
Abstract
During the past two decades, a great evolution of bispecific antibodies (BsAbs) for therapeutic applications has been made. BsAbs can bind simultaneously two different antigens or epitopes, which leads to a wide range of applications including redirecting T cells or NK cells to tumor cells, blocking two different signaling pathways, dual targeting of different disease mediators, and delivering payloads to targeted sites. Aside from approved catumaxomab (anti-CD3 and anti-EpCAM) and blinatumomab (anti-CD3 and anti-CD19), many more BsAbs are now in various phases of clinical development. Here, this review focus on the development of bispecific antibodies and their applications in tumor immune escape.
Similar content being viewed by others


Background
Although considerable evidence supports the hypothesis that immune cells play a vital role in the immune response against cancer, the ability to mount and sustain tumor-specific cellular responses in vivo remains a challenge [1]. Cancer immunotherapy, which harnesses the immune system to battle cancer [2], was named “2013’s Breakthrough of the Year” by Science [3]. Cancer immunotherapy was proposed decades ago but has only recently been realized as a promising approach to cancer treatment due to the success of immunomodulating anti-CTLA-4 and anti-PD-1 monoclonal antibodies against various of cancers [4].
In addition to immunomodulating antibodies, bispecific antibodies (BsAbs) are another promising strategy to fight cancer by directly redirecting immune cells to tumor cells. BsAbs have a long history [5], starting in the 1960s when antigen-binding fragments (Fabs) from two different polyclonal sera were re-associated into bispecific F(ab’)2 molecules [6]. A bispecific antibody is based on a conventional monoclonal antibody, and it can recognize and bind two different antigens or epitopes simultaneously. Thus, BsAbs show several advantages [1, 7,8,9]: (1) BsAbs can redirect specific immune effector cells to the proximity tumor cells to enhance tumor killing, which is not achievable with a combination monoclonal antibody strategy; (2) BsAbs can potentially increase binding specificity by interacting with two different cell-surface antigens instead of one; (3) BsAbs offer an opportunity to reduce cost in terms of development, production clinical trials, and regulatory reviews, compared to the single antibody-based agents development in combination therapies; (4) BsAbs will enable the simultaneous blocking of two different pathways that exert unique or overlapping functions in pathogenesis.
The development of BsAbs has long been hampered due to manufacturing problems such as product instability, low expression yields, and immunogenicity [10]. With the development of molecular cloning technology and antibody engineering, there are diverse bispecific antibody formats from which to choose to pursue the optimal biological activity and clinical purpose [11]. There are around 100 different bispecific antibody formats, including small molecules solely of the antigen-binding sites of two antibodies, molecules with an IgG format, and large complex molecules composed of different antigen-binding moieties usually combined with dimerization modules [9]. The engineering of monospecific antibodies to be bispecific opens up a variety of potential therapeutic applications as evidenced by the more than 30 BsAbs currently in clinical development [12]. And the BsAbs against cancers in clinical development were summarized in Table 1.
Like armed monoclonal antibodies, BsAbs do not occur naturally in human body and must be produced by either recombination DNA or cell-fusion technologies. And BsAbs are mainly produced by three methods [13]: (1) chemical conjugation, which involves chemical cross-linkers; (2) quadroma technology based on the somatic fusion of two different hybridoma cell lines; (3) genetic approaches using recombinant DNA technology. This review focuses on the development of the strategies to generate recombinant bispecific antibodies and strategies to reverse immune escape in the treatments.
Generation of BsAbs
Chemical engineering of BsAbs
Chemical conjugation of two different purified monoclonal antibodies was employed to create BsAbs by oxidative recombination firstly in 1961 [6]. Two purified monoclonal antibodies were conjugated through a cross-linker such as the bispecific antibody anti-CD3 × anti-GD2 (3F8BiAb) which was designed to redirect activated T cells to GD2-positive neuroblastomas [14]. Alternative approach is to yield Fab fragments through enzymatic digestion and reduction of desired specific purified antibodies. Bifunctional reagents, which bind to the Fab fragments, are then added to allow for heterodimer assembly by association of the Fab fragments.
