
Abstract
Background
Postoperative atrial fibrillation (POAF) constitutes a significant complication following coronary artery bypass graft surgery (CABG), potentially linked to epicardial adipose tissue (EAT). This investigation seeks to elucidate the association between POAF and EAT at the genetic level.
Methods
EAT and clinical data from patients undergoing CABG were systematically acquired, adhering to established inclusion and exclusion criteria. Patients were categorized into POAF and Non-POAF groups based on the presence or absence of POAF. High-throughput sequencing data of EAT were subjected to differential expression analysis and gene function assessment. A random selection of long noncoding RNAs (lncRNAs) underwent quantitative real-time polymerase chain reaction (qRT-PCR) for validation of the high-throughput sequencing findings. Coexpression analysis was employed to elucidate the interactions between lncRNAs and messenger RNAs (mRNAs).
Results
RNA sequencing yielded a total of 69,685 transcripts (37,740 coding and 31,945 noncoding sequences), representing 16,920 genes. Within this dataset, 38 mRNAs and 12 lncRNAs exhibited differential expression between the POAF and Non-POAF groups (P < 0.05, fold change > 1.5). The qRT-PCR results for lncRNAs corroborated the sequencing findings (P < 0.01). Functional enrichment analysis of genes and the coexpression network indicated that these differentially expressed RNAs were primarily implicated in processes such as cell growth, differentiation, signal transduction, as well as influencing tissue fibrosis and ion transmembrane transport.
Conclusions
This study unveils a potential association between myocardial fibrosis and ion channels co-regulated by mRNAs and lncRNAs, closely linked to the emergence of new-onset POAF, after accounting for clinical risk factors. This discovery holds promise for further advances in clinical and fundamental research.
Highlights
-
1.
This study describes, for the first time, the RNA expression profile of EAT in patients with non-valvular POAF
-
2.
We identified that the LncRNA expression profile of EAT was closely related to the occurrence of PAOF
-
3.
We found that the co-regulation of mRNAs and LncRNAs on myocardial fibrosis or ion channel may be closely related to the occurrence.
Similar content being viewed by others


Introduction
There has been a noteworthy rise in the number of hospitalized patients globally, suffering from atrial fibrillation, in recent years. This surge has been linked to an increased incidence of ischemic stroke, heart failure, and notably elevated mortality rates. Consequently, it has placed a substantial burden on both patients and the healthcare system [1]. Among the most prevalent complications is the emergence of new-onset atrial fibrillation following coronary artery bypass grafting (CABG), occurring in approximately 10–35% of cases [2,3,4,5,6]. New-onset postoperative atrial fibrillation (New-onset POAF) is associated with a heightened risk of all-cause mortality within 30 days post-surgery, as well as an increased likelihood of complications such as stroke, cardiac insufficiency, renal insufficiency, prolonged hospitalization, extended intensive care unit monitoring, and prolonged ventilator use. Beyond its impact on postoperative recovery, POAF places a significant financial strain on patients, their families, and society at large. Long-term studies reveal a 5 year all-cause mortality rate of 16.3% in CABG patients with atrial fibrillation, a figure significantly higher than that of non-atrial fibrillation patients [13].
The onset of atrial fibrillation is influenced by various factors, including advanced age, atrial fibrosis, electrolyte imbalances, and other variables. Research findings [7] indicate that fluctuations in autonomic nervous system activity may represent a significant mechanism contributing to the heightened susceptibility to paroxysmal atrial fibrillation in diabetic patients. A recurring challenge in the treatment of atrial fibrillation is the reoccurrence of the condition after ablation procedures. Sardu et al. [8] observed an association between elevated levels of sarcoplasmic reticulum calcium ATPase (SERCA) and a reduced risk of post-ablation atrial fibrillation recurrence. Similarly, miRNA [9] and the overexpression of inflammatory pathways [10] have been linked to the recurrence of atrial fibrillation after radiofrequency catheter ablation. These factors can be utilized to evaluate patients' responsiveness to radiofrequency catheter ablation treatment. However, Sardu et al. [10] have affirmed that anti-inflammatory therapy has not yielded success in preventing atrial fibrillation recurrence after ablation treatment.
Adipose tissue serves as a vital endocrine organ within the human body [11]. Epicardial adipose tissue (EAT) is situated on the surface of the atrium and ventricle, enclosed by the pericardium, with a prominent presence on the surface of the right atrium and anterior interventricular sulcus [12]. It resides in close proximity to cardiomyocytes and can secrete various proinflammatory molecules that exert a localized influence on myocardial cells [13]. Inflammation of EAT plays a pivotal role in adverse cardiovascular outcomes following coronary artery bypass grafting [14]. Recent research [15] has revealed that the secretion of adipo-fibrokines, such as activin A, can promote myocardial fibrosis, subsequently contributing to the development of atrial fibrillation. Furthermore, studies have confirmed the intrinsic correlation between the gene expression profile of EAT (both protein-coding and noncoding genes) and the occurrence of atrial fibrillation [16]. Risk factors associated with atrial fibrillation may activate specific genes, thereby participating in the pathological processes that lead to atrial fibrillation [17].
