
Abstract
Background
2-amino pyridine derivatives have attracted considerable interest because they are useful precursors for the synthesis of a variety of heterocyclic compounds possessing a medicinal value. In this work we aim to study both structural and electronic as well as high quality vibrational spectra for 2-amino-3-methylpyridine (2A3MP) and 2-amino-4-methylpyridine (2A4MP).
Results
Møller–Plesset perturbation theory (MP2/6-31G(d) and MP2/6-31++G(d,p) methods were used to investigate the structure and vibrational analysis of (2A3MP) and (2A4MP). Tautomerization of 2A4MP was investigated by Density Functional Theory (DFT/B3LYP) method in the gas phase. For the first time, all tautomers including NH → NH conversions as well as those usually omitted, NH → CH and CH → CH, were considered. The canonical structure (2A4MP1) is the most stable tautomer. It is 13.60 kcal/mole more stable than the next (2A4MP2). Transition state structures of pyramidal N inversion and proton transfer were computed at B3LYP/6-311++G(d,p). Barrier to transition state of hydrogen proton transfer is calculated as 44.81 kcal/mol. Transition state activation energy of pyramidal inversion at amino N is found to be 0.41 kcal/mol using the above method. Bond order and natural atomic charges were also calculated at the same level. The raman and FT-IR spectra of (2A3MP) and (2A4MP) were measured (4000–400 cm−1). The optimized molecular geometries, frequencies and vibrational bands intensity were calculated at ab initio (MP2) and DFT(B3LYP) levels of theory with 6-31G(d), 6-31++G(d,p) and 6-311++G(d,p) basis sets. The vibrational frequencies were compared with experimentally measured FT-IR and FT-Raman spectra.
Conclusion
Reconsidering the vibrational analysis of (2A3MP) and (2A4MP) with more accurate FT-IR machine and highly accurate animation programs result in new improved vibrational assignments. Sophisticated quantum mechanics methods enable studying the transition state structure for different chemical systems.
Similar content being viewed by others

Background
Pyridine and its derivatives has been the subject of investigation by several workers in the last 50 years. The structural and vibrational spectrum of pyridine and its derivatives have been extensively studied and analyzed in recent years because of their involvement in bioactivities and applications in pharmaceutical, agro chemical and many other industries (Jose and Mohan 2006; Othmer 1997; Pierrat et al. 2005). In particular it has been mentioned that 2-amino pyridine derivatives have attracted considerable interest because they are useful precursors for the synthesis of a variety of heterocyclic compounds possessing a medicinal value (Temple et al. 1987; Janssens et al. 1985; Mantlo et al. 1991; Oguchiet al. 2000). It has been reported recently that the inhibitory properties of 3-nitropyridine derivatives and their salts can be used as therapeutic or preventive agents for hepatitis B and acquired immune deficiency syndrome (AIDS) (Yoon et al. 2004). Various spectroscopic studies have been reported on methyl substituted pyridine derivatives which have cholesterol lowering properties and anti–cancer activity (Suheyla Kurkcuoglu et al. 2009; Green et al. 1963; Long and George 1963). The pyridine ring appears in a large number of natural substances such as vitamin B5, vitamin B6, pyridoxal and pyridoxamine; and drugs such as nifedipine, nichetamine and sulphapyridine (Ög˘retir et al. 2006; Ziessel 2001; Hagadorn et al. 2000; Holland et al. 2001; Marlin et al. 2001; Lipinski et al. 2001). The spectroscopic studies of N–heterocyclic molecules including substituted pyridines have become quite interesting as they are the constituents of DNA and RNA (Arivazhagan and Krishnakumar 2003; Singh and Srivastava 2009; Yadav et al. 2007). Also Pyridine and its derivatives have been studied as corrosion inhibitors (Arshadi et al. 2004; Yadav and Wadhwani 1993; Talati and Gandhi 1983; Yurt et al. 2005; Yurt et al. 2006; Ashassi-Sorkhabi et al. 2006; Lashkari and Arshadi 2004; Xiao-Ci et al. 2000).
In the literature, there are many studies reporting on the vibrational analysis of pyridine (Pongor et al. 1984; Stidham and Dilella 1979; Stidham and Dilella 1980; Dilella and Stidham 1980; Dilella 1980; Zerbi et al. 1963) and its methylated derivatives (Draeger 1983). X-ray, IR and Raman studies as well as quantum chemical calculations performed for 3 and 5-nitroderivatives of 2A4MP (Bryndal et al. 2012). DFT and ab initio computation of structure and vibrational frequencies of pyridine and its isotopomers were reported (Szafran and Koput 2001).
The geometry of isomeric pyridines were studied by X-ray crystallography (Kubiak et al. 2002), microwave spectroscopy (Ford 1975) and surface-enhanced Raman spectroscopy (Arenas et al. 1993). DFT vibrational studies of 5-bromo-2-nitropyridine (Sundaraganesan et al. 2005), pyridinium complexes (Tayyari et al. 2010), and 2-chloro-5-bromopyridine (Hiremath et al. 2010; Kishor and Bhoop 2013) were reported. Tautomerization and rotamerization of aminopyridines has been reported (Davoodnia et al. 2011; Alkorta and Elguero 2002; Pietrzycki et al. 1993). The vibration (IR and Raman) spectra of aminopyridines, methyl-substituted aminopyridines and hydrogen chlorides of aminopyridines had been determined experimentally by Spinner (Spinner 1962). However, to the best of our knowledge, according to literature survey there is no results based on quantum chemical calculations, on tautomerization, transition state structures and FT-IR/FT-Raman spectral studies on (2A3MP) and (2A4MP). Hence in the present work, we reported detailed tautomeization, transition state activation energies and interpretations of the infrared and Raman spectra based on the experimental and theoretical results. Reconsidering the vibrational analysis of (2A3MP) and (2A4MP) with more accurate FT-IR machine and highly accurate simulation programs lead to new improved vibrational assignments.