However, it is difficult to purify the bispecific heterodimers from homodimers because of the heterogeneity of the end products. And another drawback of chemical cross-linking is poor stability and decreased activity of the antibodies. To improve the purity and yield of products, a scalable method to prepare BsAbs, which was named controlled Fab-arm exchange (cFAE), was developed [15, 16]. The process involves separate expression of two parental antibodies, each containing single matched point mutations in the CH3 domains (F405L and K409R, respectively). Then the parental antibodies (IgG1-F405L-EGFR and IgG1-K409R-CD20) are mixed and subjected to controlled reducing conditions (incubated with 50 Mm 2-mercaptoethylamine-HCl for 5 h at ambient temperature) in vitro that separate the antibodies in HL half-molecules and allow reassembly and re-oxidation to form highly pure BsAbs. And this process results in generating BsAbs with a greater than 90% heterodimerization efficiency and greater than 90% yield [16]. Additionally, Suparna Paul et al. proved that the process could be accelerated by conducting the cFAE using culture supernatants from separate cultures that were producing the respective parental antibodies with matched mutations prior to purification [17], which provides an shorter processing time with potential benefits in large-scale BsAbs preparation.
Quadroma technology for BsAbs
Another early attempt to produce BsAbs employed quadroma technology. This approach is based on the somatic fusion of two different hybridoma cells producing monoclonal antibodies with the desired specificity. BsAbs produced by quadromas resemble conventional antibodies, which retain Fc-mediated effector functions such as antibody-dependent cell-mediated cytotoxicity (ADCC), complement-dependent cytotoxicity (CDC), and antibody-dependent cellular phagocytosis (ADCP) [10]. Additionally, the Fc region of BsAbs favor purification and improves solubility and stability. And because of their large size and FcRn-mediated recycling, BsAbs with IgG-like formats usually have longer serum half-lives [12].
Because quadromas express two different immunoglobulin heavy and light chains that assemble randomly, nonfunctional antibodies are also produced. However, mispaired by-products could be significantly decreased by fusing two hybridomas of different species such as mouse/rat quadroma. Catumaxomab, produced by fusion of a mouse hybridoma and rat hybridoma, is the first approved bispecific antibody in 2009 for the treatment of malignant ascites in patients with EpCAM-positive Tumors [18]. Interestingly, the resulting hybrid mouse/rat Fc portion efficiently interacted with activating human Fc receptors (FcγRI and FcγRIII), but not inhibitory ones (FcγRIIB) [19]. However, catumaxomab is a mouse-rat hybrid IgG molecule, human anti-mouse or anti-rat antibody response are observed in most patients [12]. Several other BsAbs have been produced using similar quadroma technology. For example, FBTA05 [20] (anti-CD3 and anti-CD20) and ertumaxomab [21] (anti-CD3 and anti-HER2) are in different phases of clinical development for patients with relapsed or refractory B-cell lymphoma or metastatic breast cancers, respectively.
Homodimerization of the two heavy chains of IgG is mediated by the interaction between CH3 domains. To overcome the problem of undesirable heavy-chain pairing, a strategy called “knobs-into-holes” [22] was developed. The knobs-into-holes scenario employs a “knob” mutation (T366W) and pairing “hole” mutations (T336S, L368A, Y407V) in the CH3 domains [23]. In this approach a “knob” variant was obtained by replacement of a small amino acid with a larger one in the CH3 domain, which was desired to insert into a “hole” in another CH3 domain created by replacement of a large residue with a small one [23]. The mutated CH3 domains could facilitate heterodimerization of heavy chains [23]. However, another challenge for BsAbs in this format is the problem with light chain mispairing. To circumvent this, several methods have been proposed [24]: (1) generating BsAbs with common light chains; (2) expressing the knob- and hole-containing half-molecules separately in different bacteria; (3) combining CrossMab and knobs-into-holes strategies; (4) introducing additional mutations into VH–VL and CH1–CL interfaces.