Nonetheless, the mechanisms underlying POAF, particularly the genetic factors linked to epicardial adipose gene expression profiles, remain unclear.
We posit that the RNA expression profile in EAT may bear a close association with the emergence of POAF. The objective of this study was to investigate the pivotal genes potentially implicated in the onset and progression of POAF following CABG through RNA sequencing (RNA-seq) of EAT. In addition, we aimed to predict their functions and involvement in regulatory pathways using bioinformatics analysis. These efforts seek to unveil the plausible mechanisms behind POAF, laying a theoretical foundation for the prevention and treatment of POAF post-CABG.
Materials and methods
Study population and sample
Patients who underwent coronary artery bypass surgery at the Department of Cardiac Surgery, Beijing Chaoyang Hospital, Capital Medical University, during the period from March 2021 to August 2021, were enrolled according to specified inclusion and exclusion criteria. Individuals meeting any of the exclusion criteria were excluded from the study. Inclusion criteria encompassed patients who were (1) 18 years of age or older, (2) subjected to coronary artery bypass surgery, and (3) devoid of a prior history of arrhythmia. Exclusion criteria encompassed patients who underwent (1) emergency surgery, (2) repeated bypass surgery, (3) concurrent valve surgery or other cardiac procedures, and (4) employment of cardiopulmonary bypass.
Our previous study reported a POAF incidence ranging from 11 to 16% within our institution. For the present investigation, we planned to conduct RNA-seq analysis on 10 specimens (5 in the POAF group and 5 in the Non-POAF group); therefore, a total of 50 EAT specimens were procured for this purpose.
The classical off-pump CABG technique was employed in patients with a median sternal incision, whereas patients with endoscope-assisted small incisions received left intercostal small incisions. The proximal end of the free bridge was anastomosed with the ascending aorta, and the "Heartstring non-blocking proximal anastomosis system" was consistently utilized. During the operation, ultrasonic instantaneous blood flow measurement techniques were employed to assess the blood flow in the bridging vessels.
EAT samples were collected from the surface of the right atrium, in close proximity to the superior vena cava, using a conventional scalpel (not an electric knife). This occurred after the pericardium was opened but prior to the initiation of the bypass procedure. Subsequently, these samples were promptly snap-frozen in liquid nitrogen and preserved at − 80 °C until ribonucleic acid extraction. The study protocol obtained approval from the Beijing Chaoyang Hospital Ethics Committee and adhered to the principles outlined in the Declaration of Helsinki.
Definition and detection of new-onset POAF
Continuous electrocardiogram (ECG) monitoring was carried out for 24 h in the ICU post-surgery. Once the patient was transferred to the general ward, a 12-lead ECG was promptly conducted to investigate any associated symptoms. Atrial fibrillation often manifested at various intervals during the ICU stay, with ECG revealing atrial fibrillation episodes lasting more than 30 s. In the general ward, the occurrence of atrial fibrillation was assessed based on related symptoms and the results of the 12-lead ECG. It is important to note that spontaneous conversion from atrial fibrillation to sinus rhythm within 24 h was not observed in this study, necessitating medical cardioversion.
lncRNA and mRNA expression profiling by RNA-seq
Total RNA, inclusive of long noncoding RNAs (lncRNAs), was extracted from freshly frozen EAT employing the TrIzol reagent (Invitrogen, CA, USA). Approximately 1–2 µg of total RNA from each sample underwent quantification using a NanoDrop ND-1000 instrument and qualification through agarose gel electrophoresis. RNA isolation from the total RNA (ribosomal RNA removed) was performed using an NEBNext Poly(A) mRNA Magnetic Isolation Module (New England Biolabs). This RNA was then utilized for RNA-seq library preparation, employing a KAPA Stranded RNA-Seq Library Prep Kit (Illumina). To assess the libraries, their concentration, fragment size distribution (ranging between 400 and 600 bp), and adapter dimer contamination were evaluated using an Agilent 2100 Bioanalyzer. Sequencing was conducted using the Illumina HiSeq 6000 (Illumina, CA, USA), following the manufacturer's protocols, to yield raw sequencing data for quality control purposes.
The NanoDrop ND-1000 instrument was utilized to gauge the concentration (abs 260) and assess protein contamination (ratio abs260/abs230) within the total RNA samples. Subsequent to quality control, fragments underwent trimming through the 5′ and 3′ adapters, with reads of ≤ 20 bp being filtered out using Cutadapt software. Ultimately, the processed, clean reads were mapped to the reference genome using Hisat 2 software to identify expression profiles. Scatter and volcano plots were generated to visualize the differentially expressed genes within the statistical computing and graphics environments of R, Python, or shell.
