
Abstract
Background
Hepatoblastoma is the most common hepatic malignancy in children, accounting for approximately 80% of all childhood liver tumors. KRAS and NRAS, members of the RAS gene family, are closely linked to tumorigenesis, and are frequently mutated in a variety of malignancies. They may thus play critical roles in tumorigenesis. However, there are few studies on the association between the RAS gene polymorphisms and risk of hepatoblastoma.
Methods
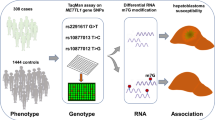
We investigated whether the polymorphisms at these genes are associated with hepatoblastoma susceptibility in a hospital-based study of 213 affected Chinese children and 958 cancer-free controls. Genotypes were determined by TaqMan assay, and association with hepatoblastoma risk was assessed based on odds ratios and 95% confidence intervals.
Results
No significant differences were observed between patients and controls in terms of age and gender frequency. All NRAS and KRAS genotypes are in Hardy–Weinberg equilibrium in the entire study population. We did not observe any significant association between hepatoblastoma risk and polymorphisms at NRAS and KRAS. The association between selected polymorphisms and hepatoblastoma risk was assessed after stratification by age, gender, and clinical stage. However, no significant association was observed even after stratification by age, gender, and clinical stage.
Conclusions
The data suggest that NRAS and KRAS polymorphisms are irrelevant to hepatoblastoma susceptibility among Chinese population.
Similar content being viewed by others


Introduction
Hepatoblastoma, an embryonic tumor, accounts for about 80% of all childhood liver malignancies and 1% of all childhood malignancies [1, 2]. The most common clinical symptoms are abdominal masses usually accompanied by fever, weight loss, anorexia, obstructive jaundice, or acute abdominal bleeding due to tumor rupture [3, 4]. Of note, more than 90% of cases are associated with elevated levels of alpha-fetoprotein, an important biomarker [5].
Unlike adult hepatic cellular carcinoma, hepatoblastoma is not related to hepatitis B virus or liver cirrhosis [6]. Individual environmental risk factors may increase risk, while premature delivery and very low birth weight are associated with increased incidence [7]. The genetic disorders Beckwith–Wiedemann syndrome and familial adenomatous polyposis are closely associated with hepatoblastoma, suggesting that genetic factors may accelerate pathogenesis [2]. In addition, genetic polymorphisms that result in loss or alteration of the function of tumor-associated proteins may increase susceptibility to tumors and subsequent prognosis [8, 9]. Hence, genome-wide association studies of hepatoblastoma risk are warranted but rarely conducted.
The RAS genes KRAS and NRAS are believed to be closely linked to tumorigenesis [10]. KRAS is located on chromosome 12p12.1, and has diverse biological functions, including in angiogenesis, epidermal growth factor receptor (EGFR) signaling to the nucleus, and cell division, differentiation, proliferation, and growth [11,12,13]. Indeed, the RAS/RAF/MAPK pathway is one of the most important downstream pathways triggered by EGFR, and one that critically depends on KRAS and NRAS expression [14, 15]. The pathway is activated when an extracellular signaling molecule binds to and alters the conformation of a membrane receptor such as EGFR, which, in turn, binds to a series of proteins related to Ras activation, e.g., Grb2, SOS, etc. Ultimately, activated Ras triggers a phosphorylation cascade via MAPK to transduce the extracellular signal to the nucleus and elicit a response.
Mutations in KRAS and NRAS may constitutively activate signaling pathways downstream of EGFR, thereby promoting aberrant cell growth [16] and differentiation, which may then lead to tumorigenesis [17, 18]. Accordingly, patients with KRAS mutations do not respond to EGFR inhibitors [19]. Mutations in KRAS occur in about 30% to 40% of the population, and cluster at codons 12–13 of exon 2, and at codons 59, 61, and 17 of exon 3 [20, 21]. On the other hand, NRAS mutations are relatively uncommon, but result in malignant proliferation and metastasis [22]. Moreover, NRAS and KRAS mutations are much more common in the elderly [23].
