
Abstract
Background
Although many causative genes have been uncovered in recent years, genetic diagnosis is still missing for approximately 50% of autosomal recessive cerebellar ataxia (ARCA) patients. Few studies have been performed to determine the genetic spectrum and clinical profile of ARCA patients in the Chinese population.
Methods
Fifty-four Chinese index patients with unexplained autosomal recessive or sporadic ataxia were investigated by whole-exome sequencing (WES) and copy number variation (CNV) calling with ExomeDepth. Likely causal CNV predictions were validated by CNVseq.
Results
Thirty-eight mutations including 29 novel ones were identified in 25 out of the 54 patients, providing a 46.3% positive molecular diagnostic rate. Ten different genes were involved, of which four most common genes were SACS, SYNE1, ADCK3 and SETX, which accounted for 76.0% (19/25) of the positive cases. The de novo microdeletion in SACS was reported for the first time in China and the uniparental disomy of ADCK3 was reported for the first time worldwide. Clinical features of the patients carrying SACS, SYNE1 and ADCK3 mutations were summarized.
Conclusions
Our results expand the genetic spectrum and clinical profiles of ARCA patients, demonstrate the high efficiency and reliability of WES combined with CNV analysis in the diagnosis of suspected ARCA, and emphasize the importance of complete bioinformatics analysis of WES data for accurate diagnosis.
Similar content being viewed by others


Background
Autosomal recessive cerebellar ataxias (ARCAs) are a heterogeneous group of rare metabolic and degenerative genetic disorders that are characterized by progressive damage of the cerebellum and/or its associated afferent tracts [1, 2]. The overall prevalence of ARCAs is estimated to be 3–5 per 100,000 in the general population [3], but each individual type may have varied prevalence among ethnic groups [2, 4]. Most ARCAs have an early onset, occurring before 30 years old, and a major clinical feature of progressive cerebellar ataxia, variably accompanied by dysarthria, ophthalmoplegia, pyramidal and extrapyramidal signs, peripheral neuropathy, cognitive impairment and other symptoms [1, 5, 6].
With advanced technologies in genetic screening, more than 90 genes have been identified in ARCAs [6]. Friedreich’s ataxia (FRDA) is the most common type of ARCA, accounting for up to 25% of all ARCAs, followed by ataxia-telangiectasia or ataxia with oculomotor apraxia (AOA) [1, 2]. It is now increasingly acknowledged that spectrin repeat-containing nuclear envelope protein (SYNE1) ataxia and autosomal-recessive spastic ataxia of Charlevoix-Saguenay (ARSACS; gene: SACS), which were long thought to be largely confined to a specific group of French-Canadian populations, are relatively frequent ARCAs distributed around the world [2, 7]. Despite the discovery of many disease-causing genes in recent years, the genetic cause of ARCAs remains elusive in more than 50% of affected individuals [2, 8, 9]. The advent of whole-exome sequencing (WES) technology has enabled efficient diagnosis with single-nucleotide variants (SNVs) and small indels [10], and new WES-based methods, such as ExomeDepth, have enabled detection of structural variation like copy number variations (CNVs) in many hereditary diseases [11]. Moreover, due to the low incidence, few studies have reported the genetic or clinical characteristic of ARCA patients in the Chinese population.
In this study, we performed WES and CNV calling for 54 unrelated Chinese patients with autosomal recessive or sporadic hereditary cerebellar ataxia (HCA) to assess the possibility and prevalence of mutations.
Methods
Subjects
All patients were consecutively enrolled from Huashan Hospital of Fudan University and the Second Affiliated Hospital of Zhejiang University School of Medicine between August 9, 2008 and May 5, 2021. The patients were evaluated and diagnosed with HCA based on Harding’s criteria [12] by at least two senior neurologists. Informed consent was signed by all participants or their guardians. This study was approved by the Ethics Committees of the above two hospitals.
Fifty-four patients meeting the following inclusion criteria were enrolled in this study: (1) having progressive cerebellar ataxia; (2) being negative for molecular analysis of mitochondrial ataxia or 10 common subtypes of spinocerebellar ataxia (SCA) (including SCA1, 2, 3, 6, 7, 8, 10, 12, 17 and DRPLA) resulting from dynamic mutations or Huntington’s disease; (3) exclusion of other identified etiologies, e.g., multiple system atrophy, Niemann–Pick disease, Parkinson’s disease, Wilson’s disease, multiple sclerosis, viral infection, alcohol or drug intoxication, or paraneoplastic syndrome; (4) autosomal recessive inheritance with the presence of patients in siblings and/or consanguineous union of the parents (n = 12); and (5) sporadic cases with onset before the age of 40 years [5, 13] (n = 42). In addition, 1000 unrelated Chinese individuals without a history of ataxia were included as the control.