Results and discussion
Geometrical features
The optimized molecular structures of 2A3MP and 2A4MP are obtained at different computational methods. The optimized structural parameters were shown in Additional file 1: Tables S1 and S2 for 2A3MP and 2A4MP in the Additional files respectively. By allowing the relaxation of all parameters, the calculations converge to optimized geometries, which correspond to true energy minima, as revealed by the lack of imaginary frequencies in the vibrational mode calculation. These molecules have three CN, two NH, five CC and six CH bonds. It was reported that the HF approximation is, in general, insufficient to study the geometry of amino group-containing compounds (Sponer and Hobza 1994a, b, c; Sponer et al. 1996). Thus MP2 with high basis sets was used to examine the molecular structure and amino group pyramidalization of 2A3MP and 2A4MP. The geometrical parameters at the MP2 methods are shown in Fig. 1 and in Additional file 1: Tables S3 and S4 for 2A3MP and 2A4MP respectively. As shown in Fig. 1, the C-Npy bond lengths are slightly different. The one closer to the amino group is shorter by a value of 0.004 and 0.005 Å for 2A3MP and 2A4MP respectively as predicted by MP2/6-31++G(d,p). This could be due to the interaction of the amino nitrogen lone pair with the C-Npy bond. This interaction leads to the increase of double bond character of the C-Npy bond and hence the shortening of the bond closer to the amino group. All methods used throughout this work prove the pyramidalization of the amino group as displayed in Additional file 1: Tables S1–S4. However, the inclusion of d-polarization functions in the amino group hydrogen is quite essential. The non-planarity of the amino group was mostly found to be much larger at the MP2 level, compared with the uncorrelated methods. Computations reveals the asymmetry of the amino group hydrogen dihedral angles, which is due to the repulsive electrostatic interactions with the neighboring hydrogens. The calculations revealed a significant difference between the correlated and uncorrelated results. It is easily noticed from Additional file 1: Tables S1–S4 that the B3LYP/6 311++G(d,p) method overestimates bond lengths, particularly the CH bonds. The CC bond distances are differing in value. The reasons for that may be the replacement of hydrogen atoms in the pyridine ring by methyl groups and the distortion of the aromatic ring hexagonal symmetry in the pyridine ring. Geometry based on MP2 and B3LYP calculation shows that the average bond lengths of CC and CH in the pyridine ring are 1.394 and 0.990 Å, for 2A3MP and 2A4MP. Comparing MP2 and B3LYP methods, the predicted N–H (amino group) bond lengths are found at 1.011 and 1.009 Å for 2A3MP and 2A4MP, respectively. The experimental value (1.001 Å), is more closer to that predicted by B3LYP method (Fukuyo et al. 1982). The optimized CN bond length is 1.347 and 1.340 Å for 2A3MP and 1.348 and 1.342 Å for 2A4MP by MP2 and B3LYP methods, respectively. Both methods predict the C–N bond length very close to each other but shorter than the measured value (1.402 Å measured for a similar aromatic ring, aniline) (Fukuyo et al. 1982). Some calculated ring angles, C2–C1–C6 and C5–C6–C1 are deviating from the perfect hexagon value (120°) at C1 and C6 atom positions for 2A3MP and the same deviation is also shown at the corresponding atom positions, C2 and C6 for 2A4MP. This could be due to the substitutions of –NH2 and –CH3 groups. This effect results from the interaction of the N lone pair of electrons with the aromatic ring.

Optimized geometry at MP2/6-31++G(d,p) showing all bond lengths
The B3LYP geometry is similar to the MP2 geometry at the same basis set. The bond distance C2–C3 in 2A3MP (1.399 Å) at MP2/6-31++G(d,p) is similar to that (1.400 Å) at B3LYP/6-31G(d,p) and the bond distance C3–C4 is the same as predicted by the two methods (1.392 Å). The effect of adding diffuse functions and polarization functions on H atoms on the predicted geometry by the energy correlated method MP2 could be observed in Additional file 1: Tables S3 and S4. The increase of the basis set size overestimates the ring C–C and C–N bond distances and underestimates the C–H distances. The exo-cyclic C–C and C–N bonds remain unchanged.
Tautomerization of 2A4MP
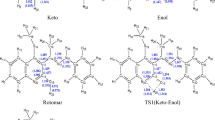
Tautomerism is crucial for the understanding of the chemical behavior of 2A4MP. All possible tautomers of 2A4MP were optimized at B3LYP/6-311++G(d,p) and shown in Fig. 2. Relative energy (kcal/mol) for 2A4MP tautomers at B3LYP/6-311++G(d,p) computational levels is presented in Table 1. The canonical structure of the 2A4MP1 is the most stable tautomer. It is 13.60 kcal/mole more stable than 2A4MP2. Comparing the cis (2A4MP3) and trans (2A4MP2) of 2(1H)-pyridinimine reveals that, the trans tautomer is 2.76 kcal/mol more stable than the cis one. This result agrees with the reported experimental and computational (at B3LYP/6-31++G(d,p) level) data of 2-aminopyridine/2(1H)-pyridinimine (Akai and Ohno 2005). The destabilization of the cis tautomer may result from the interaction of the neighboring H atoms. As could be predicted from the computed total energy values which are, corrected for zero-point energy, one may rank the stability order as 2A4MP1 > 2A4MP2 > 2A4MP3 > 2A4MP5 > 2A4MP7 > 2A4MP4 > 2A4MP6. The attachment of the movable H atom to N (2A4MP2 and 2A4MP3) is more favored than to C (2A4MP4, 2A4MP5, 2A4MP6, and 2A4MP7). Bonding of the movable H to C3 (2A4MP5 and 2A4MP7) is also more stable than bonding to C5 (2A4MP4 AND 2A4MP6) by about 4 kcal/mol.