Differential expression analysis of mRNAs and lncRNAs
The expression levels, quantified as fragments per kilobase of transcript per million mapped reads (FPKM values), were computed for known genes and transcripts using the R package ballgown. These FPKM values were derived from transcript abundances estimated by StringTie. The number of identified genes and transcripts per group was determined based on a mean FPKM in the group of ≥ 0.5. Subsequent differential expression analyses for genes and transcripts were conducted employing the R package ballgown, with criteria for filtering including a fold change (cutoff 1.5), P value (≤ 0.05), and FPKM (≥ 0.5 in one group).
qRT-PCR validation of lncRNAs
RNA obtained through the TrIzol (Invitrogen) method underwent reverse transcription to produce cDNA using SuperScript III Reverse Transcriptase (Invitrogen) with the Gene Amp PCR System 9700 (Applied Biosystems). Real-time quantitative reverse transcriptase–polymerase chain reaction (qRT-PCR) was executed using a QuantStudio 5 Real-time PCR System (Applied Biosystems). The housekeeper gene β-actin, whose expression remained relatively constant across different samples, served as the internal reference to correct for quantitative RNA concentration variations and reverse transcription efficiency errors. This allowed for the determination of relative expression levels using the 2−ΔΔCT method. Detailed primer information can be found in Additional file 1: Table S1.
Gene Ontology and pathway analysis
The GO initiative (http://www.geneontology.org) defines gene attributes across three structured categories: biological process (BP), molecular function (MF), and cellular component (CC). Pathway analysis was conducted on the differentially expressed genes based on the latest Kyoto Encyclopedia of Genes and Genomes (KEGG) database (http://www.genome.jp/kegg). It aimed to identify whether the differentially expressed mRNAs were enriched in specific biological pathways. A Fisher’s exact test was employed to evaluate the statistical significance of such enrichments between the two groups, with a P value < 0.05 indicating statistical significance.
Construction of a coexpression network
The combined analysis of lncRNA and mRNA was conducted to infer the function of lncRNA. The correlation coefficient between lncRNA and mRNA was determined based on the following criteria: Pearson’s correlation coefficient ≥ 0.8 and a P value < 0.05. Cytoscape (version 3.9.1) was employed to construct a coexpression network between lncRNA and mRNA.
Statistical analysis
Statistical analyses were performed using SPSS Statistics 26.0 (IBM). Continuous measurement data were presented as mean ± standard deviation (x ± s). Hypothesis testing was conducted using the two-independent-samples t test. Categorical data were expressed as percentages, and differences between groups for unordered binary and multicategorical variables were assessed using the χ2 test or Fisher’s exact probability method.
Results
Baseline clinical characteristics of the study population
Out of the 50 EAT samples, six were from patients with POAF. One sample did not meet the criteria for RNA quantification and quality assurance, while the remaining five samples were valid. In the Non-POAF group, 44 patients were included after 1:1 propensity score matching. The primary clinical characteristics are summarized in Table 1. Perioperative basic clinical data showed no statistically significant differences in age, prevalence of hypertension, or diabetes between the two groups. There were also no statistically significant differences in postoperative drainage volume and the number of anastomoses. However, the levels of total cholesterol (TC) (2.8 ± 1.0 vs 4.6 ± 0.5, P = 0.005) and triglycerides (TG) (1.0 ± 0.5 vs 2.3 ± 0.8, P = 0.013) were significantly lower in the POAF group compared to the Non-POAF group. The low-density lipoprotein (LDL) levels exhibited a similar trend but did not reach statistical significance (1.6 ± 0.8 vs 2.6 ± 0.8, P = 0.100).
Medication upon admission and medication upon discharge are detailed in Additional file 4: Table S4. Subgroup analyses have been conducted separately for diabetic and non-diabetic patient cohorts, and the outcomes will be presented in Additional file 3: Table S3.
lncRNA and mRNA sequencing results and differential expression
In EAT, RNA-seq yielded 69,685 transcripts, encompassing 37,740 coding sequences and 31,945 noncoding sequences, corresponding to 16,920 genes. This gene set consisted of 13,902 protein-coding genes and 3018 noncoding genes. Table 2 presents the top 30 transcripts, including both protein-coding and non-protein coding sequences. Some of these highly expressed transcripts are known to play pivotal roles in cardiovascular diseases, particularly in patients with type 2 diabetes mellitus. For example, MT-ATP6 is regulated by miR-378a and affects cardiac function by influencing ATP synthase function [18]. LncRNA RMBP has been attributed significant therapeutic value in the context of myocardial dysfunction [19].
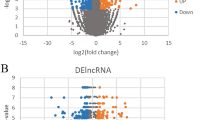
Differential expression analysis, depicted in Fig. 1, revealed 286 genes with differential expression between the POAF and Non-POAF groups. This included 11 significantly upregulated sequences and 39 significantly downregulated sequences (P < 0.05, |log2FC|≥ 0.585). Figure 1A illustrates a predominance of downregulated genes in the POAF group, while Fig. 1B underscores that despite the abundant RNA expression in EAT, the number of differentially expressed genes remained limited. In patients with POAF, gene expression of both long noncoding RNA (lncRNA) and messenger RNA (mRNA) in EAT exhibited significant differences from the Non-POAF group. Specifically, 7 mRNA expression levels were upregulated, and 31 were downregulated, along with 4 upregulated and 8 downregulated lncRNA expression levels (P < 0.05, |log2FC|≥ 0.585) (Fig. 2).