Genomic DNA extraction and whole-exome sequencing
Genomic DNA was extracted from peripheral blood samples of all patients by using the QIAamp Blood Genome Extraction Kit (Qiagen, Germany) following a standard protocol. Whole-exome Illumina sequencing of DNA was performed according to a detailed protocol described in our previous study [14]. All variants were annotated by ANNOVAR. Two public databases, the 1000 Genomes Project (http://browser.1000genomes.org) and the Exome Aggregation Consortium (http://exac.broadinstitute.org/), and our in-house WES database containing 1000 Chinese control individuals, were used to check the frequency of the variants in the general population. Three software programs, SIFT (http://sift.jcvi.org/), PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2/) and Mutation Taster (http://www.mutationtaster.org/), were used to predict the possible deleterious effects of mutations. Moreover, these variants were compared with the Human Gene Mutation Database (HGMD, Professional 2021.1, http://www.hgmd.cf.ac.uk/) to determine whether they were known or novel. Finally, the interpretation and classification of variants was performed based on the American College of Medical Genetics and Genomics (ACMG) standards [15].
CNV analysis
CNVs were called from read-depth of WES data using ExomeDepth algorithm according to the developers’ guidelines [16]. For these analyses, each test exome was compared with a set of matched, aggregate reference samples. CNV calls were annotated using AnnotSV [17]. Candidate CNVs were prioritized by exon number, Bayes factor, minor allele frequency and the ratio of observed/expected number of reads. Candidate CNVs were further proved by CNVseq, which performed a low-coverage WGS strategy and included DNA extraction, interruption, library construction and sequencing by Illumina HiSeq 2500 (Illumina, San Diego, CA). The databases ISCA, DGV, Decipher, OMIM, ClinVar and ClinGen were used to analyze the CNVs. Comprehensive assessments of CNV hazard levels were undertaken based on a frequency database and annotation information according to the ACMG standards and guidelines [18].
Affymetrix CytoScan® Dx assay
The Affymetrix CytoScan® Dx Assay was used to identify the uniparental disomy (UPD), which utilizes a high-density combined SNP and comparative genomic hybridization array platform, which assesses approximately 2,696,550 markers, including approximately 750,000 SNP markers. The whole-genome screening and analysis of chromosomal rearrangements was performed by Affymetrix CytoScan® Dx Assay according to the manufacturer’s recommendations [19].
Sanger sequencing and parent analysis
After analyzing and filtering the WES data, Sanger sequencing was performed to confirm the sequencing results and the family co-segregation pattern. The parenthood of patients with de novo variants was analyzed using 21 core short tandem repeat regions, including D19S433, D5S818, D21S11, D18S51, D6S1043, AMEL, D3S1358, D13S317, D7S820, D16S539, CSF1PO, Penta D, D2S441, vWA, D8S1179, TPOX, Penta E, TH01, D12S391, D2S1338, and FGA.
Results
Twenty-nine novel variants were identified in 25 unrelated ARCA patients
A total of 38 variants (Table 1) including 29 novel and nine known variants in 10 genes were identified in 25 out of 54 ARCA patients (chromatograms for novel variants except the chr13:23490196-24866656del are shown in Fig. 1a). All novel variants were absent from or present at extremely low frequency in both public databases and our in-house WES database. The pathogenicity of variants was consistently predicted by different in silico prediction programs. All of the novel missense variants were highly conserved among animal species (Fig. 1b). According to the ACMG standard, 17 out of the 29 novel variants were classified as pathogenic variants, 10 as likely pathogenic variants, and the remaining two (ADCK3: p.R271H, SETX: p.Y2455C) as variants of uncertain significance, whose pathogenicity needed to be confirmed by further functional studies. Six out of the nine known variants (Additional file 1: Fig. S1a) were identified as pathogenic variants and three as likely pathogenic variants.

Chromatogram, homology comparison, and genetic spectrum. a Chromatograms of 28 novel variants. The upper chromatograms each represent the normal sequence and the lower ones represent the variant. b Highlighted zones indicating 13 novel missense variants among 8 species. c Distribution of ARCA subtypes with different genetic causes in our ARCA families (n = 27, including two previously reported cases)
Therefore, 25 out of 54 (46.3%) patients were genetically diagnosed with ARCA. Previously, we reported an index patient with AOA type 2 (AOA2) [20] and an index patient with autosomal recessive spinocerebellar ataxia-27 (SCAR27) [21]. Taken together, genetic diagnoses were made for a total of 27 out of 56 (48.2%) ARCA index patients in our center. The most common subtype was ARSACS (22.2%), followed by SCAR8 (18.5%), SCAR9 (18.5%) and AOA2 (14.8%) (Fig. 1c). The clinical features of the patients identified with the genetic variants are listed in Table 2.
Identification of a de novo microdeletion and clinical features of patients with SACS mutations
After WES analysis, a novel homozygous mutation (p.K3646fs) was identified in case 1 without family history (Table 2). However, the heterozygous p.K3646fs was only confirmed in his mother and brother but not in his father after family segregation. The parent–child relationship was also established by parenthood analysis (Fig. 2a). Therefore, CNV analysis with ExomeDepth was conducted in the proband and a large deletion was detected in chromosome 13. Then, a trio-copy number variation sequencing (CNVseq) was performed and identified a 1.38 Mb deletion (chr13:23490196-24866656del) in the proband, but the corresponding chromosome in the parents was normal, which means it was a de novo large deletion (Fig. 2b). The chr13:23490196-24866656 in Genome browser includes the entire SACS gene and 5 other genes (Fig. 2c). Thus, a total of nine mutations including eight novel ones in SACS were identified in six patients (Table 2).

