
Abstract
Acokanthera schimperi is a medicinal plant traditionally used for the treatment of wounds, scabies, and malaria. Rhus glutinosa has been also utilized for the management of ectoparasites and hemorrhoids. Silica gel column chromatography separation of CH2Cl2/MeOH (1:1) extract root of A. schimperi afforded oleic acid (1), lupeol (2), dihydroferulic acid (3), acovenosigenin A- 3-O-α-L-rhamnopyranoside (4) and sucrose (5) whereas CH2Cl2/ MeOH (1:1) and MeOH roots extracts of R. glutinosa afforded β-sitosterol (6), (E)-5-(heptadec-14-en-1-yl)-4,5-dihydroxycyclohex-2-enone (7), methyl gallate (8), and gallic acid (9). The structures of the compounds were established using spectroscopic (1D and 2D NMR) and FT-IR techniques. Disc diffusin and DPPH assay were used, respectively, to evaluate the antibacterial and antioxidant potential of the extracts and isolated compounds. MeOH extract root of A. schimperi showed a modest antibacterial effect against E.coli with an inhibition zone (ZI) of 16 ± 0.0 mm compared to ciprofloxacin (ZI of 27.0 ± 0.0 mm). CH2Cl2/MeOH (1:1) and MeOH root extracts of R. glutinosa showed maximum activity against S. aureus with ZI of 17.3 ± 0.04 and 18.0 ± 0.0 mm, respectively. At 5 mg/mL, the highest activity was noted against S. aureus by 8 with ZI of 18.6 ± 0.08 mm. Dihydroferulic acid (3), methyl gallate (8), and gallic acid (9) displayed potent scavenging of DPPH radical with respective IC50 of 10.66, 7.48, and 6.08 µg/mL, compared with ascorbic acid (IC50 of 5.83 µg/mL). Molecular docking results showed that lupeol (2) exhibited strong binding energy of -7.7 and − 10 kcal/mol towards PDB ID: 4F86 and PDB ID: 3T07, respectively, compared to ciprofloxacin (-6.5 and − 7.2 kcal/mole). Towards PDB ID: 1DNU receptor, compounds 3, 8, and 9 showed minimum binding energy of -5.1, -4.8, and − 4.9 kcal/mol, respectively, compared to ascorbic acid (-5.7 kcal/mol). The Swiss ADME prediction results indicated that compounds 2, 3, 8, and 9 obeyed the Lipinksi rule of five and Veber rule with 0 violations. The in vitro antibacterial and antioxidant results supported by in silico analysis indicated that compounds 2, 3, 8, and 9 can potentially be lead candidates for the treatment of pathogenic and free radical-induced disorders.
Similar content being viewed by others

Introduction
Infectious diseases have been frequently found to be among the major causes of threat to global health [1]. The World Health Organization estimates that infectious diseases account for about 50% of all fatalities in tropical countries [2]. The misuse of conventional antimicrobial drugs results in the progress of multiple-drug resistance of pathogenic microorganisms [3]. The prolonged bacterial infectious diseases are also important for the emergency of reactive oxygen species (ROS) and are linked with oxidative stress [4]. Oxidative stress plays a critical role in the development of various pathological complications such as cardiovascular dysfunction, inflammation, carcinogenesis, drug toxicity, and neurodegenerative diseases [5]. Higher plant natural products may be a novel source of antioxidant and antibacterial chemicals with possibly unique mechanisms of action [6, 7]. Antimicrobials cause harm to pathogens by disrupting the establishment of microbial cell walls, inhibiting the synthesis of microbial proteins and nucleic acids, altering the shape and function of microbial membranes, or blocking metabolic pathways by inhibiting essential enzymes [8]. Plant-derived antioxidants may act as free radicals scavengers and combat oxidative stress by keeping a balance between antioxidants and oxidants [9]. Antioxidant compounds block the effects of free radicals which have been linked to the pathogenesis of many diseases including atherosclerosis, heart disease, cancer, and Alzheimer’s disease [10]. The antioxidant potential of the plant product is mainly associated with compounds such as flavonoids and phenolic acids, ascorbic acid, vitamin E, and different carotenoids [11]. Recently, researchers have been paying more attention to new antimicrobial and antioxidant compounds isolated from various medicinal plants which might be good sources of novel therapeutic agents [12].
Acokanthera schimperi (Apocynaceae) is a drought-resistant evergreen plant that grows in southern Yemen and East Africa including Ethiopia [13]. A. schimperi has been reported to possess traditionally for the management of wounds, malaria, scabies, and insect repellent [14]. In Ethiopia, A. schimperi locally called “Merenz” (in Amharic) and “Kararu” (in Afaan Oromo) have been used for the treatment of headaches, eye disease, skin infection, and snake bites as anti-venom [15]. The leaves and bark of the plant have been used for the treatment of skin disorders, inflammatory-related diseases, and malaria [16]. In Ethiopia, the grounded bark is mixed with water and taken orally to treat stomachache while its roots are burned on fire and fumigated to cure hepatitis [17]. There are limited studies on leaves of A. schimperi that reported the isolations of only a few triterpenoids and acaricidal coumarins from the leaves part of the plant [18, 19]. A more comprehensive study of compounds isolated from A. schimperi is needed.
Rhus glutinosa (Family Anacardiaceae) is a medicinal plant indigenous to Ethiopia and widely used for the treatment of various ailments [20, 21]. The dry/fresh stem bark of R. glutinosa is orally given for deficiency of vitamin C [22]. In Eriteria, the leaves of R. glutinosa have been used for the management of ectoparasites and scabies [23]. In Ethiopia, the dried roots and leaves of R. glutinosa are also used to treat hemorrhoids [24]. To the best of our knowledge, even though the plant possesses a wide range of therapeutic values, prior scientific investigations done on the plant are very limited. Herein, we report a comprehensive study on the isolation and characterization of chemical constituents of the roots of A. schimperi and R. glutinosa, and evaluation of their in vitro antibacterial and antioxidant activities and in silico molecular docking and ADMET analysis.
Materials and methods
Chemicals and instruments
FT- IR spectra were recorded in the KBr pellet using Thermo Scientific Spectrometer SMART iTR basic in NICOLET iS10 model within a 4000 –400 cm-1 spectral range. 1H and 13C NMR spectral data were analyzed on a JNM-ECZ 400 S (400 MHz, JEOL, Ldt. USA) spectrometer. CDCl3 and CD3OD were used as solvents for NMR analysis. TLC silica gel Merck pre-coated aluminum plate (silica gel GF254 German) was used for TLC analysis, and compounds on TLC were identified with a UV lamp (UV4AC6/2, CBIO, Bioscience, and Technologies. Ldt., China) at 254 and 365 nm and spraying with vanillin/H2SO4. Column chromatography was used to isolate compounds (silica gel mesh size of 60–120 mm as stationary phase). A double-beam UV-Vis spectrophotometer (P9-UV-Vis, VWR International Ltd, China) was used to measure absorbance at 517 nm for the DPPH assay. Siliga gel (60–120 mesh, LOBA CHEMIE PVT. LTD., India) was used for column chromatography. HPLC grade solvents such as methanol (Alpha Chemika, India) ethyl acetate (Alpha Chemika, India), Chloroform (Alpha Chemika, India), dichloromethane (LOBA CHEMIE PVT. LTD., India), and petroleum ether (60–80ºC) (LOBA CHEMIE PVT. LTD., India) were used for the isolation of compounds. 2,2-diphenyl-1-picrylhydrazyl (DPPH) was used to evaluate the radical scavenging activity of compounds. Ciprofloxacin and ascorbic acid were used as the standard drug for antibacterial and antioxidant assays respectively.
Plant material collection
The roots of A. Schimperi and R. glutinosa were collected in April 2023 from Lalibela Town, North Wollo Zone, Amhara Regional State, Ethiopia, located about 710 km north of the capital, Addis Ababa. The plants were authenticated by Mr. Melaku Wondafrash and a voucher for the specimen BA001 and BA004 was given, respectively, for A. schimperi and R. glutinosa and deposited at the National Herbarium, Department of Biology, Addis Ababa University, Addis Ababa, Ethiopia.
Extraction and isolation of compounds
Extraction and isolation of compounds from the roots of A. schimperi
Powdered roots of A. schimperi (600 g) were soaked for 72 h with 4 L of CH2Cl2/MeOH (1:1), filtered, and concentrated using a rotary evaporator at 40 °C to afford 20 g (4.9%) crude extract. The marc was soaked with methanol (3 L) for 72 h, filtered, and concentrated to afford 6 g (1%). Preliminary TLC profiles of CH2Cl2/MeOH (1:1) and MeOH extract were examined with the help of a UV cabinet (UV254 and UV365 nm lamps) and vanillin/H2SO4 as a spraying agent. CH2Cl2/MeOH (1:1) extract (15 g) was adsorbed with an equal amount of silica gel and subjected to chromatographic separation (silica gel 150 g, 60–120 mesh size as stationary phase). Column elution was done by increasing the gradient of EtOAc in petroleum ether to collect 130 fractions (each 100 mL). The TLC profile of each fraction was detected under a UV cabinet (254 and 365 nm) and visualized after spraying with vanillin/H2SO4. Fractions 8–13 (eluted with 10% EtOAc in petroleum ether) afforded compound (1, 18 mg) as a single spot. Fractions 17–23 (eluted with 10% EtOAc in petroleum ether) showed a similar spot, combined fractions, and re-chromatographed using the increasing gradient of EtOAc in petroleum ether. A total of 36 fractions were collected of which sub-fractions 19–25 (eluted with 10% EtOAc in petroleum ether) showed a single spot after spraying with vanillin/H2SO4 to give compound 2 (23 mg). Fractions 32–40 (eluted with 30% EtOAc in petroleum ether) were combined and rechromatographed using gradient increasing of EtOAc in petroleum ether. A total of 35 fractions were collected of which sub fractions 21–25 (30% EtOAc in petroleum ether) showed a single spot under UV254 lamp to afford compound 3 (26 mg). Fractions 76–79 formed a white precipitate and showed a single spot after spraying with vanillin/H2SO4 to give compound 4 (57 mg). Fractions 93–97 (eluted with 10% MeOH in EtOAc) were mixed and purified with silica gel column chromatography with increasing gradient of MeOH in EtOAc to obtain compound 5 (40 mg).