Clustering heatmaps (A) and scatterplots of differentially (B) expressed RNA-seq

Clustering heatmaps and volcano plots of differentially expressed (P < 0.05, fold change > 1.5) mRNAs (A, B) and lncRNAs (C, D)
To validate the sequencing results, six randomly selected lncRNAs, chosen from the co-expression selected group (comprising 12 downregulated and 9 upregulated lncRNAs), underwent testing via qRT-PCR. The results demonstrated a consistent trend with the sequencing findings, with a significance level of P < 0.01 (Fig. 3).

qRT-PCR validation results of randomly selected differentially expressed lncRNAs with P values less than 0.01
GO and KEGG pathway analysis of differentially expressed genes
To elucidate the specific functions and biological processes associated with differentially expressed genes, we conducted GO and KEGG pathway analyses. The GO enrichment analysis (Fig. 4) encompassed BP, CC, and MF domains. Among the downregulated genes, the enrichment scores primarily highlighted their involvement in biological processes, with the top three GO entries pertaining to the regulation of cellular processes (GO:0050794), activation of MAPK activity (GO:0000187), and the MAPK cascade (GO:0000165) in BP. In CC, they were associated with nucleoplasm (GO:0005654), cytoplasm (GO:0005737), and membrane-bounded organelles (GO:0043227). Molecular Function analyses revealed roles in protein binding activity (GO:0005515), S100 protein binding activity (GO:0044548), and growth factor activity (GO:0008083). Conversely, upregulated genes exhibited a smaller number of entries distributed nearly evenly, with the top entries across the three domains being the regulation of signal transduction (GO:0009966) in BP, the endomembrane system (GO:0012505) in CC, and RNA polymerase II cis-regulatory region sequence-specific DNA binding activity (GO:0000978) in MF.

Top 10 significantly downregulated (A–C) and upregulated (D–F) items in descending order of enrichment score are presented as DotPlot. BP, biological process; CC, cellular component; MF, molecular function
Regarding KEGG pathway analysis, eight signaling pathways were identified among the significantly downregulated differentially expressed genes (Fig. 5). These pathways included the Ras signaling pathway (hsa04014), phosphatidylinositol-3-kinase (PI3K)–Akt signaling pathway (hsa04151), Apelin signaling pathway (hsa04371), phospholipase D signaling pathway (hsa04072), cellular senescence pathway (hsa04218), repressor/activator protein 1 signaling pathway (hsa04015), actin cytoskeleton regulatory pathway (hsa04810), and MAPK signaling pathway (hsa04010). However, no related signaling pathways were identified due to the limited number of downregulated differentially expressed genes.

KEGG pathway analysis of significantly downregulated differentially expressed genes
Constructing coexpression network
Differential expression analysis revealed that 283 lncRNA transcripts exhibited differential expression (P < 0.05, |log2FC| ≥ 0.585) between the POAF and Non-POAF groups, comprising 121 significantly upregulated and 162 significantly downregulated sequences. These transcripts were sorted in descending order based on |log2FC|. The top five transcripts were selected from both the upregulated and downregulated lncRNA collections. Subsequently, we cross-referenced the results of gene functional enrichment (GO and KEGG) to identify genes potentially associated with myocardial fibrosis and ion transport and confirmed their chromosomal locations. Among the remaining lncRNA transcript sequences, those with similar chromosomal locations to the aforementioned genes were selected.
By employing these two selection methods, we identified a total of 12 downregulated and 9 upregulated lncRNA transcripts (detailed in Additional file 2: Table S2) for constructing a coexpression network. This network utilized significantly differentially expressed mRNA transcripts (P < 0.05, fold change > 1.5) to investigate the interactions between coding RNA and lncRNA (Fig. 6). This analysis involved 228 nodes (mRNA transcripts) connected to lncRNA transcripts. When sorted by correlation coefficient in descending order, the top five mRNAs were B3GALNT1-202, RPL18-201, CTSB-228, CXorf40B-201, and PPP6R3-203, with corresponding lncRNAs being ENST00000570269, ENST00000490923, ENST00000541196, and ENST00000493138, respectively.

Analysis of lncRNA and mRNA coexpression network. The circle represents the coding gene, and the triangle represents lncRNAs. Red represents upregulation, and green represents downregulation. The solid line represents a positive correlation, and the dashed line represents a negative correlation
Discussion
The occurrence of atrial fibrillation following bypass surgery is influenced by factors such as cardiac-related surgery, cardiopulmonary bypass, and perioperative shock, with advanced age being a well-established risk factor. This study, by excluding extracorporeal circulation, meticulously examined the correlation between genetic molecules and POAF in patients undergoing off-pump CABG. The consistency in age, prevalence of diabetes, and hypertension between the Non-AF and POAF groups served to minimize confounding factors, ensuring the study's reliability.
Epicardial adipose tissue infiltration into myocardial tissue plays a pivotal role in promoting myocardial fibrosis through the secretion of adipo-fibrokines, significantly impacting cardiac function [15]. In this study, EAT was obtained from the epicardial surface adjacent to the right atrium and atrioventricular node. The unique anatomical structure of epicardial fat enables it to exert paracrine-like effects on adjacent myocardial tissue, influencing both its structure and function [20]. However, due to the limitations in sample size, the following discussion should be considered preliminary, with further research awaited.