A de novo large deletion in a patient with ARSACS. a The pedigree of case 1 shows segregation of p.K3646fs in SACS and chr13:23490196-24866656del. The analysis of the repeat numbers of 21 core short tandem repeat loci in the four participants showed that the probability of the patient being the alleged parents’ biological son was 99.99%. Open symbol: unaffected; filled symbol: affected; square: male; circle: female; arrow: proband of the family. Symbol with “+/+” indicates patient. Symbols with “+/−” indicate mutation carrier. b CNVseq of the proband and his parents showed that chr13:23490196-24866656del was a de novo mutation, as indicated by the blue box. c Schematic diagram of the known genes in this deletion region (reference human Genome Build GRCh37, UCSC Assembly hg19)
Of the six patients, the mean age of disease onset was 9.5 years (1–39 years), and onset with gait disturbance was all accompanied by sensorimotor neuropathy. Weakness of limbs was present in 4 cases, skeletal abnormality in 5 cases (pes cavus in 4 cases [Fig. 3a] with 1 accompanied by flexion deformity of fingers [Fig. 3b], dental abnormalities in 1 case [Fig. 3c]), spasticity in 3 cases, listening loss in 2 cases, and mental retardation in 1 case. Radiological evaluation of the 3-year-old patient (case 1) showed a normal contract, whereas the other 5 cases showed cerebellum atrophy, thinning of the corpus callosum, bulky pons, bilateral pontine linear hypointense lesions and hyperintensities around the thalamus (Fig. 3d–f), and one had craniocerebral dysplasia (case 4). A characteristic retinal finding of case 2 and case 4 was the presence of yellow streaks of hypermyelinated fibers radiating from the edges of the optic disc and retinal nerve fiber hypertrophy, as demonstrated on ocular coherence tomography (Fig. 3g–k).

Clinical features of patients with ARSACS. a–c The special phenotypes of ARSACS: pes cavus (a), flexion deformity of fingers in case 4 (b) and abnormality of dentition in case 1 (c). d–f Classic brain magnetic resonance images in case 2. d Sagittal T2 sequence shows thinning of the corpus callosum (red arrow), superior vermian atrophy (yellow arrow), and bulky pons (blue arrow); e axial T2 shows bilateral pontine linear hypointense lesions (arrow); f axial T2 shows hyperintensities around the thalamus (arrows). g–k Typical retinal findings in case 4: Fundus photographs of the right (g) and left eyes (j) show yellow streaks of hypermyelinated fibers radiating from the edges of the optic disc; ocular coherence tomography imaging of the right (h) and left eyes (i) show thickened retinal nerve fiber layer (RNFL) (the yellow areas); k statistical graph of OCT showing thickening of the RNFL. The black line indicates oculus dexter (OD), that is, the right eye; the dotted line indicates oculus sinister (OS), the left eye. The green band represents the 5%–95% range of the normative data. (Quadrants: TEMP temporal, SUP superior, NAS nasal, INF inferior)
Characteristics of patients with SYNE1 mutations
Among the nine novel SYNE1 pathogenic variants found in five cases, four were frameshift, three were nonsense and two were splicing variants. The onset age was before 25 years in four patients and at 53 years (late onset) in one patient. All of the five cases had onset with gait disturbance, and three of them had dysarthria and horizontal nystagmus. Case 7 had the earliest onset with most complex phenotypes including psychiatric symptoms, external ophthalmoplegia and myoclonic jerks (Additional file 2: Video S1). Case 8 with a homozygous mutation (p.E3053fs) appeared to be pure cerebellar ataxia. Both case 9 and case 11 had sensorimotor neuropathy, and case 9 was also accompanied by tremor, dizziness and pes cavus. Case 10 had a 15-year history of sensorineural hearing loss and mild ataxia. She also exhibited two pathogenic mutations (Additional file 1: Fig. S1b) including c.6149-3T > G and c.1898dupA (p.E633fs) in PTPRQ (NM_001145026.1). After pedigree verification, her brother affected by impaired hearing but without ataxia, was found to carry these two mutations in PTPRQ but only one heterozygous mutation in SYNE1, and all mutations in SYNE1 and PTPRQ were derived independently from two parents with normal phenotypes (Additional file 1: Fig. S1c). Owing to the lack of magnetic resonance imaging report of case 8, the remaining four probands were estimated to have a varied degree of cerebellar atrophy.
Identification of a rare UPD and clinical features of patients with ADCK3 mutations
After WES analysis, six ADCK3 mutations including three novel ones (p.R271H, p.L320fs and p.R598H) were identified in four males with no family history of disease and one female with an affected sister. The homozygous variant (p.R410X) within ADCK3 was identified in case 12 (Table 2). However, the heterozygous p.R410X was only confirmed in his mother but not in his father, and the parental analysis was normal (Fig. 4a). The CNV analysis of the index patient was normal, too. Thus, UPD was considered in this situation. The results of Affymetrix CytoScan® Dx Assay analyses were as follows: arr [hg19] 1pterp36.11 (888,658-25,445,510) hmz; arr [hg19] 1q42.12qter (225,105,798-249,198,164) hmz, which suggested that the index patient had maternal UPD for a segment of chromosome 1 (Fig. 4b), which included the whole ADCK3 gene (Fig. 4c).