Genetic engineering of BsAbs
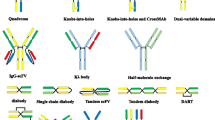
By using molecular cloning technology, BsAbs can be constructed with some or all of the constant domains of an antibody. BsAbs in this group can be divided into two categories: IgG-like formats and non-IgG-like formats. As mentioned above, IgG-like formats are BsAbs bearing an Fc region, which retain Fc-mediated effector functions. These formats roughly include “knob into hole” IgG, crossMab, ortho-Fab IgG, DVD-Ig, two in one IgG, IgG-scFv and scFv2-Fc [12].
Here, we focus on the non-IgG-like formats of BsAbs. The smaller size of such antibodies offers a better tumor tissue penetration over IgG-like formats. In this format, the variable domains of each parental monoclonal antibody and the linkers are cloned and linked to form a single-chain bispecific antibody. These bispecific antibodies represent many formats, including tandem scFvs, diabody format, single-chain diabodies, tandem diabodies (TandAbs), dual-affinity retargeting molecules (DARTs), dock-and-lock (DNL), and nanobodies [24].
Among these bispecific antibody constructs, tandem scFvs and diabodies have been intensively studied. Tandem scFvs are two scFv fragments linked by an extra peptide linker such as glycine-serine repeat motifs [13]. The most frequently-used domain order is VLA-linker1–VHA-linker2–VHB-linker3–VLB (VL and VH derive from the single chain antibody fragment; A and B represent the parental monoclonal antibody A and B) [25]. The length of the linker1 and linker3 determines the polymerization situation of scFv, while the linker2 determines the movement flexibility between two scFvs. The short linker prevents intra-chain but not inter-chain pairing of the VL and VH domains. And the long flexible linker permits antigen-binding sites to rotate freely. Like one of the best known bispecific T cells engagers (BiTEs) blinatumonmab, two longer linkers are placed between the light chain and heavy chain, and a short linker is used to bridge the two scFvs in tandem format [26]. Although it is difficult to be produced in E. coli, BiTEs are well-expressed in mammalian cells. In the diabody format, the VH of the first antibody is linked to the VL of the second antibody, while the VL of the first antibody is linked to the VH of the second antibody. In addition, this format has been improved by adding an inter-chain disulfide bond between the two polypeptides in order to decrease the amount and stability of homodimers, which were called DARTs [27].
Compared with IgG-like formats, scFv-based BsAbs have many advantages including less immunogenic, ease of manufacturing, and enhanced tissue penetration. However, their short half-lives due to their small sizes and lack of Fc region become a drawback in the clinical applications. For example, blinatumomab is administrated over a 28-day continuous infusion using a mini-pump in order to maintain a steady drug concentration [28], which results in inconvenience for patients and an increased possibility of treatment-related adverse event. To overcome this drawback, a tetravalent TandAbs format is developed. TandAbs contain two pairs of VL and VH domains connected in a single polypeptide chain [29]. Upon expression, two polypeptide products dimerize in a head-to-tail fashion, forming homodimers with large molecular weight (~105 kDa) [30]. AFM11 is a tetravalent bispecific TandAb targeting CD19 and CD3 with a half-life ranging from 18.4 to 22.9 h after intravenous administration in mice [29]. Because of its excellent preclinical outcomes, AFM11 is entering clinical trials and currently recruiting patients with relapsed and/or refractory CD19 positive B-cell NHL (ClinicalTrials.gov: NCT02106091) and patients with relapsed or refractory adult B-precursor ALL (ClinicalTrials.gov: NCT02848911). Another TandAb AFM13 (anti-CD30/anti-CD16A) is designed for the treatment of CD30-positive malignant lymphoma by redirecting and activating NK cells. AFM13 is now entering phase II clinical trials in patients with relapsed of refractory Hodgkin Lymphoma (ClinicalTrials.gov: NCT02321592).
In addition to multimerization, the serum half-life of single-chain-based BsAbs can be extended by other available strategies including conjugating a single polyethylene glycol (PEG) chain [31], fusion with human serum albumin (HSA) [32], or fusion with an Fc fragment [33].