Optimized structures of common and rare tautomers of 2A4MP
Transition state for hydrogen transfer
The transition state structure for proton transfer within the amine-imine tautomerism was calculated at B3LYP/6-311++G(d,p) and confirmed by the presence of one imaginary frequency. The transition state structure for proton transfer is shown in Fig. 3. The imaginary frequency value for hydrogen transfer is (−1897 cm−1). The curvature of the potential energy surface (PES) for the proton transfer reaction is measured by the second derivative of energy with respect to geometry. The magnitude of the imaginary frequency is a measure of the curvature of the transition state region along the reaction coordinate. For the proton transfer related reactions, a small imaginary frequency should correspond to a low barrier and a large imaginary frequency to a high barrier. The higher values of the imaginary frequency of the proton transfer reactions are because the vibrational frequencies are mass-weighted eigenvalues of the force constant matrix (second derivative matrix). Therefore, light atoms e.g. the H atom, is associated with higher vibrational frequency transition state. The activation energy of the proton transfer process is computed as 44.81 kcal/mol at B3LYP/6-311++G(d,p) level of theory. At the DFT/B3LYP/6-311++G** level, the calculated energy barrier for 2-hydroxypyridine → 2-pyridone conversion is about 34.00 kcal/mol (Hazra and Chakraborty 2006). It is believed that examination of key molecular orbitals (like HOMO) may provide insight into molecular geometry. The HOMO energy of the transition state is calculated to be -135.76 kcal/mol. The difference in HOMO energy between the transition state and the ground state is found to be 3.53 kcal/mol. The low-lying HOMO is interacting with the moving proton in such a way that facilitate the transfer of the proton and hence the lower the activation barrier. The geometry of transition state structures are given in Additional file 1: Table S5. The transferred proton shows a longer bond distance (1.395 Å) compared to that of the minimum structure bond distance (1.009 Å). The amino group in the transition state is planar as evidenced from the dihedral angles D(1,6,8,15), D(1,6,8,16), D(5,6,8,15) and D(5,6,8,16) as shown in Table S5.

Optimized transition state structures for pyramidal inversion and proton transfer
Pyramidal inversion barrier at amino-N
The inversion of pyramidal centers is an important process, and takes place very rapidly in the case of amines. The inversion barrier at the N atom is calculated as the difference of total energy between the planar transition state structure (saddle point) and the unconstrained optimized pyramidal structure (local minimum). The transition state structure of pyramidal inversion is displayed in Fig. 3. The activation energy of pyramidal inversion is calculated to be 0.41 kcal/mol at B3LYP/6-33++G(d,p). The rapid rate of inter-conversion in the case of 2-aminopyridine with activation energy of 0.41 kcal/mol is much less than in the case of other pyramidal compounds of elements from the second row and higher (Montgomery 2013). The imaginary frequency of pyramidal inversion at the transition state structure is computed as −346 cm−1. The experimental vibration is reported as 289–375 cm−1 in the gas phase (McKean 1989). The computed inversion to barrier is 0.41 kcal/mol which is less than the computed value of 0.88 kcal/mol at HF/6-31G(d) (Bludský et al. 1996) and the estimated value near 0.70 kcal/mol for 2-aminopyridine (Barrow et al. 1976).
The HOMO energy of the pyramidal optimized structure and the planar transition state are calculated as −139.29 and −126.32 kcal/mol respectively. The difference of HOMO energies is 12.98 kcal/mol. The smaller the difference the more rapid the inversion. The HOMO contains the lone electron pair and one would expect that this orbital should be an sp3 orbital in the pyramidal structure and a p orbital in the planar case. The HOMO energy difference in NMe3 is reported 23.81 kcal/mol, and the inversion barrier is 8.38 kcal/mol (Montgomery 2013). It is found that, the HOMO energy increases moving from the pyramidal ground state to the planar transition state.
Natural charges and bond order of 2A4MP1
One of the most important electronic properties is the population of electronic density on atoms constituting the molecule. A good way to explore the distribution of charge within a system is to calculate the natural charge. The values of the NPA charges for all atoms in 2A4MP1 were computed at B3LYP/6-311+G(d,p). NPA charges for 2A4MP1 are given in Additional file 1: Table S6. In Gaussian, NBO analysis calculates atomic charges, by summing occupancy of natural atomic orbitals. In 2A4MP positive charges are accommodated on some carbon atoms (C2, C4 and C6) and H atoms. These carbon atoms (C4 and C6) are either directly bonded to the more electronegative N atoms or in the meta position (C2) with regard to the amino group. N atoms and some carbon atoms (C1 and C7) accommodate negative charge. N8 is highest negatively charged atom (−0.792).
Bond order of 2A4MP1 molecule is computed at the level B3LYP/6-311+G(d,p). The spin-corrected Mayer bond order in the natural atomic basis (NAO), is shown in Additional file 1: Table S7. Since 2A4MP1 is spin unpolarized system, the spin-corrected Mayer bond order in the NAO basis equals the “Wiberg bond index in the NAO basis” as implemented in Gaussian NBO Version 3.1. As can be seen from Additional file 1: Table S7, the highest calculated bond order is for the bond C3–C4 (1.463) and the smallest computed bond order for bonded atom pairs is N8-H15 (0.831). The bond order between C1–C2 is 1.458 indicating a partial double bond and that between C1–C7 is 1.036 indicating a pure single bond. The bond order C6–N5 is 1.369 which suggests a partial double bond while the bond order of the C6–N8 bond is 1.145 showing some partial double bond character which results from N lone pair delocalization.