A maternal uniparental disomy in a patient with SCAR9. a The pedigree of case 12 shows the segregation of p.R410X in ADCK3. The analysis of the repeat numbers of 21 core short tandem repeat loci in the three participants showed that the probability of the patient being the alleged parents’ biological son was 99.99%. b Uniparental disomy of chromosomes 1p and 1q detected by Affymetrix CytoScan® Dx Assay in the patient. c Schematic diagram of ADCK3 in this UPD region (1pterp36.11 and 1q42.12qter; reference human Genome Build GRCh37, UCSC Assembly hg19)
The mean age at onset was 16.2 years (range 2–32 years). All patients showed ataxia signs, and four also displayed dysarthria. Cases 12, 13 and 16 had onset with gait disturbance, case 12 displayed pure ataxia, while cases 13 and 16 had additional cognitive impairment. Cases 14 and 15 presented onset with uncontrollable hand shaking, followed later by head shaking. Both were identified to have the p.S616fs mutation, but the mutation was heterozygous in case 14 and homozygous in case 15. Case 15 had additional dysphagia and sensorimotor neuropathy. Magnetic resonance imaging revealed that all five patients had cerebellar atrophy, and one also had atrophy of the brainstem. Case 15 was of the Dong ethnic minority and had started supplementation with 300 mg/day of CoQ10. At 1-year follow-up, he showed clinical improvement, which was more evident in the tremor and gait items. His total Scale for the Assessment and Rating of Ataxia scores were 10 at baseline and 4.5 after 1 year; similarly, his International Cooperative Ataxia Rating Scale score decreased from 21 to 9, and his non-ataxia score decreased from 4 to 1 (Additional file 3: Table S1). A homozygous mutation was identified by Sanger sequencing in his 24-year-old sister, who had no clinical symptoms yet.
Discussion
To date, few studies have been conducted to investigate all causative genes and the clinical features of ARCA in the Chinese population. In this study, the prevalence of ARCA was systematically investigated in 54 unrelated autosomal recessive/sporadic ataxia patients by WES analysis and CNV calling, which is the largest cohort in China to date. Thirty-eight mutations, including 29 novel mutations in 10 genes related to ARCA, were identified in 25 unrelated patients. Among them, the de novo microdeletion in SACS was reported for the first time in the Chinese population and the UPD of ADCK3 was reported for the first time worldwide.
In this study, the diagnostic yield was 46.3% (25 of 54 patients). This rate is at a relatively high level compared to those previously reported using exome sequencing [5, 22,23,24,25,26,27]. Marelli et al. also performed a mini-exome and read-depth-based CNV analysis in 33 ataxic patients and identified pathogenic variants in 14 cases (42%) including CNV in 2 patients [28]. The differences in diagnostic rate were most likely due to the differences in source populations (ethnic and geographic origin), sample size, the inclusion criteria used and study methodology. One of the major highlights of the present study is the combination of WES and CNV analysis, and the results truly demonstrate that structural variation might not be extremely rare in ARCA. With the development of high throughput sequencing technology and bioinformatics algorithms, many established approaches for CNV calling in WES data have been available and easily usable for biologists and geneticists to test structural variations, such as ExomeDepth, CovCopCan, IonCopy, DeviCNV, and Cov’Cop [29]. Thus, we suggest that tools for detecting structural variations such as CNVs should be used routinely for NGS data analysis in order to increase the rate of positive diagnosis.
In this study, 12 autosomal recessive ataxia and 42 sporadic ataxia families were included. Mutations in ARCA causative genes were identified in eight autosomal recessive ataxia and 17 sporadic ataxia families. Combined with our previous studies, the number of ARCA families with definite diagnosis in our center was 27. Further, we demonstrate that ARSACS (gene: SACS; n = 6, 22.2%), SCAR8 (gene: SYNE1; n = 5, 18.5%), SCAR9 (gene: ADCK3; n = 5, 18.5%) and AOA2 (gene: SETX; n = 4, 14.8%) are the most common recessive ataxia subtypes in the Chinese population. In a previously reported Chinese ARCA cohort, the ARCA-causing genes were identified in 19 out of 26 probands, including AOA2 (n = 4, 15.4%), Niemann-Pick disease (n = 3, 11.5%), one ARSACS and one SCAR8 [27]. SACS and SYNE1 mutations have been observed mainly in Quebec and Canada, where ARSACS and SCAR8 are the second and third most common hereditary ataxia, respectively [7]. FRDA has been reported as the most frequent ARCA in Caucasians but is much rare in Chinese [30], thus the identification of dynamic mutations about FRDA was not undertaken in our study. Although the aetiology of ARCA in Chinese is different from the reported patterns in Caucasians presumably due to the different genetic backgrounds and ethnicities, there are still some similarities which may contribute to a better understanding of the epidemiology and mechanism of ARCA.