BsAbs in tumor immune escape
A CD33/CD3-bispecific BiTE construct called AMG330 has been designed to target acute myeloid leukemia (AML) [34]. In preclinical studies, AMG330 was shown to be very effective in recruiting and activating autologous T cells [34]. However, reduced T-cell activation and decreased tumor cell lysis was observed in some patient cases [35]. Recently, a study from C. Krupka, et al. [36] showed that although PD-1 and PD-L1 were not expressed at a relevant level at time of diagnosis, their expression was induced by AMG330-mediated T-cell activation in primary AML patient samples. They also demonstrated that blockade of the PD-1/PD-L1 interaction augmented lysis of AML cells by AMG330. Thus, the use of bispecific antibodies especially which leads to strong T-cell activation and production of proinflammatory cytokines might also trigger tumor cells to employ immunosuppressive strategies to escape antibody-mediated tumor cell lysis.
Aside from the PD-1/PD-L1 axis, the CD47/SIRPα interaction should also be noted. CD47 is a ubiquitously expressed immune checkpoint receptor that is usually up-regulated in cancers [37]. CD47 could interact with its counter-receptor SIRPα on macrophages and other myeloid cells to inhibit tumor cell phagocytosis and trigger immune evasion [38, 39]. For this reason, IgG-based bispecific antibodies were created to block CD47 combining tumor targeting. These bispecific antibody constructs including anti-CD47/CD20 [40], anti-CD47/CD19 [41], and anti-CD47/MSLN [41]. By neutralizing CD47, tumor cells could be efficiently killed effector cells mediated by their Fc portion.
As a subclass of growth factor receptors, receptor tyrosine kinases (RTKs) play a vital role in oncogenesis [42]. Although several monospecific antibodies targeted RTKs have been approved for cancer treatment, simultaneously blocking two RTKs with BsAbs may offer better therapeutic potential than monoclonal antibodies. The Her family of RTKs has four members, HER1/EGFR, HER2, HER3, and HER4. HER2-HER3 heterodimerization leads to breast cancer cell proliferation and is involved in transformation [43]. Then MM-111, a BsAb with two scFv fused to modified HSA in phase 2 study, was developed to bind to both of HER2 and HER3 [44]. Another BsAb in phase 1 study is MM-141, which bind HER3 as well as insulin-like growth factor-1R (IGF1R) with a scFv-IgG format. Thus, simultaneous inhibition of two RTKs could be a valid strategy to overcome escape of tumors. And several other BsAbs that inhibit two RTKs are included in Table 1, such as RG7597 and LY3164530.
On the other hand, tumor cells may down-regulate the antibody’s target antigen and escape recognition during the treatment, which is another major escape mechanism. Multiple clinical trials have shown that anti-CD19 chimeric antigen receptor T cells (CART19) have curative potential against relapsed B-cell malignancies [45]. However, a recent trial of CD19 CAR T-cell therapy revealed that 90% of patients acquired a complete response, but 11% of those patients eventually relapsed with CD19-negative tumors [46]. The probability of antigen escape by spontaneous mutation and selective expansion of antigen-negative tumor cells decreases with each additional antigen that can be recognized by the CAR T cells. Therefore, a potential strategy against antigen escape is to combine bispecific antibodies to generate T cells that could recognize multiple antigens. The first bispecific CAR T cells, which could simultaneously recognize both of CD19 and CD20, were developed to prevent antigen escape by malignant B cells [47].
Conclusions
As the next generation of strategies for cancer therapy, bispecific antibodies have acquired much attention owing to their unique mechanism of action. Although just only two of BsAbs have gained marketing approval, numerous designed BsAbs are now being tested in clinical trials [24] not only for cancers, but also for other diseases.
Future advances in BsAbs technology will be focused on the development of new platforms which encompass the entire process from discovery and preclinical studies to clinical material production. On the other hand, the discovery of new targets is also urgently needed to increase efficacy and reduce adverse effects of bispecific antibodies. The complexity of tumors should also be in consideration during the process of cancer therapy. Thus, bispecific antibodies might probably be combined with other therapeutics such as checkpoint antibodies, IDO inhibitors, or vaccines.