Indeed, diabetic patients are at a higher risk of experiencing poorer outcomes after CABG surgery. We conducted separate subgroup analyses for diabetic and non-diabetic patient cohorts, but no significant differences were observed between these groups. Interestingly, our study [21] revealed a higher prevalence of SGLT2 inhibitor usage among diabetic patients in the Non-AF group. It is noteworthy that anti-diabetic medications, including SGLT2 inhibitors, known for their application in non-diabetic patients to enhance cardiovascular outcomes, may mitigate inflammatory distress and improve post-CABG results.
mRNA function
Numerous significantly upregulated or downregulated coding genes in EAT are implicated in cardiovascular diseases, specifically myocardial remodeling and lipid metabolism, which are closely associated with atrial fibrillation occurrence.
For instance, PMEPA1 (ENSG00000124225.15_3, log2FC = – 1.36, P = 0.036) displayed a positive correlation with inflammation from tumor-infiltrating immune cells [22]. The delivery of the PMEPA1 gene can inhibit activin A activity, a molecule highly expressed in EAT that promotes myocardial/atrial fibrosis [15] in vitro. It can deactivate the activin/myostatin–SMAD3 pathway, thereby regulating muscle mass [23].
In our study, we identified that ENSG00000113578.17_2 [fibroblast growth factor 1 (FGF1)] and ENSG00000241186.9_3 [teratocarcinoma-derived growth factor-1 (TDGF1)] were enriched in multiple pathways associated with cell fibrosis, epithelial and endothelial cell migration (GO:0044344, GO:0071774, GO:0010595, GO:0010634, GO:000187, and GO:000165).
The KEGG results indicated that FGF1 played a crucial role in regulating the PI3K–Akt pathway (hsa04151), potentially inducing FGF1's neuroprotective effects against glutamate toxicity by inhibiting GSK-3β [24]. The interaction between GNG2 and MRAS inhibited Akt (a core component of the PI3K–Akt pathway) activity in an MRAS-dependent manner [25].
The PI3K–Akt pathway encompasses lipid kinases crucial for growth and metabolism, exerting a significant regulatory role in cardiac protection during stressful conditions [26]. This includes hepatic fibrosis [27], pulmonary fibrosis [28], and fibrosis within the cardiovascular system [29]. The PI3K–Akt signaling pathway has been implicated in the regulation of cardiac fibrosis occurrence and pathological progression [30] by governing cell survival, apoptosis, growth, and cardiac contractility, ultimately leading to heart failure [31]. Inhibition of cardiac PI3K–Akt signaling has been linked to an increased risk of atrial fibrillation [32]. Mice with reduced PI3K activation exhibited compromised cardiac function, more pronounced atrial enlargement, and fibrosis, thereby significantly increasing susceptibility to atrial fibrillation [33]. PI3K plays a pivotal cardioprotective role [34], and its increased activity can extend mouse lifespan by 15% to 20% [26].
The POAF group displayed lower total triglyceride and total cholesterol levels compared to the Non-POAF group. This suggests that while lower blood lipid levels are protective against coronary heart disease, they may be associated with an increased risk of POAF—a seemingly paradoxical phenomenon. A prior retrospective clinical study conducted in our center and several clinical investigations [35,36,37] have reported a negative correlation between preoperative LDL levels and POAF occurrence. However, the underlying mechanism behind this association remains unclear.
FGF1 is significantly downregulated and closely linked to lipid metabolism [38]. An upsurge in FGF1 levels corresponds to increases in TC, TG, and LDL levels [39], and there exists a positive correlation between FGF1 mRNA expression and cholesterol levels [40]. In our study, the lower blood lipid levels observed in the POAF group may be closely related to the downregulated FGF1 mRNA.
lncRNA transcripts
LncRNAs, which exceed 200 nucleotides in length [41], typically function as transcriptional or posttranscriptional regulators involved in chromatin remodeling and splicing. They are associated with various pathological processes [41].
We established a coding–noncoding gene coexpression (CNC) network to investigate the potential role of differentially expressed lncRNA transcripts in the POAF group. The top 10 mRNA transcripts with the highest upregulated and downregulated differential expression ratios displayed significant correlations with lncRNAs within the CNC network, and these mRNA transcripts were under the regulatory influence of lncRNA transcripts.
Several differentially expressed mRNA transcripts were concurrently regulated by multiple lncRNA transcripts. For instance, COL6A3-209 exhibited downregulation in tandem with the decreased expression of lncRNAs ENST00000527306 (HOOK3-204) and ENST00000515719 (CLPTM1L-215). COL6A3 is known to promote the formation of collagen fibers, consequently inducing myocardial fibrosis and expediting atrial remodeling [42]. Furthermore, mRNA transcripts such as IARS-206 [43], TDGF1 [44], and CNN3-202 [45], regulated by two or more lncRNAs, demonstrated close associations with myocardial or hepatic fibrosis.
