
Abstract
Purpose
To build models combining circulating microRNAs (miRNAs) able to identify women with breast cancer as well as different types of breast cancer, when comparing with controls without breast cancer.
Method
miRNAs analysis was performed in two phases: screening phase, with a total n = 40 (10 controls and 30 BC cases) analyzed by Next Generation Sequencing, and validation phase, which included 131 controls and 269 cases. For this second phase, the miRNAs were selected combining the screening phase results and a revision of the literature. They were quantified using RT-PCR. Models were built using logistic regression with LASSO penalization.
Results
The model for all cases included seven miRNAs (miR-423-3p, miR-139-5p, miR-324-5p, miR-1299, miR-101-3p, miR-186-5p and miR-29a-3p); which had an area under the ROC curve of 0.73. The model for cases diagnosed via screening only took in one miRNA (miR-101-3p); the area under the ROC curve was 0.63. The model for disease-free cases in the follow-up had five miRNAs (miR-101-3p, miR-186-5p, miR-423-3p, miR-142-3p and miR-1299) and the area under the ROC curve was 0.73. Finally, the model for cases with active disease in the follow-up contained six miRNAs (miR-101-3p, miR-423-3p, miR-139-5p, miR-1307-3p, miR-331-3p and miR-21-3p) and its area under the ROC curve was 0.82.
Conclusion
We present four models involving eleven miRNAs to differentiate healthy controls from different types of BC cases. Our models scarcely overlap with those previously reported.
Similar content being viewed by others
Background
Breast cancer is the most common type of cancer in women and a major cause of cancer death in developed countries [1]. Epidemiological research has identified several risk factors (age at menarche, parity, age at first part, age at menopause), most of them associated with estrogen production [2, 3]. Several risk factors are related to lifestyle (smoking, alcohol consumption, being overweight or obesity), although they appear to be less important than that of risk factors associated with reproductive life and estrogen production [4]. Known risk factors may explain approximately 40% of breast cancer risk.
Screening using mammograms for early diagnosis, is strongly subject to debate as observational studies suggest that its influence on breast cancer mortality is low [5]. However, identifying women at high risk of breast cancer who could benefit from different early diagnosis protocols and personalized screening is crucial. In this way, with the advent of Next Generation Sequencing (NGS) techniques, up to 313 low-penetrance genetic variants have been identified as related to breast cancer [6] and polygenic tests have been commercialized to identify women at high risk of breast cancer, although their clinical relevance is uncertain. On the other hand, the current recommendation of the St. Gallen consensus [7] is to use gene expression signatures to decide the adjuvant treatment of cancers in early stages, except in those with low clinical risk.
MicroRNAs (miRNAs) are the main class of small non-coding RNAs. Their main function is to regulate the gene expression at messenger RNA (mRNA) level [8]. In fact, it has been estimated that miRNAs regulate the expression of 30% of protein-coding genes functioning as targets of epigenetic changes or as regulators of epigenetic modifiers [8, 9]. A single miRNA can interact with quite a few mRNAs, which can have an impact on the expression of many genes at the same time [9]. More than 60% of human mRNAs contain one miRNA binding site [10]. The biological activity of individual miRNAs has been extensively studied, and the importance of their complex regulation function in many biological process has been demonstrated [8, 9]. They are involved in such vital processes for example cell proliferation, differentiation, invasion, migration, or apoptosis [9].
Any alteration in miRNAs activity (alteration in expression or in the interaction with another miRNA, for example) could be related to a variety of human diseases, including cancer [8, 9, 11]. Besides, comparing normal and tumoral tissue, miRNAs are often dysregulated in the last one [9]. In addition, it has been seen that dysregulated miRNAs could act as oncogenes (oncomiRs) or tumor suppressors [8]. However, nowadays the mechanism for the dysregulation of miRNAs in cancer is not clear but, it is possible that multiple mechanisms are at play [11]. Nonetheless, miRNAs are a good tool to diagnosis and predict prognosis in cancer patients analyzing relative miRNA expression profiles between normal and tumoral samples [9]. Although they occur in tissues, several studies have shown that tumor-specific miRNAs can be detected in the bloodstream, so in recent years interest in circulating miRNAs as non-invasive markers of disease and prognosis has been growing [12]. Specially circulating miRNAs have become potential diagnostic biomarkers in cancer given that they can easily be detected and are very robust against degradation [9]. Currently 38,589 entries from 271 organisms (1917 entries from humans) have been registered in the miRbase miRNA database [13].
Many studies carried out over the last decade have demonstrated that the dysregulation of miRNAs is present in different types of cancer, including breast cancer [10]. Breast cancer is a heterogeneous disease that involves the alteration of multiple oncogenic biological pathways and/or genetic alterations [10]. These alterations can be made by miRNAs, so researchers have performed miRNAs analysis to identify their role. For example, 64 miRNAs were identified as candidate tumor suppressor in BC cells [10]
Despite all this, the number of studies carried out on human samples is not high and much less in large cohorts. However, the identification of miRNAs as specific biomarkers would enhance early diagnosis, and personalized treatment, helping to improve breast cancer survival [10]. For this reason, the main objective in this analysis is to identify miRNAs signatures able to differentiate between controls and breast cancer cases and between controls and different types of breast cancer, using blood samples collected at recruitment in a case–control study.
Methods
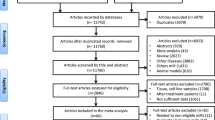
MCC-Spain is a case–control study that recruited 1738 cases of incident breast cancer in women between 2008 and 2013 as well as 1910 controls without breast cancer in 10 Spanish provinces. All cancers had been diagnosed with pathological analysis. Later, cases were follow-up until 2018 to ascertain their vital status and whether they were disease-free or not. The recruitment phase [14] and the follow-up [15, 16] have been described elsewhere. All participants signed the informed consent. The protocol of MCC-Spain was approved by the ethics committees of the participating institutions. Information about ethics and the availability of data are offered at http://www.mccspain.org. In addition, the database was registered in the Spanish Agency for Data Protection (no. 2102672171).
For the purpose of this article, breast cancer cases were classified in three categories: (A) cases diagnosed by screening (i.e., mammogram performed in asymptomatic women), as recorded at recruitment. (B) Cases diagnosed in symptomatic women who remained disease-free after the follow-up. (C) Cases diagnosed in symptomatic women who did not remain disease-free after the follow-up, but without metastases.