Extraction and isolation of compounds from the roots of R. glutinosa
Powdered roots of R. glutinosa (600 g) were soaked with 4 L of CH2Cl2/MeOH (1:1) for 72 h, filtered, and concentrated using a rotary evaporator at 40 °C to afford 15 g (2.5%) crude extract. The marc was soaked with methanol (3 L) for 72 h, filtered, and concentrated to afford 7 g (1.16%) crude extract. Preliminary TLC profiles of both crude extracts showed similar spots under a UV lamp and hence combined. The combined crude extract (16 g) was adsorbed with an equal amount of silica gel and subjected to silica gel column chromatographic separation with increasing gradient of EtOAc in petroleum ether as eluent to afford 150 fractions (100 mL each). The TLC profile of each fraction was analyzed under UV cabinet at 254 and 365 nm wave length. Fractions 11–16 (eluted in 10% EtOAc in Petroleum ether) showed a single spot to afford compound 6 (23 mg). Fractions 26–33 (eluted in 20% EtOAc in petroleum ether) were combined and re-chromatographed using an increasing gradient of EtOAc in petroleum ether. A total of 32 fractions were collected of which sub-fractions 17–22 showed a single spot under the UV254 lamp to afford compound 7 (17 mg). Fractions 53–62 (eluted with 50% EtOAc in petroleum ether) were combined and purified by silica gel column chromatography using gradient increasing of EtOAc in petroleum ether to collect a total of 62 fractions (10 mL each). Sub-fractions 28–34 (eluted with 50% EtOAc in petroleum ether) and sub-fractions 49–53 (eluted with 60% EtOAc in petroleum ether) showed a single spot under the UV254 lamp to afford compounds 8 (40 mg) and 9 (70 mg), respectively.
Antibacterial activity
The antibacterial potential of the crude extracts and isolated compounds were evaluated using Mueller Hinton agar medium following the standard procedure reported in the literature [25] against Gram-negative bacteria (E. coli and P. aeruginosa) and Gram-positive bacteria (S. aureus and S. pyogenes). In brief, a few colonies (3–5) of bacterial strains with similar morphologies were properly introduced into a liquid medium (saline solution) using a sterilized inoculating loop. The turbidity of the suspension was adjusted to 0.5 McFarland barium sulfate standards (108 CFU/mL). The experiment was carried out at two-fold serial solutions of 200, 100, and 50 mg/mL of CH2Cl2/MeOH (1:1) and MeOH crude extracts, and 5, 2.5, and 1.25 mg/mL of the isolated compounds using DMSO as the diluting agent. Bacterial suspensions were applied onto a petri plate containing MHA. Whatman No 1 paper discs (6 mm) were applied on the surface of the inoculated MHA and about 20 µl each test sample was applied onto the disks. The samples were tested in triplicates and expressed as mean ± SD. Ciprofloxacin (30 µg) was used as the reference drug.
Radical scavenging activity
The radical scavenging activities of crude extracts of roots of A. schimperi and R glutinosa and isolated compounds were evaluated following DPPH assay as described by Sasikumar et al., [26] using ascorbic acid as standard. Two-fold serial dilution solutions of 1000, 500, 250, 125, and 62.5 µg/mL were prepared from 2 mg/mL stock solutions. Then, 4 mL of DPPH (4 mg/100 mL in methanol) was added to 1 mL of each serially prepared sample and kept in the dark for 30 min. The absorbance was measured using a UV-Vis spectrophotometer at 517 nm. The percentage of inhibition of DPPH radical was calculated as;
where Acontrol is the absorbance of DPPH, and Asample is the absorbance of the sample with DPPH solution. The inhibitory concentration, the concentration needed to scavenge 50% of DPPH radical (IC50) was calculated from the Prism dose-response curve (GraphPad, Prism version 4.0 for Windows, GraphPad Software, San Diego, CA, USA) obtained by plotting the percentage of inhibition versus the concentrations [27].
Molecular docking analysis
The present study aimed to support the in vitro study via in silico molecular docking analysis by predicting the mode of binding orientation and affinity of the isolated compounds towards the antibacterial and antioxidant target proteins. Based on the in vitro analysis, compounds with promising antibacterial activity (2, 3, 8, and 9) were docked against proteins such as DNA gyrase B of E. coli (PDB ID: 4F86), pyruvate kinase of S. aureus (PDB ID: 3T07), and S. pyogenes 10,782 streptopain (PDB ID: 6UKD) receptors. The selection of antibacterial target protein towards the corresponding docked compound was chosen following the findings obtained in vitro results. The promising DPPH radical scavenging activity of compounds noted by 3, 8, and 9 observed in the experimental analysis also inspired us to predict the binding pattern and affinity of these compounds towards human myeloperoxidase (PDB ID: 1DNU), an important antioxidant target protein.
Protein and ligand preparation
The 3D crystal structures of the selected target proteins (PDB ID: 4F86, PDB ID: 3T07, PDB ID: 6UKD, and PDB ID:1DNU) were downloaded from the RCSB Protein Databank (RCSB PDB: Homepage) and saved in the PDB format. The protein preparation was done using AutoDock 4.2.6 (MGL tools 1.5.6) following the standard protocol [28] by removing the co-crystallized ligand, water molecules, and co-factors. Polar hydrogens and Kollman charges were added to the protein and a pdbqt format file was generated using the AutoDockTools 1.5.6 software. The chemical structures of the studied compounds (2, 3, 8, and 9) were drawn using ChemOffice tool (Chem Draw 16.0) assigned with proper 2D orientation, and the energy of each molecule was minimized using ChemBio3D. The energy-minimized ligand molecules were saved in pdbqt format using the AutoDock Vina tool to carry out the docking simulation.
Docking simulations
The graphical user interface program was used to set the grid box for docking simulations. The grid was set so that it surrounds the region of interest in the macromolecule. Accordingly, the 3D sizes of the structure-based design (SBD) site sphere of each protein were retrieved as follows; (PDB ID: 4F86 (1.979, 8.744, 62.387), PDB ID: 3T07 (0.013, 0.147, -0.003) PDB ID: 6UKD (-25.515, -13.743, 13.171) and PDB ID:1DNU (27.535, 2.689, 13.523) Å for x, y and z dimensions, respectively. The AutoDock Vina searched for the best-docked conformation between isolated compounds and target proteins. Nine different conformations were generated for each ligand in the docking process using the Lamarckian genetic algorithm (LGA) program. The conformations with the most favorable (least) binding free energy were selected for analyzing the interactions between the target receptor and ligands by Discovery Studio visualizer and PyMOL. Finally, the ligand interactions, hydrogen bonds, residual amino acid interactions, 2D and 3D structural orientations, and image preparations were run using the Biovia Discovery Studio Visualizer 2021 [29, 30].
In-silico pharmacokinetics analysis
The pharmacokinetic properties of the studied compounds (2, 3, 8, 9) were assessed to estimate their potential as drug candidates. This estimation was carried out based on the standard protocol adopted by the Lipinski rule [31]. Pro Tox II was employed to predict the toxicities, toxicological endpoints, and LD50 of the isolated compounds [32].
Results and discussion
Characterization of compounds from the root of A. schimperi
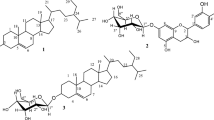
CH2Cl2/MeOH (1:1) roots extract of A. schimperi afforded five compounds (1–5) (Fig. 1). 1H NMR spectrum (SI-Figure S1A, SI-Table S1) of compound 1 ranged from δ 0.88 to 5.33. The signals displayed at δ 5.33 (m) and 2.34 (2 H, t) were attributed, respectively, to olefinic and methylene protons α- to an acyl group. The signals observed at δ 1.63 (2 H, m) and 2.02 (4 H, m) were recognized for methylene (H-3) and allylic methylene protons (H-8 and H-9), respectively. The signals displayed at δ 1.25 (24 H, 12 x CH2, brs) and δ 0.88 (3 H, t) were assigned to the long-chain methylene protons and terminal methyl protons, respectively [33]. The 13C NMR and DEPT-135 spectra (SI-Figure S1B&C, SI-Table S1) revealed a total of 18 well-resolved carbons. The signal observed at δ 180.4 was an indicative of the presence of a carboxylic acid. The peaks appeared at δ 129.9 and 129.6 were recognized for olefinic methine carbons. The presence of 14 methylene carbons ranged from δ 22.7 to 34.0, pointing downward in the DEPT-135 spectrum, and one terminal methyl carbon displayed at δ 14.1 indicated the presence of unsaturated 18-carbon fatty acid. The above spectra data and comparing it with the literature report indicated that compound 1 is identified as oleic acid [34].
The peaks on the 1H NMR spectrum (SI-Figure S2 A, SI-Table S2) of compound 2 range from δ 0.74 to 4.67. The 1H NMR showed the presence of seven singlet methyl protons resonated at δ 0.74, 0.77, 0.81, 0.92, 0.94, 1.01, and 1.66. Doublet protons observed at δ 4.55 (d, J = 2 Hz) and 4.67 (d, J = 2 Hz) were attributed to diastereotopic protons (2 H, H-α-29, and H-β-29) suggesting the presence of a lupine-type triterpenoid skeleton [35]. The peaks displayed at δ 3.19 (dd, 1H) and 2.36 (1H) were characteristic peaks for H-3 and H-19 of the lupeol skeleton. The 13C NMR and DEPT-135 spectra (SI-Figure S2B& C, SI-Table S2) revealed a total of 30 well-resolved carbons. The 13C NMR spectrum suggests the presence of seven methyls, eleven methylenes, six methines, and six quaternary carbons. The appearance of olefinic peaks displayed at δ 109.4 (C-29) and 151.0 (C-20) and oxy-methine at δ 79.1 (C-3) confirmed that compound 2 was a lupane-type triterpenoid. Based on the above spectral information and comparison with literature reports, compound 2 was recognized as lupeol which has been reported to possess antioxidant, antimicrobial, anti-tumor, and anti-carcinogenic activities [36].
