
Abstract
Background
Accurate evaluation of SNP effects is important for genome wide association studies and for genomic prediction. The genetic architecture of quantitative traits differs widely, with some traits exhibiting few if any quantitative trait loci (QTL) with large effects, while other traits have one or several easily detectable QTL with large effects.
Methods
Body weight in broilers and egg weight in layers are two examples of traits that have QTL of large effect. A commonly used method for genome wide association studies is to fit a mixture model such as BayesB that assumes some known proportion of SNP effects are zero. In contrast, the most commonly used method for genomic prediction is known as GBLUP, which involves fitting an animal model to phenotypic data with the variance-covariance or genomic relationship matrix among the animals being determined by genome wide SNP genotypes. Genotypes at each SNP are typically weighted equally in determining the genomic relationship matrix for GBLUP. We used the equivalent marker effects model formulation of GBLUP for this study. We compare these two classes of models using egg weight data collected over 8 generations from 2,324 animals genotyped with a 42 K SNP panel.
Results
Using data from the first 7 generations, both BayesB and GBLUP found the largest QTL in a similar well-recognized QTL region, but this QTL was estimated to account for 24 % of genetic variation with BayesB and less than 1 % with GBLUP. When predicting phenotypes in generation 8 BayesB accounted for 36 % of the phenotypic variation and GBLUP for 25 %. When using only data from any one generation, the same QTL was identified with BayesB in all but one generation but never with GBLUP. Predictions of phenotypes in generations 2 to 7 based on only 295 animals from generation 1 accounted for 10 % phenotypic variation with BayesB but only 6 % with GBLUP. Predicting phenotype using only the marker effects in the 1 Mb region that accounted for the largest effect on egg weight from generation 1 data alone accounted for almost 8 % variation using BayesB but had no predictive power with GBLUP.
Conclusions
In conclusion, In the presence of large effect QTL, BayesB did a better job of QTL detection and its genomic predictions were more accurate and persistent than those from GBLUP.
Similar content being viewed by others


Background
The choice of methods for accurate evaluation of SNP effects in the presence of large effect QTL can be critical for genome wide association studies (GWAS) and for genomic selection (GS). Mixture models, sometimes known as variable selection models, assume some SNP effects are zero. One such mixture model, method BayesB [1], has been shown to adequately estimate marker effects in simulated data [2, 3] but comparisons based on real data where true effects are unknown are less straightforward. In a recent paper using broiler body weight data, Wang et al. [4] conclude that compared to single step GBLUP procedures “BayesB appears to overly shrink regions to zero, while overestimating the amount of genetic variation attributed to the remaining SNP effects”. Egg weight is one trait in layers for which multiple studies report detecting a QTL with large effect [5–8] at a consistent position on chromosome 4. The objective of this study was to demonstrate that in the presence of such large effect QTL, mixture models like BayesB and BayesC [9, 10] are superior to GBLUP for detecting and quantifying the effects of QTL, and that mixture models provide more accurate genomic estimates of breeding values (GEBV) that are more persistent over generations, using brown egg layer data.
Methods
Average egg weight data of 3–5 eggs collected at 42–45 wk of age from a brown egg layer line, described in detail by Wolc et al. [8], were used. Only records of animals that were genotyped and had individual performance data were retained, resulting in the following numbers of observations in each of 8 consecutive generations: 295, 323, 294, 360, 290, 252, 300 and 210. All 2,324 animals were genotyped with a custom made 42 K Illumina SNP chip, of which 24,383 segregating SNPs were retained after quality control (removing SNPs with minor allele frequency <0.025, proportion of missing genotypes >0.05, and parent-offspring mismatches >0.05). Build WUGSC 2.1/galGal3 (http://moma.ki.au.dk/genome-mirror/cgi-bin/hgGateway?db=galGal3) was used to identify the genomic locations of markers. Data were analyzed using mixture models that assume π = 99 % of SNPs have no effect, namely BayesB [1], which assumes a different unknown variance for each SNP, BayesC [9, 10], which assumes equal unknown variance for all SNPs. Performance of the mixture models were compared to two ridge regression methods that fit all SNPs in the model (i.e. π = 0), assuming equal variance of SNP effects. The latter methods were two parameterizations of BayesC0, one with the same degrees of freedom (4, 10) for the genetic and residual scale factors as used for BayesB and BayesC, which results in the variance components being estimated jointly from the data and the prior, and one with large values for the degrees of freedom for the genetic and residual scale factor priors (100, 100), which is equivalent to GBLUP [11] in treating the equal variance of SNP effects as known. This model was referred to as GBLUP throughout the paper. The fixed class effect of hatch within generation was fitted in addition to random marker effects. All methods were implemented in the GenSel software [12], which uses Gibbs sampling MCMC algorithms. For inference on parameters we used a chain of 35,000 samples with the first 5,000 discarded as burn in. As a reference for accuracy of prediction, pedigree-based analysis was used with the phenotypes of genotyped individuals and their pedigree which included 26,300 individuals from generations −2 (2 generations prior to the first generation with data) to generation 8.