Biological samples
Blood samples were obtained at recruitment from both cases and controls. Blood was centrifuged at 3000 g for 20 min at 10 °C followed by further centrifugation of the supernatant at 15000 g for 10 min at 10 C to remove cell debris. Serum was stored at – 80 °C until use.
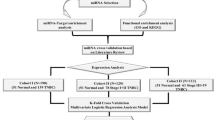
miRNAs analysis was performed in two phases: the first phase is the screening phase and it consisted in the library preparation and Next Generation Sequencing for a small number of patients, the second phase is the validation phase and it consisted in a quantitative real-time PCR (qRT-PCR) for a larger number of patients. All experiments were conducted at QIAGEN Genomic Services.
Screening phase
Ten control women and ten women belonging to each type of case were randomly selected for the screening phase (total n = 40, 10 controls, 30 cases—all of them coming from the Cantabria node and considering the three categories aforementioned) (Table 1). RNA was isolated from serum samples using the miRNeasy Serum/Plasma kit (QIAGEN) by QIAGEN Genomic Services according to manufacturer’s instructions. The library preparation was done using QIAseq miRNA Library Kit (QIAGEN), followed by quality control assessment using either Bioanalyzer2100 (Agilent) or TapeStation4200 (Agilent). A total of 200µl total RNA were converted into miRNA NGS libraries. Adapters containing Unique Molecular Index (UMIs) were ligated to the RNA, to eliminate library amplification bias. The RNA was converted to cDNA and amplified using PCR. Then, the samples were purified. The library pool was quantified using qPCR and sequenced on a NextSeq500 (Illumina). After, FASTQ files for each sample were generated. Cutadapt was used to correct PCR bias with UMI information, Bowtie2 was used for mapping the reads to Homo sapiens miRNA entries from miRbase (v22.1) and EdgeR statistical software package (Bioconductor) was used to do the differential expression analysis. Reads for each miRNA were normalized with the trimmed mean of M-values (TMM) method [17] and converted to a log2 scale to obtain delta Cq values (dCq). For this phase, the annotation of the obtained sequences was performed using the reference genome CRCh37 from the organism Homo sapiens and the annotation reference miRbase_v22.1.
All the samples used in this phase have been subjected to quality controls such as: UMI collapsing (Reads need to have a unique sequence/UMI combination), high quality score and read length of > 15, be mappable to the genome CRCh37 and pass background filtering based on read numbers (removing low copy reads). If the samples do not meet these criteria, they are removed from the dataset.
Validation phase
400 participants were randomly selected for this phase. The criterion employed was the same that in screening phase but considering that the date of blood sample collection was prior to the start date of treatment, and coming from either of the 10 Spanish provinces in MCC-Spain study. This phase included 131 controls, 102 screening-diagnosed cases, 102 disease-free cases and 65 non-disease-free cases (Table 1). The latter figure in the last group was lower than in the others because of the small total number of non-disease-free cases in the whole cohort. Fifty miRNAs were analyzed in the validation phase; they were chosen out of the results in the screening phase or for their presence in signatures already published in 2020 or 2021 [18,19,20,21,22,23]. Additional file 3: Table S1 displays the selected miRNAs and the rationale for their selection. In this phase, the serum was thawed on ice and centrifuged at 3000×g for 5 min in a 4 °C microcentrifuge. An aliquot of 200 µl per sample was transferred to a FluidX tube and 60 µl of Buffer RPL containing 1 µg carrier-RNA per 60 µl Buffer RPL and RNA spike-in template mixture was added to the sample and mixed for 1 min and incubated for 7 min at room temperature, followed by addition of 20 µl Buffer RPP. Total RNA was extracted from the samples using the miRNeasy Serum/Plasma Advanced kit. The purified total RNA was eluted in a final volume of 50 µl. The experiments were conducted by QIAGEN Genomic Services one more time. Later, 2 µl RNA was reverse transcribed in 10 µl reactions using the miRCURY LNA RT Kit (QIAGEN). cDNA was diluted 50 × and assayed in 10 µl PCR reactions. Each miRNA was assayed once by qPCR on the miRCURY LNA miRNA Custom PCR Panel using miRCURY LNA SYBR Green master mix (QIAGEN). Probes without RNA template from the RT step were included as negative controls and profiled like the samples. The amplification was performed in a LightCycler® 480 Real-Time PCR System (Roche) in 384 well plates. The amplification curves were analyzed using the Roche LC software, both for determination of Cq (by the 2nd derivative method) and for melting curve analysis. All data was normalized to the average of custom defined assays, namely let-7d-5p and let-7i-5p, detected in all samples. As in the screening phase, reads for each miRNA were normalized with the TMM method [17] and converted to a log2 scale.
Once again, the samples used in this phase has been subjected to quality control. Data from individual reactions have been removed from the data set based on the following criteria: (i) More than one melting temperature of the amplified product; (ii) Melting temperature deviating from database values; (iii) Low amplification efficiency.
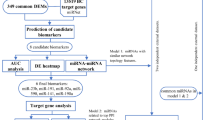
Statistical analysis
The screening phase was analyzed by comparing dCq in controls with each type of case using the Student-t test, without any adjustment. Its results are displayed as log fold change (log FC), p-value and false discovery rate (FDR) using the Benjamini–Hochberg method (Benjamini and Hochberg 1995). LogFC positive indicate the miRNA is upregulated in cases and logFC negative that it is downregulated; the higher the absolute value of logFC, the more difference between cases and controls.