Vibrational assignments
2A3MP and 2A4MP molecules belong to C1 point group symmetry, having 16 atoms with 42 normal modes of vibrations which are active in both Raman scattering and IR absorption. The vibrational analysis of fundamental modes with FT-IR and FT- Raman experimental frequencies are tabulated in Table 2 for 2A3MP and 2A4MP. Vibrational frequencies using HF, B3LYP and MP2 methods with different basis sets, for 2A3MP and 2A4MP were reported in Tables 3 and 4 respectively. The experimental Infrared and Raman spectra in solid phase were shown in Figs. 4 and 5 for 2A3MP and Figs. 6 and 7 for 2A4MP along with calculated IR and Raman spectra by HF and B3LYP methods with 6-31++G(d,p) and 6-311++G(d,p) basis sets.

Comparison of experimental and calculated IR spectra of 2-amino-3-methylpyridine

Comparison of experimental and calculated Raman spectra of 2-amino-3-methylpyridine

Comparison of experimental and calculated IR spectra of 2-amino-4-methylpyridine

Comparison of experimental and calculated Raman spectra of 2-amino-4-methylpyridine
C–H vibrations
Aromatic C–H stretching vibrations normally occur at 3100–3000 cm−1 (Sharma 1994). The 2A3MP and 2A4MP are di-substituted aromatic systems. The expected three C–H symmetric stretching vibrations correspond to mode nos. 3, 4 and 5 in Table 2.
The calculated frequencies of the C-H symmetric stretching vibrations using B3LYP/6-311++G(d,p) in 2A3MP at 3067, 3032 and 3022 cm−1 are in good agreement with the experimental data. Similar observations are also found in the case of 2A4MP at 3135, 3064 and 3051 cm−1. The C–H in-plane bending and C–H out-of-plane bending vibrations are normally found in the range 1300–1000 and 1000–750 cm−1, respectively in aromatic compounds and are very useful for characterization purposes (Prabavathi and Krishnakumar 2009; Krishnakumar and Prabavathi 2008; Altun et al. 2003; Singh and Pandey 1974).
In the case of 2A3MP three modes are associated mainly with the C–H in-plane-bending vibrations: mode nos. 19, 21 and 22. These modes are at the following wavenumbers 1257, 1137 and 1079 cm−1 and at 1250, 1125 and 1037 cm−1 in the case of 2A4MP. The C–H out-of-plane bending vibrations correspond mainly to the mode nos. 28, 29 and 30. The calculated frequencies of the C–H out-of-plane bending vibrations in 2A3MP at 852, 755 and 748 cm−1 and in 2A4MP at 828, 780 and 746 cm−1 show very good agreement with experimental data.
NH2 vibrations
There are six internal modes of vibrations in the NH2 group. These are the symmetric stretching, the anti-symmetric stretching, the scissoring, the rocking, the wagging and torsional mode. The NH2 group has two (N–H) stretching vibrations, one being asymmetric and another symmetric. The –NH2 symmetric and asymmetric stretches in the range (3311–3475 cm−1) (mode 1) and (mode 2) are in good agreement with experimental value of (3470–3300 cm−1) in both 2A3MP and 2A4MP molecules. The computed –NH2 scissoring vibration mode no. (9) at 1624 and 1643 cm−1 for 2A3MP and 2A4MP, respectively by B3LYP/6-311++G(d,p) level are in excellent agreement with the expected characteristic value, 1600 cm−1 (Yadav and Sing 1985; Wiberg and Shrake 1973). This band observed in FT-IR at 1623 cm−1 and in FT-Raman at 1618 cm−1 for 2A3MP and at 1646 cm−1 in both FT-IR and FT-Raman spectra for 2A4MP. The –NH2 wagging computed at 520 and 457 cm−1 (mode 35) in both 2A3MP and 2A4MP molecules using B3LYP/6-311++G(d,p) level have been observed at 524 and 525 cm−1 in FT-IR and FT-Raman spectra, respectively for 2A3MP and in the case of 2A4MP observed at 455 cm−1 in both FT-IR and FT-Raman spectra.
Methyl group vibrations
Nine modes of vibration (Sundaraganesan and Doinic 2007) are assigned to each CH3 group; the symmetrical and asymmetrical stretching, in plane stretching modes, deformation modes (symmetrical and asymmetrical), in-plane and out-of-plane rocking, and twisting modes. Stretching frequency of methyl C–H usually occurs at lower frequencies (2800–3000 cm−1) than those of the aromatic ring normally occurs at 3000–3100 cm−1. Stretching vibrations of methyl C-H, νsym and νasym, generally found in the ranges 2850–2940 and 2970–3010 cm−1 (Sathyanarayana 2004; Lithivinov 1992; Furic et al. 1992). The reported spectra of 2-methyl pyridine, shows the CH3 stretching in the range 2900–3000 cm−1, the in-plane deformations around 1370–1450 cm−1 and the rocking around 990–1040 cm−1 (Furic et al. 1992). The measured wavenumbers of asymmetric and symmetric stretching vibrations of CH3 group for both molecules are listed in Table 2. For the methyl substituted benzene derivatives the symmetric and asymmetric deformation vibrations νdef of methyl groups normally appear in the region 1370–1390 and 1440–1465 cm−1, respectively (Arenas et al. 1997; Lin-Vien et al. 1991; Diem 1993). The rocking mode of vibrations of the CH3 group usually appear in the region 1010–1070 cm−1 (Silverstein et al. 1981). In the present study, this band appears at 1035 cm−1 in FT-IR and at 1031 cm−1 in FT-Raman (mode no. 23) for 2A3MP. The same vibration in 2A4MP is shown in FT-IR at 1035 cm−1 and at 1033 cm−1 in FT-Raman. The theoretically calculated values by MP2 and B3LYP method agree well with the experimental values. CH3 torsional mode is expected below 400 cm−1, the computed bands at 150 cm−1 in 2A3MP and 141 cm−1 in 2A4MP are assigned to this mode (mode no. 42).