The marker effects were estimated using whole genome data from all of the first 7 generations, as well as from each generation separately. Accuracy of prediction was assessed as the correlation between GEBV and hatch-corrected phenotype in the validation generation, which was generation 8, or in each successive generation when training only used the 295 individuals from generation 1. Accuracy of prediction was quantified using marker effects from the whole genome, or using only estimates of effects of markers located in the largest QTL region, which was defined as the 1 Mb window that accounted for the largest proportion of variance (the effects of the 1 Mb markers were extracted from solutions of a model fitting all SNPs simultaneously). Persistence of predictions was quantified in terms of the correlations in successive post-training generations.
In order to quantify potential bias in the estimation of the largest effect by BayesB, the SNP that explained the greatest proportion of variance in the 1 Mb window that contributed the largest proportion of variance was fitted as a fixed effect in an animal model using ASReml [13] and the regression coefficients for predicting the 8th generation of hatch-corrected phenotypes were calculated and compared to the corresponding regression from BayesB – a regression coefficient of phenotype on the SNP effect estimate greater than 1 would indicate overshrinkage of the SNP effect; this means that the observed difference in phenotypes is larger than expected based on marker solutions.
Results and discussion
Detection of QTL
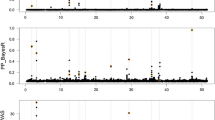
The 1 Mb windows that contributed the greatest proportion of genetic variance for each genomic method are shown in Table 1. Using all data from the first 7 generations detected a very large QTL on chromosome 4 at 78 Mb (4_78) using either of the mixture models, and at 4_77 using the models that fitted all loci simultaneously. The effect attributed to this QTL has been shown in other data to be associated with differential expression of the cholecystokinin receptor A (CCKAR) gene [14], which is located around 2 Mb away at ~75.6 Mb. That gene appears to increase orexigenic drive by reducing feedback from the gut to the brain, signaled by CCK [14]. The precise location or nature of the causal mutation responsible for these expression differences is not known. Although it is conceivable that SNPs in 4_78 are in high long-range LD with variants in CCKAR, the 4_78 QTL region on chromosome 4 also includes the LCORL and NCAPG genes, which have been observed to be associated with growth or stature in numerous mammalian GWAS studies [15–18]. So, although this effect could involve long-range LD, or a long-range enhancer acting on CCKAR, it could alternatively be the effect of a different gene, conserved across birds and mammals. Regardless of the cause, a positive genetic correlation has been reported between body weight and yolk weight and between body weight and egg weight for which this QTL is a major contributor [8].
The 4_78 window accounted for 23.7 % of the genetic variance for egg weight when using BayesB, and 14.4 % when using BayesC. In small datasets such as used here, BayesC is expected to result in smaller estimates of variance than BayesB, because BayesC shrinks all SNP effects according to the same variance ratio, whereas BayesB shrinks the effect of each SNP according to the ratio of the residual variance to the unique variance estimate for that SNP. The sampling of the SNP effects variances involves a function of the square of the sample of the SNP effect [19] that results in little shrinkage of large effect loci and considerable shrinkage of small effect loci. In contrast, the two methods that simultaneously fitted all 24,383 equal variance loci using 2,114 animals in the first 7 generations detected the largest QTL 1 Mb upstream at 4_77 and attributed less than 1 % genetic variance to that window.