KRAS and NRAS mutations are common in a variety of malignancies, including colorectal, pancreatic, and lung cancer [24, 25]. For example, such mutations are found in 20–50% and 1–6% of colorectal cancers, respectively [26]. Mutations in KRAS are also an early event in the development of pancreatic ductal adenocarcinoma, and are present in more than 90% of cases [27]. Further, KRAS mutations are found in about 22.5% to 36.0% of non-small cell lung cancers, of which about 97% are located in intron 12 and 13 [28]. On the other hand, NRAS mutations that are potentially targetable by therapy have been detected in small-cell lung cancer [29]. RAS mutations have also been detected in a small number of neuroblastoma patients. Of note, such mutations can be targeted effectively with everolimus, which is already on the market [30, 31]. Collectively, the growing body of evidence suggests that RAS mutations are present and may play important roles in a variety of solid tumors, including in the breast, cervix, small intestine, liver, and other organs [32]. Nevertheless, the relationship between RAS polymorphisms and hepatoblastoma has not been investigated. In this study, we analyzed the association between NRAS and KRAS polymorphisms and hepatoblastoma risk.
Results
Characteristics of the study population
The demographic characteristics of 213 hepatoblastoma patients and 958 controls recruited in Guangdong, Henan, Shaanxi, and Shannxi are listed in Additional file 1: Tables S1, S2. No significant differences were observed between patients and controls in terms of age and gender frequency, both as a single cohort or in each province.
Association between hepatoblastoma risk and NRAS and KRAS polymorphisms
Genotypes at the NRAS polymorphism rs2273267 A > T are listed in Table 1 for hepatoblastoma patients and controls, along with those at the KRAS polymorphisms rs12587 G > T, rs7973450 A > G, and rs7312175 G > A. All NRAS and KRAS genotypes are in accordance with Hardy–Weinberg equilibrium in the entire study population. We did not observe any significant association between hepatoblastoma risk and polymorphisms at NRAS and KRAS. On the contrary, we found that subjects carrying the genotypes rs12587 TT, rs7973450 AG/GG, and rs7312175 GA/AA, alone or in combination, have a marginally lower risk of hepatoblastoma that is not statistically significant (adjusted odds ratio [OR] = 0.91; 95% confidence interval [CI] 0.67–1.25; P = 0.561), even though these genotypes are considered to indicate cancer risk.
Association of NRAS and KRAS polymorphisms with hepatoblastoma risk after demographic stratification
The association between select polymorphisms and hepatoblastoma risk was assessed after stratification by age, gender, and clinical stage (Tables 2, 3). However, no significant association was observed between hepatoblastoma risk and the NRAS rs2273267 A > T polymorphism in children aged more than 17 months (adjusted OR = 1.42, 95% CI 0.68–2.96, P = 0.350) or younger (adjusted OR = 1.23, 95% CI 0.62–2.43, P = 0.556, Table 2). Gender was also not linked to hepatoblastoma risk (adjusted OR = 1.84, 95% CI 0.90–3.77, and P = 0.094 for females, and adjusted OR = 0.97, 95% CI 0.47–1.97, and P = 0.925 for males). In addition, there was no significant correlation between stage I + II patients and the genotypes AA/AT and TT (adjusted OR = 1.77, 95% CI 0.94–3.32, P = 0.075), nor between such genotypes and stage III + IV patients (adjusted OR = 1.66, 95% CI 0.73–3.80, P = 0.229).
Further analysis also showed that hepatoblastoma risk was not significantly associated with the KRAS polymorphisms rs12587 G > T, rs7973450 A > G, and rs7312175 G > A in children aged more than 17 months (P = 0.179, P = 0.286, and P = 0.383) or younger (P = 0.998, P = 0.486, and P = 0.189), nor in females (P = 0.963, P = 0.916, and P = 0.344) and males (P = 0.231, P = 0.750, and P = 0.765). There was also no significant correlation between stage I + II patients and the genotypes AA/AT and TT (adjusted OR = 1.06, 95% CI 0.69–1.64, P = 0.784), nor between such genotypes and stage III + IV patients (adjusted OR = 0.83, 95% CI 0.47–1.48, P = 0.532).