In the validation phase, we built logistic regression models with LASSO penalization [24]. LASSO penalization allows the inclusion of many regressors in the analysis while producing final parsimonious models with few regressors (miRNAs readings, in our study), as LASSO shrinks the coefficient of the less relevant to 0. To do this, we began with a model with all 50 miRNAs included in the validation phase. In the LASSO procedure, the regularization parameter λ was obtained via cross-validation. Then, the final models after LASSO were validated with tenfold cross-validation. Additionally to dCq, fold change and p-value, results of the validation phase are displayed as odds ratios adjusted for the remaining miRNAs in the model. The discrimination ability of each model was measured with the cross-validated mean area under the ROC curve, which is reported with its bootstrap bias corrected 95% confidence interval. In the logistic regression models, the interpretation of odds ratios would be as follows: miRNAs upregulated in cases will have odds ratios higher than 1, while miRNAs downregulated in cases will have them lower than 1. As the unit of analysis is dCq, an odds ratio of, say, 1.5 would mean that comparing two women whose dCq differ in a unity, the woman with higher dCq would have 1.5 higher odds of being a case than the woman with lower dCq. As sensitivity analysis, we reran the obtained logistic regression models with different subsets of cases: (a) breast cancers with oestrogen receptors (n = 212), (b) breast cancers with progesterone receptors (n = 187), (c) breast cancers ErbB2 positive (n = 50) and (d) triple negative breast cancers (n = 33). Of note, there is some overlapping degree between groups (a), (b) and (c). Results from this analysis are reported as area under the ROC curve with its 95% confidence interval.
All statistical analyses were carried out with the package Stata 16/SE (StataCorp, College Station, TX, US). Cross-validation was performed with the user command cvauroc[25].
Biological functions of the selected miRNAs
The Database for Annotation, Visualization and Integrated Discovery (DAVID) version 2021 [26, 27] was used to analyse the biological functions of miRNA genes. For this purpose, sub-databases of GOTERM_BP_DIRECT, GOTERM_CC_DIRECT and GOTERM_MF_DIRECT (Gene Ontology, GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analyses were combined using DAVID online tool. In this way, we identified general miRNA functions related to regulation of gene expression (RISC complex, miRNA-mediated gene silencing and inhibition of translation, post-transcriptional gene silencing exerted by miRNAs, miRNA binding to 3′UTR regions, as well as positive and negative regulation of gene expression.
Results
Screening phase
The results of the quality control in this phase shown, on average, 2.4 million UMI-corrected reads were obtained for each sample and the average percentage of mappable reads was 59.3%.
The 25 miRNAs showing more difference between controls and each type of case are reported in Additional file 3: Table S2 (controls vs. cases diagnosed by screening), Additional file 3: Table S3 (controls vs. disease-free cases) and Additional file 3: Table S4 (controls vs. non-disease-free cases.) There is little overlapping among these three Tables as shown in the Venn diagram in Fig. 1: Only two miRNAs (miR-29b-3p and miR-31-5p) appeared in all three, seven miRNAs came out when comparing controls vs. both cases diagnosed by screening and disease-free cases (miR-29b-3p, miR-31-5p, miR-34-3p, miR-143-5p, miR-150-3p, miR-195-5p, miR-376c-3p). Six overlapped when analyzing controls vs. cases diagnosed by screening and non-disease-free cases (miR-15b-3p, miR-206, miR-542-3p, miR-625-5p, miR-6513-3p and miR-7850-5p); and only 5 appeared when studying controls vs. both disease-free and non-disease-free cases (miR-136-3p, miR-184, miR-203a, miR-376a-3p and miR-4669).

Venn diagram displaying the overlapping of miRNAs in three different comparisons: controls vs. cases diagnosed via screening, controls vs. disease-free cases and controls vs. cases with active disease in the follow-up
Validation phase
The results of the crude analysis in the validation phase appear as volcano plots in Additional file 1: Fig. S1. In each quadrant of the figure, only the miRNAs selected for the below described models are highlighted with their name.
The model comparing controls with all cases is reported in Table 2. It includes seven miRNAs: miR-423-3p, miR-139-5p, miR-324-5p and miR-1299 are upregulated (odds ratio > 1) and miR-101-3p, miR-186-5p and miR-29a-3p were downregulated in cases. The whole model has an area under the ROC curve = 0.7205 (Bootstrap bias corrected 95% CI: 0.6637–0.7773) (Fig. 2a).

Cross-validated (solid red) and tenfold (dashed) ROC curves of the four final models. A Controls vs. all cases. B Controls vs. cases detected via screening. C Controls vs. disease-free cases in the follow up. D Controls vs. cases with active disease in the follow-up
When comparing controls with cases detected by screening, only miR-101-3p was selected for the model, this miRNA being downregulated in cases (odds ratio = 0.52, 95% confidence interval: 0.36, 0.77) (Table 3.) The area under the ROC curve was 0.6370 (0.5209–0.6791) (Fig. 2b).
Five miRNAs were selected for the model when comparing controls vs. disease-free cases. Two were downregulated (miR-101-3p and miR-186-5p) and three were upregulated (miR-423-3p, miR-142-3p and miR-1299) (Table 4.) The area under the ROC curve was 0.7075 (0.6180–0.7690) (Fig. 2c.)
The model comparing controls with cases with active disease in the follow-up included six miRNAs. miR-101-3p was strongly downregulated (odds ratio = 0.22, 95% confidence interval: 0.12, 0.43), while miR-423-3p, miR-139-5p, miR-1307-3p, miR-331-3p and miR-21-3p were upregulated in cases (Table 5.) The area under the ROC curve was 0.7835 (0.6946–0.8415) (Fig. 2d.) Results from the sensitivity analysis carried out according to the receptors present in each cancer are provided in Additional file 1: Table S5. This table should be cautiously interpreted as the sensitivity analysis could not be carried-out by cross-validation, so its results could be overfitted. Altogether, results using ErbB2-positive cases or triple negative cases tend to reach higher values in the area under the ROC curve, although confidence intervals widely overlap with those obtained with oestrogen or progesterone-positive receptors. Next, we explore the function of the 11 selected miRNAs using the DAVID bioinformatic tool. The results are displayed in Additional file 3: Table S6 (functional annotation Table for each miRNA) and Additional file 2: Figs. S2 (summary of functions involved). Six out of 11 miRNAs were involved in cancer. In particular, several functions related to angiogenesis were predicted, including positive regulation of sprouting angiogenesis, endothelial cell migration and vascular endothelial cell proliferation. Two miRNAs were associated with interleukin-1 response and with the negative regulation of beta-amyloid formation, which has been linked to cancer progression. Some biological functions related to extracellular vesicles and the extracellular space were pointed out, consistent with the fact that these miRNAs were isolated from serum. Additional file 1: Table S7 presented the miRNA sequences and their corresponding accession numbers obtained from miRBase. For a comprehensive understanding of our sample, Additional file 1: Table S8 offered a detailed description of the 269 breast cancer cases included. Furthermore, Additional file 1: Table S9 provided intricate details about the characteristics of both the cases and controls.
