
Abstract
As chimeric antigen receptor (CAR) T cells have displayed an unprecedented efficacy in the treatment of CD19-positive malignances, it is believed that this cell therapy will be a milestone in the history of mankind’s conquering of cancer. However, there are some issues that restrict CAR T cells from reaching their optimal anti-tumor capacity, especially in the treatment of solid tumors. Inhibitory cytokines, immune checkpoint molecules, hypoxia and other adverse factors have been reported to be involved in this process. To obtain better efficacy in the treatment of leukemia and solid tumors, we need to continuously upgrade CAR T cell technology by incorporating novel functional elements into CAR T cells to overcome these restrictions. In this review, we summarize recent advances regarding this topic.
Similar content being viewed by others

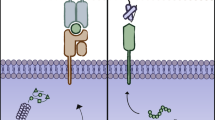
Background
Benefiting from the remarkable therapeutic outcome of chimeric antigen receptor (CAR) T cell treatment of refractory/relapsed B cell malignancy, CAR T cells are transitioning from the lab to the cancer ward. Several CAR-T products are in the advanced stage of clinical development [1,2,3]. CARs are recombinant receptors that generally consist of scFv, hinge, transmembrane domain, costimulatory molecule and CD3 ζ chain. Current research on CAR T cells mainly focuses on exploring new scFvs that are expressed at high levels on tumor cell surfaces rather than in healthy tissue and investigating an appropriate co-stimulatory intensity that enhances CAR T cell killing capacity and persistence [4,5,6]. However, there are some issues that restrict CAR T cells from reaching an optimal anti-tumor capacity, especially in the treatment of solid tumors [7]. Such as inhibitory cytokines, immune checkpoint molecules, hypoxia and other adverse factors hindering CAR T cell from efficiently expanding and killing tumor cells. Some researchers are attempting to combine some novel elements into CAR vectors to make CAR T cell slip the leash of aforementioned restrictions and thereby exhibiting optimal antitumor capacity (Fig. 1). In this review, we summarize the progresses of the incorporation of novel functional elements to improve CAR T cells, which have obtained prominent results.

Adverse factors including inhibitory cytokines, immune checkpoint molecules and hypoxia restrict CAR T cells from reaching an optimal anti-tumor capacity in the tumor microenvironment
Cytokines
IL-12 is a heterodimeric inflammatory cytokine produced by activated antigen-presenting cells (APCs), neutrophils, and macrophages. Scientific research has shown that a hostile tumor microenvironment can be significantly regulated by IL-12 through multiple mechanisms, including reactivation of anergic tumor-infiltrated lymphocytes (TILs), inhibition of Treg-mediated suppression of effector T cells, recruitment of NK cells to the tumor site, and inhibition of IL-10 and transforming growth factor-β (TGF-β) secretion by tumor-associated macrophages (TAM). In a novel syngeneic tumor model, Pegram et al. [8] demonstrated that tumor elimination requires both CD4+ and CD8+ T cell subsets, autocrine IL-12 stimulation, and subsequent IFN-γ secretion by CAR T cells. Therefore, they modified CD19-targeted CAR T cells to constitutively secrete IL-12, and the results showed that this treatment was able to safely eradicate established disease in the absence of prior conditioning and to acquire intrinsic resistance to Treg-mediated inhibition. You et al. [9] modified CAR T cell targeting MUC1 antigen to co-express IL12 for the treatment of seminal vesicle cancer in a phase I clinical trial. Interestingly, they found that the co-expression of IL-12 may contribute to TIA-1 expression in tumors after MUC1 CAR T cell treatment. IL-4 is an immunosuppressive cytokine in the tumor microenvironment that can promote fibrogenesis, support tumor growth and protect malignant cells from immune destruction. To protect CAR-PSCA T cells from the inhibitory effects of IL4, Somala Mohammed transgenically expressed a custom inverted cytokine receptor (ICR) in which the IL-4 receptor ectodomain was fused to the IL-7 receptor endodomain, switching the inhibitory effects to promoting effects to ultimately result in potent and sustained anti-tumor effects [10]. To achieve the selective expansion of CAR T cells, Whilding et al. [11] co-expressed an IL-4-responsive fusion gene (4αβ), which fused the IL-4 receptor α ectodomain to the shared human IL-2/IL-15 receptor β transmembrane and endodomain regions. Binding of IL-4 led to the delivery of a potent and selective growth signal in 4αβ+ CAR T cells.