C–CH3 vibrations
In 2A3MP, the band observed in FT-IR at 1196 cm−1 and at 1188 cm−1 in FT -Raman is assigned to C–CH3 stretching vibration. It is found that there is a good agreement between this experimental value and the calculated value mode no. 20 at 1195 cm−1 using B3LYP/6-311++G(d,p) method. A similar band is predicted at 1175 cm−1 for 2A4MP shows good agreement with the experimental value at 1177 cm−1 in FT-IR and FT-Raman. In methyl substituted benzenes, the C-CH3 in-plane-bending vibrations give rise to absorptions in the range 390–260 cm−1. The C–CH3 in-plane bending vibration mode no. 39 is assigned at 288 and 282 cm−1 for 2A3MP and 2A4MP, respectively and out-of-plane bending vibration mode No. 40 is assigned at 255 cm−1 for 2A3MP and 202 cm−1 for 2A4MP. These assignments are in good agreement with the literature (Hameka and Jensen 1996).
C–N, C=N vibrations
The identification of C–N vibrations is very difficult because of the interference of many bands in the area where the vibration of this bond happens. For the aromatic amines, the C-N stretching appears in the region 1266–1382 cm−1 (Silverstein et al. 1981). This band observed, in 3,5-dibromopyridine, at 1410 cm−1 in FT-IR and 1412 cm−1 in FT-Raman. The band observed at 1368 cm−1 in benzamide is assigned to C–N stretching (Shanmugam and Sathyanarayana 1984). In the current study, a band appeared at 1282 and 1264 cm−1 for the stretching vibration of the C=N bond in the FT-IR and FT-Raman, respectively for 2A3MP and at 1268 and 1270 cm−1 for the stretching vibration of the C=N bond in the FT-IR and FT-Raman, respectively for 2A4MP. These values are consistent with the theoretically calculated values (mode No. 18).
Conclusion
In this work, the structure and vibrational analysis of 2A3MP and 2A4MP were investigated by MP2/6-31G(d) and MP2/6-31++G(d,p) methods. Tautomerization of 2A4MP was studied by DFT/B3LYP method in the gas phase. In this study, all possible tautomers including NH → NH conversions as well as, NH → CH and CH → CH, were considered. The most stable tautomer is the canonical structure, 2A4MP1. The umbrella effect transition state structure of the pyramidal N and transition structure of the proton transfer were computed at B3LYP/6-311++G(d,p). Activation energy of the hydrogen proton transfer and pyramidal inversion at amino N is calculated as 44.81 and 0.41 kcal/mol respectively. Bond order and natural atomic charges were also calculated.
The vibrational spectral analysis was carried out using FT-IR and FT- Raman spectroscopy for 2A3MP and 2A4MP. The computations were performed at ab initio (MP2) and DFT (B3LYP) levels of theory with 6-31++G(d,p) and 6-311++G(d,p) basis sets to get the optimized geometry and vibrational wavenumbers of the normal modes of the title compounds. The complete vibrational assignments of wavenumbers were made on the basis of potential energy distribution and using Gauss View software. The difference between the observed and scaled wave number values of the most of the fundamentals is very small.
Experimental details
The compounds 2A3MP and 2A4MP were purchased from Sigma–Aldrich with a stated purity of 99 % and were used without further purification. FT-IR spectra of 2A3MP and 2A4MP have been recorded in the region 4000–400 cm−1 using a Thermo Nicolet Nexus 870 FT-IR instrument. The instrument is equipped with a KBr beam splitter and an In GaAs detector. The spectral resolution is ±2 cm−1. The Raman spectra were measured using a dispersive Nexus 870 FT-Raman instrument. The instrument is equipped with Nd:YAG laser source operating at 1.064 μm line widths with 200 mW powers. The spectra were recorded with scanning speed of 30 cm−1 min−1 of spectral width 2 cm−1.
Computational details
All electronic structure calculations were performed using the Gaussian03 suite of programs (Pittsburgh 2003). Geometry optimizations for all compounds and tautomers have been performed using Møller–Plesset perturbation theory (MP2/6-31G(d) and MP2/6-31++G(d,p) methods and DFT at the B3LYP functional in conjunction with the 6-31++G(d,p) and 6-311++G(d,p) basis set. Geometries have been first optimized with full relaxation on the potential energy surfaces at HF/6-31++G(d,p) and HF/6-311++G(d,p) levels. The geometry was then re-optimized at B3LYP/6-31++G(d,p), B3LYP/6-311++G(d,p), (MP2/6-31G(d) and MP2/6-31++G(d,p) levels. For each stationary point, we carried out vibrational frequency calculation at the same level to characterize their nature as minima or transition states and to correct energies for zero-point energy and thermal contribution. The transition states for tautomerization have been located using the eigenvalue-following (EF) optimization procedure as implemented in the Gaussian programs. The nature of the transition states was confirmed by the presence of one negative eigenvalue in the Hessian matrix. The vibrational modes were examined by using the GAUSS-VIEW program (Frisch et al. 2000). In this study two different scaling factors (Sundaraganesan et al. 2005) viz. 0.89 for HF and 0.96 for B3LYP were used to correct the theoretical harmonicity error. Partial charge distributions were calculated using the natural population analysis (NPA) method.