Discussion
In this study on circulating miRNAs in breast cancer, we found models able to differentiate controls from BC cases and controls from different types of BC cases, namely cases detected by screening, cases which are disease-free in the follow-up and cases that are not disease-free in the follow-up. Although there is some degree of overlapping between the different models, it is remarkable that their calibration (i.e., their ability to discriminate between cases and controls) increases with the severity of the cancer, as shown by their areas under the ROC curve: 0.6327 to distinguish between controls and cases diagnosed by screening, 0.7345 to differentiate between controls and disease-free cases in the follow-up and 0.8216 to distinguish between controls and cases with active disease.
A total of eleven miRNAs were selected in our four models. Three miRNAs appear as downregulated (miR-101-3p, miR-186-5p and miR-29a-3p) and eight as upregulated in cases (miR-423-3p, miR-139-5p, miR-324-5p, miR-1299, miR-142-3p, miR-1307-3p, miR-331-3p and miR-21-3p).
miRNAs downregulated in breast cancer
miR-101-3p is consistently downregulated in our four models. miR-101-3p has been described as downregulated in women with BC [28, 29]. It promotes BC cell apoptosis by targeting JAK2 (Janus kinase 2) [30] and inhibits BC growth by targeting CXCR7 (CXC chemokine receptor 7) [28] and STMN1 (Stathmin1) [29]. Harati et al. [31, 32] observe that the miR-101-3p is downregulated in metastatic breast cancer cells in comparison with less invasive cells due to the COX-2 (cyclooxygenase-2) induction. Liu et al. [33] consider that the miR-101-3p inhibits the expression of AMPK (AMP-activated protein kinase) in triple negatives breast cancer, whose dysfunction has been linked to breast cancer; while Zhao et al. [34] reflect that the overexpression of this miRNA could induce changes in the macrophages, increasing cellular proliferation and migration.
miR-186-5p appears as downregulated in our models comparing controls with all BC cases and with disease-free cases, in agreement with Giussani et al. [20]. This miRNA seem to inhibit CXCL13 (C-X-C motif chemokine ligand 13) and is associated with tumor staging and size [35]. Another way of action was raised by Hamurcu et al. [36]. They contemplate that the FOXM1 (Forkhead Box 1), which is upregulated in breast cancer cells, exerts its oncogenic effects acting over the miRNA expression. In this work, one of the miRNAs with altered expression is miR-186-5p whose upregulation is associated with the development and progression of breast cancer [36]
miR-29a-3p only appears downregulated in the model comparing controls with all BC cases. Previous results on miR-29a-3p are contradictory. While Wu et al. (2019) found a tumorigenesis role via downregulation of the histone H4K20 trimethylation, Wu et al. [37] and Li et al. [38] found it was downregulated in BC. In addition, some authors [39, 40] indicate that when the miRNA is sponged by a circRNA such as ACAP2 (circACAP2) [39] or PVT1 (Pvt1 oncogene) [40], cellular invasion, proliferation or migration increased.
miRNAs upregulated in breast cancer
miR-423-3p is upregulated in three out of four models of ours: controls vs. all BC cases, controls vs. disease-free cases and controls vs. non-disease-free cases in the follow-up. Consistent with these results, Murria et al. [41] found that the miRNA hyperexpression is associated with estrogen or progesterone receptor positive breast cancers. In addition, the same authors [42] found that this miRNA is part of a signature, together another nine (being miR-423-3p the best differentiated), that allows discriminated hereditary and non-hereditary breast cancers. It has been experimentally observed that miR-423-3p promotes cell proliferation in BC cell lines, and its silencing leads to a decrease in cell proliferation [43]. Consistent with these results, the same authors [41] found that the miRNA hyperexpression is associated to estrogen or progesterone receptor positive breast cancers. However, it shows a lower expression in triple negative breast cancers [41]. No reference against our results was found.
Contrary to our results, miR-139-5p had previously found downregulated in BC [22, 44]. We have found no other article in agreement with our results and have no explanation for this disagreement.
Furthermore, miR-324 is upregulated when compared controls vs. all BC cases and in the comparison of controls vs. screening. In the bibliography, miR-324-5p was found upregulated in BC cases in Giusani et al. [20], Kuo et al. [45], Hong et al. [46], Lou et al. [47], and Turashvili et al. [48]. All of them have demonstrated that its upregulation is associated with worse prognosis, especially in triple negative breast cancer cancers [46,47,48]. Lou et al. [47] proposed a possible mechanism for this miRNA. They analyzed the GPX3 (Glutathione peroxidase 3) in BC and found that its low expression increased cell proliferation and this could be due to the release of miR-324-5p inhibition.
miR-1299 inhibits tumor cell proliferation, invasion and metastasis [49] and, so, it was found downregulated by Liu et al. [50]. This result concurs with its role in other cancers and contradicts our result which shows it as upregulated in BC. In fact, Sant et al. [51] propose that the ciRS-7 sponge the miR-1299 in triple negative breast cancer cells, leading to increase the migration and invasion cells. In the same way, Zhang et al. [52] conclude that the circ-UBR1 sponge also the miR-1299, being able to inhibit the apoptosis and facilitating the proliferation cell and metastasis.
Several authors have reported that miR-142-3p is downregulated in BC and exerts a protective role via inhibiting BC cell invasiveness [53] or targeting HMGA2 (high mobility group AT-hook 2) and inducing apoptosis [54]. These results contradict our finding of miR-142-3p as upregulated in BC. However, some authors support our results: Jusoh et al. [55] found that this miRNA was upregulated in breast cancer patients as compared to the miRNA expression of healthy subjects. In addition, Naseri et al. [56] consider that this miRNA is upregulated in many types of breast cancer resulting in the hyperproliferation of cancer cells in vitro and mammary glands in vivo.