SRCAP, which experienced downregulation alongside the decreased expression of lncRNAs ENST00000527306 (HOOK3-204) and ENST00000521308 (CCDC69-205), has the potential to influence the levels of sarcoplasmic reticulum Ca2+ regulatory proteins and cardiac Ca2+ homeostasis. The loss of its core subunit Znhit1 has been linked to postnatal arrhythmias and rapid heart failure in mice [46]. Notably, ENST00000521308 (CCDC69-205) exhibited the most significant downregulation (log2FC = – 2.60, P = 0.002) among all significantly differentially expressed lncRNA transcripts.
Limitations
The limited sample size in this study may affect the reliability of the results. Larger studies are necessary to validate these findings. Although we have explored the potential relationship between lncRNAs and coding RNAs, further experimental and clinical verifications are essential to establish a causal link and elucidate specific action pathways.
Conclusions
This study represents the first examination of the RNA expression profile of EAT in patients with nonvalvular POAF. Gene function and coexpression network analyses have unveiled the likelihood of co-regulation between mRNAs and lncRNAs in myocardial fibrosis or ion channel regulation, which may be closely associated with the onset of POAF. This genetic perspective has identified potential targets for POAF, and the discovery of key lncRNAs may pave the way for further clinical and fundamental research.
Availability of data and materials
The datasets generated and analyzed in this study can be accessed in the Gene Expression Omnibus (GEO) under the accession number [GSE222739, https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE222739]."
Abbreviations
- POAF:
-
Postoperative atrial fibrillation
- CABG:
-
Coronary artery bypass grafting
- BMI:
-
Body mass index
- BNP:
-
Brain Natriuretic Peptide
- GLU:
-
Glucose
- HbA1C:
-
Glycosylated Hemoglobin
- TC:
-
Total Cholesterol
- HDL:
-
High Density Lipoprotein
- LDL:
-
Low Density Lipoprotein
- TG:
-
Triglyceride
- Lpa:
-
Apolipoprotein A
- BNU:
-
Blood Urea Nitrogen
- Scr:
-
Serum creatinine
- LVEDD:
-
Left ventricular end-diastolic dimension
- LVEF:
-
Left ventricular ejection fraction
- LAD:
-
Left atrial diameter
- RAD:
-
Right atrial diameter
- ICU:
-
Intensive care unit
References
Shen J, Lall S, Zheng V, Buckley P, Damiano RJ, Schuessler RB. The persistent problem of new-onset postoperative atrial fibrillation: a single-institution experience over two decades. J Thorac Cardiovasc Surg. 2011;141:559–70.
Lee SH, Kang DR, Uhm JS, Shim J, Sung JH, Kim JY, et al. New-onset atrial fibrillation predicts long-term newly developed atrial fibrillation after coronary artery bypass graft. Am Heart J. 2014;167:593-600.e1.
Creswell LL, Schuessler RB, Rosenbloom M, Cox JL. Hazards of postoperative atrial arrhythmias. Ann Thorac Surg. 1993;56:539–49.
Filardo G, Hamilton C, Hebeler RF, Hamman B, Grayburn P. New-onset postoperative atrial fibrillation after isolated coronary artery bypass graft surgery and long-term survival. Circ Cardiovasc Qual Outcomes. 2009;2:164–9.
El-Chami MF, Kilgo P, Thourani V, Lattouf OM, Delurgio DB, Guyton RA, et al. New-onset atrial fibrillation predicts long-term mortality after coronary artery bypass graft. J Am Coll Cardiol. 2010;55:1370–6.
Mathew JP, Fontes ML, Tudor IC, Ramsay J, Duke P, David Mazer C, et al. A Multicenter risk index for atrial fibrillation after cardiac surgery. https://jamanetwork.com/
Rizzo MR, Sasso FC, Marfella R, Siniscalchi M, Paolisso P, Carbonara O, et al. Autonomic dysfunction is associated with brief episodes of atrial fibrillation in type 2 diabetes. J Diabetes Complications. 2015;29:88–92.
Sardu C, Santulli G, Guerra G, Trotta MC, Santamaria M, Sacra C, et al. Modulation of SERCA in patients with persistent Atrial Fibrillation treated by Epicardial Thoracoscopic Ablation: the CAMAF study. J Clin Med. 2020;9:544.
Sardu C, Santamaria M, Paolisso G, Marfella R. microRNA expression changes after atrial fibrillation catheter ablation. Pharmacogenomics. 2015;16:1863–77.
Sardu C, Santulli G, Santamaria M, Barbieri M, Sacra C, Paolisso P, et al. Effects of alpha lipoic acid on multiple cytokines and biomarkers and recurrence of Atrial Fibrillation within 1 year of catheter ablation. Am J Cardiol. 2017;119:1382–6.
Kita S, Maeda N, Shimomura I. Interorgan communication by exosomes, adipose tissue, and adiponectin in metabolic syndrome. J Clin Investig. 2019;129:4041–9.
Gaborit B, Sengenes C, Ancel P, Jacquier A, Dutour A. Role of epicardial adipose tissue in health and disease: a matter of fat¿. Compr Physiol. 2017;7:1051–82.
Wong CX, Ganesan AN, Selvanayagam JB. Epicardial fat and atrial fibrillation: current evidence, potential mechanisms, clinical implications, and future directions. Eur Heart J. 2017;38:1294–302.