References
Akai N, Ohno K, Aida M (2005) Photochemistry of p-toluidine in a low-temperature argon matrix: infrared spectrum and geometrical structure of 4-methylaniino radical. Chem Phys Lett 413:306–310
Alkorta I, Elguero J (2002) Influence of intermolecular hydrogen bonds on the tautomerism of pyridine derivatives. J Org Chem 67(5):1515–1519
Altun A, Golcuk K, Kumru M (2003) Structure and vibrational spectra of p-methylaniline: Hartree-Fock, MP2 and density functional theory studies. J Mol Struct (Theochem) 637:155–639
Arenas JF, Montanez MA, Otero JC, Marcos JI (1993) Surface enhanced Raman spectra of 2-cyanopyridine and picolinamide. J Mol Struct 293:341–344
Arenas JF, Tocón IL, Otero JC, Marcos JI (1997) Vibrational spectrum of 2-methylpyridine. J Mol Struct 410:443–446
Arivazhagan M, Krishnakumar V (2003) Vibrational and normal coordinate analysis of thymine hydrochloride. Indian J Pure Appl Phys 41:341–345
Arshadi MR, Lashgari M, Parsafar GhA (2004) Mater. Cluster approach to corrosion inhibition problems: interaction studies. Mater Chem Phys 86:311–314
Ashassi-Sorkhabi H, Shabani B, Aligholipour B, Seifzadeh D (2006) The effect of some Schiff bases on the corrosion of aluminum in hydrochloric acid solution. Appl Surf Sci 252:4039–4047
Barrow RF, Long DA, Sheridan J (1976) Molecular spectroscopy
Bludský O, Šponer J, Leszczynski J, Špirko V, P Hobza J (1996) Amino groups in nucleic acid bases, aniline, aminopyridines, and aminotriazine are nonplanar: results of correlated ab initio quantum chemical calculations and anharmonic analysis of the aniline inversion motion. J Chem Phys 105:11042
Bryndal I, Kucharska E, Sasiadek W, Wandas M, Lis T, Lorenc J, Hanuza J (2012) Molecular and crystal structures, vibrational studies and quantum chemical calculations of 3 and 5-nitroderivatives of 2-amino-4-methylpyridine. Spectrochim Acta A Mol Biomol Spectrosc 96:952–962
Davoodnia A, Attar P, Morsali A, Eshghi H, Hoseini NT, Khadem S (2011) Experimental and theoretical studies on the tautomerism in 2-aminopyridines and 2(1H)-pyridinones: synthesis of 2-amino-4-aryl-3-cyano-6-(3,4-dimethoxyphenyl)pyridines and 4-Aryl-3-cyano-6-(3,4-dimethoxyphenyl)-2(1H)-pyridinones. Bull Korean Chem Soc 32(6):1873–1878
Diem M (1993) Introduction to modern vibrational spectroscopy. Wiley, New York
Dilella DP (1980) Vibrational spectra of C 2ν deuterium substituted pyridines. 3—pyridine-3,5-d 2 and pyridine-3,4,5-d 3. J Raman Spectrosc 9:239
Dilella DP, Stidham HD (1980) Vibrational spectra of C 2 v deuterium substituted pyridines. 2—Pyridine, pyridine-4-d, pyridine-2,6-d 2 and pyridine-2,4,6-d 3. J Raman Spectrosc 9:90
Draeger JA (1983) Methylpyridines: vibrational assignments and an approximate force field. Spectrochim Acta A Mol Biomol Spectrosc 39:809
Ford RG (1975) The microwave spectra and dipole moments of the cyanopyridines. J Mol Spectrosc 58:178–184
Frisch MJ, Nielsen AB, Holder AJ (2000) Gaussview users manual. Gaussian Inc., Pittsburg
Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Montgomery, Jr JA, Vreven T, Kudin KN, Burant JC, Millam JM, Iyengar SS, Tomasi J, Barone V, Mennucci B, Cossi M, Scalmani G, Rega N, Petersson GA, Nakatsuji H, Hada M, Ehara M, Toyota K, Fukuda R, Asegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Klene M, Li X, Knox JE, Hratchian HP, Cross JB, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Ayala PY, Morokuma K, Voth GA, Salvador P, Dannenberg JJ, Zakrzewski VG, Dapprich S, Daniels AD, Strain MC, Farkas O, Malick DK, Rabuck AD, Raghavachari K, Foresman JB, Ortiz JV, Cui Q, Baboul AG, Clifford S, Cioslowski J, Stefanov BB, Liu G, Liashenko A, Piskorz P, Komaromi I, Martin RL, Fox DJ, Keith T, Al-Laham MA, Peng CY, Nanayakkara A, Challacombe M, Gill PMW, Johnson B, Chen W, Wong MW, Gonzalez C, Pople JA (2003) Gaussian 03, Revision E.01, Gaussian, Inc., Pittsburgh
Fukuyo M, Hirotsu K, Higuchi T (1982) The structure of aniline at 252 K. Acta Crystallogr B 38:640–643
Furic K, Mohacek V, Bonifacic M, Stefanic I (1992) Raman spectroscopic study of H 2 O and D 2 O water solutions of glycine. J Mol Struct 267:39–44
Green JHS, Kynaston W, Paisley HM (1963) Far infra-red spectroscopy of benzene derivatives. Spectrochim Acta A Mol Biomol Spectrosc 19:807
Hagadorn JR, Que L Jr, Tolman WB (2000) N-Donor effects on carboxylate binding in mononuclear iron(II) Complexes of a sterically hindered benzoate ligand. Inorg Chem 39:6086–6090
Hameka HF, Jensen JO (1996) Theoretical studies of the methyl rotational barrier in toluene. J Mol Struct (Theochem) 362(3):325–330
Hazra MK, Chakraborty T (2006) 2-Hydroxypyridine ↔ 2-pyridone tautomerization: catalytic influence of formic acid. J Phys Chem A 110(29):9130–9136
Hiremath CS, Yenagi J, Tonannavar J, Sundius T (2010) Ab initio/DFT electronic structure calculations, spectroscopic studies and normal coordinate analysis of 2-chloro-5-bromopyridine. Spectrochim Acta A Mol Biomol Spectrosc 918–926
