, Vardi A, et al. Endothelial and complement activation as predictors of survival in adult allogeneic hematopoietic cell transplantation. HemaSphere. 2021;5(1):e487.
El-Bietar J, Warren M, Dandoy C, Myers KC, Lane A, Wallace G, et al. Histologic features of intestinal thrombotic microangiopathy in pediatric and young adult patients after hematopoietic stem cell transplantation. Biol Blood Marrow Transplant. 2015;21(11):1994–2001.
Gavriilaki E, Sakellari I, Karafoulidou I, Pasteli N, Batsis I, Mallouri D, et al. Intestinal thrombotic microangiopathy: a distinct entity in the spectrum of graft-versus-host disease. Int J Hematol. 2019;110(5):529–32.
Jodele S, Laskin BL, Goebel J, Khoury JC, Pinkard SL, Carey PM, et al. Does early initiation of therapeutic plasma exchange improve outcome in pediatric stem cell transplant-associated thrombotic microangiopathy? Transfusion. 2013;53(3):661–7.
Campistol JM, Arias M, Ariceta G, Blasco M, Espinosa L, Espinosa M, et al. An update for atypical haemolytic uraemic syndrome: diagnosis and treatment. A consensus document. Nefrologia. 2015;35(5):421–47.
Gavriilaki E, Brodsky RA. Complementopathies and precision medicine. J Clin Invest. 2020;130(5):2152–63.
Defitelio prescribing information. https://pp.jazzpharma.com/pi/defitelio.en.USPI.pdf. Accessed 16 Mar 2021.
Defitelio EPAR product information. https://www.ema.europa.eu/en/documents/product-information/defitelio-epar-product-information_en.pdf. Accessed 16 Mar 2021.
Uderzo C, Fumagalli M, De Lorenzo P, Busca A, Vassallo E, Bonanomi S, et al. Impact of thrombotic thrombocytopenic purpura on leukemic children undergoing bone marrow transplantation. Bone Marrow Transplant. 2000;26(9):1005–9.
Corti P, Uderzo C, Tagliabue A, Della Volpe A, Annaloro C, Tagliaferri E, et al. Defibrotide as a promising treatment for thrombotic thrombocytopenic purpura in patients undergoing bone marrow transplantation. Bone Marrow Transplant. 2002;29(6):542–3.
Uderzo C, Bonanomi S, Busca A, Renoldi M, Ferrari P, Iacobelli M, et al. Risk factors and severe outcome in thrombotic microangiopathy after allogeneic hematopoietic stem cell transplantation. Transplantation. 2006;82(5):638–44.
Martínez-Muñoz ME, Forés R, Lario A, Bautista G, Bueno JL, de Miguel C, et al. Use of defibrotide to treat adult patients with transplant-associated thrombotic microangiopathy. Bone Marrow Transplant. 2019;54(1):142–5.
Yeates L, Slatter MA, Bonanomi S, Lim F, Ong SY, Dalissier A, et al. Use of defibrotide to treat transplant-associated thrombotic microangiopathy: a retrospective study of the Paediatric Diseases and Inborn Errors Working Parties of the European Society of Blood and Marrow Transplantation. Bone Marrow Transplant. 2017;52(5):762–4.
Devadas SK, Toshniwal M, Bagal B, Khattry N. Successful treatment of transplant associated thrombotic microangiopathy (TA-TMA) with low dose defibrotide. Indian J Hematol Blood Transfus. 2018;34(3):469–73.
Gavriilaki E, de Latour RP, Risitano AM. Advancing therapeutic complement inhibition in hematologic diseases: PNH and beyond. Blood. 2021. https://doi.org/10.1182/blood.2021012860.
Thomas TC, Rollins SA, Rother RP, Giannoni MA, Hartman SL, Elliott EA, et al. Inhibition of complement activity by humanized anti-C5 antibody and single-chain Fv. Mol Immunol. 1996;33(17–18):1389–401.
Jodele S, Fukuda T, Vinks A, Mizuno K, Laskin BL, Goebel J, et al. Eculizumab therapy in children with severe hematopoietic stem cell transplantation-associated thrombotic microangiopathy. Biol Blood Marrow Transplant. 2014;20(4):518–25.
Sevindik OG, Alacacioglu I, Katgi A, Solmaz SM, Acar C, Piskin O, et al. Renal and neurological response with eculizumab in a patient with transplant associated thrombotic microangiopathy after allogeneic hematopoietic progenitor cell transplantation. Case Rep Hematol. 2015;2015:425410.