Immune checkpoint molecules
As an evasion mechanism, many tumors are able to express various immune checkpoint molecules, resulting in the exhaustion of T cells that cannot prevent tumor progression. Emerging clinical data have highlighted the importance of the PD-L1/PD-1 immune inhibitory axis, and immune checkpoint blockers targeting both PD1 and PD-L1 have obtained great success in cancer therapy [12, 13]. Suarez et al. [14] developed a new combination immunotherapy that consists of human anti-carbonic anhydrase IX (CAIX)-targeted CAR T cells engineered to secrete human antibodies at the tumor site. Local anti-PD-L1 antibody delivery led to a fivefold reduction in tumor growth and a 50–80% reduction in tumor weight when compared with the anti-CAIX CAR T cells alone in a humanized mice model of clear cell renal cell carcinoma (ccRCC). What was more interesting was that because the isotype of the anti-PD-L1 antibody was IgG1, it had the potential to mediate ADCC and was able to recruit NK cells to the tumor site. Tanoue et al. [15] designed a new immunotherapy strategy for the treatment of prostate cancer; this strategy comprised an oncolytic adenovirus (Onc.Ad), a helper-dependent adenovirus (HDAd) that expressed a PD-L1 blocking mini-antibody, and HER2.CAR T cells. The results demonstrated that this combinatory therapy enhanced the anti-tumor effect compared with treatment with either HER2.CAR T cells alone or with HER2.CAR T cells plus Onc.Ad, and the benefits of locally produced PD-L1 mini-body could not be replaced by the infusion of anti-PD-L1 IgG. To overcome PD-L1 immunosuppressive effects on adoptively transferred T cells, Prosser et al. [16] converted PD-1 to a T cell costimulatory receptor by substituting its transmembrane and intracellular domains with the CD28 domain. Rather than becoming exhausted upon engagement of PDL1+ tumors, adoptively transferred T cells modified to express this PD1:CD28 chimera exhibited enhanced functional attributes. In addition, several research groups have tried to rescue CAR T from exhaustion through gene editing. For example, Ren et al. [17] used the one-shot CRISPR protocol to generate allogeneic universal T cells, simultaneously editing four gene loci, to decrease the expression of both PD1 and CTLA-4. Liu et al. [18] also constructed PD1 knockout universal CAR T cells.
Hypoxia
Hypoxia is a hallmark of a hostile tumor microenvironment, and tumor cells have high levels of oxidative stress and reactive oxygen species (ROS) production that substantially impair the antitumor activity of adoptively transferred T cells. Ligtenberg et al. [19] presented a strategy to render antitumor T cells more resilient toward ROS by co-expressing catalase along with a tumor-specific CAR to increase their anti-oxidative capacity by metabolizing H2O2. CAR T cells engineered to co-express catalase (CAR-CAT) showed increased levels of intracellular catalase and sharp decreases in ROS accumulation while maintaining their antitumor activity despite high H2O2 levels. More importantly, CAR T cells co-expressing catalase substantially protected bystander effector cells from oxidative stress-mediated repression. The lung is an oxygen-rich environment that frequently permits colonization by metastatic tumor cells. The prolyl hydroxylase domain (PHD) functions as an intracellular sensor of oxygen. Clever et al. [20] found that T cell intrinsic expression of PHD proteins maintained local tolerance against innocuous antigens in the lung. Meanwhile, PHD proteins also restrained the responses of the pulmonary-type helper Th-1 and CD8+ T cells, promoted Treg cell induction, and finally intensively enabled the colonization of circulating tumor cells. Inhibition of PHD proteins by dimethyloxalylglycine (DMOG) increased Th1 differentiation and IFN-γ production compared with vehicle-treated cultures. DMOG-treated TRP-1 CD4+ T cells mediated superior clearance of established subcutaneous tumors and lung metastases of melanoma. Based on the hypoxic tumor microenvironment, Juillerat et al. [21] developed an oxygen-sensitive CAR by introducing an oxygen-sensitive subdomain of HIF1α to a CAR scaffold. CAR begins to be expressed only when CAR T cells infiltrate a low oxygen environment, and expression is rapidly switched down once the cells get away from the hypoxic environment.