Sardu C, D’Onofrio N, Torella M, Portoghese M, Loreni F, Mureddu S, et al. Pericoronary fat inflammation and Major adverse cardiac events (MACE) in prediabetic patients with acute myocardial infarction: effects of metformin. Cardiovasc Diabetol. 2019;18:126.
Venteclef N, Guglielmi V, Balse E, Gaborit B, Cotillard A, Atassi F, et al. Human epicardial adipose tissue induces fibrosis of the atrial myocardium through the secretion of adipo-fibrokines. Eur Heart J. 2015;36:795–805.
Zhao L, Ma Z, Guo Z, Zheng M, Li K, Yang X. Analysis of long non-coding RNA and mRNA profiles in epicardial adipose tissue of patients with atrial fibrillation. Biomed Pharmacother. 2020;121:109634.
Chilukoti RK, Giese A, Malenke W, Homuth G, Bukowska A, Goette A, et al. Atrial fibrillation and rapid acute pacing regulate adipocyte/adipositas-related gene expression in the atria. Int J Cardiol. 2015;187:604–13.
Durr AJ, Hathaway QA, Kunovac A, Taylor AD, Pinti MV, Rizwan S, et al. Manipulation of the miR-378a/mt-ATP6 regulatory axis rescues ATP synthase in the diabetic heart and offers a novel role for lncRNA Kcnq1ot1. Am J Physiol Cell Physiol. 2022;322:C482–95.
Han Y, Cai Y, Lai X, Wang Z, Wei S, Tan K, et al. lncRNA RMRP prevents mitochondrial dysfunction and cardiomyocyte apoptosis via the miR-1-5p/hsp70 axis in LPS-induced sepsis mice. Inflammation. 2020;43:605–18.
Shi X, Shao X, Liu B, Lv M, Pandey P, Guo C, et al. Genome-wide screening of functional long noncoding RNAs in the epicardial adipose tissues of atrial fibrillation. Biochim Biophys Acta Mol Basis Dis. 2020;1866: 165757.
Sardu C, Massetti M, Testa N, Di ML, Castellano G, Turriziani F, et al. Effects of sodium-glucose transporter 2 inhibitors (SGLT2-I) in patients with ischemic heart disease (IHD) treated by coronary artery bypass grafting via MiECC: inflammatory burden, and clinical outcomes at 5 years of follow-up. Front Pharmacol. 2021;12: 777083.
Qiu D, Hu J, Hu J, Yu A, Othmane B, He T, et al. PMEPA1 is a prognostic biomarker that correlates with cell malignancy and the tumor microenvironment in bladder cancer. Front Immunol. 2021;12: 705086.
Hagg A, Kharoud S, Goodchild G, Goodman CA, Chen JL, Thomson RE, et al. TMEPAI/PMEPA1 is a positive regulator of skeletal muscle mass. Front Physiol. 2020;11: 560225.
Hashimoto M, Sagara Y, Langford D, Everall IP, Mallory M, Everson A, et al. Fibroblast growth factor 1 regulates signaling via the glycogen synthase kinase-3beta pathway. Implicat Neuroprot J Biol Chem. 2002;277:32985–91.
Zhao A, Li D, Mao X, Yang M, Deng W, Hu W, et al. GNG2 acts as a tumor suppressor in breast cancer through stimulating MRAS signaling. Cell Death Dis. 2022;13:260.
McMullen JR, Amirahmadi F, Woodcock EA, Schinke-Braun M, Bouwman RD, Hewitt KA, et al. Protective effects of exercise and phosphoinositide 3-kinase(p110alpha) signaling in dilated and hypertrophic cardiomyopathy. Proc Natl Acad Sci USA. 2007;104:612–7.
Wu L, Zhang Q, Mo W, Feng J, Li S, Li J, et al. Quercetin prevents hepatic fibrosis by inhibiting hepatic stellate cell activation and reducing autophagy via the TGF-β1/Smads and PI3K/Akt pathways. Sci Rep. 2017;7:9289.
Hu X, Xu Q, Wan H, Hu Y, Xing S, Yang H, et al. PI3K-Akt-mTOR/PFKFB3 pathway mediated lung fibroblast aerobic glycolysis and collagen synthesis in lipopolysaccharide-induced pulmonary fibrosis. Lab Invest. 2020;100:801–11.
Qin W, Cao L, Massey IY. Role of PI3K/Akt signaling pathway in cardiac fibrosis. Mol Cell Biochem. 2021;476:4045–59.
Xue Y, Zhang M, Liu M, Liu Y, Li L, Han X, et al. 8-Gingerol ameliorates myocardial fibrosis by attenuating reactive oxygen species, apoptosis, and autophagy via the PI3K/Akt/mTOR signaling pathway. Front Pharmacol. 2021;12: 711701.
Yang J, Savvatis K, Kang JS, Fan P, Zhong H, Schwartz K, et al. Targeting LOXL2 for cardiac interstitial fibrosis and heart failure treatment. Nat Commun. 2016;7:13710.