Holland JM, Kilner CA, Thornton-Pett M, Halcrow MA (2001) Steric effects on the electronic and molecular structures of nickel(II) and cobalt(II) 2,6-dipyrazol-1-ylpyridine complexes. Polyhedron 20:2829–2840
Janssens F, Torremans J, Janssen M, Stokbroekx RA, Luyckx M, Janssen PAJ (1985) New antihistaminic N-heterocyclic 4-piperidinamines. 3. Synthesis and antihistaminic activity of N-(4-piperidinyl)-3H-imidazo[4,5-b]pyridin-2-amines. J Med Chem 28:1943–1947
Jose SP, Mohan S (2006) Vibrational spectra and normal co-ordinate analysis of 2-aminopyridine and 2-amino picoline. Spectrochim Acta A Mol Biomol Spectrosc 64:240–245
Kishor A, Bhoop S (2013) Theoretical studies of vibrational spectral modes and HOMO, LUMO studies of some synthetic organic compounds. Res J Chem Sci 3(10):36–43
Krishnakumar V, Prabavathi N (2008) Simulation of IR and Raman spectral based on scaled DFT force fields: a case study of 2-amino 4-hydroxy 6-trifluoromethylpyrimidine, with emphasis on band assignment. Spectrochim Acta A Mol Biomol Spectrosc 71:449–457
Kubiak R, Janczak J, Sledz M (2002) Crystal structures of 2- and 3-cyanopyridine. J Mol Struct 610:59–64
Lashkari M, Arshadi MR (2004) DFT studies of pyridine corrosion inhibitors in electrical double layer: solvent, substrate, and electric field effects. Chem Phys 299:131–137
Lin-Vien D, Colthup NB, Fateley WG, Grasselli JG (1991) the handbook of infrared and raman characteristic frequencies of organic molecules. Academic Press, Boston
Lipinski R, Chruscinski L, Mlynarz P, Boduszek B, Kozlowski H (2001) Coordination abilities of amino-phosphonate derivatives of pyridine. Inorg Chim Acta 322:157–161
Lithivinov G (1992) Proceedings of the 13th International Conference on Raman Spectroscopy, Weizburg, Germany
Long DA, George WO (1963) Spectroscopic and thermodynamic studies of pyridine compounds—V: vibrational spectra and assignments for γ-Picoline and γ-picoline-d7. Spectrochim Acta A Mol Biomol Spectrosc 19:1777
Mantlo NB, Chakravarty PK, Ondeyka DL, Siegl PK, Chang RS, Lotti VJ, Faust KA, Schorn TW, Chen TB (1991) Potent, orally active imidazo[4,5-b]pyridine-based angiotensin II receptor antagonists. J Med Chem 34:2919–2922
Marlin DS, Olmstead MM, Mascharak PK (2001) Structure—spectroscopy correlation in distorted five-coordinate Cu(II) complexes: a case study with a set of closely related copper complexes of pyridine-2,6-dicarboxamide ligands. Inorg Chem 40:7003–7008
McKean DC, Coats AM (1989) Infrared spectra of B(OMe)3, ClB(OMe)2 and Cl2BOMe species, isolated CH stretching frequencies and bond strengths. Spectrochem Acta 45A:409–419
Montgomery CD (2013) Factors affecting energy barriers for pyramidal inversion in amines and phosphines: a computational chemistry lab exercise. J Chem Educ 90(5):661–664
Öğretir C, Zöğüt D, Yarligan S, Arslan T (2006) Quantum chemical studies on acidity–basicity behaviours of some substituted pyridine derivatives. J Mol Struct (THEOCHEM) 759:73–78
Oguchi M, Wada K, Honma H, Tanaka A, Kaneko T, Sakakibara S, Ohsumi J, Serizawa N, Fujiwara T, Horikoshi H, Fujita T (2000) Molecular design, synthesis, and hypoglycemic activity of a series of thiazolidine-2,4-diones. J Med Chem 43:3052–3066
Othmer K (1997) Encyclopedia of chemical technology, 4th edn, pp 20
Pierrat P, Gros PC, Fort Y (2005) Solid phase synthesis of pyridine-based derivatives from a 2-chloro-5-bromopyridine scaffold. J Comb Chem 7:879–886
Pietrzycki W, Sepiol J, Tomasik P, Brzozka L (1993) Tautomerism and rotamerism in 2-methylamino-, 2-anilino-, 2-acetamido-, and 2-benzamido-pyridines. Bulletin des Societes Chimiques Belges 102(11–12):709–717
Pongor G, Pulay P, Fogarasi G, Boggs JE (1984) Theoretical prediction of vibrational spectra. I. The in-plane force field and vibrational spectra of pyridine. J Am Chem Soc 106:2765–2769
Prabavathi N, Krishnakumar V (2009) Scaled quantum chemical calculations and FTIR, FT-Raman spectral analysis of 2-methylpyrazine. Spectrochim Acta A Mol Biomol Spectrosc 72:743–747
Sathyanarayana DN (2004) Vibrational spectroscopy theory and applications, 2nd edn. New Age International (P) Limited, New Delhi
Shanmugam R, Sathyanarayana D (1984) Molecular and vibrational structure of 2-mercapto pyrimidine. Spectrochim Acta A Mol Biomol Spectrosc 40:764
Sharma YR (1994) Elementary organic spectroscopy—principles and chemical applications. S. Chande and Company Ltd., New Delhi
Silverstein M, Clayton Basseler G, Morill C (1981) Spectrometric identification of organic compound. Wiley, New York
Singh SJ, Pandey SM (1974) Molecular Structure and vibrational analysis of 1-bromo-2-chlorobenzene using ab initio HF and density functional theory (B3LYP) calculations. Indian J Pure Appl Phys 12:300–304
Singh HJ, Srivastava P (2009) Computational studies on the structure and vibrational spectra of 2-hydroxy-5-methyl-3-nitropyridine. Indian J Pure Appl Phys 47:557