Fernandez C, Lario A, Fores R, Cabrera R. Eculizumab treatment in a patient with hematopoietic stem cell transplantation-associated thrombotic microangiopathy and steroid-refractory acute graft versus host disease. Hematol Rep. 2015;7(4):6107.
de Fontbrune FS, Galambrun C, Sirvent A, Huynh A, Faguer S, Nguyen S, et al. Use of eculizumab in patients with allogeneic stem cell transplant-associated thrombotic microangiopathy: a study from the SFGM-TC. Transplantation. 2015;99(9):1953–9.
Bohl SR, Kuchenbauer F, von Harsdorf S, Kloevekorn N, Schönsteiner SS, Rouhi A, et al. Thrombotic microangiopathy after allogeneic stem cell transplantation: a comparison of eculizumab therapy and conventional therapy. Biol Blood Marrow Transplant. 2017;23(12):2172–7.
Gavriilaki E, Sakellari I, Bousiou Z, Batsis I, Mallouri D, Masmanidou M, et al. Complement inhibition with eculizumab in adult transplant-associated thrombotic microangiopathy: opening the Pandora’s box. Transplant Cell Ther. 2021;27(3 Supplement):S270–1.
Scordo M, Giralt S. Compliments to complement blockade for TA-TMA. Blood. 2020;135(13):981–3.
Rondeau E, Scully M, Ariceta G, Barbour T, Cataland S, Heyne N, et al. The long-acting C5 inhibitor, ravulizumab, is effective and safe in adult patients with atypical hemolytic uremic syndrome naïve to complement inhibitor treatment. Kidney Int. 2020;97(6):1287–96.
Lee JW, de Fontbrune FS, Lee WLL, Pessoa V, Gualandro S, Füreder W, et al. Ravulizumab (ALXN1210) vs eculizumab in adult patients with PNH naive to complement inhibitors: the 301 study. Blood. 2019;133(6):530–9.
Ravulizumab in thrombotic microangiopathy after hematopoietic stem cell transplant. https://clinicaltrials.gov/ct2/show/NCT04543591. Accessed 16 Nov 2021.
Study of ravulizumab in pediatric participants with HSCT-TMA. https://clinicaltrials.gov/ct2/show/NCT04557735. Accessed 16 Nov 2021.
Akari Therapeutics. https://www.akaritx.com/2019/12/23/akari-therapeutics-announces-initiation-of-pivotal-phase-iii-trial-of-nomacopan-in-pediatric-hematopoietic-stem-cell-transplant-related-thrombotic-microangiopathy-hsct-tma-following-the-opening-of-i/. Accessed 5 May 2021.
Goodship THJ, Pinto F, Weston-Davies WH, Silva J, Nishimura J-I, Nunn MA, et al. Use of the complement inhibitor Coversin to treat HSCT-associated TMA. Blood Adv. 2017;1(16):1254–8.
Nomacopan (rVA576) in transplant associated thrombotic microangiopathy. https://clinicaltrials.gov/ct2/show/NCT04784455. Accessed 16 Nov 2021.
van Emmerik LC, Kuijper EJ, Fijen CA, Dankert J, Thiel S. Binding of mannan-binding protein to various bacterial pathogens of meningitis. Clin Exp Immunol. 1994;97(3):411–6.
Safety and efficacy study of OMS721 in patients with thrombotic microangiopathies. https://clinicaltrials.gov/ct2/show/NCT02222545. Accessed 16 Nov 2021.
Khaled SK, Boelens JJ, Cairo MS, Champlin R, Duarte RF, Giralt SA, et al. Narsoplimab (OMS721), a MASP-2 inhibitor, for the treatment of adult hematopoietic stem cell transplant-associated thrombotic microangiopathy (HSCT-TMA). Transplant Cell Ther. 2021;27(3 Supplement):S24–6.
de la Cámara R. Vaccinations. In: Carreras E, Dufour C, Mohty M, Kröger N, editors. The EBMT handbook: hematopoietic stem cell transplantation and cellular therapies. Cham: Springer International Publishing; 2019. p. 207–19. https://doi.org/10.1007/978-3-030-02278-5_29.
Mahler MB, Taur Y, Jean R, Kernan NA, Prockop SE, Small TN. Safety and immunogenicity of the tetravalent protein-conjugated meningococcal vaccine (MCV4) in recipients of related and unrelated allogeneic hematopoietic stem cell transplantation. Biol Blood Marrow Transplant. 2012;18(1):145–9.