In total, 14 ARSACS patients from 10 families have been reported in China (Additional file 4: Table S2), including the 6 probands in this study, confirming that nonsense or frameshift mutations in SACS are the most common genetic cause in Chinese patients. In our study, a majority of those patients who had at least one truncation variant appeared to have a typical ARSACS clinical presentation with childhood onset of symptoms. But one patient who harbored a homozygous non-frameshift deletion variant (p.3758_3759del), exhibited an atypical disease presentation with an absence of spasticity or pyramidal signs and onset in adulthood, and this same variant has been reported in two heterozygous patients, both with early onset (1 and 13 years old) and typical triad symptoms [31, 32]. Thus, whether the truncation variant in SACS is linked to a typical clinical manifestation of ARSACS is an issue that requires further exploration. Even this de novo large deletion containing SACS was the second report in the world [33], the CNV in SACS has already been reported in many populations including Belgian, French, Italian, Canadian, German and Chinese [33,34,35,36,37,38,39]. Therefore, presence of CNV must be considered if no or only one heterozygous mutation had been identified in those patients with an ARSACS phenotype suggested by means of clinical presumption or auxiliary examination.
Defects in SYNE1 are associated with adult-onset, slowly progressive, relatively pure cerebellar ataxia with only a few extracerebellar symptoms (SCAR8), and almost all reported variants that cause this phenotype are protein truncations [7]. Previous studies showed that SYNE1 ataxia accounted for 5.3% (23/434), 6% (7/116), and 10.26% (4/39) of recessive and sporadic ataxia patients in two European combined cohorts and one Brazilian cohort [7, 40, 41]. In the present study, 9.3% (5/54) of ataxia patients had biallelic truncating variants in SYNE1, demonstrating that SCAR8 is also a common subtype of recessive ataxia in China. Two SCAR8 patients (cases 8 and 10) both had symptoms of pure cerebellar ataxia, while the hearing loss of case 10 was caused by mutations in PTPRQ. Moreover, the remaining three patients exhibited variable additional extracerebellar neurological symptoms (peripheral polyneuropathy, mental retardation, dizziness, pes cavus, external ophthalmoplegia, myoclonic jerks) and non-neurologic dysfunctions (psychiatric symptoms). The reported nine SCAR8 Chinese patients included three presenting with pure cerebellar ataxia and six presenting with variable ataxia syndrome [27, 42, 43]. Thus, in the Chinese population, pure cerebellar ataxia only accounted for 35.7% (5/14) of SCAR8 cases, while the other 64.3% (9/14) of patients showed complex ataxia phenotypes with a wide range of noncerebellar abnormalities. This further supports the concept that SYNE1 ataxia is a multisystemic neurodegenerative disease, as proposed by Synofzik et al. [7].
A total of 65 pathogenic mutations in ADCK3 have been reported around the world (HGMD, Professional 2021.1), and SCAR9 has also been reported as a common subtype of recessive ataxia [26]. However, cases of SCAR9 were rarely reported among the Chinese population before [44]. Here, we report for the first time that SCAR9 also had a high frequency in China. The homozygous p.S616fs in ADCK3 has been reported in two siblings from a consanguineous family of Pakistani origin, and both siblings presented with cerebellar ataxia, myoclonus, tremor and dysarthria at age of 10 and 14, respectively [45]. However, in our study, two patients harboring p.S616fs in heterozygous or homozygous form both presented with prominent tremor and mild ataxia symptoms, but onset at adolescence and adulthood, respectively. Moreover, the homozygous patient presented with additional dysphagia and peripheral neuropathy, and his sister was identified to have the mutation but was still asymptomatic at the age of 24. Thus, the clinical presentation of SCAR9 may be highly variable, even in patients with the same mutation from one family. Our study also proved that supplementation with COQ10 is significantly helpful for SCAR9 patients, even though this therapy has different curative effects in several studies [45,46,47]. The UPD of ADCK3 identified in our study is the first report worldwide, which not only enriches the genotypic spectrum of SCAR9, but also emphasizes the importance of a detailed analysis of family segregation.
The absence of diagnosis for 29 patients in the cohort may be explained by the following reasons. First, it is difficult for exome capture to fully cover all coding regions of the genome, especially regions rich with GC. Second, large genomic rearrangements and trinucleotide expansions cannot be reliably detected from exome-capture data, even though there are some CNV-detecting tools developed, and based on the read-depth of NGS data, such as ExomeDepth used in this study, we cannot easily detect inversion or translocation [34]. Third, it is also likely that some causal variants are outside the coding regions and adjacent splice sites [48]. Finally, insights into the functional consequences of the variants are missing, as some synonymous mutations may be causative, too [49]. Some of these issues can be addressed by whole-genome sequencing, which is, however, expensive and needs complex bioinformatics analyses. In addition, mutations in yet unknown genes of ARCA may play a key role in these unclear disorders.