Discussion
Hepatoblastoma is a rare pediatric embryonic tumor with incidence of about 1/1,000,000 [33], and is often associated with chromosomal abnormalities, especially at chromosome 2, 11, 18, and 20 [34]. However, the relative risk of hepatoblastoma is 2280 times higher in children with Beckwith–Wiedemann syndrome, indicating that aberrations in chromosome 11 play an important role in pathogenesis [35]. Similarly, the risk is 1220-fold higher in children with familial adenomatous polyposis, implying that lesions in chromosome 5 are also involved [36]. Of note, somatic mutation of the tumor suppressor APC, which is located on chromosome 5, is present in 67–89% of sporadic hepatoblastoma. Such mutations occur at the 5′ half of the gene, and generally considered to be at or near base pair 1309 [37]. Finally, some genes that are typically imprinted and differentially methylated are already abnormally methylated even before the development of hepatoblastoma, suggesting that methylation at these sites is related to pathogenesis [38].
RAS is a membrane-bound GTP/GDP-binding protein and an important proto-oncogene in intracellular EGFR signaling [39]. Accordingly, it is an essential regulator of cell proliferation and angiogenesis, and regarded as a molecular switch that senses and transmits extracellular stimuli of proliferation, growth, differentiation, and related processes [40]. Indeed, RAS genes, including KRAS, HRAS, and NRAS, are all implicated in tumorigenesis. For example, activating mutations in RAS may cause continuous growth, dedifferentiation of cells, and tumor development [41].
Currently, the relationship between KRAS mutations and clinical outcomes is not fully elucidated. On one hand, Chang et al. [42] found that KRAS mutations are associated with tumor size, degree of differentiation, lymph node metastasis, and poor prognosis. Similarly, Zhang et al. [43] found that KRAS mutations were significantly more frequent in Chinese patients with mucinous colorectal adenocarcinomas and well-differentiated colorectal cancers, implying that KRAS mutations in such patients are causative but different from those patients in Western countries. Our data also show that hepatoblastoma risk in Chinese patients is not significantly associated with polymorphisms in NRAS and KRAS, even after stratification by age, gender, and clinical stage.
We note that although synergistic interactions between environmental and genetic factors contribute to the development of hepatoblastoma, we did not collect data on parental exposure to hazards, diets, and lifestyles. In addition, our cohort is certainly not representative of the whole Chinese population. Nevertheless, the findings are probably not generalizable to other races. Finally, the sample size is relatively small, and thus has limited statistical power. These issues should be avoided as much as possible in future studies to better investigate the relationship between hepatoblastoma risk and NRAS and KRAS polymorphisms.
Conclusions
We find that NRAS and KRAS polymorphisms are irrelevant to hepatoblastoma susceptibility among Chinese population. Moreover, further investigations of polymorphisms that might mediate the risk of hepatoblastoma would help gain a better understanding of the pathogenesis and improve prognosis.
Materials and methods
Study population
The cohort consisted of 213 hepatoblastoma cases diagnosed by histopathology in Guangdong, Henan, Shaanxi, and Shanxi. There are no direct blood relationships among cases, and 958 cancer-free children were included as controls (Additional file 1: Table S1). Written informed consent was obtained from legal guardians, and the protocol was approved by the institutional review board at Guangzhou Women’s and Children’s Medical Center.
DNA extraction and genotyping
NRAS and KRAS polymorphisms were genotyped in blinded fashion using TaqMan real-time PCR [44,45,46,47]. Assays were repeated for 10% of randomly selected samples, and results were 100% concordant with original genotypes.
Statistical analysis
The demographic characteristics of and genotype frequency distribution in cases and controls were compared by χ2 test. Deviation from Hardy–Weinberg equilibrium was tested in control subjects using χ2 goodness-of-fit test. Odds ratios and 95% confidence intervals were calculated to assess the association between hepatoblastoma risk and NRAS and KRAS polymorphisms. Age, gender, and clinical stages were compared by χ2 test and logistic regression among patients with different genotypes. Polymorphic loci were evaluated using dominant, recessive, and additive models, and corresponding P values, relative risk odds ratios, and 95% confidence intervals were calculated. All statistical analyses were performed in SAS version 9.4 (SAS Institute, Cary, NC), with P values < 0.05 considered as statistically significant.