1H NMR spectrum (SI-Figure S3A, SI-Table S3) of compound 3 showed the presence of one set of ABX spin system aromatic protons observed at δ 6.82 (1H, d, J = 2.0, H-2), 6.69 (1H, d, J = 8.0 Hz, H-5) and 6.67 (1H, dd, J = 8.0, 2.0 Hz, H-6). Two sets of triplet protons integrated for two protons each of methylene groups were resonated at δ 2.63 (2H, t, H-7) and 2.87 (2H, t, H-8). The signal displayed at δ 3.84 (3H, s, H-9’’) is recognized as a methoxy proton. The COSY spectrum (SI-Figure S3B, SI-Table S3) showed 1H-1H cross connectivity between H-5 (δ 6.70) with H-6 (δ 6.67) and proton displayed at δ 2.65 (H-7) with δ 2.88 (H-8). The 13C NMR and DEPT-135 spectra (SI-Figure S3 C & D, SI-Table S3) displayed a total of ten well-resolved carbons. The signals appeared at δ 30.4 and 35.9 disclosed the presence of two aliphatic methylene carbons. The presence of three sp2 methine carbons was observed at δ 111.0, 114.4, and 120.9. The signals displayed at δ 132.2, 144.1, and 146.5 were assignable for sp2 quaternary carbons. The peaks displayed at δ 55.9 and 178.1 were attributed, respectively, to methoxy and a carboxylic group. The HSQC spectrum (SI-Figure S3E, SI-Table S3) demonstrated 1J C-H bond correlations of protons observed at δ 6.82 (H-2), 6.69 (H-5), 6.67 (H-6), 3.84, 2.65(H-7), and 2.88 (H-8) with δ 111.0 (C-2), 114.4 (C-5), 120.9 (C-6), 55.9 (C-3a), 30.9 (C-7), 36.1(C-8), respectively. Based on the above spectroscopic data and comparing with the reported literature, compound 3 was identified as dihydroferulic acid, previously reported from Suaeda japonica [37].
The FT-IR spectrum (SI-Figure S4A) of compound 4 displayed a broad band at 3368 cm− 1, indicating the presence of O-H stretching vibration. Intense bands appeared at 2925 and 1745 cm− 1 suggesting the presence of C-H of stretching methyl and C = O stretching of an ester group, respectively. The weak band showed at 1616 cm− 1 and a medium band at 1046 cm− 1 were attributed to C = C and C-O stretching vibration, respectively. The peaks at 1446 cm− 1 indicated the C-H in-plane bending vibration of sp3 hydrocarbons. The peaks on the 1H NMR spectrum (SI-Figure S4B, SI-Table S4) ranges from δ 0.74–5.88. The presence of a doublet proton observed at δ 4.90 (1 H, d, J = 17.6, H-21α), δ 4.98 (1 H, d, J = 17.6, H-21β,), and an olefinic singlet proton displayed at δ 5.90 (1 H, s, H-22) was indicative of a characteristic feature of the cardenolide skeleton [38]. The signals displayed at δ 0.74 (H-18) and 0.94 (H-19) were recognized for two angular methyl singlet protons of the steroidal skeleton. The peaks resonated at δ 4.77 (s), and 3.29 (3H, s) were recognized for anomeric and methoxy protons, respectively. 13C NMR and DEPT-135 spectra (SI-Figure S4C&D, SI-Table S4) showed a total of 30 well-resolved carbons. The presence of oxy-methylene carbon observed at δ 73.9, olefinic carbons δ 116.5 and δ 177.0, and carbonyl carbon at δ 175.9 confirms the presence of butenolide ring [39]. The peaks displayed at δ 15.0, and 15.5 were attributed, respectively, to C-18 and C-19 peaks for two steroidal angular methyl carbons. The signals displayed at δ 84.8 and 54.4 were assignable to sp3 quaternary and a methoxy carbon, respectively. The presence of rhamnose sugar moiety was confirmed due to signals of oxy-methine carbons observed at δ 98.2, 75.0, 69.6, 68.4, 67.5, and methyl carbon at δ 15.5. The abovementioned spectral information and comparison with published data suggested that compound 4 was recognized as a covenosigenin A- 3-O-α-L-rhamnopyranoside reported from leaves of A. oblongifolia [40].
1H NMR of the spectrum (SI-Figure S5A, SI-Table S5) of compound 5 ranges from δ 3.30 to 5.24. The signals observed at δ 5.24 (d, J = 5.8), 5.17 (d, 4.08), 4.80 (dd), 4.52 (m), 3.88 (t), 3.78 (d), 3.65 (t), and 3.30 (t) were recognized for sp3 oxy-methine protons. The peaks shown at δ 3.41 (H-10), 4.45 (H-11), and 5.08 (H-12) were assigned to oxy-methylene protons. The 13C and DEPT-135 spectra (SI-Figure S5B &C, SI-Table S5) revealed a total of 12 well-resolved carbons. The signal observed at δ 104.3 was attributed to anomeric carbon. Eight sp3 oxy-methine carbons were observed at δ 92.1, 82.7, 77.4, 74.6, 73.2, 72.9, 71.9, and 70.2. The signals that appeared at δ 63.5, 62.6, and 61.8 were recognized as three oxy-methylene carbons. The above spectroscopic data was similar to the spectroscopic data of sucrose reported in the literature [41].
Characterization of compounds from the roots of R. glutinosa
The combined CH2Cl2/MeOH (1:1) and MeOH root extracts of R. glutinosa led to the isolation of four compounds 6–9 (Fig. 1). The FT-IR spectrum (SI-Figure S6A) of compound 6 showed a broad band at 3413 cm− 1 suggesting the O-H stretching vibration of an alcohol group. The bands observed at 2935 and 2860 cm− 1 showed the asymmetric and symmetric C-H stretching vibration of sp3 hydrocarbons. The bands displayed at 1464 and 1383.16 cm− 1 represent the C-H bending vibration of methyl groups. Weak bands observed at 1650, and 1048 cm− 1 were assigned to C = C and C-O stretching vibration, respectively. The 1H NMR spectrum (SI-Figure S6B, SI-Table S6) exhibited two sets of singlet protons integrated as three protons each resonated at δ 0.66 (H-18) and 1.01 (H-19) were attributed to two angular methyl protons and peaks shown at δ 5.33 (1H, t), 3.53 (1H, m), δ 2.32 (1H, d) and 2.02 (1H, m) recognized to an olefinic proton (H-6), sp3 methine proton bearing a hydroxyl group (H-3) and methylene protons of H-4 and H-7, respectively, was indicative of the characteristic signals for β-sitosterol skeleton [42]. 13C NMR and DEPT-135 spectra (SI-Figure S6C & 6D, SI-Table S6) displayed a total of 29 well-resolved peaks of which six methyl, nine methine, eleven methylene, and three quaternary carbons. The peaks observed at δ 140.0 and 121.7 belong to sp2 quaternary and sp2 methine olefinic carbons respectively, whereas the signal appeared δ 71.9 belongs to sp3 methine-bearing hydroxyl group (C-3). Two angular methyl carbons appeared at δ 11.8 (C-19) and 19.3 (C-18). The above spectral data are in good agreement with spectroscopic data of β-sitosterol, previously reported from Baccaurea macrocarpa species [43].
1H NMR spectrum (SI-Figure S7A, SI-Table S7) of compound 7 demonstrated the presence of olefinic protons resonated at δ 6.11, d (J = 7.9), δ 6.88 (m) and 5.33 (2H m, H-16’ &H-17’). The peaks displayed at δ 2.53 (4 H, m) and 2.00 (4 H, m) were recognized to be allylic protons for (H-6 &H-4) and (H-15’ & H-18’), respectively. A multiplet peaks observed at δ 1.22 (H-2’ to H-13’) were assignable of methylene protons. The peak observed at δ 0.86 (3H, t) was attributed to terminal methyl protons. The COSY spectrum (SI-Figure S7B, SI-Table S7) showed 1H-1H connectivity of protons displayed at δ 6.88 (H-3) with δ 6.11 (H-2) and δ 2.53 (H-4) and vice versa. COSY spectrum also confirms the 1H-1H cross connectivity of olefinic protons (H-16’ and H-17’) with their respective neighboring allylic protons of H-15’ and H-18’, respectively. 13C NMR spectrum supported by its corresponding DEPT − 135 spectrum (SI-Figure S7C & D, SI-Table S7) revealed a total of 25 well-resolved carbons. The signal resonated at δ 198.2 was recognized for the presence of a ketone group. The signals observed at δ 146.7, 131.6, 129.6, and 129.4 were assigned to sp2 olefinic carbons. The presence of sp3 oxy-methine carbon was displayed at δ 74.2. The signals observed at δ 38.3, and 50.5 were attributed to the α-carbon (C-6) to the carbonyl carbon, and allylic carbons (C-4), respectively. The signals resonated at δ 42.2 and 14.1 were assigned to the carbon attached to oxy-methine carbon (C-1’) and terminal methyl (C-19’) carbons, respectively. Based on the above spectroscopic data and comparison with the literature reports, compound 7 is identified as (E)-5-(heptadec-14-en-1-yl)-4,5-dihydroxycyclohex-2-enone, previously isolated from the root bark of Lannea acida [44].
The FT-IR spectrum (SI-Figure S8A) of compound 8 showed a broad band observed at 3280, cm− 1 representing the O-H stretching vibration, and the intense band that appeared at 1701 cm− 1 was assignable to C = O strtching of an ester group. The bands displayed at 2931 was attributed to the C-H stretching vibration of methyl group. The presence of C = C stretching vibration of aromatic carbons was observed at 1608, and 1450 cm− 1. The band displayed at 1009 cm-1 was recognized for C-O stretching vibration. 1H NMR spectrum of (SI-Figure S8B) showed the presence of aromatic protons displayed at δ 6.90 (2H, s, H-2/6) and methoxy proton at δ 3.70 (3 H, s). The broad singlet peak observed at δ 9.16 was recognized for the protons belonging to the hydroxyl group. The 13C NMR and DEPT-135 spectra (SI-Figure S8C & D) revealed a total of six well-resolved carbons. The presence of a symmetry sp2 methine carbons (C-2/C-6) was displayed at δ 109.0. The signals observed at δ 119.8 (C-1), 138.9 (C-4), and 146.1 (C-3/C-5) were recognized to be sp2 quaternary carbons. The presence of a methoxy carbon resonated at δ 52.0 and α-β unsaturated carbonyl δ 166.9 was indicative of the presence of an ester group. Based on the above combined spectral data and comparing it with the literature report, compound 8 was identified as methyl gallate [45].