Conclusions
In summary, this is so far the largest WES analysis and CNV calling study to explore the genetic background and describe clinical characteristics of ARCA in a Chinese population. We identified 38 mutations including 29 novel ones in 25 unrelated Chinese ARCA patients. We also reported for the first time the UPD of ADCK3 in the world and the de novo microdeletion of SACS in China. Our results expand the genetic spectrum and clinical profiles of ARCA patients, demonstrate the high efficiency and reliability of WES combined with CNV analysis in diagnosing suspected ARCA, and emphasize the importance of complete bioinformatics analysis of WES data in making an accurate diagnosis. Further functional studies will help to determine the pathogenicity of novel variants and to better understand the pathogenetic mechanisms of these complicated diseases.
Availability of data and materials
The data presented in this study are available on request from the corresponding author. The data are not publicly available due to privacy reasons.
Abbreviations
- ACMG:
-
American College of Medical Genetics and Genomics
- AOA:
-
Ataxia with oculomotor apraxia
- ARCA:
-
Autosomal recessive cerebellar ataxia
- ARSACS:
-
Autosomal-recessive spastic ataxia of Charlevoix-Saguenay
- CNV:
-
Copy number variation
- FRDA:
-
Friedreich’s ataxia
- HCA:
-
Hereditary cerebellar ataxia
- HGMD:
-
Human gene mutation database
- SNV:
-
Single-nucleotide variant
- SYNE1 :
-
Spectrin repeat-containing nuclear envelope protein
- WES:
-
Whole-exome sequencing
- UPD:
-
Uniparental disomy
References
Anheim M, Tranchant C, Koenig M. The autosomal recessive cerebellar ataxias. N Engl J Med. 2012;366(7):636–46.
Synofzik M, Puccio H, Mochel F, Schols L. Autosomal recessive cerebellar ataxias: paving the way toward targeted molecular therapies. Neuron. 2019;101(4):560–83.
Manto M, Gandini J, Feil K, Strupp M. Cerebellar ataxias: an update. Curr Opin Neurol. 2020;33(1):150–60.
Anheim M, Fleury M, Monga B, Laugel V, Chaigne D, Rodier G, et al. Epidemiological, clinical, paraclinical and molecular study of a cohort of 102 patients affected with autosomal recessive progressive cerebellar ataxia from Alsace, Eastern France: implications for clinical management. Neurogenetics. 2010;11(1):1–12.
Coutelier M, Hammer MB, Stevanin G, Monin ML, Davoine CS, Mochel F, et al. Efficacy of exome-targeted capture sequencing to detect mutations in known cerebellar ataxia genes. JAMA Neurol. 2018;75(5):591–9.
Rossi M, Anheim M, Durr A, Klein C, Koenig M, Synofzik M, et al. The genetic nomenclature of recessive cerebellar ataxias. Mov Disord. 2018;33(7):1056–76.
Synofzik M, Smets K, Mallaret M, Di Bella D, Gallenmüller C, Baets J, et al. SYNE1 ataxia is a common recessive ataxia with major non-cerebellar features: a large multi-centre study. Brain. 2016;139(Pt 5):1378–93.
Mallaret M, Renaud M, Redin C, Drouot N, Muller J, Severac F, et al. Validation of a clinical practice-based algorithm for the diagnosis of autosomal recessive cerebellar ataxias based on NGS identified cases. J Neurol. 2016;263(7):1314–22.
Parodi L, Coarelli G, Stevanin G, Brice A, Durr A. Hereditary ataxias and paraparesias. Curr Opin Neurol. 2018;31(4):462–71.
Chen CX, Dong HL, Wei Q, Li LX, Yu H, Li JQ, et al. Genetic spectrum and clinical profiles in a southeast Chinese cohort of charcot-marie-tooth disease. Clin Genet. 2019;96(5):439–48.
Zampaglione E, Kinde B, Place EM, Navarro-Gomez D, Maher M, Jamshidi F, et al. Copy-number variation contributes 9% of pathogenicity in the inherited retinal degenerations. Genet Med. 2020;22(6):1079–87.
Harding AE. Clinical features and classification of inherited ataxias. Adv Neurol. 1993;61:1–14.
Synofzik M, Németh AH. Recessive ataxias. Handb Clin Neurol. 2018;155:73–89.
Dong HL, Wei Q, Li JQ, Li HF, Bai G, Ma H, et al. Genetic spectrum of MCM3AP and its relationship with phenotype of charcot-marie-tooth disease. J Peripher Nerv Syst. 2020;25(2):107–11.
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for molecular pathology. Genet Med. 2015;17(5):405–24.
Plagnol V, Curtis J, Epstein M, Mok KY, Stebbings E, Grigoriadou S, et al. A robust model for read count data in exome sequencing experiments and implications for copy number variant calling. Bioinformatics. 2012;28(21):2747–54.
Geoffroy V, Herenger Y, Kress A, Stoetzel C, Piton A, Dollfus H, et al. AnnotSV: an integrated tool for structural variations annotation. Bioinformatics. 2018;34(20):3572–4.
Riggs ER, Andersen EF, Cherry AM, Kantarci S, Kearney H, Patel A, et al. Technical standards for the interpretation and reporting of constitutional copy-number variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics (ACMG) and the Clinical Genome Resource (ClinGen). Genet Med. 2019;22(2):245–57.