In conclusion, new formats and producing methods of bispecific antibodies should be persistently developed. And continued persistence is needed in the anti-cancer battle.
Abbreviations
- ADCC:
-
antibody-dependent cell-mediated cytotoxicity
- ADCP:
-
antibody-dependent cellular phagocytosis
- AML:
-
acute myeloid leukemia
- ALL:
-
acute lymphoblastic leukemia
- BCL:
-
B cell lymphoma
- BiTE:
-
bispecific T cell engager
- BsAbs:
-
bispecific antibodies
- CAR:
-
chimeric antigen receptor
- CDC:
-
complement-dependent cytotoxicity
- cFAE:
-
controlled Fab-arm exchange
- DARTs:
-
dual-affinity retargeting molecules
- DNL:
-
dock-and-lock
- Fab:
-
antigen-binding fragment
- HSA:
-
human serum albumin
- NHL:
-
non-Hodgkin’s lymphoma
- NK:
-
natural killer cells
- PEG:
-
polyethylene glycol
- RTK:
-
receptor tyrosine kinase
- scFv:
-
single- chain variable fragment
References
Choi BD, Cai M, Bigner DD, Mehta AI, Kuan C-T, Sampson JH. Bispecific antibodies engage T cells for antitumor immunotherapy. Expert Opin Biol Ther. 2011;11:843–53.
Yang Y. Cancer immunotherapy: harnessing the immune system to battle cancer. J Clin Invest. 2015;125:3335–7.
Couzin-Frankel J. Cancer Immunother. Science. 2013;342:1432.
Le Mercier I, Lines JL, Noelle RJ. Beyond CTLA-4 and PD-1, the generation Z of negative checkpoint regulators. Front Immunol. 2015;6:418.
Riethmüller G. Symmetry breaking: bispecific antibodies, the beginnings, and 50 years on. Cancer Immun. 2012;12:12.
Nisonoff A, Rivers M. Recombination of a mixture of univalent antibody fragments of different specificity. Arch Biochem Biophys. 1961;93:460–2.
Dhimolea E, Reichert JM. World bispecific antibody summit, September 27–28, 2011, Boston, MA. mAbs. 2012;4:4–13.
Kontermann R. Dual targeting strategies with bispecific antibodies. mAbs. 2012;4:182–97.
Brinkmann U, Kontermann RE. The making of bispecific antibodies. mAbs. 2017;9:182–212.
Spiess C, Zhai Q, Carter PJ. Alternative molecular formats and therapeutic applications for bispecific antibodies. Mol Immunol. 2015;67:95–106.
Yang F, Wen W, Qin W. Bispecific antibodies as a development platform for new concepts and treatment strategies. Int J Mol Sci. 2016;18:48.
Kontermann RE, Brinkmann U. Bispecific antibodies. Drug Discov Today. 2015;20:838–47.
Chames P, Baty D. Bispecific antibodies for cancer therapy: the light at the end of the tunnel? mAbs. 2009;1:539–47.
Yankelevich M, Kondadasula SV, Thakur A, Buck S. Cheung N-KV, Lum LG. Anti-CD3 × anti-GD2 bispecific antibody redirects T-cell cytolytic activity to neuroblastoma targets. Pediatr Blood Cancer. 2012;59:1198–205.
Labrijn AF, Meesters JI, de Goeij BECG, van den Bremer ETJ, Neijssen J, van Kampen MD, et al. Efficient generation of stable bispecific IgG1 by controlled Fab-arm exchange. Proc Natl Acad Sci. 2013;110:5145–50.
Gramer MJ, van den Bremer ET, van Kampen MD, Kundu A, Kopfmann P, Etter E, et al. Production of stable bispecific IgG1 by controlled Fab-arm exchange: scalability from bench to large-scale manufacturing by application of standard approaches. mAbs. 2013;5:962–73.
Paul S, Connor J, Nesspor T, Haytko P, Boakye K, Chiu ML, et al. An efficient process of generating bispecific antibodies via controlled Fab-arm exchange using culture supernatants. Protein Expr Purif. 2016;121:133–40.