The FT-IR spectrum (SI-Figure S9A) of compound 9 showed the presence of O-H and C = O stretching vibration at 3345, and 1680 cm− 1, respectively. The vibrational stretching of C = C of aromatic carbons were observed at 1600, 1550, and 1446 cm− 1 The bands observed at 1320, and 1052 cm− 1 were assigned to -C-O- stretching vibrations. 1H NMR spectrum (SI-Figure S9B) showed the presence of singlet aromatic proton integrated for two protons observed at δ 6.87 (2H, s, H-2/6). The peaks observed at δ 8.80, and 9.15 were assigned to the protons of the hydroxyl group. The signal displayed at δ 11.80 was attributed to the proton belonging to the carboxyl group. The 13C NMR spectrum supported its DEPT-135 spectrum (SI-Figure S9C&D) showed a total of five well-resolved carbons. The presence of sp2 methine carbon (C-2/C-6) was observed at δ 109.0. The signals observed at δ 119.8 (C-1), 138.9 (C-4), and 146.1 (C-3/C-5) were recognized as sp2 quaternary carbons. The peak observed at δ 168.0 was attributed to the presence α-β unsaturated carboxylic acid. The above spectroscopic information and comparison with the literature reported indicated that compound 9 is identical to gallic acid [45].

Structure of compounds isolated from roots of A. schimperi (1–5) and roots of R. glutinosa (6–9)
Biological activity
Antibacterial activity
The antibacterial activity of the CH2Cl2/MeOH and MeOH root extracts of A. schimperi and R. glutinosa and isolated compounds (2, 3, 4, 6, 7, 8, and 9) were evaluated against E. coli, P. aeruginosa, S. aureus, and S. pyogenes using disc diffusion assay. The CH2Cl2/MeOH (1:1) and MeOH extracts of the root of A. schimperi and R. glutinosa exhibited low to promising antibacterial activity following dose-dependent approaches against the tested pathogens (Table 1). The CH2Cl2/MeOH (1:1) and MeOH extract of the root of A. schimperi showed significant antibacterial activity against E.coli and S. aureus with respective ZI of 16.0 ± 0 and 15.6 ± 0.47 mm at 200 mg/mL, compared to ciprofloxacin (27.0 ± 0 to 31 ± 0.0 mm at 30 µg/disc). MeOH extract also exhibited modest antibacterial activity against S. pyogenes and S. aureus with ZI of 13.0 ± 0.0 and 14.3 ± 0.8 mm, respectively. A similar study carried out by Taye et al., (2011) indicated that MeOH extract leaves of A. schimperi showed moderate antibacterial activity with ZI of 14 mm against against S. pyogenes and S. aureus 500 mg/mL [46]. According to Tadeg et al., (2005) investigation, the MeOH extract of leaves of A. schimperi demonstrated potent antibacterial activity against S. aureus and P. aeruginosa with inhibition of 24 ± 0.5 and 17.0 ± 0.0 mm at 100 mg/mL [47]. CH2Cl2/MeOH extract root of A. schimperi also demonstrated modest activity against E. coli and S. pyogenes with ZI of 13.3 ± 0.8 and 12.6 ± 0.47 mm, respectively. A related investigation conducted by Kenubih et al., (2021) demonstrated that the CH2Cl2 extract leaves of A. schimepri exhibited significant antibacterial activity against E. coli, P. aeruginosa and S. aureus with ZI of 16.67, 15.67, and 15.0 mm, respectively [14]. The CH2Cl2/MeOH (1:1) and MeOH extracts root of R. glutinosa ZI displayed significant antibacterial effects against S. aureus with 17.3 ± 0.8 and 18.0 ± 0.0 mg/mL, respectively, at 200 mg/mL compared to ciprofloxacin (31 ± 0.0 at 30 µg/disc) (Table 1). MeOH extract also showed significant activity against E. coli and S. pyogenes with ZI of 16.3 ± 0.8 and 16.0 ± 0.0 mm, respectively. CH2Cl2/MeOH (1:1) extract displayed a significant antibacterial action against S. pyogenes and E. coli with ZI of 15.6 ± 0.47 and 15.0 ± 0.0 mm, respectively. The present finding revealed that both plant extracts had significant antibacterial activity against all bacteria tested with varying degrees of activity. The differences in the antibacterial potential of the CH2Cl2/MeOH (1:1) and MeOH extracts against the tested bacteria could be attributed to the differences in the chemical constituents and the concentration of the secondary metabolites present in the corresponding crude extracts [14].
The antibacterial activity of the isolated compounds (2, 3, 4, 6, 7, 8, and 9) demonstrated modest to promising activity towards the tested pathogens (Table 2). Lupeol (2) displayed the highest antibacterial activity against E.coli and S. aureus with ZI of 16.6 and 17.3 mm respectively, compared to ciprofloxacin (27.0–31.0 ± 0.0 mm). The finding was in good agreement with the literature report demonstrated that lupeol (2) exhibited antibacterial activity against E. coli and S. aureus with ZI of 19 and 20 mm respectively at 3 mg/mL [48]. Lupeol (2) also showed modest antibacterial activity P. aeruginosa and S. pyogenes with ZI of 12.3 and 11.6 mm respectively. Musa et al., (2024) investigated on evaluation of the antibacterial potency of lupeol (2) using the broth dilution method and the authors found that the lupeol (2) exhibited antibacterial activity against E.coli and S. aureus with MIC values of 2.5 and 5 mg/mL respectively [49]. Dihydroferulic acid (3) displayed the highest antibacterial activity against S. pyogenes and E. coli with ZI of 14.6 and 12 mm respectively. Compound 4 demonstrated significant antibacterial activity S. pyogenes, S. aureus, and P. aeruginosa with ZI of 15.3, 13.6, and 14.6 mm respectively (Table 2). The antibacterial activity outcome of the studied compounds from the root of A. schimperi indicated that lupeol (2) showed the best antibacterial activity compared to other compounds (3 and 4) which implies that lupeol (2) could be the active constituent of the A. schimperi. The observed maximum activity of lupeol (2) may be associated with its nature of lipophilicity and it readily penetrates the cell membrane and exerts its biological effect [50].
β-sitosterol (6) showed modest antibacterial activity against S. aureus with an inhibition zone of 11.0 mm, compared to ciprofloxacin (31 ± 0.0 mm). A similar study investigated by Sen et al., (2012) indicated that β- sitosterol (6) exhibited moderate antibacterial activity against E. coli and S. aureus with ZI of 13 and 14 mm respectively [51]. According to Melaku et al, (2022) investigation, β- sitosterol (6) had antibacterial activity against S. aureus, K. pneumoniae, and E. coli with ZI of 12 mm, 11 mm, and 10 mm respectively [52]. Compound 7 exhibited potent activity against E. coli and S. pyogenes with ZI of 15.6 and 16 mm respectively. Compound 7 and methyl gallate (8) also demonstrated significant antibacterial effects against P. aeruginosa with ZI of 14.3 and 15.6 mm respectively compared to ciprofloxacin (ZI of 29.0 ± 0.0). Methyl gallate (8) showed potent antibacterial action against S. aureus and E. coli ZI of 18.6 and 16.3 mm respectively. Gallic acid (9) noted the best antibacterial activity S. pyogenes and S. aureus with ZI of 16 and 17.6 mm respectively (Table 2). A related study investigated by Al-Zahrani (2019) revealed that methyl gallate (8) and gallic acid (9) exhibited antibacterial activity against methicillin-resistant and susceptible methicillin S. aureus strains with MIC values of ranges 3.5 to 25 µg/mL [53]. A study investigated by Pinho and his coworkers (2014) showed that gallic acid presented potent antibacterial activity against S. epidermidis, S. aureus, and K. pneumonia with MIC values of 0.0098, 0.0195, 0.00975 mg/mL, and MBC values of 0.0039, 0.039 and 0.0039 mg/mL respectively [54]. Phenolic compounds such as methyl gallate (8) and gallic acid (9) disrupt the bacterial cell wall by hyperacidification of the plasma membrane via proton donation and acidification of the intracellular cytosolic which inhibits the enzyme H+-ATPase, which is required for the synthesis of ATP [55].
The present study demonstrated that gram-positive bacteria were more susceptible than gram-negative bacteria. The susceptibility of gram-positive bacteria could be associated with the structure of the cell membrane of the bacteria, that gram-positive bacteria lack the outer membrane and have a thick layer of peptidoglycan that surrounds the plasma membrane to protect bacteria from the harsh environment where they livein [56]. The present finding revealed that the crude extracts and isolated compound of roots of R. glutinosa demonstrated moderate to promising antibacterial activities against the tested pathogens proving the therapeutic values of the plant recorded by traditional medicinal healers.
Radical scavenging activity
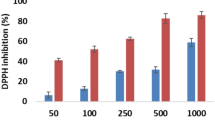
Plants contain abundant antioxidants which provide to defense mechanism against free radicals-associated disorders. Natural antioxidants have the power to stop chain reactions and prevent oxidation processes by eliminating radical intermediates and oxidizing themselves [57]. Recently, the radical scavenging and antioxidant capacity of plant natural products have drawn a lot of interest due to their potential to lower the risk of cardiovascular disease and cancer in humans [58]. In the present study, the antioxidant activity of CH2Cl2/MeOH (1:1) and MeOH extracts, and isolated compounds that have enough amount for activity (2, 3, 4, 6, 7, 8, and 9) of the roots of A. schimperi and R. glutinosa was determined following the DPPH assay using compared with ascorbic acid. The dark purple solution of DPPH was turned to yellow when the DPPH radical subsequently received an electron or a hydrogen radical from an antioxidant and the observed color change was measured by UV-Vis spectrophotometry at 517 nm [59]. The crude extracts and isolated compounds of (2, 3, 4) of the root of A. schimperi studied compounds demonstrated moderate to strong quenching of DPPH free radicals (Table 3; Fig. 2). CH2Cl2/MeOH (1:1) and MeOH extracts of the roots of A. schimperi showed modest inhibition of DPPP radical with IC50 values of 43.0 and 32.89 µg/mL respectively, compared with ascorbic acid (IC50 value 5.83 µg/Ml). A related study investigated by Alemu et al., (2020) demonstrated that the methanol extract of leaves of A. schimperi exhibited 74.85% DPPH radical inhibition at 200 mg/mL [60]. Dihydroferulic acid (3) displayed strong scavenging of DPPH radical with an IC50 value of 10.66 µg/mL, compared with ascorbic acid (IC50 value of 5.83 µg/mL). Lupeol (2) and compound (3) also displayed medium inhibition of DPPH radical with respective IC50 values of 53.97 and 47.54 µg/mL. The observed antioxidant potential of lupeol (2) showed close agreement in published literature indicating that lupeol established scavenging of DPPH radical with an IC50 value of 44.3 µg/mL [61]. Dihydroferulic acid (3) displayed higher antioxidant activity compared to the crude extracts and other compounds (2 and 4) of A. schimperi. A similar study investigated by Deters et al., (2008) demonstrated that dihydroferulic acid (3) exhibited scavenging of DPPH radical with IC50 values of 19.5 µM [62]. The observed promising antioxidant potential of dihydroferulic acid (3) could be associated with the nature of the phenolic compound that could easily donate hydrogen to stabilize the DPPH radicals [63].