Chemotherapeutic drugs and kinase inhibitors
Chemotherapeutic drugs and kinase inhibitors play important roles in clinical cancer treatment. In order to elicit stronger antitumor activity, some researchers are attempting to combine CAR T cell therapy with these drugs. Fraietta et al. [22] found that T cells from chronic lymphocytic leukemia (CLL) patients usually exhibit deficiencies in proliferation, and this proliferative defect is completely reversed after long-term ibrutinib therapy. Ibrutinib is a first-in-class irreversible inhibitor of Bruton tyrosine kinase (BTK), which can irreversibly inhibit the IL-2 inducible T cell kinase (ITK). Ibrutinib inhibits Th2-polarized CD4 T cells, thus skewing T cells toward a Th1 anti-tumor immune response. Therefore, Fraietta et al. combined ibrutinib with CTL019 for treatment of CLL, and the results demonstrated that Ibrutinib increased the expansion of CTL019 and decreased PD-1 and CD200 expression from CLL patients ex vivo. Moreover, they found that continuous ibrutinib treatment could enhance CTL019 efficacy in drug-resistant ALL and CLL mouse models. Ruella et al. [23] also added Ibrutinib to CTL019 for the treatment of mantle cell lymphoma; the results demonstrated that 80–100% of mice in the CTL019+ ibrutinib arm and 0–20% of mice in the CTL019 arm remained in long-term remission (p < 0.05). Prostaglandin E2 (PGE2) and adenosine are two other critical immunosuppressive mediators that are present in solid tumors. PGE2 and adenosine activate protein kinase A (PKA), which then inhibits T cell receptor (TCR) activation. This inhibition process requires PKA to localize to the immune synapse by binding to the membrane protein ezrin. Newick et al. [24] manufactured anti-mesothelin CAR T cells that co-expressed a small peptide called the “regulatory subunit I anchoring disruptor” (RIAD). RIAD can prevent the association of PKA with ezrin, thus eliminating the negative effects of PKA on TCR activation. Lenalidomide is a synthetic derivative of thalidomide, which has multifaceted immunomodulatory efficacies. The most outstanding function of lenalidomide is to restore and facilitate immune synapse formation between T cells and antigen presenting cells. Kuramitsu et al. [25] added lenalidomide to EGFRvIII (epidermal growth factor receptor variant III)-targeted CAR T cells against glioblastoma multiforme (GBM). An interesting and novel finding of their study was that lenalidomide enhanced immunological synapse formation between the effector cells and the target cells and then enhanced the persistent antitumor activity of CAR T cells. The interaction between HVEM (TNFRSF14) and BTLA (B and T lymphocyte attenuator) is lost in most follicular lymphomas due to an HVEM mutation. HVEM deficiency induces a tumor-supportive microenvironment. Boice et al. [26] modified anti-CAD19 CAR T cells to become in vivo micro-pharmacies for the local delivery of soluble HVEM, and these modified CAR-T cells showed enhanced therapeutic activity against xenografted lymphomas.
Conclusion
In the above studies, in order to achieve better anti-tumor efficacy in the CAR T cell therapy, CAR T cells were modified to contain another single functional elements. The results demonstrate that incorporation of functional elements can significantly enhance the anti-tumor efficacy of CAR T cells. However, CAR T cells will encounter a more complicated and wicked tumor microenvironment after infuse into patients. Therefore, in order to make CAR T cells display the best anti-tumor capacity in clinic, it is better to simultaneously incorporate multiple functional elements into CAR T cells. For example, CAR T cells co-express PD1:CD28 chimera and IL12 can break the restriction imposed by immune inhibitory molecules and by the lack of cytokine signals. Expand CAR T cells with DMOG in vitro is also an option to make CAR T cells better overcome the hypoxic tumor microenvironment. By combining these approaches, CAR T cells can resist several adverse factors and finally we obtain more powerful CAR T cells.