References
von Schweinitz D. Hepatoblastoma: recent developments in research and treatment. Semin Pediatr Surg. 2012;21:21–30.
Devi LP, Kumar R, Handique A, Kumar M. Hepatoblastoma—a rare liver tumor with review of literature. J Gastrointest Cancer. 2014;45(Suppl 1):261–4.
Stocker JT, Ishak KG. Hepatoblastoma. Semin Diagn Pathol. 1994;11:136–43.
Herzog CE, Andrassy RJ, Eftekhari F. Childhood cancers: hepatoblastoma. Oncologist. 2000;5:445–53.
El Asmar A, El Rassi Z. Hepatoblastoma in childhood, long term survival achieved: 2 case reports and literature review. Int J Surg Case Rep. 2016;21:55–8.
Reyes JD, Carr B, Dvorchik I, Kocoshis S, Jaffe R, Gerber D, et al. Liver transplantation and chemotherapy for hepatoblastoma and hepatocellular cancer in childhood and adolescence. J Pediatr. 2000;136:795–804.
Oue T, Kubota A, Okuyama H, Kawahara H, Nara K, Kawa K, et al. Hepatoblastoma in children of extremely low birth weight: a report from a single perinatal center. J Pediatr Surg. 2003;38:134–7.
Whibley C, Pharoah PD, Hollstein M. p53 polymorphisms: cancer implications. Nat Rev Cancer. 2009;9:95–107.
Wang BG, Yi DH, Liu YF. TLR3 gene polymorphisms in cancer: a systematic review and meta-analysis. Chin J Cancer. 2015;34:272–84.
Hill KS, Wang X, Roberts ER, Kim J, Messina M, Kim M. The importance of the RASA1/R-Ras/Ral-A signaling axis in melanoma tumorigenesis. Cancer Res. 2018;78:B12.
Alamo P, Gallardo A, Di Nicolantonio F, Pavón MA, Casanova I, Trias M, et al. Higher metastatic efficiency of KRas G12V than KRas G13D in a colorectal cancer model. FASEB J. 2015;29:464–76.
Valtorta E, Misale S, Sartore-Bianchi A, Nagtegaal ID, Paraf F, Lauricella C, et al. KRAS gene amplification in colorectal cancer and impact on response to EGFR-targeted therapy. Int J Cancer. 2013;133:1259–65.
Sakamoto H, Shimizu J, Horio Y, Ueda R, Takahashi T, Mitsudomi T, et al. Disproportionate representation of KRAS gene mutation in atypical adenomatous hyperplasia, but even distribution of EGFR gene mutation from preinvasive to invasive adenocarcinomas. J Pathol. 2010;212:287–94.
Bouali S, Chrétien AS, Ramacci C, Rouyer M, Becuwe P, Merlin JL. PTEN expression controls cellular response to cetuximab by mediating PI3K/AKT and RAS/RAF/MAPK downstream signaling in KRAS wild-type, hormone refractory prostate cancer cells. Oncol Rep. 2009;21:731–5.
Molina JR, Adjei AA. The Ras/Raf/MAPK pathway. J Thorac Oncol. 2006;1:7–9.
Kalikaki A, Koutsopoulos A, Trypaki M, Souglakos J, Stathopoulos E, Georgoulias V, et al. Comparison of EGFR and K-RAS gene status between primary tumours and corresponding metastases in NSCLC. Br J Cancer. 2008;99:923–9.
Milano G, Etienne-Grimaldi MC, Dahan L, Francoual M, Spano JP, Benchimol D, et al. Epidermal growth factor receptor (EGFR) status and K-Ras mutations in colorectal cancer. Ann Oncol. 2008;19:2033–8.
Gallegos Ruiz MI, Floor K, Rijmen F, Grünberg K, Rodriguez JA, Giaccone G. EGFR and K-ras mutation analysis in non-small cell lung cancer: comparison of paraffin embedded versus frozen specimens. Cell Oncol. 2007;29:257–64.
Allegra CJ, Jessup JM, Somerfield MR, Hamilton SR, Hammond EH, Hayes DF, et al. American Society of Clinical Oncology provisional clinical opinion: testing for KRAS gene mutations in patients with metastatic colorectal carcinoma to predict response to anti-epidermal growth factor receptor monoclonal antibody therapy. J Clin Oncol. 2009;27:2091–6.