Webb BD, Scharf RJ, Spear EA, Edelmann LJ, Stroustrup A. Evaluation of the affymetrix CytoScan(®) Dx assay for developmental delay. Expert Rev Mol Diagn. 2015;15(2):185–92.
Lu C, Zheng YC, Dong Y, Li HF. Identification of novel senataxin mutations in Chinese patients with autosomal recessive cerebellar ataxias by targeted next-generation sequencing. BMC Neurol. 2016;16(1):179.
Dong HL, Cheng HL, Bai G, Shen Y, Wu ZY. Novel GDAP2 pathogenic variants cause autosomal recessive spinocerebellar ataxia-27 (SCAR27) in a Chinese family. Brain. 2020;143(6):e50.
Fogel BL, Lee H, Deignan JL, Strom SP, Kantarci S, Wang X, et al. Exome sequencing in the clinical diagnosis of sporadic or familial cerebellar ataxia. JAMA Neurol. 2014;71(10):1237–46.
Shakya S, Kumari R, Suroliya V, Tyagi N, Joshi A, Garg A, et al. Whole exome and targeted gene sequencing to detect pathogenic recessive variants in early onset cerebellar ataxia. Clin Genet. 2019;96(6):566–74.
Pyle A, Smertenko T, Bargiela D, Griffin H, Duff J, Appleton M, et al. Exome sequencing in undiagnosed inherited and sporadic ataxias. Brain. 2015;138(Pt 2):276–83.
Sawyer SJ, Schwartzentruber J, Beaulieu CL, Dyment D, Smith A, Warman Chardon J, et al. Exome sequencing as a diagnostic tool for pediatric-onset ataxia. Hum Mutat. 2014;35(1):45–9.
Sun M, Johnson AK, Nelakuditi V, Guidugli L, Fischer D, Arndt K, et al. Targeted exome analysis identifies the genetic basis of disease in over 50% of patients with a wide range of ataxia-related phenotypes. Genet Med. 2019;21(1):195–206.
Guan RY, Wu JJ, Ding ZT, Wang J, Sun YM. Clinical and genetic findings in a cohort of Chinese patients with autosomal recessive spinocerebellar ataxia. Clin Genet. 2020;97(3):532–5.
Marelli C, Guissart C, Hubsch C, Renaud M, Villemin JP, Larrieu L, et al. Mini-exome coupled to read-depth based copy number variation analysis in patients with inherited ataxias. Hum Mutat. 2016;37(12):1340–53.
Derouault P, Chauzeix J, Rizzo D, Miressi F, Magdelaine C, Bourthoumieu S, et al. CovCopCan: an efficient tool to detect copy number variation from amplicon sequencing data in inherited diseases and cancer. PLoS Comput Biol. 2020;16(2):e1007503.
Zeng JS, Wang JL, Zeng S, He M, Zeng XF, Zhou Y, et al. Friedreich’s Ataxia (FRDA) is an extremely rare cause of autosomal recessive ataxia in Chinese Han population. J Neurol Sci. 2015;351(1–2):124–6.
Rezende Filho FM, Parkinson MH, Pedroso JL, Poh R, Faber I, Lourenço CM, et al. Clinical, ophthalmological, imaging and genetic features in Brazilian patients with ARSACS. Parkinsonism Relat Disord. 2019;62:148–55.
Sun W, Yu M, Zhuo YJ, Wang ZX, Yuan Y. Novel spastic ataxia of Charlevoix-Saguenay gene compound heterozygous mutations in late onset autosomal recessive spastic ataxia of Charlevoix-Saguenay. Chin J Neurol. 2017;50(11):831–6.
Breckpot J, Takiyama Y, Thienpont B, Van Vooren S, Vermeesch JR, Ortibus E, et al. A novel genomic disorder: a deletion of the SACS gene leading to spastic ataxia of Charlevoix-Saguenay. Eur J Hum Genet. 2008;16(9):1050–4.
Miressi F, Faye PA, Pyromali I, Bourthoumieux S, Derouault P, Husson M, et al. A mutation can hide another one: Think structural variants! Comput Struct Biotechnol J. 2020;18:2095–9.
Terracciano A, Casali C, Grieco GS, Orteschi D, Di Giandomenico S, Seminara L, et al. An inherited large-scale rearrangement in SACS associated with spastic ataxia and hearing loss. Neurogenetics. 2009;10(2):151–5.
Prodi E, Grisoli M, Panzeri M, Minati L, Fattori F, Erbetta A, et al. Supratentorial and pontine MRI abnormalities characterize recessive spastic ataxia of Charlevoix-Saguenay. A comprehensive study of an Italian series. Eur J Neurol. 2013;20(1):138–46.
Dougherty SC, Harper A, Al Saif H, Vorona G, Haines SR. A chromosomal deletion and new frameshift mutation cause ARSACS in an African-American. Front Neurol. 2018;9:956.
Vill K, Müller-Felber W, Gläser D, Kuhn M, Teusch V, Schreiber H, et al. SACS variants are a relevant cause of autosomal recessive hereditary motor and sensory neuropathy. Hum Genet. 2018;137(11–12):911–9.