Abbreviations
- CAR:
-
chimeric antigen receptor
- scFv:
-
single chain fragment variable
- Treg:
-
regulatory T cells
- PD1:
-
programmed death 1
- APC:
-
antigen-presenting cell
- TAM:
-
tumor-associated macrophage
- ICR:
-
inverted cytokine receptor
- CAIX:
-
carbonic anhydrase IX
- ccRCC:
-
clear cell renal cell carcinoma
- ADCC:
-
antibody dependent cellular cytotoxicity
- Onc.Ad:
-
oncolytic adenovirus
- HDAd:
-
helper-dependent adenovirus
- ROS:
-
reactive oxygen species
- PHD:
-
prolyl hydroxylase domain
- DMOG:
-
dimethyloxalylglycine
- CLL:
-
chronic lymphocytic leukemia
- BTK:
-
bruton tyrosine kinase
- ITK:
-
inducible T cell kinase
- PGE2:
-
prostaglandin E2
- PKA:
-
protein kinase A
- RIAD:
-
regulatory subunit I anchoring disruptor
- EGFRvIII:
-
epidermal growth factor receptor variant III
- GBM:
-
glioblastoma multiforme
- BTLA:
-
B and T lymphocyte attenuator
References
Davila ML, Riviere I, Wang X, Bartido S, Park J, Curran K, Chung SS, Stefanski J, Borquez-Ojeda O, Olszewska M, et al. Efficacy and toxicity management of 19-28z CAR T cell therapy in B cell acute lymphoblastic leukemia. Sci Transl Med. 2014;6(224):224ra225.
Kalos M, Levine BL, Porter DL, Katz S, Grupp SA, Bagg A, June CH. T cells with chimeric antigen receptors have potent antitumor effects and can establish memory in patients with advanced leukemia. Sci Transl Med. 2011;3:95ra73.
Cai B, Guo M, Wang Y, Zhang Y, Yang J, Guo Y, Dai H, Yu C, Sun Q, Qiao J, et al. Co-infusion of haplo-identical CD19-chimeric antigen receptor T cells and stem cells achieved full donor engraftment in refractory acute lymphoblastic leukemia. J Hematol Oncol. 2016;9:131.
Dai H, Wang Y, Lu X, Han W. Chimeric antigen receptors modified T-cells for cancer therapy. J Natl Cancer Inst. 2016;108(7):djv439.
Wang Z, Wu Z, Liu Y, Han W. New development in CAR-T cell therapy. J Hematol Oncol. 2017;10:53.
Qin L, Lai Y, Zhao R, Wei X, Weng J, Lai P, Li B, Lin S, Wang S, Wu Q, et al. Incorporation of a hinge domain improves the expansion of chimeric antigen receptor T cells. J Hematol Oncol. 2017;10:68.
Zhang C, Liu J, Zhong JF, Zhang X. Engineering CAR-T cells. Biomark Res. 2017;5:22.
Pegram HJ, Lee JC, Hayman EG, Imperato GH, Tedder TF, Sadelain M, Brentjens RJ. Tumor-targeted T cells modified to secrete IL-12 eradicate systemic tumors without need for prior conditioning. Blood. 2012;119:4133–41.
You F, Jiang L, Zhang B, Lu Q, Zhou Q, Liao X, Wu H, Du K, Zhu Y, Meng H, et al. Phase 1 clinical trial demonstrated that MUC1 positive metastatic seminal vesicle cancer can be effectively eradicated by modified Anti-MUC1 chimeric antigen receptor transduced T cells. Sci China Life Sci. 2016;59:386–97.
Ford-Jones EL, Mindorff CM, Langley JM, Allen U, Navas L, Patrick ML, Milner R, Gold R. Epidemiologic study of 4684 hospital-acquired infections in pediatric patients. Pediatr Infect Dis J. 1989;8:668–75.
Whilding LM, Parente-Pereira AC, Zabinski T, Davies DM, Petrovic RM, Kao YV, Saxena SA, Romain A, Costa-Guerra JA, Violette S, et al. Targeting of aberrant alphavbeta6 integrin expression in solid tumors using chimeric antigen receptor-engineered T cells. Mol Ther. 2017;25:259–73.
Westin JR, Chu F, Zhang M, Fayad LE, Kwak LW, Fowler N, Romaguera J, Hagemeister F, Fanale M, Samaniego F, et al. Safety and activity of PD1 blockade by pidilizumab in combination with rituximab in patients with relapsed follicular lymphoma: a single group, open-label, phase 2 trial. Lancet Oncol. 2014;15:69–77.
Ansell SM, Lesokhin AM, Borrello I, Halwani A, Scott EC, Gutierrez M, Schuster SJ, Millenson MM, Cattry D, Freeman GJ, et al. PD-1 blockade with nivolumab in relapsed or refractory Hodgkin’s lymphoma. N Engl J Med. 2015;372:311–9.
Suarez ER, de Chang K, Sun J, Sui J, Freeman GJ, Signoretti S, Zhu Q, Marasco WA. Chimeric antigen receptor T cells secreting anti-PD-L1 antibodies more effectively regress renal cell carcinoma in a humanized mouse model. Oncotarget. 2016;7:34341–55.