McMullen JR, Boey EJH, Ooi JYY, Seymour JF, Keating MJ, Tam CS. Ibrutinib increases the risk of atrial fibrillation, potentially through inhibition of cardiac PI3K-Akt signaling. Blood. 2014;124:3829–30.
Pretorius L, Du XJ, Woodcock EA, Kiriazis H, Lin RCY, Marasco S, et al. Reduced phosphoinositide 3-kinase (p110alpha) activation increases the susceptibility to atrial fibrillation. Am J Pathol. 2009;175:998–1009.
Weeks KL, Gao X, Du XJ, Boey EJH, Matsumoto A, Bernardo BC, et al. Phosphoinositide 3-kinase p110α is a master regulator of exercise-induced cardioprotection and PI3K gene therapy rescues cardiac dysfunction. Circ Heart Fail. 2012;5:523–34.
Kara K, Gronewold J, Neumann T, Mahabadi AA, Weimar C, Lehmann N, et al. B-type natriuretic peptide predicts stroke of presumable cardioembolic origin in addition to coronary artery calcification. Eur J Neurol. 2014;21:914–21.
Li ZZ, Du X, Guo XY, Tang RB, Jiang C, Liu N, et al. Association between blood lipid profiles and atrial fibrillation: a case-control study. Med Sci Monit. 2018;24:3903–8.
Chua SK, Shyu KG, Lu MJ, Lien LM, Lin CH, Chao HH, et al. Clinical utility of CHADS2 and CHA2DS 2-VASc scoring systems for predicting postoperative atrial fibrillation after cardiac surgery. J Thorac Cardiovasc Surg. 2013;146:919-926.e1.
Stefanon B, Colitti M. Original Research: Hydroxytyrosol, an ingredient of olive oil, reduces triglyceride accumulation and promotes lipolysis in human primary visceral adipocytes during differentiation. Exp Biol Med. 2016;241:1796–802.
Zhang L, Cheng H, Yue Y, Li S, Zhang D, He R. TUG1 knockdown ameliorates atherosclerosis via up-regulating the expression of miR-133a target gene FGF1. Cardiovasc Pathol. 2018;33:6–15.
Georgi N, Cillero-Pastor B, Eijkel GB, Periyasamy PC, Kiss A, van Blitterswijk C, et al. Differentiation of mesenchymal stem cells under hypoxia and normoxia: lipid profiles revealed by time-of-flight secondary ion mass spectrometry and multivariate analysis. Anal Chem. 2015;87:3981–8.
Hobuß L, Bär C, Thum T. Long non-coding RNAs: at the heart of cardiac dysfunction? Front Physiol. 2019;10:30.
Mesquita RFDS, Paul MA, Valmaseda A, Francois A, Jabr R, Anjum S, et al. Protein kinase Cε-calcineurin cosignaling downstream of toll-like receptor 4 downregulates fibrosis and induces wound healing gene expression in cardiac myofibroblasts. Mol Cell Biol. 2014;34:574–94.
Kopajtich R, Murayama K, Janecke AR, Haack TB, Breuer M, Knisely AS, et al. Biallelic IARS mutations cause growth retardation with prenatal onset, intellectual disability, muscular hypotonia, and infantile hepatopathy. Am J Hum Genet. 2016;99:414–22.
Karkampouna S, van der Helm D, Scarpa M, van Hoek B, Verspaget HW, Goumans MJ, et al. Oncofetal protein CRIPTO is involved in wound healing and fibrogenesis in the regenerating liver and is associated with the initial stages of cardiac fibrosis. Cells. 2021;10:3325.
Maddala R, Mongan M, Xia Y, Rao PV. Calponin-3 deficiency augments contractile activity, plasticity, fibrogenic response and Yap/Taz transcriptional activation in lens epithelial cells and explants. Sci Rep. 2020;10:1295.
Shi Y, Fan W, Xu M, Lin X, Zhao W, Yang Z. Critical role of Znhit1 for postnatal heart function and vacuolar cardiomyopathy. JCI Insight. 2022;7: e148752.
Acknowledgements
Not applicable.
Funding
This study received no external funding.
Author information
Authors and Affiliations
Contributions
Yuanshu Peng was responsible for specimen collection, paper writing, specific experimental procedures, and data analysis. Lei Zhao and Pixiong Su were responsible for project design, quality control, and initial draft review.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
All specimens were collected following informed consent, which included disclosure for future research purposes. This investigation adhered to the principles of the Declaration of Helsinki and received approval from the Ethics Committee of Beijing Chaoyang Hospital.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Table S1.
Primer lists were used for Quantitative real-time polymerase chain reaction.
Additional file 2: Table S2.
Results of differential expression analysis of LncRNA selected for co-expression analysis.
Additional file 3: Table S3.
Demographic and clinical characteristics of patient in subgroup.
Additional file 4: Table S4.
Medication on admission and medication on discharge for the patient cohort.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Peng, Y., Su, P. & Zhao, L. Long noncoding RNA and messenger RNA profiling in epicardial adipose tissue of patients with new-onset postoperative atrial fibrillation after coronary artery bypass grafting. Eur J Med Res 29, 134 (2024). https://doi.org/10.1186/s40001-024-01721-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40001-024-01721-x