Using Bayes B for training only on data from any one of the first 7 generations involving at most 360 animals, detected the largest QTL at 4_78 in all but generation 7. Using BayesC detected the largest QTL in 4_78 in all but generations 6 or 7. The estimated effects of the 4_78 QTL were always smaller than obtained with training on all 7 generations, and were never larger for BayesC than for BayesB. The effect of the variance ratio in shrinking SNP effects reduces as the size of the dataset increases [12], and is expected to result in consistent window variances for BayesB and BayesC if the dataset is sufficiently large. In contrast, the two methods that fitted all SNPs never detected the largest effect SNP on chromosome 4 when data from only a single generation was used. Thus, the mixture models were clearly better at detecting large effect QTL than models such as GBLUP, especially in small datasets.
Accuracy of prediction
The accuracies of prediction of the animals in the most recent generation 8 based on training on data from all the 7 previous generations or from any one of the previous 7 generations are in Table 2. For every method, the most accurate predictions were obtained when training on the much larger dataset comprising all 7 generations. The poorest accuracy was for pedigree based BLUP (PBLUP), which had accuracy of 0.27 for predicting phenotype, while the mixture models had accuracy of 0.57 and outperformed the models that fitted all SNPs, which had an average accuracy of 0.50.
Training on only a single generation to predict generation 8 had no predictive power for PBLUP, unless the training generation was only 1 or 2 generations distant from generation 8 (i.e. generation 6 or 7). Averaged across all 7 individual training generations, PBLUP resulted in accuracy of 0.06, which would account for less than 1 % of phenotypic variance. For the genomic methods, accuracy decreased with distance between the training and validation population, confirming published results [20], but all methods had some predictive power in generation 8 based on training in generation 1. On average, training in any one generation had accuracy of 0.43 for BayesB and 0.39 for BayesC, compared to less than 0.27 for the models that fitted all SNPs with an equal variance ratio. These findings confirm previous studies reporting that BayesB outperforms GBLUP for genomic prediction in the presence of large effect QTL using both simulated [21] and real data [22].
Persistence of the accuracy in genomic predictions
One of the appeals of genomic selection is its application to traits that are difficult or costly to measure. Ideally, one would train in a suitable population of individuals with phenotypic and genomic information, and then apply the resulting prediction equation in successive generations of selection candidates without investing the time and effort for ongoing phenotypic measurement. The accuracy of this scenario for this case with a QTL of large effect is demonstrated in Table 3, where training was undertaken using only 295 animals from generation 1 and accuracies of prediction were obtained for each successive generation. Not surprisingly, the predictive ability of PBLUP was greatest in the immediate next generation after training but had no real predictive power in subsequent generations. The mixture models showed the most promising results, with BayesB and BayesC accounting for an average of 10 and 9 % phenotypic variance across generations 2 to 8. The models that fitted all SNPs accounted for on average 6 % of variance. In the last validation generation, 7 generations distant from training, the mixture models were still accounting for 10 % of phenotypic variation, whereas the models fitting all SNPs accounted for only 4 %.
Contribution of the largest QTL to the accuracy of genomic prediction
Genes with large effects can obviously contribute a greater than average contribution to predictive ability. Genomic predictions based on training in generation 1 using all SNPs but computing GEBV only using estimates of effects for SNPs in the window that contributed the most variance, as identified in the second row of Table 1, were used to compute the persistence of accuracy over generations, similar to what was done for whole genome GEBV in Table 3. The results are in Table 4 and show that 7 % of phenotypic variance can be predicted by the largest QTL region when using BayesB or BayesC and training on the 295 animals from generation 1, whereas the models that fitted all SNPs had no predictive ability. The latter is not surprising, as Table 1 had demonstrated that these methods resulted in inconsistent locations and small estimates of the effect of the largest QTL. The predictions using BayesB and BayesC indicate a slight trend of increasing accuracy, reflecting the fact that the frequency of the chromosome 4 QTL appears to be increasing slightly over these successive generations [8].