Seimetz D, Lindhofer H, Bokemeyer C. Development and approval of the trifunctional antibody catumaxomab (anti-EpCAM × anti-CD3) as a targeted cancer immunotherapy. Cancer Treat Rev. 2010;36:458–67.
Zeidler R, Mysliwietz J, Csánady M, Walz A, Ziegler I, Schmitt B, et al. The Fc-region of a new class of intact bispecific antibody mediates activation of accessory cells and NK cells and induces direct phagocytosis of tumour cells. Br J Cancer. 2000;83:261–6.
Buhmann R, Michael S, Juergen H, Horst L, Peschel C, Kolb H-J. Immunotherapy with FBTA05 (Bi20), a trifunctional bispecific anti-CD3x anti-CD20 antibody and donor lymphocyte infusion (DLI) in relapsed or refractory B-cell lymphoma after allogeneic stem cell transplantation: study protocol of an investigator-driven, open-label, non-randomized, uncontrolled, dose-escalating Phase I/II-trial. J Transl Med. 2013;11:160.
Jäger M, Schoberth A, Ruf P, Hess J, Lindhofer H. The trifunctional antibody ertumaxomab destroys tumor cells that express low levels of human epidermal growth factor receptor 2. Cancer Res. 2009;69:4270.
Shatz W, Chung S, Li B, Marshall B, Tejada M, Phung W, et al. Knobs-into-holes antibody production in mammalian cell lines reveals that asymmetric afucosylation is sufficient for full antibody-dependent cellular cytotoxicity. mAbs. 2013;5:872–81.
Ridgway JB, Presta LG, Carter P. “Knobs-into-holes” engineering of antibody CH3 domains for heavy chain heterdimerization. Protein Eng. 1996;9:617–21.
Fan G, Wang Z, Hao M, Li J. Bispecific antibodies and their applications. J Hematol Oncol J Hematol Oncol. 2015. doi:10.1186/s13045-015-0227-0.
Löffler A, Kufer P, Lutterbüse R, Zettl F, Daniel PT, Schwenkenbecher JM, et al. A recombinant bispecific single-chain antibody, CD19 × CD3, induces rapid and high lymphoma-directed cytotoxicity by unstimulated T lymphocytes. Blood. 2000;95:2098.
May MB, Glode A. Blinatumomab: a novel, bispecific, T-cell engaging antibody. Am J Health Syst Pharm. 2016;73:e6–13.
Walseng E, Nelson CG, Qi J, Nanna AR, Roush WR, Goswami RK, et al. Chemically programmed bispecific antibodies in diabody format. J Biol Chem. 2016;291:19661–73.
Le Jeune C, Thomas X. Potential for bispecific T-cell engagers: role of blinatumomab in acute lymphoblastic leukemia. Drug Des Dev Ther. 2016;10:757–65.
Reusch U, Duell J, Ellwanger K, Herbrecht C, Knackmuss SH, Fucek I, et al. A tetravalent bispecific TandAb (CD19/CD3), AFM11, efficiently recruits T cells for the potent lysis of CD19 + tumor cells. mAbs. 2015;7:584–604.
Kipriyanov SM, Moldenhauer G, Schuhmacher J, Cochlovius B, Von der Lieth C-W, Matys ER, et al. Bispecific tandem diabody for tumor therapy with improved antigen binding and pharmacokinetics1. J Mol Biol. 1999;293:41–56.
Fishburn CS. The Pharmacology of PEGylation: balancing PD with PK to generate novel therapeutics. J Pharm Sci. 2008;97:4167–83.
Merlot AM, Kalinowski DS, Kovacevic Z, Jansson PJ, Lane DJ, Huang ML-H, et al. Making a case for albumin—a highly promising drug-delivery system. Future Med Chem. 2015;7:553–6.
Kontermann RE. Strategies for extended serum half-life of protein therapeutics. 226 Chem Biotechnol. Pharm Biotechnol. 2011;22:868–76.