Antioxidant activity of CH2Cl2/MeOH (1:1), and MeOH extracts and compounds (2, 3 and 4) from the roots of A. schimperi
The DPPH radical scavenging activity of CH2Cl2/MeOH and MeOH extract and isolated compounds (6, 7, 8 and 9) of the roots R. glutinosa demonstrated moderate to promising DPPH radical scavenging activity (Fig. 3; Table 4). The CH2Cl2/MeOH (1:1) and MeOH extract showed strong scavenging of DPPH radical with respective IC50 values of 20.08 and 13.66 µg/mL respectively. β- sitosterol (6) displayed minimum scavenging of DPPH radical with IC50 values of 114.5 µg/mL and compound 7 showed medium inhibition of DPPH radical with IC50 values of 66.86 µg/mL. Methyl gallate (8) and gallic acid (9) demonstrated strong quenching of DDPH radical with respective IC50 values of 7.48 and 6.08 µg/mL, compared to ascorbic acid (IC50 value of 5.83 µg/mL). A similar investigation studied by Habtemariam (2011) indicated that gallic acid (9) showed 50% inhibition of DPPH radical at 6.26 ± 0.83 µg/mL [64]. According to the study conducted by Ekaprasada et al., (2009), methyl gallate (8) exhibited potent scavenging of DPPH radical with an IC50 value of 1.02 µg/mL [65]. Another study studied by Bhadoriya et al., (2012) indicated that gallic acid (8) displayed 91.99 ± 0.59 inhibition of DPPH radical at 50 µg/mL [66]. The findings indicated that methyl gallate (8) and gallic acid (9) showed the highest DPPH scavenging activity than the crude extracts and other isolated compounds suggesting that these compounds could be the active antioxidant constituents of the roots of R. glutinosa. The presence of the phenolic hydroxyl group has been comprehensively reported as the main structural descriptor for the antioxidant activity of polyphenols, due to the hydrogen atom ability to scavenge free radicals [67]. Gallic acid (9) has been demonstrated as the chief antioxidant component responsible for the efficient antiradical and anticancer properties of several plant extracts [68]. The present finding confirms that the crude extracts and isolated compounds of the roots of A. schimperi and R. glutinosa possess antibacterial and antioxidant activity indicating that the chemical constituents of the studied plant cloud have a therapeutic value of pathogenic microorganism and free radical-inducing disorders.

Antioxidant activity of CH2Cl2/MeOH (1:1), and MeOH extracts and isolated compounds (6, 7, 9 and 10), of the root of R. glutinosa
Molecular docking analysis
Most pharmaceuticals or biologically active compounds work by interacting with targets on proteins. Due to many drugs that can target multiple proteins, target identification is crucial in drug development and biomedical studies [69]. Molecular docking is an effective method for determining the possible drug candidates by predicting the binding affinity of small molecules to target protein. Docking analysis is used to screen a large database of small molecules to identify those with a high affinity for binding to a particular protein [70].
Drug targets are proteins or enzymes whose activities affect the survival of a disease-causing organism and therefore inhibiting the activity of these enzymes is harmful to the survival of the microorganisms [71]. For instance, the S. aureus protein kinase catalyzes the last rate-limiting step of glycolysis, which is essential to the metabolism of carbohydrates. Pharmacological inhibition of PK may cause MRSA to have impaired metabolism so it could be a promising target for novel antibiotics [72]. Similarly, the DNA gyrase of E. coli is an important enzyme for bacterial DNA replication and transcription to maintain topology and integrity [73]. Therefore, it is essential to use bacterial DNA gyrase as a target for antibacterial drugs [74]. On the other hand, streptopain, a cysteine protease secreted by S. pyogenes is an essential enzyme for full host infectivity due to its ability to cleave host proteins (plasminogen, fibrinogen), antimicrobial peptide, and antibodies [75]. Streptopain represents a potential virulence factor target, and its inhibition could enhance complement-mediated host defense functions during S. pyogenes infection [76]. Human myeloperoxidase (MPO) is an important enzyme found in human neutrophils, whose main role is to maintain defenses against invading pathogens [77]. MPO can catalyze the H2O2-mediated oxidation of chloride to the powerful oxidizing agent hypochlorous acid (HOCl), which leads to the oxidation (degradation) of biomolecules of pathogens in the phagosome. The initial product of MPO-H2O2-Cl− system is the potent antimicrobial oxidant hypochlorous acid/hypochlorite (HOCl/OCl). However, recent evidence claims that excessive HOCl production can result in oxidative stress, tissue damage, initiation and propagation of acute and chronic vascular inflammatory disease. HOCl can initiate modification reactions targeting lipids, DNA, and proteins through halogenation, nitration, and oxidative crosslinking [78, 79]. MPO-derived chlorinated compounds are specific markers for inflammation progression, which attracted considerable intrest in the development of therapeutically useful MPO inhibitors and HOCl scavengers. Therefore inhibition of the MPO target is an important mechanism for the treatment of free radical-induced disorders [78].
In the present study, the predication of the binding modes and affinity of compounds (2, 3, 8, and 9) were investigated towards DNA gyrase of E. coli (PDB ID: 4F86), protein kinase of S. aureus (PDB ID: 3T07), S. pyogenes streptopain (PDB ID: 6UKD) and antioxidant target, human myeloperoxidase (PDB ID: 1DNU) target proteins. The choice of the protein targets toward their respective docked compounds was conducted based on the findings obtained from the experimental antibacterial and antioxidant activity results. For each ligand-protein complex analysis, a total of nine different conformations were generated using the Lamarckian genetic algorithm (LGA) program, and the conformations with the most favorable (least) binding free energy were selected for analyzing the interactions between the target receptor and ligands. A lower binding energy, or more negative free energy of binding, generally signifies greater stability and binding affinity between the compound and the receptors [80]. The molecular docking projection outcomes showed that lupeol (2) and methyl gallate (8) demonstrated strong binding affinity towards DNA gyrase B of E. coli (PDB ID: 4F86) receptor with the binding energy of -7.7 and − 6.8 kcal/mol respectively, compared to ciprofloxacin (binding energy of -6.5 kcal/mol) (SI-Figure S10, SI-Table S8). The complex formed between lupeol (2) and PDB ID: 4F86 receptor was stabilized by Vander Waals interactions with the active site of ASP-A32, GLY-A33, THR-A34, GLU-A193, ARG-190, LYS-A189, PHE-A41 and pi-alkyl interaction through ILE-A186 amino acid. The ligand-protein complex formed by methyl gallate (8) and PDB ID: 4F86 receptor was stabilized mainly by hydrogen bond and Vander Waal interactions. Methyl gallate (8) interacts with the PDB ID: 4F86 receptor through hydrogen bonds via the active sites of GLY-I117, GLY-I118, and HIS -I56 amino acids. Residual amino acids MET-I120, SER-I120, HIS-I58, ASN-I180, GLU-I181, SER-I182, GLN-I140 GLY-I1115, GLY-I113, HIS-I57, CYS-I114, ARG-I116, and TYR-I55 were involved through Vander Waal interactions (SI-Table S8).
Towards pyruvate kinase of S. aureus (PDB ID: 3T07) protein, the strongest binding energy was recorded by lupeol (2) with a binding energy of -10.0 kcal/mol, compared to -7.2 kcal/mol for ciprofloxacin (SI-Figure S11, SI-Table S9). The complex formed by lupeol (2) and PDB ID: 3T07 receptor was stabilized by hydrogen bond and Vander Waals interactions. Lupeol (2) forms one hydrogen bond interaction through the active site ASP-D346 amino acid in its stable complex. The active site of amino acids ASP-A261, ALA-D337, ARG-A264, ASP-D338, GLN-D338, LYS-D342, LEU-D343, and ARG-A347 were involved through Vander Waals interaction. Both methyl gallate (8) and gallic acid (9) demonstrated a significant binding energy of -5.9 kcal/mol compared to ciprofloxacin (-7.2 kcal/mol). Methyl gallate (8) interacts with the protein target by ASP-A303, LYS-A260, ASN-A299, ASP-D303, and TYR-D302 through hydrogen bonds. Residual amino acids TYR-A302, ASN-D-299, and LYS-D260 were involved through Vander Waals interactions (SI-Figure S11). Gallic acid (9) also forms four hydrogen bond interactions through the active site THR-C353, ASN-D369, and THR-D366 (2 H -bonds) amino acids in its stable complex with pyruvate kinase of S. aureus (PDB ID: 3T07) receptor. Vander Waals interactions were also involved through residual amino acids THR-C348, HIS-C365, HIS-D365, SER-C365, SER-C362, SER-D362, MET-D347, ALA-C358, and LEU-D370 amino acids (SI-Table S9).
Towards, S. pyogenes 10,782 streptopain (PDB ID: 6KUD) receptor, gallic acid (9) exhibited a promising binding free energy of -5.7 kcal/mol, compared to ciprofloxacin (-6.8 kcal/mol) (SI-Figure S12, SI-Table S10). Gallic acid (9) forms four hydrogen bond interactions towards the PDB ID: 6KUD receptor through the active site of SER-A308 (2 H-bonds), GLN-A332, and GLN-A390 amino acids. Residual amino acids SER-A390, SER-A285, ARG-A307, and ASN-A306 were contributed through Vander Waal interaction in its ligand-protein complex stabilization. Similarly, residual amino acid TYR-A389 contributed by pi-pi stack and hydrophobic interaction in its stable ligand-protein complex.