Smit VT, Boot AJ, Smits AM, Fleuren GJ, Cornelisse CJ, Bos JL. KRAS codon 12 mutations occur very frequently in pancreatic adenocarcinomas. Nucleic Acids Res. 1988;16:7773–82.
Loupakis F, Ruzzo A, Cremolini C, Vincenzi B, Salvatore L, Santini D, et al. KRAS codon 61, 146 and BRAF mutations predict resistance to cetuximab plus irinotecan in KRAS codon 12 and 13 wild-type metastatic colorectal cancer. Br J Cancer. 2009;101:715–21.
Belli C, De Brasi C, Larripa I. Rapid detection of exon 1 NRAS gene mutations using universal heteroduplex generator technology. Hum Mutat. 2003;21:132–7.
Malumbres M, Barbacid M. RAS oncogenes: the first 30 years. Nat Rev Cancer. 2003;3:459–65.
Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S, et al. Mutations of the BRAF gene in human cancer. Nature. 2002;417:949–54.
Emel’ianova MA, Amosenko FA, Chudinov AV, Surzhikov SA, Kazubskaia TP, Liubchenko LN, et al. Detection of KRAS mutations in tumor cells using biochips. Mol Biol. 2011;45:863–70.
Vaughn CP, Zobell SD, Furtado LV, Baker CL, Samowitz WS. Frequency of KRAS, BRAF, and NRAS mutations in colorectal cancer. Genes Chromosomes Cancer. 2011;50:307–12.
Kulemann B, Liss AS, Warshaw AL, Seifert S, Bronsert P, Glatz T, et al. KRAS mutations in pancreatic circulating tumor cells: a pilot study. Tumour Biol. 2016;37:7547–54.
Mack PC, Holland WS, Redman M, Lara PN Jr, Snyder LJ, Hirsch FR, et al. KRAS mutation analysis in cetuximab-treated advanced stage non-small cell lung cancer (NSCLC): SWOG experience with S0342 and S0536. J Clin Oncol. 2009;27:8022.
Kodaz H, Taştekin E, Erdoğan B, Hacıbekiroğlu İ, Tozkır H, Gürkan H, et al. KRAS mutation in small cell lung carcinoma and extrapulmonary small cell cancer. Balkan Med J. 2016;33:407–10.
Kiessling MK, Curioni-Fontecedro A, Samaras P, Lang S, Scharl M, Aguzzi A, et al. Targeting the mTOR complex by everolimus in NRAS mutant neuroblastoma. PLoS ONE. 2016;11:e0147682.
Pylayeva-Gupta Y, Grabocka E, Bar-Sagi D. RAS oncogenes: weaving a tumorigenic web. Nat Rev Cancer. 2011;11:761–74.
Karnoub AE, Weinberg RA. Ras oncogenes: split personalities. Nat Rev Mol Cell Biol. 2008;9:517–31.
Roebuck DJ, Perilongo G. Hepatoblastoma: an oncological review. Pediatr Radiol. 2006;36:183–6.
Tomlinson GE, Douglass EC, Pollock BH, Finegold MJ, Schneider NR. Cytogenetic evaluation of a large series of hepatoblastomas: numerical abnormalities with recurring aberrations involving 1q12-q21. Genes Chromosomes Cancer. 2005;44:177–84.
Kim SY, Jung SH, Kim MS, Han MR, Park HC, Jung ES, et al. Genomic profiles of a hepatoblastoma from a patient with Beckwith–Wiedemann syndrome with uniparental disomy on chromosome 11p15 and germline mutation of APC and PALB2. Oncotarget. 2017;8:91950–7.
Lee SH, Shin MS, Lee JY, Park WS, Kim SY, Jang JJ, et al. In vivo expression of soluble Fas and FAP-1: possible mechanisms of Fas resistance in human hepatoblastomas. J Pathol. 1999;188:207–12.
Hirschman BA, Pollock BH, Tomlinson GE. The spectrum of APC mutations in children with hepatoblastoma from familial adenomatous polyposis kindreds. J Pediatr. 2005;147:263–6.