In our results, hsa-miR-1307-3p was significantly upregulated in non-disease-free survival patients compared to controls. In the bibliography, Han et al. [57] found that the upregulation of this miRNA correlates with a poor prognosis (lower survival rate) given that this miRNA seems to stimulate cell proliferation. Shimomura et al. [58], comparing patients with breast cancer and non-breast cancer serums, conclude that a combination of five miRNA (miR-1246, miR-1307-3p, miR-4634, miR-6861-5p and miR-6875-5p) is able to detect breast cancer. Its possible mechanism has been proposed by Han et al. and Shimomura et al. who consider that the miR-1307-3p contributes to BC development and progression by targeting SMYD4 (SET and MYND domain containing 4) [57, 58]
miR-331 was overexpressed in women in metastatic BC, not only when comparing with healthy controls, but also when comparing to women with non-invasive luminal-A BC [59]. Likewise, miR-331 was overexpressed in BC with lymph node metastasis, higher TNM stage and poor prognosis [60]. These publications are consistent with our results. In addition, Pane et al. [61], using omic data integration and machine learning, anticipated that five miRNAs (mir-323a-3p, mir-323b-3p, mir-331-3p, mir-381-3p, and mir-1301-3p) could target in EGFR (epidermal growth factor receptor) family to develop breast cancer in the patients (among other tumors).
In our results, miR-21-3p was significantly upregulated in non-disease-free survival patients compared to controls. This is consistent with Amirfallah et al. [62], who found that its upregulation is associated with metastasis and a short disease-free survival. In addition, they found that the overexpression of this miRNA is associated with a poor prognosis. Ouyang et al. [63] also support the results. They identified 5 upregulated miRNAs (miR-155-5p, miR-21-3p, miR-181a-5p, miR-181b-5p, and miR-183-5p) when comparing the miRNAs profile expression between triple negative breast cancer and normal breast tissues. Aure et al. [64] also observed that the overexpression of three miRNAs associated with copy number gain (miR-21-3p, miR-148b-3p and miR-151a-5p) increases proliferation of breast cancer cell lines. Regarding its mechanism, some authors consider that miR-21 promotes cell proliferation and suppression of apoptosis by targeting SMAD7 (SMAD—Mothers Against decapentaplegic homolog- family member 7), PDCD4 (programmed cell death 4) and PTEN (phosphatase and tensin homolog) [65], eventually leading to increased proliferation and invasiveness of some BC [66].
As shown in both the background and the discussion sections, results on miRNA role in BC are far from homogeneous. While the role of some miRNAs (namely, miR-21, miR-101-3p, miR-186-3p, miR-331, miR-423-3p, miR-1307-3p) appears to be coherent across the literature, results on others (miR-29a-3p, miR139-5p, miR-1299 miR-142-3p) are contradictory and no clear conclusion could be reached. A similar statement could be made regarding combinations of miRNAs in models/signatures: miRNAs selected vary from model to model, making the results unreliable. For instance, only one out of five miRNAs included in the model by Shimomura et al. [58] was selected in any of our models (miR-1307-3p); Kahraman et al. (2018) [67] developed a model with seven miRNAs, but only one of them (miR-101-3p) was selected in ours; and Giussani et al. [20] obtained signatures using five miRNAs, but none was selected in our analysis. By-the-way, signatures developed by Shimomura et al. [58] Kahraman et al. [67] and Giussani et al. [20] do not share any miRNA with each other [20, 58, 67].
Explanations for this result variability would include [1] differences in statistical or lab procedures; in this regard, to select miRNAs on their crude statistical significance or using methods such as stepwise regression, which is known to inflate alpha error, could even involuntarily lead to p-hacking or cherry picking. [2] Random variability -somehow associated with the frequently small sample sizes-; and [3] true biological variability, which could be associated with diversity in the genetic background in patients studied in different countries or continents or to biological differences according to the intrinsic subtype of BC included in each study.
Our study has some limitations. Firstly, the selection of miRNAs for the validation phase was only partially based on the screening phase results, but also on previously published studies. When doing it, the authors chose miRNAs associated with BC in most recent studies (i.e., published in 2020 and 2021), but at the end the selection has some degree of subjectivity. In this way, the selection of miRNAs using their p-value in the screening phase could have led to missing some miRNAs that could have been associated with BC cases in the multivariate setting. Secondly, although beginning with a cohort of 1738 BC women, the final sample size was relatively small; this is especially true for the group of women with active disease in the follow-up, which was strongly limited out of the progressive improvement in diagnosing and treating BC. Thirdly, the discriminative power of our models is moderate as shown in areas under the ROC curve ranging 0.637 to 0.783. The study has also some strengths. Firstly, women included in the analysis were diagnosed in 10 different Spanish provinces and 23 Spanish hospitals, which guarantees some clinical variability. Secondly, our models were obtained using regression with penalization. This method (LASSO) allows for selecting parsimonious models (i.e., models with few regressors) while controlling the alpha error and avoiding the intervention of the researchers in selecting the finally included miRNAs. Moreover, LASSO is considered to outperform regression methods (e.g., stepwise) that select variables using the criticized p-value. Thirdly, we have a variety of cases (diagnosed by screening, disease-free in the follow-up and with active disease in the follow-up), which allows us to develop different models for diverse types of cases.
Conclusion
Summarizing, we present four models involving eleven miRNAs to differentiate healthy controls from different types of BC cases. Our models scarcely overlap with those previously reported. Whether the lack of reproducibility of miRNA signatures in BC is due to methodological issues, random variability or true biological variability requires a joint analysis of data from different studies, eventually via creation of international consortia.
Availability of data and materials
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
References
Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2021;71:209–49.
Golubnitschaja O, Debald M, Yeghiazaryan K, Kuhn W, Pešta M, Costigliola V, et al. Breast cancer epidemic in the early twenty-first century: evaluation of risk factors, cumulative questionnaires and recommendations for preventive measures. Tumour Biol. 2016;37:12941–57.
Lambertini M, Santoro L, Del Mastro L, Nguyen B, Livraghi L, Ugolini D, et al. Reproductive behaviors and risk of developing breast cancer according to tumor subtype: a systematic review and meta-analysis of epidemiological studies. Cancer Treat Rev. 2016;49:65–76.
Hankinson S, Tamimi R, Hunter D. Breast cancer textbook of cancer epidemiology. Oxford: Oxford University Press; 2008.