Dihydroferulic acid (3), methyl gallate (8), and gallic acid (9) displayed minimum binding energy of -5.1, -4.8, and − 4.9 kcal/mol towards human peroxidase (PDB ID: 1DNU) receptor compared to ascorbic acid (binding energy − 5.7 kcal/mol) (SI-Figure S13, SI-Table S11). As shown in SI-Figure S13, dihydroferulic acid (3) forms two hydrogen bond interactions via GLN-C121 and ASP-C142 amino acids. Residual amino acids TRP-A47, PRO-A49, PHE-C126, CYS-119, CYS-c143, ILE-C130, and PRO-C127, LYS-C129, ALA-C141 were involved through hydrogen/pi-alkyl and Vander Waals interactions respectively. Methyl gallate (8) interacts with hydrogen bonds through the active site of ALA-C141 amino acid in its ligand-protein complex towards PDB ID: 6UKD receptor. Residual amino acids VAL-C120, GLN-C121, ILE-C130, 1EU-C128, CYS-134, ASP-C142 (Vander Waals interaction), and LYS-C129, PRO-C127 (hydrophobic interactions) were also contributed towards the stabilization of ligand-protein complex. Gallic acid (9) forms hydrogen bonding interactions through the active site of TYR-A47, CYS-C119, and ALA-C14 amino acids. Gallic acid (9) interacts with PDB ID: 6UKD receptor through residual amino acids PRO-A49, GLY-46, PRO-C127, CYS-143, ASP-C142 (Vander Waals interactions) and LYS-C129 (hydrophobic interaction) in its ligand-protein complex. The docking prediction result indicates that compounds (3, 8, and 9) may serve as an antioxidant agent by inhibiting the PDB ID: 1DNU receptor and then by decreasing the production of excess ROS in host cells.
Drug likeness, ADME, and toxicity profiles of compounds
Drug-likeness is a prediction that evaluates whether a given organic molecule possesses characteristics that make it suitable for use as an orally active medication [31]. The drug-likeness and pharmacokinetics properties of the isolated compounds with high-ranking binding energy scores (2, 3, 8, and 9) were assessed using the Swiss-ADME online tool. Swiss ADME is an authenticated free web tool for investigating and estimating the physicochemical parameter, lipophilicity, water solubility, pharmacokinetics, and drug-likeness following the principles of Lipinski’s rule-of-five and Veber’s rule [81]. The Lipinski rule of Five (RO5) principles states that bioactive compounds with a molecular weight of less than 500, logP of less than 5, less than 5 hydrogen bond donors (nHBD), and less than 10 hydrogen bond acceptors (nHBA) are more likely to have good oral bioavailability and be orally active [82]. According to the Veber rule a drug-like molecule should have a total polar surface area (TPSA) should be less than 140 Å and the number of rotatable bond values should be less than 10 [83]. The present finding revealed that all compounds under investigation (2, 3, 8, and 9) have a molecular weight in the acceptable range (MW ≤ 500) implying they can be readily absorbed in the gastrointestinal tract [84] (SI-Table S12). The nHBA and nHBD for all tested compounds were found to be within Lipinski’s limit range. This implies that the compounds are expected to be well absorbed or permeable from the gastrointestinal tract when they are administrated. The TPSA value of the analyzed compounds was recorded in the range from 20.23 to 97.99 Å and is much below the limit of 140 Å, suggesting a high probability of favorable oral bioavailability for drug-like candidates [85] (SI-Table S12).
The number of rotatable bonds is also an important parameter to estimate the drug-likeness potential of a drug candidate and is linked with the bioavailability and flexibility of lead compounds. Hence, all studied compounds fell within the acceptable range (nRB ≤ 10), indicative of their potential permeability and oral bioavailability. Overall, all the analyzed compounds meet the criterion of Lipinski’s rule and Veber’s rule with 0 violations, suggesting the potential to be orally administered drugs (SI-Table S12). The skin permeability value (Kp, in cm/s) of the analyzed compounds ranges from − 1.90 to -6.84 cm/s suggesting low skin permeability and is within the range of broad-spectrum antibiotic ciprofloxacin (-9.09 cm/s) [86]. The lipophilicity (log P) values of all studied compounds ranges from 0.21 to 4.86 fell under the Lipinski rule and suggested optimal lipophilicity of the compound. The Swiss-ADME screening outcome revealed that except lupeol (2), all compounds under investigation were likely to have high gastrointestinal absorption. This implies that compound (3, 8, and 9) compounds have the potential to be absorbed in the gastrointestinal tract upon oral administration [87] (SI-Table S12). The BBB permeability test results indicated that except for compound 3, all compounds under investigation lacked BBB permeability, revealing these compounds were expected to have a high level of CNS safety [88]. The ADME estimation results demonstrated that all compounds under investigation are not found to be substrates of permeability glycoprotein (P-gp), implying favorable drug absorption [89].
Cytochrome P450 monooxygenase plays a fundamental part in drug mobilization and elimination in biological systems. Inhibition of isoforms of this enzyme system by drugs could result in poor elimination, resulting in drug-induced toxicity. Therefore it is important to note that a drug candidate should have minimal inhibitory effect against various enzyme isoforms [90]. The present finding indicated that none of the tested compounds were found to be inhibitors of CYP1A2, CYP2C19, CYP2C9, CYP2C9, and CYP2D6 isozymes. Moreover, except gallic acid (9) none of the analyzed compounds were inhibitors of CYP23A4, suggesting that the compounds would be well metabolized in the liver and readily eliminated from the organism [91]. The results of the Pro Tox II toxicity analysis of the studied compounds (2, 3, 8, and 9) found to have hepatotoxicity, mutagenicity, immunotoxicity, and cytotoxicity. Except for gallic acid (9), all tested compounds were found to be noncarcinogenic. The obtained toxicity profile of the analyzed compounds suggested that the compounds might be good candidates for drug development. The acute oral toxicity (LD50) predictions indicated that all the tested compounds were categorized into class 4 (300 < LD50 ≤ 2000), indicating slight toxicity levels (SI-Table S12).
In conclusion, the CH2Cl2/MeOH (1:1) extract of the roots of A. schimperi afforded five (1–5) compounds. Of which, dihydroferulic acid (3), acovenosigenin A- 3-O-α-L-rhamnopyranoside (4), and sucrose (5) were reported here for the first time from this species. CH2Cl2/ MeOH (1:1) and MeOH extract of R. glutinosa afforded four (6–9), all reported here for the time from this species. The CH2Cl2/MeOH (1:1) and MeOH crude extracts and isolated compounds of A. schipmeri and R. glutinosa showed modest to promising antibacterial activity against the tested bacteria strains. Among them, lupeol (2) methyl gallate (8) and gallic acid (9) displayed promising activity against E.coli, S. aureus and S. payogens with an inhibition zone ranges of 16.6 ± 0.09 to 18.6 ± 0.08 mm at 5 mg/mL. Dihydroferulic acid (3), methyl gallate (8), and gallic acid (9) also demonstrated promising scavenging of DPPH radical with IC50 values of 10.66, 7.48, and 6.08 µg/mL, respectively, compared to ascorbic acid (IC50 value 5.83 µg/mL). The in vitro antibacterial and antioxidant activities of the crude extracts and isolated compounds of A. schimperi and R. glutinosa supported by the considerable minimum binding free energy of the studied compounds towards the respective target proteins prove that the chemical constituents of the studied plants could serve as antimicrobial agents by blocking the essential cellular reactions required for the survival of the bacterial and the treatment of free radical-induced disorders. Further comprehensive in vivo study is needed to explore the plants’ full therapeutic potential and confirm the efficacy and safety of crude extracts and isolated compounds.
Data availability
All data generated or analyzed during this study are included in this published article and supporting information.
The NMR spectral data of compounds, Tables, and Figures used in this work are included as Supplementary Material.