Aigner M, Feulner J, Schaffer S, Kischel R, Kufer P, Schneider K, et al. T lymphocytes can be effectively recruited for ex vivo and in vivo lysis of AML blasts by a novel CD33/CD3-bispecific BiTE antibody construct. Leukemia. 2013;27:1107–15.
Laszlo GS, Gudgeon CJ, Harrington KH, Dell’Aringa J, Newhall KJ, Means GD, et al. Cellular determinants for preclinical activity of a novel CD33/CD3 bispecific T-cell engager (BiTE) antibody, AMG 330, against human AML. Blood. 2014;123:554–61.
Krupka C, Kufer P, Kischel R, Zugmaier G, Lichtenegger FS, Köhnke T, et al. Blockade of the PD-1/PD-L1 axis augments lysis of AML cells by the CD33/CD3 BiTE antibody construct AMG 330: reversing a T-cell-induced immune escape mechanism. Leukemia. 2016;30:484–91.
Jaiswal S, Jamieson CHM, Pang WW, Park CY, Chao MP, Majeti R, et al. CD47 is upregulated on circulating hematopoietic stem cells and leukemia cells to avoid phagocytosis. Cell. 2009;138:271–85.
Majeti R, Chao MP, Alizadeh AA, Pang WW, Jaiswal S, Gibbs KD Jr, et al. CD47 is an adverse prognostic factor and therapeutic antibody target on human acute myeloid leukemia stem cells. Cell. 2009;138:286–99.
Brown EJ, Frazier WA. Integrin-associated protein (CD47) and its ligands. Trends Cell Biol. 2001;11:130–5.
Piccione EC, Juarez S, Liu J, Tseng S, Ryan CE, Narayanan C, et al. A bispecific antibody targeting CD47 and CD20 selectively binds and eliminates dual antigen expressing lymphoma cells. mAbs. 2015;7:946–56.
Dheilly E, Moine V, Broyer L, Salgado-Pires S, Johnson Z, Papaioannou A, et al. Selective blockade of the ubiquitous checkpoint receptor CD47 is enabled by dual-targeting bispecific antibodies. Mol Ther. 2017;25:523–33.
Gschwind A, Fischer OM, Ullrich A. The discovery of receptor tyrosine kinases: targets for cancer therapy. Nat Rev Cancer. 2004;4:361–70.
Holbro T, Beerli RR, Maurer F, Koziczak M, Barbas CF, Hynes NE. The ErbB2/ErbB3 heterodimer functions as an oncogenic unit: ErbB2 requires ErbB3 to drive breast tumor cell proliferation. Proc Natl Acad Sci USA. 2003;100:8933–8.
McDonagh CF, Huhalov A, Harms BD, Adams S, Paragas V, Oyama S, et al. Antitumor activity of a novel bispecific antibody that targets the ErbB2/ErbB3 oncogenic unit and inhibits heregulin-induced activation of ErbB3. Mol Cancer Ther. 2012;11:582.
Ruella M, Maus MV. Catch me if you can: leukemia escape after CD19-directed T cell immunotherapies. Comput Struct Biotechnol J. 2016;14:357–62.
Maude SL, Frey N, Shaw PA, Aplenc R, Barrett DM, Bunin NJ, et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med. 2014;371:1507–17.
Zah E, Lin M-Y, Silva-Benedict A, Jensen MC, Chen YY. T cells expressing CD19/CD20 Bispecific chimeric antigen receptors prevent antigen escape by malignant B cells. Cancer Immunol Res. 2016;4:498–508.
Authors’ contributions
XZ drafted the manuscript; YY, DF, and DX revised the manuscript. All authors read and approved the final manuscript.
Acknowledgements
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Funding
This work was supported by the National Natural Science Foundation of China (Grant Nos. 30971291, 81400176, 81572993), and CAMS Initiative for Innovative Medicine (Grant Nos. 2016-I2 M-1-007, 2016-I2 M-3-013).
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Zhang, X., Yang, Y., Fan, D. et al. The development of bispecific antibodies and their applications in tumor immune escape. Exp Hematol Oncol 6, 12 (2017). https://doi.org/10.1186/s40164-017-0072-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40164-017-0072-7