Autier P, Boniol M, Koechlin A, Pizot C, Boniol M. Effectiveness of and overdiagnosis from mammography screening in the Netherlands: population based study. BMJ. 2017. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5712859
Kapoor PM, Mavaddat N, Choudhury PP, Wilcox AN, Lindström S, Behrens S, et al. Combined associations of a polygenic risk score and classical risk factors with breast cancer risk. J Natl Cancer Instit. 2021;113:329–37.
Curigliano G, Burstein HJ, Winer P, et al. De-escalating and escalating treatments for early-stage breast cancer the St gallen international expert on the primary therapy of early breast cancer 2017. Ann Oncol. 2017;28:1700–12.
Saliminejad K, KhorramKhorshid HR, SoleymaniFard S, Ghaffari SH. an overview of microRNAs: biology, functions, therapeutics, and analysis methods. J Cell Physiol. 2019;234:5451–65.
Shi Y, Liu Z, Lin Q, Luo Q, Cen Y, Li J, et al. MiRNAs and cancer: key link in diagnosis and therapy. Genes. 2021. https://doi.org/10.3390/genes12081289.
Mu H, Zhang W, Qiu Y, Tao T, Wu H, Chen Z, et al. miRNAs as potential markers for breast cancer and regulators of tumorigenesis and progression (Review). Int J Oncol. 2021;58:16.
Hill M, Tran N. miRNA interplay: mechanisms and consequences in cancer. Dis Models Mechan. 2021. https://doi.org/10.1242/dmm.047662.
Allegra A, Alonci A, Campo S, Penna G, Petrungaro A, Gerace D, et al. Circulating microRNAs: new biomarkers in diagnosis, prognosis and treatment of cancer. Int J Oncol. 2012;41:1897–912.
miRBase. https://www.mirbase.org/
Castaño-Vinyals G, Aragonés N, Pérez-Gómez B, Martín V, Llorca J, Moreno V, et al. Population-based multicase-control study in common tumors in Spain (MCC-Spain): rationale and study design. Gac Sanit. 2015;29:308–15.
Alonso-Molero J, Molina AJ, Jiménez-Moleón JJ, Pérez-Gómez B, Martin V, Moreno V, et al. Cohort profile: the MCC-Spain follow-up on colorectal, breast and prostate cancers: study design and initial results. BMJ Open. 2019;9: e031904.
Gomez-Acebo I, Dierssen-Sotos T, Palazuelos-Calderon C, Perez-Gomez B, Amiano P, Guevara M, et al. Tumour characteristics and survivorship in a cohort of breast cancer: the MCC-Spain study. Breast Cancer Res Treat. 2020;181:667–78.
Robinson MD, Oshlack A. A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. 2010;11:R25.
Escuin D, López-Vilaró L, Mora J, Bell O, Moral A, Pérez I, et al. Circulating microRNAs in early breast cancer patients and its association with lymph node metastases. Front Oncol. 2021;11: 627811.
Galvão-Lima LJ, Morais AHF, Valentim RAM, Barreto EJSS. miRNAs as biomarkers for early cancer detection and their application in the development of new diagnostic tools. Biomed Eng Online. 2021;20:21.
Giussani M, Ciniselli CM, De Cecco L, Lecchi M, Dugo M, Gargiuli C, et al. Circulating miRNAs as novel non-invasive biomarkers to aid the early diagnosis of suspicious breast lesions for which biopsy is recommended. Cancers. 2021;13:4028.
Jang JY, Kim YS, Kang KN, Kim KH, Park YJ, Kim CW. Multiple microRNAs as biomarkers for early breast cancer diagnosis. Mol Clin Oncol. 2021;14:31.
Li X, Zou W, Wang Y, Liao Z, Li L, Zhai Y, et al. Plasma-based microRNA signatures in early diagnosis of breast cancer. Mol Genet Genomic Med. 2020;8: e1092.
Liu X, Chen F, Tan F, Li F, Yi R, Yang D, et al. Construction of a potential breast cancer-related miRNA-mRNA regulatory network. Biomed Res Int. 2020;2020:6149174.
Tibshirani R. Regression shrinkage and selection via the lasso. J Roy Stat Soc: Ser B. 1996;58:267–88.
Luque-Fernandez MA, Redondo-Sánchez D, Maringe C. cvauroc: command to compute cross-validated area under the curve for ROC analysis after predictive modeling for binary outcomes. Stand Genomic Sci. 2019;19:615–25.
Huang DW, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4:44–57.
Huang DW, Sherman BT, Lempicki RA. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucl Acids Res. 2009;37:1–13.
Li J-T, Jia L-T, Liu N-N, Zhu X-S, Liu Q-Q, Wang X-L, et al. MiRNA-101 inhibits breast cancer growth and metastasis by targeting CX chemokine receptor 7. Oncotarget. 2015;6:30818–30.
Wang R, Wang H-B, Hao CJ, Cui Y, Han X-C, Hu Y, et al. MiR-101 is involved in human breast carcinogenesis by targeting stathmin1. PLoS ONE. 2012;7: e46173.
Wang L, Li L, Guo R, Li X, Lu Y, Guan X, et al. miR-101 promotes breast cancer cell apoptosis by targeting Janus kinase 2. Cell Physiol Biochem. 2014;34:413–22.
Harati R, Mohammad MG, Tlili A, El-Awady RA, Hamoudi R. Loss of mir-101-3p promotes transmigration of metastatic breast cancer cells through the brain endothelium by inducing COX-2/MMP1 signaling. Pharmaceuticals. 2020;13:1–19.
Harati R, Mabondzo A, Tlili A, Khoder G, Mahfood M, Hamoudi R. Combinatorial targeting of microRNA-26b and microRNA-101 exerts a synergistic inhibition on cyclooxygenase-2 in brain metastatic triple-negative breast cancer cells. Breast Cancer Res Treat. 2021;187:695–713.
Liu P, Ye F, Xie X, Li X, Tang H, Li S, et al. mir-101-3p is a key regulator of tumor metabolism in triple negative breast cancer targeting AMPK. Oncotarget. 2016;7:35188–98.
Zhao Y, Yu Z, Ma R, Zhang Y, Zhao L, Yan Y, et al. lncRNA-Xist/miR-101-3p/KLF6/C/EBPα axis promotes TAM polarization to regulate cancer cell proliferation and migration. Mol Ther—Nucl Acids. 2021;23:536–51.