Bias in SNP effects estimation
The within generation estimates of SNP effects from different training data sets using BayesB were compared to fixed effect estimates from a single marker model, fitting an animal model using full pedigree to correct for family relationships in ASReml. For every data set, Table 5 demonstrates that the single marker model estimated larger SNP effects than BayesB. However, as more data were used (all 7 generations) the shrinkage of the BayesB estimate was reduced. These results are consistent with the downward bias (regression coefficient larger than 1) when predicting generation 8 phenotypes using predicted merit based upon SNP estimates from the 1 Mb window with the largest effects using either the single SNP animal model or BayesB. We therefore conclude that in this data there is no evidence for overestimation of the largest effect using the single SNP animal model and therefore, in contrast to the claims of [4], no evidence of overestimation using BayesB.
Conclusions
The mixture models known as BayesB or BayesC more accurately identified the QTL region than the two models that fitted all markers, BayesC0 or the SNP equivalent of GBLUP. Thus, mixture models should be preferred for traits with large effect QTL over models such as GBLUP, which fit all SNP effects assuming equal SNP variance for both genome wide association studies and for genomic prediction. We anticipate that the same finding will be true for models that include information on non-genotyped individuals. That is, single-step mixture models such as proposed in Fernando et al. [23] are expected to outperform single-step GBLUP [24] in the presence of large-effect QTL, until such a time that SNPs comprising randomly-chosen genome-wide markers can be replaced with SNPs in high or perfect linkage disequilibrium with causal mutations that are fitted as fixed effects or weighted according to their contribution to genetic variance.
References
Meuwissen THE, Hayes BJ, Goddard ME. Prediction of total genetic value using genome-wide dense marker maps. Genetics. 2001;157:1819–29.
Mucha S, Pszczoła M, Strabel T, Wolc A, Paczyńska P, Szydłowski M. Comparison of analyses of the QTLMAS XIV common dataset. II: QTL analysis. BMC Proc. 2011;5 Suppl 3:S2.
Zeng J, Pszczola M, Wolc A, Strabel T, Fernando RL, Garrick DJ, et al. Genomic breeding value prediction and QTL mapping of QTLMAS2011 data using Bayesian and GBLUP methods. BMC Proc. 2012;6 Suppl 2:S7.
Wang H, Misztal I, Aguilar I, Legarra A, Fernando RL, Vitezica ZG, et al. Genome-wide association mapping including phenotypes from relatives without genotypes in a single-step (ssGWAS) for 6-week body weight in broiler chickens. Front Genet. 2014;5:134.
Tuiskula-Haavisto M, Honkatukia M, Vilkki J, de Koning DJ, Schulman NF, Maki-Tanila A. Mapping of quantitative trait loci affecting quality and production traits in egg layers. Poult Sci. 2002;81:919–27.
Sasaki O, Odawara S, Takahashi H, Nirasawa K, Oyamada Y, Yamamoto R, et al. Genetic mapping of quantitative trait loci affecting body weight, egg character and egg production in F2 intercross chickens. Anim Genet. 2004;35:188–94.
Schreiweis MA, Hester PY, Settar P, Moody DE. Identification of quantitative trait loci associated with egg quality, egg production, and body weight in an F2 resource population of chickens. Anim Genet. 2006;37:106–12.
Wolc A, Arango J, Settar P, Fulton JE, O’Sullivan NP, Preisinger R, et al. Genome-wide association analysis and genetic architecture of egg weight and egg uniformity in layer chickens. Anim Genet. 2012;43 Suppl 1:87–96.
Kizilkaya K, Fernando RL, Garrick DJ. Genomic prediction of simulated multibreed and purebred performance using observed fifty thousand single nucleotide polymorphism genotype. J Anim Sci. 2010;88:544–51.
Habier D, Fernando RL, Kizilkaya K, Garrick DJ. Extension of the Bayesian alphabet for genomic selection. BMC Bioinformatics. 2011;12:186.