Rumbajan JM, Maeda T, Souzaki R, Mitsui K, Higashimoto K, Nakabayashi K, et al. Comprehensive analyses of imprinted differentially methylated regions reveal epigenetic and genetic characteristics in hepatoblastoma. BMC Cancer. 2013;13:608.
Soung YH, Lee JW, Kim SY, Seo SH, Park WS, Nam SW, et al. Mutational analysis of EGFR and K-RAS genes in lung adenocarcinomas. Virchows Arch. 2005;446:483–8.
Bos JL. Ras oncogenes in human cancer: a review. Cancer Res. 1989;49:4682–9.
Downward J. Targeting RAS signalling pathways in cancer therapy. Nat Rev Cancer. 2003;3:11–22.
Chang YS, Yeh KT, Chang TJ, Chai C, Lu HC, Hsu NC, et al. Fast simultaneous detection of K-RAS mutations in colorectal cancer. BMC Cancer. 2009;9:179.
Zhang J, Zheng J, Yang Y, Lu J, Gao J, Lu T, et al. Molecular spectrum of KRAS, NRAS, BRAF and PIK3CA mutations in Chinese colorectal cancer patients: analysis of 1,110 cases. Sci Rep. 2015;5:18678.
Chang J, Tian J, Yang Y, Zhong R, Li J, Zhai K, et al. A rare missense variant in TCF7L2 associates with colorectal cancer risk by interacting with a GWAS-identified regulatory variant in the MYC enhancer. Cancer Res. 2018;78:5164–72.
Chang J, Tian J, Zhu Y, Zhong R, Zhai K, Li J, et al. Exome-wide analysis identifies three low-frequency missense variants associated with pancreatic cancer risk in Chinese populations. Nat Commun. 2018;9:3688.
Chang J, Zhong R, Tian J, Li J, Zhai K, Ke J, et al. Exome-wide analyses identify low-frequency variant in CYP26B1 and additional coding variants associated with esophageal squamous cell carcinoma. Nat Genet. 2018;50:338–43.
Li J, Chang J, Tian J, Ke J, Zhu Y, Yang Y, et al. A rare variant P507L in TPP1 interrupts TPP1-TIN2 interaction, influences telomere length, and confers colorectal cancer risk in Chinese population. Cancer Epidemiol Biomark. 2018;27:1029–35.
Authors’ contributions
TY, JL, JZ, YX, SL and HX conceptualized and designed the study, drafted the initial manuscript, and reviewed and revised the manuscript. YW, TT, JY, JP, CH and YY designed the data collection instruments, collected data, performed preliminary data analyses, and reviewed and revised the manuscript. YZ and JH conceptualized and designed the study, coordinated and supervised data collection, and critically reviewed the manuscript for important intellectual content. All authors approved the final manuscript as submitted and agree to be accountable for all aspects of the work. All authors read and approved the final manuscript.
Acknowledgements
None.
Competing interests
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential competing interests.
Availability of data and materials
Data and material will be available upon corresponding author approval. All datasets [GENERATED/ANALYZED] for this study are included in the manuscript and the additional files.
Consent for publication
All authors are agree to publish.
Ethics approval and consent to participate
The institutional review board at Guangzhou Women’s and Children’s Medical Center approved current study.
Funding
This study was funded by National Natural Science Foundation of China [Grant Numbers 81602199, 81802333], by Guangzhou Science Technology and Innovation Commission [Grant Number 201607010395], and by Natural Science Foundation of Guangdong Province, China [Grant Number 2016A030313496, 2018A030310053]. The funding bodies did not participate in study design, in data collection, analysis, and interpretation, and in writing the manuscript.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Author information
Authors and Affiliations
Corresponding authors
Additional file
Additional file 1: Table S1.
Frequency distribution of select variables in hepatoblastoma patients and cancer-free controls. Table S2. Demographic characteristics of the study population.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Yang, T., Wen, Y., Li, J. et al. NRAS and KRAS polymorphisms are not associated with hepatoblastoma susceptibility in Chinese children. Exp Hematol Oncol 8, 11 (2019). https://doi.org/10.1186/s40164-019-0135-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40164-019-0135-z