Wang F, Yuan C, Liu B, Yang Y-F, Wu H-Z. Syringin exerts anti-breast cancer effects through PI3K-AKT and EGFR-RAS-RAF pathways. J Transl Med. 2022;20:310.
Hamurcu Z, Sener EF, Taheri S, Nalbantoglu U, Kokcu ND, Tahtasakal R, et al. MicroRNA profiling identifies Forkhead box transcription factor M1 (FOXM1) regulated miR-186 and miR-200b alterations in triple negative breast cancer. Cell Signal. 2021;83: 109979.
Wu Z, Huang X, Huang X, Zou Q, Guo Y. The inhibitory role of Mir-29 in growth of breast cancer cells. J Exp Clin Cancer Res. 2013;32:98.
Li W, Yi J, Zheng X, Liu S, Fu W, Ren L, et al. miR-29c plays a suppressive role in breast cancer by targeting the TIMP3/STAT1/FOXO1 pathway. Clin Epigenet. 2018;10:64.
Zhao B, Song X, Guan H. CircACAP2 promotes breast cancer proliferation and metastasis by targeting miR-29a/b-3p-COL5A1 axis. Life Sci. 2020;244: 117179.
Wang J, Huang K, Shi L, Zhang Q, Zhang S. CircPVT1 promoted the progression of breast cancer by regulating MiR-29a-3p-mediated AGR2-HIF-1α pathway. Cancer Manag Res. 2020;12:11477–90.
Murria R, Palanca S, de Juan I, Alenda C, Egoavil C, Seguí FJ, et al. Immunohistochemical, genetic and epigenetic profiles of hereditary and triple negative breast cancers. Relevance in personalized medicine. Am J Cancer Res. 2015;5:2330–43.
MurriaEstal R, PalancaSuela S, de Juan JI, Egoavil Rojas C, García-Casado Z, Juan Fita MJ, et al. MicroRNA signatures in hereditary breast cancer. Breast Cancer Res Treat. 2013;142:19–30.
Zhao H, Gao A, Zhang Z, Tian R, Luo A, Li M, et al. Genetic analysis and preliminary function study of miR-423 in breast cancer. Tumour Biol. 2015;36:4763–71.
Sun H, Dai J, Chen M, Chen Q, Xie Q, Zhang W, et al. miR-139-5p was identified as biomarker of different molecular subtypes of breast carcinoma. Front Oncol. 2022;12: 857714.
Kuo W-T, Yu S-Y, Li S-C, Lam H-C, Chang H-T, Chen W-S, et al. MicroRNA-324 in human cancer: miR-324-5p and miR-324-3p have distinct biological functions in human cancer. Anticancer Res. 2016;36:5189–96.
Hong H-C, Chuang C-H, Huang W-C, Weng S-L, Chen C-H, Chang K-H, et al. A panel of eight microRNAs is a good predictive parameter for triple-negative breast cancer relapse. Theranostics. 2020;10:8771–89.
Lou W, Ding B, Wang S, Fu P. Overexpression of GPX3, a potential biomarker for diagnosis and prognosis of breast cancer, inhibits progression of breast cancer cells in vitro. Cancer Cell Int. 2020;20:1–15.
Turashvili G, Lightbody ED, Tyryshkin K, SenGupta SK, Elliott BE, Madarnas Y, et al. Novel prognostic and predictive microRNA targets for triple-negative breast cancer. FASEB J. 2018;32:5937–54.
Kaiyuan D, Lijuan H, Xueyuan S, Yunhui Z. The role and underlying mechanism of miR-1299 in cancer. Future Sci OA. 2021;7:693.
Liu L-H, Tian Q-Q, Liu J, Zhou Y, Yong H. Upregulation of hsa_circ_0136666 contributes to breast cancer progression by sponging miR-1299 and targeting CDK6. J Cell Biochem. 2019;120:12684–93.
Sang M, Meng L, Liu S, Ding P, Chang S, Ju Y, et al. Circular RNA ciRS-7 maintains metastatic phenotypes as a ceRNA of miR-1299 to target MMPs. Mol Cancer Res. 2018;16:1665–75.
Zhang L, Sun D, Zhang J, Tian Y. Circ-UBR1 facilitates proliferation, metastasis, and inhibits apoptosis in breast cancer by regulating the miR-1299/CCND1 axis. Life Sci. 2021;266: 118829.
Schwickert A, Weghake E, Brüggemann K, Engbers A, Brinkmann BF, Kemper B, et al. microRNA miR-142-3p inhibits breast cancer cell invasiveness by synchronous targeting of WASL, integrin alpha v, and additional cytoskeletal elements. PLoS ONE. 2015;10: e0143993.
Mansoori B, Duijf PHG, Mohammadi A, Safarzadeh E, Ditzel HJ, Gjerstorff MF, et al. MiR-142-3p targets HMGA2 and suppresses breast cancer malignancy. Life Sci. 2021;276: 119431.
Jusoh AR, Mohan S, Lu Ping T, Tengku Din TADAA, Haron J, Romli R, et al. Plasma circulating mirnas profiling for identification of potential breast cancer early detection biomarkers. Asian Pac J Cancer Prev. 2021;22:1375–81.
Naseri Z, KazemiOskuee R, Jaafari MR, Forouzandeh M. Exosome-mediated delivery of functionally active miRNA-142-3p inhibitor reduces tumorigenicity of breast cancer in vitro and in vivo. Int J Nanomed. 2018;13:7727–47.
Han S, Zou H, Lee J-W, Han J, Kim HC, Cheol JJ, et al. miR-1307-3p stimulates breast cancer development and progression by targeting SMYD4. J Cancer. 2019;10:441–8.
Shimomura A, Shiino S, Kawauchi J, Takizawa S, Sakamoto H, Matsuzaki J, et al. Novel combination of serum microRNA for detecting breast cancer in the early stage. Cancer Sci. 2016;107:326–34.
McAnena P, Tanriverdi K, Curran C, Gilligan K, Freedman JE, Brown JAL, et al. Circulating microRNAs miR-331 and miR-195 differentiate local luminal a from metastatic breast cancer. BMC Cancer. 2019;19:436.