Tanoue K, Shaw AR, Watanabe N, Porter C, Rana B, Gottschalk S, Brenner M, Suzuki M. Armed oncolytic adenovirus-expressing PD-L1 mini-body enhances antitumor effects of chimeric antigen receptor T cells in solid tumors. Cancer Res. 2017;77(8):2040–51.
Prosser ME, Brown CE, Shami AF, Forman SJ, Jensen MC. Tumor PD-L1 co-stimulates primary human CD8(+) cytotoxic T cells modified to express a PD1:CD28 chimeric receptor. Mol Immunol. 2012;51:263–72.
Ren J, Zhang X, Liu X, Fang C, Jiang S, June CH, Zhao Y. A versatile system for rapid multiplex genome-edited CAR T cell generation. Oncotarget. 2017;8:17002–11.
Liu X, Zhang Y, Cheng C, Cheng AW, Zhang X, Li N, Xia C, Wei X, Liu X, Wang H. CRISPR-Cas9-mediated multiplex gene editing in CAR-T cells. Cell Res. 2017;27:154–7.
Ligtenberg MA, Mougiakakos D, Mukhopadhyay M, Witt K, Lladser A, Chmielewski M, Riet T, Abken H, Kiessling R. Coexpressed catalase protects chimeric antigen receptor-redirected T cells as well as bystander cells from oxidative stress-induced loss of antitumor activity. J Immunol. 2016;196:759–66.
Clever D, Roychoudhuri R, Constantinides MG, Askenase MH, Sukumar M, Klebanoff CA, Eil RL, Hickman HD, Yu Z, Pan JH, et al. Oxygen sensing by T cells establishes an immunologically tolerant metastatic niche. Cell. 2016;166(1117–1131):e1114.
Juillerat A, Marechal A, Filhol JM, Valogne Y, Valton J, Duclert A, Duchateau P, Poirot L. An oxygen sensitive self-decision making engineered CAR T-cell. Sci Rep. 2017;7:39833.
Fraietta JA, Beckwith KA, Patel PR, Ruella M, Zheng Z, Barrett DM, Lacey SF, Melenhorst JJ, McGettigan SE, Cook DR, et al. Ibrutinib enhances chimeric antigen receptor T-cell engraftment and efficacy in leukemia. Blood. 2016;127:1117–27.
Ruella M, Kenderian SS, Shestova O, Fraietta JA, Qayyum S, Zhang Q, Maus MV, Liu X, Nunez-Cruz S, Klichinsky M, et al. The addition of the BTK inhibitor ibrutinib to Anti-CD19 chimeric antigen receptor T cells (CART19) improves responses against mantle cell lymphoma. Clin Cancer Res. 2016;22:2684–96.
Newick K, O’Brien S, Sun J, Kapoor V, Maceyko S, Lo A, Pure E, Moon E, Albelda SM. Augmentation of CAR T-cell trafficking and antitumor efficacy by blocking protein kinase A localization. Cancer Immunol Res. 2016;4:541–51.
Kuramitsu S, Ohno M, Ohka F, Shiina S, Yamamichi A, Kato A, Tanahashi K, Motomura K, Kondo G, Kurimoto M, et al. Lenalidomide enhances the function of chimeric antigen receptor T cells against the epidermal growth factor receptor variant III by enhancing immune synapses. Cancer Gene Ther. 2015;22:487–95.
Boice M, Salloum D, Mourcin F, Sanghvi V, Amin R, Oricchio E, Jiang M, Mottok A, Denis-Lagache N, Ciriello G, et al. Loss of the HVEM tu mor suppressor in lymphoma and restoration by modified CAR-T cells. Cell. 2016;167(405–418):e413.
Authors’ contributions
LQ—design, investigation, drafting of manuscript, critical review and editing. RZ—contributor of manuscript, critical review and editing. PL—conceptualization, design, critical review and editing, supervision. All authors read and approved the final manuscript.
Acknowledgements
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Availability of data and materials
Data sharing not applicable to this article as no datasets were generated or analyzed during the current study.
Consent for publication
Not applicable.
Ethics approval and consent to participate
Not applicable.
Funding
No relevant funding or disclosures to be made.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Qin, L., Zhao, R. & Li, P. Incorporation of functional elements enhances the antitumor capacity of CAR T cells. Exp Hematol Oncol 6, 28 (2017). https://doi.org/10.1186/s40164-017-0088-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40164-017-0088-z