Spinner E (1962) The vibration spectra and structures of the hydrochlorides of aminopyridines. J Chem Soc 3119–3126
Sponer J, Hobza P (1994a) Nonplanar geometries of DNA bases. Ab initio second-order Moeller-Plesset study. J Phys Chem 98:3161
Sponer J, Hobza P (1994b) Bifurcated hydrogen bonds in DNA crystal structures. An ab initio quantum chemical study. J Am Chem Soc 116:709
Sponer J, Hobza P (1994c) Sequence dependent intrinsic deformability of the DNA base amino groups. An ab initio quantum chemical analysis. J Mol Struct Theochem 304:35
Sponer J, Leszczynski J, Hobza P (1996) Interactions of DNA bases and the structure of DNA. In: Leszczynski J (ed) A Nonempirical Ab Initio Study With Inclusion Of Electron Correlation in computational chemistry—reviews of current trends. World Scientific, Singapore, p 185
Stidham HD, Dilella DP (1979) Vibrational perturbation: a chemical aid to assignment. I—Fermi resonance in the ν8 region of isotopic pyridines. J Raman Spectrosc 8:180
Stidham HD, Dilella DP (1980) Vibrational spectra of C 2ν deuterium substituted pyridines. 4—pyridine-2,3,5,6-d 4 and pyridine-d 5. J Raman Spectrosc 9:247
Suheyla Kurkcuoglu G, Kavlak I, Arslan T, Ogretir C (2009) Theoretical studies on the molecular structure and vibrational spectra of some dimethyl substituted pyridine derivatives. J Mol Model 15:79–90
Sundaraganesan N, Doinic Joshua B (2007) Vibrational spectra and fundamental structural assignments from HF and DFT calculations of methyl benzoate. Spectrochim Acta A Mol Biomol Spectrosc 68:771–777
Sundaraganesan N, Ilakiamani S, Saleem H, Wojciechowski PM, Michalska D (2005a) FT-Raman and FT-IR spectra, vibrational assignments and density functional studies of 5-bromo-2-nitropyridine. Spectrochim Acta A Mol Biomol Spectrosc 61:2995–3001
Sundaraganesan N, Illakiamani S, Saleem H, Wojciechowski PM, Michalska D (2005b) FT-Raman and FT-IR spectra, vibrational assignments and density functional studies of 5-bromo-2-nitropyridine. Spectrochim Acta A Mol Biomol Spectrosc 61:2995–3001
Szafran M, Koput J (2001) Ab initio and DFT calculations of structure and vibrational spectra of pyridine and its isotopomers. J Mol Struct 565–566:439–448
Talati JD, Gandhi DK (1983) N-heterocyclic compounds as corrosion inhibitors for aluminium-copper alloy in hydrochloric acid. Corros Sci 23:1315
Tayyari SF, Mahdizadeh SJ, Holakoei S, Wang YA (2010) Vibrational assignment and proton tunneling in pyridine–pyridinium complexes. J Mol Struct 971:39–46
Temple C Jr, Rose JD, Comber RN, Rener GA (1987) Synthesis of potential anticancer agents: imidazo[4,5-c]pyridines and imidazo[4,5-b]pyridines. J Med Chem 30(10):1746–1751
Wiberg KB, Shrake A (1973) A vibrational study of cyclohexane and some of its isotopic derivatives-III. A vibrational analysis of cyclohexane, cyclohexane-d 12, cyclohexane-1,1,4,4-d 4 and cyclohexane-1,1,2,2,4,4,5,5-d 8. Spectrochim Acta A Mol Biomol Spectrosc 29:583
Xiao-Ci Y, Hong Z, Ming-Dao L, Hong-Xuan R, Lu-An Y (2000) Quantum chemical study of the inhibition properties of pyridine and its derivatives at an aluminum surface. Corr Sci 42:645–653
Yadav RA, Sing IS (1985) Hydrogen bonding in o- and m-ethyl phenols. Ind J Pure Appl Phys 23:626–627
Yadav PNS, Wadhwani R (1993) Trans SAEST 28(3): 134
Yadav BS, Ali KI, Yadav P (2007) FTIR and laser Raman spectra of 2-hydroxy-5-methyl-3-nitro pyridine. Indian J Pure Appl Phys 45:979–983
Yoon SJ, Lee SW, Kim ND, Park YK, Lee GH, Kim JW, Park SJ, Park HJ, Shin DH (2004) US Pat 6, pp 795
Yurt A, Bereket G, Ogretir C (2005) Quantum chemical studies on inhibition effect of amino acids and hydroxy carboxylic acids on pitting corrosion of aluminium alloy 7075 in NaCl solution. J Mol Struct (THEOCHEM) 725:212–215
Yurt A, Ulutas S, Dal H (2006) Electrochemical and theoretical investigation on the corrosion of aluminium in acidic solution containing some Schiff bases. Appl Surf Sci 253:919–925
Zerbi G, Crawford B, Overend J (1963) Normal coordinates of the planar vibrations of pyridine and its deuteroisomers with a modified UreyBradley force field. J Chem Phys 38:127
Ziessel R (2001) Schiff-based bipyridine ligands. Unusual coordination features and mesomorphic behaviour. Coord Chem Rev 216:195–223
Acknowledgements
The author gratefully acknowledges the Princess Nourah bint Abdulrahman University Vice Rectorate of Postgraduate Studies and Scientific Research.
Competing interests
The author declares that he has no competing interests.
Author information
Authors and Affiliations
Corresponding author
Additional file
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Al-Otaibi, J.S. Tautomerization, molecular structure, transition state structure, and vibrational spectra of 2-aminopyridines: a combined computational and experimental study. SpringerPlus 4, 586 (2015). https://doi.org/10.1186/s40064-015-1363-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40064-015-1363-2