Jiang F, Zhang L, Liu Y, Zhou Y, Wang H. Overexpression of miR-331 indicates poor prognosis and promotes progression of breast cancer. Oncol Res Treat. 2020;43:441–8.
Pane K, Zanfardino M, Grimaldi AM, Baldassarre G, Salvatore M, Incoronato M, et al. Discovering common miRNA signatures underlying female-specific cancers via a machine learning approach driven by the cancer hallmark ERBB. Biomedicines. 2022;10:1306.
Amirfallah A, Knutsdottir H, Arason A, Hilmarsdottir B, Johannsson OT, Agnarsson BA, et al. Hsa-miR-21-3p associates with breast cancer patient survival and targets genes in tumor suppressive pathways. PLoS ONE. 2021;16:1–18.
Ouyang M, Li Y, Ye S, Ma J, Lu L, Lv W, et al. MicroRNA profiling implies new markers of chemoresistance of triple-negative breast cancer. PLoS ONE. 2014;9: e96228.
Aure M, Leivonen S-K, Fleischer T, Zhu Q, Overgaard J, Alsner J, et al. Individual and combined effects of DNA methylation and copy number alterations on miRNA expression in breast tumors. Genome Biol. 2013;14:R126.
Maryam M, Naemi M, Hasani SS. A comprehensive review on oncogenic miRNAs in breast cancer. J Genet. 2021;100:15.
Gong C, Nie Y, Qu S, Liao J-Y, Cui X, Yao H, et al. miR-21 induces myofibroblast differentiation and promotes the malignant progression of breast phyllodes tumors. Cancer Res. 2014;74:4341–52.
Kahraman M, Röske A, Laufer T, Fehlmann T, Backes C, Kern F, et al. MicroRNA in diagnosis and therapy monitoring of early-stage triple-negative breast cancer. Sci Rep. 2018;8:11584.
Acknowledgements
Some samples included in this study were provided by the Basque Biobank www.biobancovasco.org and the Biobanco La Fe (B.0000723) and they were processed following standard operation procedures with the appropriate approval of the Ethical and Scientific Committees.
Funding
This study was partially funded by the “Accion Transversal del Cancer,” approved on the Spanish Ministry Council on the 11 October 2007, Instituto de Salud Carlos III-FEDER (PI08/1770, PI08/0533, PI08/1359, PS09/00773, PS09/01286, PS09/01903, PS09/02078, PS09/01662, PI11/01889, PI11/02213, PI12/00488, PI12/01270, PI12/00715, PI14/01219, PI14/0613, and PI17/01388, PI18/00171), Fundación Marqués de Valdecilla (API 10/09), the ICGC International Cancer Genome Consortium CLL [The ICGC CLL-Genome Project was funded by Spanish Ministerio de Economía y Competitividad (MINECO) through the Instituto de Salud Carlos III (ISCIII) and Red Temática de Investigación del Cáncer (RTICC) del ISCIII (RD12/0036/0036)], the Junta de Castilla y León (LE22A10-2), the Consejería de Salud of the Junta de Andalucía (PI-0571-2009, PI-0306-2011, and salud201200057018tra), the Conselleria de Sanitat of the Generalitat Valenciana (AP_061/10), the Recercaixa (2010ACUP 00310), the Regional Government of the Basque Country, the Consejería de Sanidad de la Región de Murcia, by the European Commission grants FOOD-CT-2006-036224-HIWATE, the Spanish Association Against Cancer (AECC) Scientific Foundation, by the Catalan Government—Agency for Management of University and Research Grants (AGAUR) grants 2017SGR723 and 2014SGR850, the Fundación Caja de Ahorros de Asturias, and the University of Oviedo. ISGlobal acknowledges support from the Spanish Ministry of Science and Innovation through the “Centro de Excelencia Severo Ochoa 2019–2023” Program (CEX2018-000806-S) and support from the Generalitat de Catalunya through the CERCA Program. AP-C was supported by the MINECO (Ministry of Economy in Spain) Grant no. PRE2019-089038, fellowship.
Author information
Authors and Affiliations
Contributions
IGA, JL, JAM, MDM and TDS performed the data acquisition. IGA, JL, JAM and TDS performed the data curation. IGA and JL carried funding acquisition. IGA, JL and TDS performed the supervision of the study. IGA, JL and TDS wrote the first draft of the manuscript. All authors contributed to the study conception and design, commented on previous versions of the manuscript and read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The studies involving human participants were reviewed and approved by CEIC 2018.280 and 2008/3123/I. The patients/participants provided their written informed consent to participate in this study.
Consent for publication
Not applicable.
Competing interests
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1:
Figure S1. Volcano plots in the validation phase. A Controls vs. all cases. B Controls vs. cases detected via screening. C Controls vs. disease-free cases in the follow up. D Controls vs. cases with active disease in the follow-up
Additional file 2:
Figure S2. Summary of functions involved in the 11 miRNAs selected in the models, according to DAVID bioinformatic tool.
Additional file 3:
Table S1. Rationale for selecting miRNAs for the validation phase. When the rationale was based on the screening phase, the main results leading to the selection is indicated as log (fold change) and p value. When the rationale was based on previously reported results, the reference in cited. Table S2. Screening phase: comparison between controls and cases diagnosed by screening. Only the 25 most differentially expressed miRNAs are shown. Table S3. Screening phase: comparison between controls and disease-free cases. Only the 25 most differentially expressed miRNAs are shown. Table S4. Screening phase: comparison between controls and non-disease-free cases. Only the 25 most differentially expressed miRNAs are shown. Table S5. Performance of each logistic regression model according to the cancer receptor status: area under the ROC curve and 95% confidence interval. Table S6. Functional annotation table of the 11 miRNAs selected in the models, according to DAVID bioinformatic tool. Table S7. miRNA sequences and accession numbers for the validation phase obtained from miRBase. Table S8. Description of the 269-breast cancer included in the sample. Table S9. Characteristics of the breast cancer patients and population-based controls.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Gómez-Acebo, I., Llorca, J., Alonso-Molero, J. et al. Circulating miRNAs signature on breast cancer: the MCC-Spain project. Eur J Med Res 28, 480 (2023). https://doi.org/10.1186/s40001-023-01471-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40001-023-01471-2