References
Ugboko HU, Nwinyi OC, Oranusi SU, Fatoki TH, Omonhinmin CA (2020) Antimicrobial importance of medicinal plants in Nigeria. Sci World J 2020:1–10
Mahady GB (2005) Medicinal plants for the prevention and treatment of bacterial infections. Curr Pharm Design 11(19):2405–2427
Unal EL, Mavi A, Kara AA, Cakir A, Şengül M, Yildirim A (2008) Antimicrobial and antioxidant activities of some plants used as remedies in Turkish traditional medicine. Pharm Biol 46(3):207–224
Nagaraju Jalli SSK, Hnamte S, Pattnaik S, Paramanantham P, Siddhardha B (2019) Antioxidant and anti-infective potential of Ethanolic Extract of Eriobotrya bengalensis (Roxb.) Hook. f.: Phytochemicals Investigation and Molecular Docking studies. J Pure Appl Microbiol 13(1):361–370
Kaneria M, Baravalia Y, Vaghasiya Y, Chanda S (2009) Determination of antibacterial and antioxidant potential of some medicinal plants from Saurashtra region, India. IndianJ Pharm Sci 71(4):406
Kengne IC, Feugap LDT, Njouendou AJ, Ngnokam CDJ, Djamalladine MD, Ngnokam D, Voutquenne-Nazabadioko L, Tamokou J-D-D (2021) Antibacterial, antifungal and antioxidant activities of whole plant chemical constituents of Rumex abyssinicus. BMC ComplementMed Th s 21(1):1–14
Ahmadi M, Bahador N, Khodavandi A (2022) Phenolic compounds, antioxidants, and antibacterial activity of some native medicinal plants against Pseudomonas aeruginosa. Pharm Biomed Res 8(4):259–268
Ullah I, Gul S, Khan RU, Khan MI, Rehman HU, Ahmad N, Aziz-ud-Din SM, Jawad YI, Rehman SU (2017) Antibacterial and antioxidant activity analysis of some wild medicinal plants. J Entomol Zool Stud 5(6):1771–1775
Abdul Qadir M, Shahzadi SK, Bashir A, Munir A, Shahzad S (2017) Evaluation of phenolic compounds and antioxidant and antimicrobial activities of some common herbs. Int J Analy Chem 2017:1–6
Ifesan B, Fashakin J, Ebosele F, Oyerinde A (2013) Antioxidant and antimicrobial properties of selected plant leaves. Euro J Med Plants 3(3):465–473
Safari M, Ahmady-Asbchin S (2019) Evaluation of antioxidant and antibacterial activities of methanolic extract of medlar (Mespilus germanica L.) leaves. Biotechnol Biotechnolo Equip 33(1):372–378
Soumia K, Tahar D, Lynda L, Saida B, Chabane C, Hafidha M (2014) Antioxidant and antimicrobial activities of selected medicinal plants. J Coastal Life Med 2(6):478–483
Elkhateeb WA, El-Ghwas DE, Ahmed SA, Elnahas MO, Daba GM (2023) Ceropegia rupicola, Acokanthera schimperi, and Jatropha variegate Antimicrobial, cytotoxicity and phytochemical. Res J Pharm Technol 16(4):1833–1842
Kenubih A, Belay E, Lemma K (2021) Evaluation of the antimicrobial activity of leaf extracts of Acokanthera schimperi against various disease-causing bacteria. J Experi Pharm 889–899
Abrha B, Krishna Chaithanya K, Gopalakrishnan V, Hagos Z, Hiruy M, Devaki K (2018) Phytochemical screening and in vitro antioxidants activities of ethanolic extract of Acokanthera schimperi leaves. J Pham Res 12(5):660
Chaithanya KK, Hagos Z, Malarvizhi D, Poornima K, Gopalakrishnan V (2018) Chromatographic fingerprinting analysis of secondary metabolites present in ethanolic extract of Acokanthera schimperi leaves by high-performance thin-layer chromatography technique. Drug Invention Today 10(1)
Birhan Y, Kitaw S, Alemayehu Y, Mengesha N (2017) Ethnobotanical study of medicinal plants used to treat human diseases in Enarj Enawga district, East Gojjam zone, Amhara region, Ethiopia. SM J Med Plant Stud 1(1):1–9
Guliye AY (2015) Acaricidal coumarins from the medicinal plant. Acokanthera schimperi Archi De Medi 1(1):3
Matebie WA, Zhang W, Zhang S, Xie G (2019) Triterpenoids from Acokanthera schimperi in Ethiopia. Rec Nat Prod 13(3):182–188
Enyew A, Asfaw Z, Kelbessa E, Nagappan R (2014) Ethnobotanical study of traditional medicinal plants in and around Fiche District, Central Ethiopia. Curr Res J Biol Sci 6(4):154–167
Fassil A, Gashaw G (2019) An ethnobotanical study of medicinal plants in chiro district, West Hararghe, Ethiopia. Afr J Plant Sci 13(11):309–323
Tamene S (2020) Ethnobotanical study of indigenous knowledge on medicinal plant uses and threatening factors around the Malga District, Southern Ethiopia. Int J Biodiver Conser 12(3):215–226
Yemane B, Medhanie G, Reddy K (2018) Survey of some common medicinal plants used in Eritrean folk medicine. J Ethnomed 112:865–876
Gidey Y (2010) Use of traditional medicinal plants by indigenous people in Mekele town, capital city of Tigray regional state of Ethiopia. J Med Plants Res 4(17):1799–1804
Balouiri M, Sadiki M, Ibnsouda SK (2016) Methods for in vitro evaluating antimicrobial activity: A review. J Pharm Analy 6(2):71–79
Sasikumar J, Erba O, Egigu MC (2020) In vitro antioxidant activity and polyphenolic content of commonly used spices from Ethiopia. Heliyon 6(9)
Tundis R, Bonesi M, Menichini F, Loizzo MR, Conforti F, Statti G, Pirisi FM, Menichini F (2012) Antioxidant and anti-cholinesterase activity of Globularia meridionalis extracts and isolated constituents. Nat Prod Communi 7(8):1934578X1200700814
Uttu AJ, Sallau MS, Ibrahim H, Iyun ORA (2023) Isolation, characterization, and docking studies of campesterol and β-sitosterol from Strychnos innocua (Delile) root bark. J Taibah Univer Med Sci 18(3):566–578
Hussein K, Eswaramoorthy R, Melaku Y, ANNİSA ME (2021) Antibacterial and Antioxidant Activity of Isoflavans from the Roots of Rhynchosia ferruginea and In Silico Study on DNA Gyrase and Human Peroxiredoxin. Int J Secon Metabol 8(4):321–336
Ahmed SA, Abdelrheem DA, El-Mageed HA, Mohamed HS, Rahman AA, Elsayed KN, Ahmed SA (2020) Destabilizing the structural integrity of COVID-19 by caulerpin and its derivatives along with some antiviral drugs: an in silico approaches for a combination therapy. Stru Chem 31:2391–2412
Lipinski CA, Lombardo F, Dominy BW, Feeney PJ (1997) Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adva Drug Delive Rev 23(1–3):3–25
Oshevire DB, Mustapha A, Alozieuwa BU, Badeggi HH, Ismail A, Hassan ON, Ugwunnaji PI, Ibrahim J, Lawal B, Berinyu EB (2021) In-silico investigation of curcumin drug-likeness, gene-targets and prognostic relevance of the targets in panels of human cancer cohorts. GSC Biol Pharm Sci 14(1):037–047
Abba J, Ekwumemgbo P, Dallatu Y, Ndukwe G (2021) Isolation and characterization of the methanolic extract of the stem of Adenanthos sericeus (Woolly Bush). Nigerian Res J Chem Sci 9(1):309–317
Sintayehu N, Adane L (2020) Phytochemical screening and isolation of fatty acid and fatty acid esters of triterpene from root extract of Vernonia auriculifera grown in Sidama zone, Southern Ethiopia. J Pharmacogn Phytochem 9(1):1702–1710
Akwu N, Naidoo Y, Singh M, Thimmegowda SC, Nundkumar N, Lin J (2020) Isolation of lupeol from Grewia lasiocarpa stem bark: Antibacterial, antioxidant, and cytotoxicity activities. Biodiver J Biol Diver 21(12):5684–5690
Shwe HH, Win KK, Moe TT, Myint AA, Win T (2019) Isolation and structural characterization of lupeol from the stem bark of Diospyros ehretioides Wall. IEEE-SEM 7(8):140–144
Cho J-Y, Yang X, Park K-H, Park HJ, Park S-Y, Moon J-H, Ham K-S (2013) Isolation and identification of antioxidative compounds and their activities from Suaeda japonica. Food Sci Biotechnol 22:1547–1557
Thakur A, Moyo P, Van Der Westhuizen CJ, Yang HO, Maharaj V (2021) A novel cardenolide glycoside isolated from Xysmalobium undulatum Reduces levels of the Alzheimer’s Disease-Associated β-Amyloid Peptides Aβ42 in vitro. Pharma 14(8):743
Pecio Ł, Hassan EM, Omer EA, Gajek G, Kontek R, Sobieraj A, Stochmal A, Oleszek W (2019) Cytotoxic cardenolides from the leaves of Acokanthera oblongifolia. Planta Med 85(11/12):965–972
Abd-Alla HI, Soltan MM, Hassan AZ, Taie HA, Abo-Salem HM, Karam EA, El-Safty MM, Hanna AG (2021) Cardenolides and pentacyclic triterpenes isolated from Acokanthera oblongifolia leaves: their biological activities with molecular docking study. Z Naturforschung C 76(7–8):301–315
Bhat AH, Alia A, Mustafa Rather G, Kumar B (2019) Isolation and characterization of a water-soluble disaccharide from methanolic extract of Arisaema utile and Its evaluation for antioxidant and antifungal activity. Int J Pharma Biol Archive 10(2):83–89
Kamboj A, Saluja AK (2011) Isolation of stigmasterol and β-sitosterol from petroleum ether extract of aerial parts of Ageratum conyzoides (Asteraceae). Int J Pharm Sci 3(1):94–96
Pusparohmana W, Safitry R, Marliana E, Kusuma I (2020) Isolation and characterization of stigmasterol and β-sitosterol from wood bark extract of Baccaurea macrocarpa Miq. Mull. Arg. Rasayan J Chem 13(4):2552
Saraux N, Bruna L, Ebrahimi SN, Karimou S, Christen P, Cuendet M (2023) Antiproliferative activity of compounds isolated from the root bark of Lannea acida in multiple myeloma cell lines. Phytochem 209:113641
Esmat A, Al-Abbasi FA, Algandaby MM, Moussa AY, Labib RM, Ayoub NA, Abdel-Naim AB (2012) Anti-inflammatory activity of Pistacia khinjuk in different experimental models: isolation and characterization of its flavonoids and galloylated sugars. J Med food 15(3):278–287
Taye B, Giday M, Animut A, Seid J (2011) Antibacterial activities of selected medicinal plants in traditional treatment of human wounds in Ethiopia. Asian Paci J Trop Biomed 1(5):370–375
Tadeg H, Mohammed E, Asres K, Gebre-Mariam T (2005) Antimicrobial activities of some selected traditional Ethiopian medicinal plants used in the treatment of skin disorders. J Ethnopharmacol 100(1–2):168–175
Ekalu A, Ayo Rg-O, Habila J, Hamisu İ (2019) Bioactivity of phaeophytin A, α-amyrin and lupeol from Brachystelma togoense Schltr. J Turkish Chem Soci Sect A: Chem 6(3):411–418
Musa NM, Sallau MS, Oyewale AO, Ali T (2024) Antimicrobial activity of lupeol and β-amyrin (Triterpenoids) isolated from the rhizome of Dolichos pachyrhizus Harm. Adv J Chem :1–14