Jodele S, Dandoy CE, Danziger-Isakov L, Myers KC, El-Bietar J, Nelson A, et al. Terminal complement blockade after hematopoietic stem cell transplantation is safe without meningococcal vaccination. Biol Blood Marrow Transplant. 2016;22(7):1337–40.
Dalle JH, Giralt SA. Hepatic veno-occlusive disease after hematopoietic stem cell transplantation: risk factors and stratification, prophylaxis, and treatment. Biol Blood Marrow Transplant. 2016;22(3):400–9.
Lewis C, Kim HT, Roeker LE, Cutler C, Koreth J, Nikiforow S, et al. Incidence, predictors, and outcomes of veno-occlusive disease/sinusoidal obstruction syndrome after reduced-intensity allogeneic hematopoietic cell transplantation. Biol Blood Marrow Transplant. 2020;26(3):529–39.
Senzolo M, Germani G, Cholongitas E, Burra P, Burroughs AK. Veno occlusive disease: update on clinical management. World J Gastroenterol. 2007;13(29):3918–24.
Elbahlawan L, Srinivasan A, Morrison RR. A critical care and transplantation-based approach to acute respiratory failure after hematopoietic stem cell transplantation in children. Biol Blood Marrow Transplant. 2016;22(4):617–26.
Spitzer TR. Engraftment syndrome following hematopoietic stem cell transplantation. Bone Marrow Transplant. 2001;27(9):893–8.
Chang L, Frame D, Braun T, Gatza E, Hanauer DA, Zhao S, et al. Engraftment syndrome after allogeneic hematopoietic cell transplantation predicts poor outcomes. Biol Blood Marrow Transplant. 2014;20(9):1407–17.
Harris AC, Young R, Devine S, Hogan WJ, Ayuk F, Bunworasate U, et al. International, multicenter standardization of acute graft-versus-host disease clinical data collection: a report from the Mount Sinai Acute GVHD International Consortium. Biol Blood Marrow Transplant. 2016;22(1):4–10.
Schoemans HM, Lee SJ, Ferrara JL, Wolff D, Levine JE, Schultz KR, et al. EBMT-NIH-CIBMTR Task Force position statement on standardized terminology & guidance for graft-versus-host disease assessment. Bone Marrow Transplant. 2018;53(11):1401–15.
Jodele S, Laskin BL, Dandoy CE, Myers KC, El-Bietar J, Davies SM, et al. A new paradigm: diagnosis and management of HSCT-associated thrombotic microangiopathy as multi-system endothelial injury. Blood Rev. 2015;29(3):191–204.
Soliris prescribing information. https://alexion.com/Documents/Soliris_USPI.pdf. Accessed 5 May 2021.
Eculizumab to treat thrombotic microangiopathy/atypical hemolytic uremic syndrome-associated multiple organ dysfunction syndrome in hematopoietic stem cell transplant recipients. https://clinicaltrials.gov/ct2/show/NCT03518203. Accessed 16 Nov 2021.
Acknowledgements
Medical writing was provided by Charlotte A. Osborne, PhD, CMPP, of Omeros Corporation. Editorial assistance and figure support were provided by PharmaScribe, LLC and funded by Omeros Corporation.
Funding
Funding was provided by Omeros Corporation for medical writing, editorial assistance, and figure support. Omeros Corporation participated in preparation and review of the manuscript, and the decision to submit the article for publication.
Author information
Authors and Affiliations
Contributions
All authors have made contributions to the conception and design of the manuscript, acquisition of the data, or analysis and interpretation of the data; and drafting the article or revising it critically for important intellectual content. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
EG has consulted for Alexion and Omeros Corporation. VTH has consulted for Alexion, Janssen, Jazz, and Omeros Corporation. WS has consulted for Omeros Corporation. TD is an employee of Omeros Corporation. MD and TF declare no conflicts of interest. SJ holds United States Patent No: US 10,815,296 B2, is conducting an NIH-funded study with the study drug provided by Alexion Pharmaceuticals at no charge to study subjects, and has received travel support and lecture honoraria from Omeros Corporation.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Gavriilaki, E., Ho, V.T., Schwaeble, W. et al. Role of the lectin pathway of complement in hematopoietic stem cell transplantation-associated endothelial injury and thrombotic microangiopathy. Exp Hematol Oncol 10, 57 (2021). https://doi.org/10.1186/s40164-021-00249-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40164-021-00249-8