Strandén I, Garrick DJ. Technical note: derivation of equivalent computing algorithms for genomic prediction and reliabilities of animal merit. J Dairy Sci. 2008;92:2971–5.
Garrick DJ, Fernando RL. “Implementing a QTL detection study (GWAS) using genomic prediction methodology”. In: Gondro C, van der Werf J, Hayes B, editors. Genome-wide association studies and genomic prediction, methods in molecular biology, vol. 1019. New York: Springer Science+Business Media; 2013. p. 275–98.
Gilmour AR, Gogel BJ, Cullis BR, Thompson R: ASReml user guide release 3.0. VSN 2009, Hemel Hempstead, HP1 1ES, UK.https://www.vsni.co.uk/downloads/asreml/release3/UserGuide.pdf
Dunn IC, Meddle SL, Wilson PW, Wardle CA, Law AS, Bishop VR, et al. Decreased expression of the satiety signal receptor CCKAR is responsible for increased growth and body weight during the domestication of chickens. Am J Physiol Endocrinol Metab. 2013;304:909–21.
Sovio U, Bennett AJ, Millwood IY, Molitor J, O’Reilly PF, Timpson NJ, et al. Genetic determinants of height growth assessed longitudinally from infancy to adulthood in the Northern Finland birth cohort 1966. PLoS Genet. 2009;5:e1000409.
Lindholm-Perry AK, Sexten AK, Kuehn LA, Smith TPL, King DA, Shackelford SD, et al. Association, effects and validation of polymorphisms within the NCAPG-LCORL locus located on BTA6 with feed intake, gain, meat and carcass traits in beef cattle. BMC Genet. 2011;12:103.
Makvandi-Nejad S, Hoffman GE, Allen JJ, Chu E, Gu E, Chandler AM, et al. Four loci explain 83 % of size variation in the horse. PLoS ONE. 2012;7:e39929.
Liu R, Sun Y, Zhao G, Wang F, Wu D, Zheng M, et al. Genome-wide association study identifies loci and candidate genes for body composition and meat quality traits in Beijing-You chickens. PLoS ONE. 2013;8:e61172.
Fernando RL, Garrick DJ. Bayesian methods applied to GWAS”. In: Gondro C, van der Werf J, Hayes B, editors. Genome-wide association studies and genomic prediction, methods in molecular biology, vol. 1019. New York: Springer Science+Business Media; 2013. p. 237–74.
Habier D, Fernando RL, Dekkers JCM. The impact of genetic relationship information on genome-assisted breeding values. Genetics. 2007;177:2389–97.
Daetwyler HD, Pong-Wong R, Villanueva B, Woolliams JA. The impact of genetic architecture on genome-wide evaluation methods. Genetics. 2010;185:1021–31.
Hayes BJ, Pryce J, Chamberlain AJ, Bowman PJ, Goddard ME. Genetic architecture of complex traits and accuracy of genomic prediction: coat colour, milk-fat percentage, and type in Holstein cattle as contrasting model traits. PLoS Genet. 2010;6:e1001139.
Fernando RL, Dekkers JCM, Garrick DJ. A class of Bayesian methods to combine large numbers of genotyped and non-genotyped animals for whole-genome analyses. Genet Sel Evol. 2014;46:50.
Legarra A, Christensen OF, Aguilar I, Misztal I. Single Step, a general approach for genomic selection. Livestock Sci. 2014;166:54–65.
Acknowledgement
This study was supported by Hy-Line Int. and the EW group. The authors thank the anonymous referees for their constructive comments.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors confirm they have read Biomed Central’s guidance on competing interests and declare no competing interests.
Authors’ contributions
AW undertook the analysis and wrote the first draft. JA, PS, JEF and NPO conducted the experiment and collected the data. DJG, RF, AW and JCMD conceived the study and contributed to the methods. All authors read and approved the final manuscript.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Wolc, A., Arango, J., Settar, P. et al. Mixture models detect large effect QTL better than GBLUP and result in more accurate and persistent predictions. J Animal Sci Biotechnol 7, 7 (2016). https://doi.org/10.1186/s40104-016-0066-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40104-016-0066-z