Chaudhari L, Jawale BA, Sharma S, Sharma H, Kumar C, Kulkarni PA (2012) Antimicrobial activity of commercially available essential oils against Streptococcus mutans. J Contemp Dent Pract 13(1):71–74
Sen A, Dhavan P, Shukla KK, Singh S, Tejovathi G (2012) Analysis of IR, NMR and antimicrobial activity of β-sitosterol isolated from Momordica charantia. Sci Secure J Biotechnol 1(1):9–13
Melaku Y, Getahun T, Addisu M, Tesso H, Eswaramoorthy R, Ankita G (2022) Molecular docking, antibacterial and antioxidant activities of compounds isolated from Ethiopian plants. Int J Secon Metabol 9(2):208–328
Al-Zahrani SH (2012) Antibacterial activities of gallic acid and gallic acid methyl ester on methicillin-resistant Staphylococcus aureus. J Am Sci 8(2):7–13
Pinho E, Ferreira IC, Barros L, Carvalho AM, Soares G, Henriques M (2014) Antibacterial potential of northeastern Portugal wild plant extracts and respective phenolic compounds. BioMed Res Int 2014:1–8
Kwon Y-I, Apostolidis E, Labbe R, Shetty K (2007) Inhibition of Staphylococcus aureus by phenolic phytochemicals of selected clonal herbs species of Lamiaceae family and likely mode of action through proline oxidation. Food Biotechnol 21(1):71–89
Jubeh B, Breijyeh Z, Karaman R (2020) Resistance of gram-positive bacteria to current antibacterial agents and overcoming approaches. Molecules 25(12):2888
Nwozo OS, Effiong EM, Aja PM, Awuchi CG (2023) Antioxidant, phytochemical, and therapeutic properties of medicinal plants: a review. Int J Food Prop 26(1):359–388
Lee SC, Kwon YS, Son KH, Kim HP, Heo MY (2005) Antioxidative constituents from Paeonia lactiflora. Archi Pharm Res 28:775–783
Santiago LA, Mayor ABR (2014) Lupeol: an antioxidant triterpene in Ficus pseudopalma Blanco (Moraceae). Asian Paci J Trop Biomed 4(2):109–118
Alemu BK, Misganaw D, Mengistu G (2020) Wound healing effect of Acokanthera schimperi schweinf (Apocynaceae) methanol leaf extract ointment in mice and its in-vitro antioxidant activity. Cli Pharmacol Adva Appli 213–222
Gurupriya S, Cathrine L, Ramesh J (2018) In vitro antidiabetic and antioxidant activities of lupeol isolated from the methanolic extract of Andrographis echioides leaves. J Pharmacogn Phytochem 7(4):768–775
Deters M, Knochenwefel H, Lindhorst D, Koal T, Meyer HH, Hänsel W, Resch K, Kaever V (2008) Different curcuminoids inhibit T-lymphocyte proliferation independently of their radical scavenging activities. Pharm Res 25:1822–1827
El-Sayed MM, El-Hashash MM, Mohamed HR, Abdel-Lateef EE-S (2015) Phytochemical investigation and in vitro antioxidant activity of different leaf extracts of Salix mucronata Thunb. J Appl Pharm Sci 5(12):080–085
Habtemariam S (2011) Methyl-3-O-methyl gallate and gallic acid from the leaves of Peltiphyllum peltatum: isolation and comparative antioxidant, prooxidant, and cytotoxic effects in neuronal cells. J Med food 14(11):1412–1418
Ekaprasada MT, Nurdin H, Ibrahim S, Dachriyanus D (2009) Antioxidant activity of methyl gallate isolated from the leaves of Toona sureni. Indo J Chem 9(3):457–460
Bhadoriya U, Sharma P, Solanki SS (2012) In vitro free radical scavenging activity of gallic acid isolated from Caesalpinia decapetala wood. Asian Pac J Trop Disease 2:S833–S836
Lourençon TV, de Lima GG, Ribeiro CS, Hansel FA, Maciel GM, da Silva K, Winnischofer SM, de Muniz GI, Magalhães WL (2021) Antioxidant, antibacterial and antitumoural activities of kraft lignin from hardwood fractionated by acid precipitation. Int J Biol Macromol 166:1535–1542
Badhani B, Sharma N, Kakkar R (2015) Gallic acid: A versatile antioxidant with promising therapeutic and industrial applications. Rsc Adva 5(35):27540–27557
Huang H, Zhang G, Zhou Y, Lin C, Chen S, Lin Y, Mai S, Huang Z (2018) Reverse screening methods to search for the protein targets of chemopreventive compounds. Front Chem 6:138
Muhammed MT, Aki-Yalcin E (2024) Molecular docking: principles, advances, and its applications in drug discovery. Lett Drug Des Disco 21(3):480–495
Asiamah I, Obiri SA, Tamekloe W, Armah FA, Borquaye LS (2023) Applications of molecular docking in natural products-based drug discovery. Sci Afri 20:e01593
El Sayed MT, Sarhan AE, Ahmed E, Khattab RR, Elnaggar M, El-Messery SM, Shaldam MA, Hassan GS (2020) Novel pyruvate kinase (pk) inhibitors: new target to overcome bacterial resistance. Chem Select 5(11):3445–3453
Assefa T, Tesso H, Ramachandran VP, Guta L, Demissie TB, Ombito JO, Eswaramoorthy R, Melaku Y (2023) In silico molecular docking analysis, cytotoxicity, and antibacterial activities of aonstituents of Fruits of Cucumis dipsaceus. ACS omega 9(1):1945–1955
Bibiso M (2022) Antibacterial activity, phytochemical and molecular docking analysis of Croton macrostachyus root extracts growing in Wolaita, Ethiopia. Res J Pharmacogno 9(4):1–11
Lane MD, Seelig B (2016) Highly efficient recombinant production and purification of streptococcal cysteine protease streptopain with increased enzymatic activity. Protein Express Purif 121:66–72
Woehl JL, Kitamura S, Dillon N, Han Z, Edgar LJ, Nizet V, Wolan DW (2020) An irreversible inhibitor to probe the role of Streptococcus pyogenes cysteine protease SpeB in evasion of host complement defenses. ACS Chem Biol 15(8):2060–2069
Rivera-Antonio A, Rosales-Hernández MC, Balbuena-Rebolledo I, Santiago-Quintana JM, Mendieta-Wejebe JE, Correa-Basurto J, García-Vázquez JB, García-Báez EV, Padilla-Martínez II (2021) Myeloperoxidase inhibitory and antioxidant activities of (E)-2-hydroxy-α-aminocinnamic acids obtained through microwave-assisted synthesis. Pharma 14(6):513
Malle E, Furtmüller P, Sattler W, Obinger C (2007) Myeloperoxidase: a target for new drug development? Br J Pharmacol 152(6):838–854
Soubhye J, Chikh Alard I, Aldib I, Prévost M, Gelbcke M, De Carvalho A, Furtmüller PG, Obinger C, Flemmig S Jr (2017) Discovery of novel potent reversible and irreversible myeloperoxidase inhibitors using virtual screening procedure. J Med Chem 60(15):6563–6586
Olaokun OO, Zubair MS (2023) Antidiabetic activity, molecular docking, and ADMET properties of compounds Iisolated from bioactive ethyl acetate fraction of Ficus lutea leaf extract. Molecules 28(23):7717
Cheng F, Li W, Zhou Y, Shen J, Wu Z, Liu G, Lee PW, Tang Y(20112) admetSAR: a comprehensive source and free tool for assessment of chemical ADMET properties. J Chem Inf Model 52: 3099–3105
Rai M, Singh AV, Paudel N, Kanase A, Falletta E, Kerkar P, Heyda J, Barghash RF, Singh SP, Soos M (2023) Herbal concoction unveiled: a computational analysis of phytochemicals’ pharmacokinetic and toxicological profiles using novel approach methodologies (NAMs). Curr Res Toxicol 5:100118
Veber DF, Johnson SR, Cheng H-Y, Smith BR, Ward KW, Kopple KD (2002) Molecular properties that influence the oral bioavailability of drug candidates. J Med Chem 45(12):2615–2623
Maliehe TS, Tsilo PH, Shandu JS (2020) Computational evaluation of ADMET properties and bioactive score of compounds from Encephalartos ferox. Pharmacognosy J 12(6)
Mahmud S, Paul GK, Biswas S, Kazi T, Mahbub S, Mita MA, Afrose S, Islam A, Ahaduzzaman S, Hasan MR (2022) phytochemdb: a platform for virtual screening and computer-aided drug designing. Database 2022:baac002
Anza M, Endale M, Cardona L, Cortes D, Eswaramoorthy R, Zueco J, Rico H, Trelis M, Abarca B (2021) Antimicrobial activity, in silico molecular docking, ADMET and DFT analysis of secondary metabolites from roots of three Ethiopian medicinal plants. Adv Appl Bioinform Chem :117–132
Daisy P, Suveena S, Rajalakshmi M, Lilly V (2011) Ligand based virtual screening on natural compounds for discovering active ligands. Der Pharm Chem 3:51–57
Imieje VO, Zaki AA, Metwaly AM, Mostafa AE, Elkaeed EB, Falodun A (2021) Comprehensive in silico screening of the antiviral potentialities of a new humulene Glucoside from Asteriscus hierochunticus against SARS-CoV-2. J Chem2021:1–14
Degfie T, Ombito JO, Demissie TB, Eswaramoorthy R, Dekebo A, Endale M (2022) Antibacterial and antioxidant activities, in silico molecular docking, ADMET and DFT analysis of compounds from roots of Cyphostemma cyphopetalum. Adv Appl Bioinform Chem :79–97
Lynch T, Price A (2007) The effect of cytochrome P450 metabolism on drug response, interactions, and adverse effects. Am Fam phys 76(3):391–396
Ononamadu CJ, Ibrahim A (2021) Molecular docking and prediction of ADME/drug-likeness properties of potentially active antidiabetic compounds isolated from aqueous-methanol extracts of Gymnema sylvestre and Combretum micranthum. BioTechnologia 102(1):85
Acknowledgements
We appreciate Adama Science and Technology University for funding part of the research through grant number ASTU/AS-R/001/2020.
Funding
Part of the work was funded by Adama Science and Technology University through grant number ASTU/AS-R/001/2020.
Author information
Authors and Affiliations
Contributions
BA, YM and ME designed the experiments. BA conducted the isolation, biological activity assay, and elucidation of the structures and drafted the manuscript. YM and ME supervised the laboratory experimental work. BA, MW, KS, NA, TN, and KS conducted molecular docking studies. MH conducted NMR spectral analysis. AD, NA, TN, MW, MH, YM, and ME participated in structural elucidation and interpretation of the docking analysis. BA and KS conducted biological activity. AD, NA, TN, MW, MH, YM, KS and ME participated in the manuscript’s critical review. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflicts of interest regarding this work or the publication of this paper.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Abera, B., Melaku, Y., Shenkute, K. et al. In vitro antibacterial, antioxidant, in silico molecular docking and ADEMT analysis of chemical constituents from the roots of Acokanthera schimperi and Rhus glutinosa. Appl Biol Chem 67, 77 (2024). https://doi.org/10.1186/s13765-024-00930-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13765-024-00930-6