Liu L, Li XB, Zi XH, Shen L, Hu ZhM, Huang ShX, et al. A novel hemizygous SACS mutation identified by whole exome sequencing and SNP array analysis in a Chinese ARSACS patient. J Neurol Sci. 2016;362:111–4.
Mademan I, Harmuth F, Giordano I, Timmann D, Magri S, Deconinck T, et al. Multisystemic SYNE1 ataxia: confirming the high frequency and extending the mutational and phenotypic spectrum. Brain. 2016;139(Pt8):e46.
Gama MT, Houle G, Noreau A, Dionne-Laporte A, Dion PA, Rouleau GA, et al. SYNE1 mutations cause autosomal-recessive ataxia with retained reflexes in Brazilian patients. Mov Disord. 2016;31(11):1754–6.
Peng Y, Ye W, Chen Z, Peng HR, Wang PZ, Hou X, et al. Identifying SYNE1 ataxia with novel mutations in a chinese population. Front Neurol. 2018;9:1111.
Duan XH, Hao Y, Cao ZH, Zhou C, Zhang J, Wang RB, et al. Autosomal recessive cerebellar ataxia type 1: phenotypic and genetic correlation in a cohort of chinese patients with SYNE1 variants. Cerebellum. 2021;20(1):74–82.
Liu G, Ma DY, Li JH, Luo CY, Sun Y, Zhang JJ, et al. A novel COQ8A missense variant associated with a mild form of primary coenzyme Q10 deficiency type 4. Clin Biochem. 2020;84:93–8.
Liu YT, Hersheson J, Plagnol V, Fawcett K, Duberley KE, Preza E, et al. Autosomal-recessive cerebellar ataxia caused by a novel ADCK3 mutation that elongates the protein: clinical, genetic and biochemical characterisation. J Neurol Neurosurg Psychiatry. 2014;85(5):493–8.
Chang A, Ruiz-Lopez M, Slow E, Tarnopolsky M, Lang AE, Munhoz RP. ADCK3-related coenzyme Q10 deficiency: a potentially treatable genetic disease. Mov Disord Clin Pract. 2018;5(6):635–9.
Horvath R, Czermin B, Gulati S, Demuth S, Houge G, Pyle A, et al. Adult-onset cerebellar ataxia due to mutations in CABC1/ADCK3. J Neurol Neurosurg Psychiatry. 2012;83(2):174–8.
Dong Y, Ni W, Chen WJ, Wan B, Zhao GX, Shi ZQ, et al. Spectrum and classification of ATP7B variants in a large cohort of Chinese patients with Wilson’s disease guides genetic diagnosis. Theranostics. 2016;6(5):638–49.
Hunt RC, Simhadri VL, Iandoli M, Sauna ZE, Kimchi-Sarfaty C. Exposing synonymous mutations. Trends Genet. 2014;30(7):308–21.
Acknowledgements
We would like to thank all participants for their support and willingness to participate in this study.
Funding
This work was supported by grants from the National Natural Science Foundation of China (82071260 to Z.-Y. W.) and the Research Foundation for Distinguished Scholar of Zhejiang University (188020-193810101/089 to Z.-Y. W.).
Author information
Authors and Affiliations
Contributions
HLC: methodology, data curation, formal analysis, supervision, writing-original draft preparation. YRS: methodology, data curation. YD: formal analysis, supervision. HLD: formal analysis, supervision. LY: data curation, formal analysis. YM: methodology. YS: formal analysis. ZYW: conceptualization, resources, data curation, formal analysis, supervision, writing-reviewing and editing. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The study was approved by the Ethics Committee of Huashan Hospital of Fudan University (protocol code 2008-071, 2008-03-05) and the Second Affiliated Hospital of Zhejiang University School of Medicine (protocol code 2015-048, 2015-08-11). Written informed consents for the study have been obtained from all participants or their legal guardians.
Consent for publication
When including data of individual participants, consent for publication was obtained from them or their legal guardians.
Competing interests
The authors declare that they have no competing interests.
Supplementary Information
Additional file 1: Figure S1.
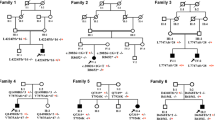
Reported mutations identified in our ARCA patients. a Sequencing chromatograms of 9 reported ARCA-related mutations. The upper sequence in each frame represents the normal sequence, whereas the lower one represents the variant. b Sequencing chromatograms of PTPRQ mutations in case 10. c The pedigree of case 10 with ataxia and hearing loss. Open symbol: unaffected; filled symbol with black or red: affected with ataxia or hearing loss; square: male; circle: female. Genotype data are shown underneath the symbols. Arrowhead: proband of the family.
Additional file 2: Video S1. Myoclonic jerks of face in case 7 with SYNE1 mutations.
Additional file 3: Table S1.
The scale score follow-up of case 15 with ADCK3 mutation after supplementation with COQ10.
Additional file 4: Table S2.
Clinical features of ARSACS patients in China.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Cheng, HL., Shao, YR., Dong, Y. et al. Genetic spectrum and clinical features in a cohort of Chinese patients with autosomal recessive cerebellar ataxias. Transl Neurodegener 10, 40 (2021). https://doi.org/10.1186/s40035-021-00264-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40035-021-00264-z